

Abstract
Background
Hypokalemia is known to promote ventricular arrhythmias, especially in combination with Class III antiarrhythmic drugs like dofetilide. Here we evaluated the underlying molecular mechanisms.
Methods and Results
Arrhythmias were recorded in isolated rabbit and rat hearts or patch-clamped ventricular myocytes exposed to hypokalemia (1.0-3.5 mmol/l) in the absence or presence of dofetilide (1 μmol/l). Spontaneous early afterdepolarizations (EADs) and ventricular tachycardia/fibrillation (VF/VF) occurred in 50% of hearts at 2.7 mmol/l [K] in the absence of dofetilide, and 3.3 mmol/l [K] in its presence. Pre-treatment with the CaMKII inhibitor KN-93, but not its inactive analogue KN-92, abolished EADs and hypokalemia-induced VT/VF, as did the selective late Na current (INa) blocker GS-967. In intact hearts, moderate hypokalemia (2.7 mmol/l) significantly increased tissue CaMKII activity. Computer modeling revealed that EAD generation by hypokalemia (with or without dofetilide) required Na-K pump inhibition to induce intracellular Na and Ca overload with consequent CaMKII activation enhancing late INa and the L-type Ca current. K current suppression by hypokalemia and/or dofetilide alone in the absence of CaMKII activation were ineffective at causing EADs.
Conclusions
We conclude that Na-K pump inhibition by even moderate hypokalemia plays a critical role in promoting EAD-mediated arrhythmias by inducing a positive feedback cycle activating CaMKII and enhancing late INa. Class III antiarrhythmic drugs like dofetilide sensitize the heart to this positive feedback loop.
Keywords: Arrhythmia, antiarrhythmia agents, potassium, signal transduction, long QT syndrome
Introduction
Hypokalemia is a commonly encountered complication of diuretic therapy in clinical settings 1 and is known to promote ventricular arrhythmias2-4 as well as potentiate the pro-arrhythmic effects of Class III (K channel blocking) antiarrhythmic drugs. Hypokalemia directly reduces repolarization reserve by decreasing K conductances, including the inward rectifier K current IK1, the rapid component of the delayed rectifier K current IKr and the transient outward current Ito1, all of which exhibit a strong allosteric dependence on extracellular K concentration [K]o5, 6. Therefore, it is not surprising that these effects act synergistically with K channel blocking drugs to promote early afterdepolarization (EAD)-mediated arrhythmias including torsades de pointes (TdP), polymorphic ventricular tachycardia (VT) and fibrillation (VF).
However, suppression of K currents alone may not be sufficient to induce EAD-mediated arrhythmias. Anderson and colleagues7, 8 previously implicated CaMKII activation by the K channel blocker clofilium as playing a key factor in EAD formation by showing that clofilium activated CaMKII activity in isolated rabbit hearts. Moreover, CaMKII inhibitors abolished clofilium-induced EADs. Recently, Yang et al9 reported that various class III antiarrhythmic drugs such as dofetilide contribute to EAD formation by activating the late Na current (INa) via phosphoinositide 3-kinase inhibition.
A sometimes overlooked effect of hypokalemia is Na-K pump inhibition, which may contribute to intracellular Na and Ca loading at even moderately reduced serum [K] levels (2.7 mmol/l) 10. Na-K pump inhibition also reduces repolarization reserve suppressing outward pump current11. We hypothesized that Na-K pump inhibition by clinically relevant moderate hypokalemia may play an important role in activating CaMKII to promote EADs and ventricular arrhythmias even in the absence of a Class III antiarrhythmic drug. Indeed, EADs induced by moderate hypokalemia in isolated mouse hearts were suppressed by both nifedipine and CaMKII inhibition12. To test this hypothesis, we exposed rabbit and rat hearts and isolated myocytes to hypokalemia in the absence and presence of the Class III antiarrhythmic drug dofetilide. We show that moderate hypokalemia, either alone or in the presence of dofetilide, causes sufficient Na-K pump inhibition and intracellular Ca accumulation to activate CaMKII, which then plays an obligatory role in causing EADs and EAD-mediated VT/VF by enhancing late INa. These findings may provide insight into why the failing heart, with chronically increased levels of activated CaMKII13, is pre-sensitized to the pro-arrhythmic effects of Class III antiarrhythmic drugs even when serum [K] is normal. In addition, our findings suggest that targeting late INa to break the CaMKII-facilitated positive feedback cycle promoting EAD formation may be a promising antiarrhythmic strategy.
Methods
The research protocol (ARC protocol #2003-063-31) was approved by the UCLA Institutional Animal Care and Use Committee following American Heart Association guidelines.
Intact heart optical mapping experiments
Hearts removed from anesthetized New Zealand White rabbits (4-6 month old) and male Fisher344 rats (3-4 month old) were mounted in Langendorff fashion, perfused at 37°C with Tyrode's solution containing (in mmol/l): NaCl 125, NaHCO3 24, KCl 5.4, NaH2PO4 1.8, MgCl2 0.5, CaCl2 1.8, glucose 5.5, albumin 100 mg/l, pH 7.4 gassed with 95%O2-5%CO2. The added KCl was reduced for hypokalemia. Hearts were stained with RH237 for voltage mapping, using cytochalasin D (5 μmol/l) to eliminate motion artifact14, 15. Extracellular electrograms were recorded continuously using unipolar Ag-AgCl electrodes, and in some experiments, intracellular membrane potential was recorded from epicardial myocytes using glass microelectrodes.
Isolated ventricular myocyte patch clamp studies
Myocytes were enzymatically isolated from 3- to 4-month-old New Zealand white male rabbits and used within 8 hours for whole-cell patch clamp studies as described previously16. The pipette solution contained (in mmol/l) K-aspartate 110, KCl 30, NaCl 5, HEPES 10, EGTA 0.0-0.1, MgATP 5, creatine phosphate 5, and cAMP 0.1 (pH 7.2 adjusted with KOH). Cells were superfused at 37°C with standard Tyrode's solution containing (in mmol/l) NaCl 136, KCl 5.4 (reduced in hypokalemia experiments), NaH2PO4 0.33, CaCl2 1.8, MgCl2 1, HEPES 10, and glucose 10 (pH 7.4 adjusted with NaOH) unless otherwise indicated. To inhibit CaMKII, KN-93 (or KN-92 as control) was added to the superfusate, or CaMKII inhibitor peptide AIP (2 μM) was added to the patch electrode solution and allowed to equilibrate for 20-30 mins after patch rupture before experimental intervention. To selectively block late INa, GS-967 (l μmol/l) was added to the superperfusate17.
CaMKII assay
After an equilibration period of 10-15 min with standard Tyrode's solution (5.4 mmol/l [K]), isolated rabbit hearts were perfused for 30 min with either 5.4 or 2.7 mmol/l [K]. Left ventricular tissue was then fast frozen in liquid nitrogen and stored at −80°C. Protein lysates were analyzed for CaMKII activity (CycLex CaMKII assay kit, MBL International, Woburn, MA, USA).
Computer modeling
Simulations were carried out using a rabbit ventricular myocyte AP model18, with the following governing equation for voltage (V):
where Cm=1 μF/cm2, total ionic current density Iion = INa + Ito,f + Ito,s + ICa,L + IKs + IKr + IK1 + INCX + INaK, and Isti is the stimulation current density. The downstream effects of CaMKII activation on ICa,L and INa were simulated as described in the Online Supplement.
Statistical Analyses
The conventional percentile bootstrap-resampling approach with 10,000 replications was used for estimating Confidence Intervals (CI) 19. Non-parametric statistical tests (Wilcoxon rank-sum test for CaMKII activity differences, Wilcoxon signed rank test for APD differences and Fisher's exact test for differences in VT/VF in unpaired hearts in the absence and presence of dofetilide) were used to assess P values. Kaplan-Meier curves were constructed to compare the time to onset of VT/VF at different [K], using the pair-wise log rank test with Bonferroni correction for multiple comparisons to estimate P values.
Results
Moderate hypokalemia induces EAD-mediated VT/VF in isolated rabbit and rat hearts
Exposure of isolated rabbit hearts to moderate hypokalemia (2.7 mmol/l) prolonged action potential (AP) duration at 90% repolarization (APD90) from 207 ms [95% CI: 204-210 ms] to 260 ms [CI: 258-268 ms] (n=10, P=0.005), associated with a 13% [CI: 10-17%] increase in AP amplitude (n=5) consistent with hyperpolarization of the resting membrane potential. No significant arrhythmias were observed initially, but after delays of 28, 33 and 80 min, 3 out of 10 hearts developed episodes of VT/VF arising spontaneously from sinus rhythm. Simultaneous intracellular microelectrode and pseudo-ECG recordings showed that the VF was initiated by EAD-mediated triggered activity causing focal VT with cycle length (CL) 112 ms [CI: 105-123 ms] that spontaneously emerged from sinus rhythm (mean CL 486 ms [CI: 445-528 ms]), as illustrated in Fig. 1A. Focal VT degenerated to VF (mean CL 66 ms [CI: 61-70 ms]) within 1-2 s after its onset, consistent with EAD-mediated mixed focal-reentrant VF described previously20. To determine if VT/VF during moderate hypokalemia was unique to rabbits as a species, we also exposed isolated rat hearts to the same level of moderate hypokalemia (2.7 mmol/l). Hypokalemia prolonged APD90 from 102 ms [CI: 93-111 ms] to 141 ms [CI: 131-152 ms] (n=10, P=0.005). No significant arrhythmias occurred immediately, but after a mean delay of 44 min [CI: 30-58 min] (range 18-86 min), 14 out of 23 hearts developed episodes of spontaneous VT/VF. Simultaneous microelectrode and pseudo-ECG recordings (n=6) showed that the VF was initiated by EAD-mediated triggered activity with a mean CL of 82 ms [CI: 71-93 ms] emerging spontaneously from sinus rhythm (mean CL 375 ms [CI: 317-433 ms], as illustrated in Fig. 1B. Triggered activity causing focal VT degenerated to VF within 1-2 sec, with a mean CL during VF of 48 ms [CI: 37-59 ms] during VF. Delayed afterdepolarizations (DADs) were also occasionally observed and triggered single beats, but no sustained arrhythmias (Fig. 1B, arrows). During optical mapping, the onset of EAD-mediated triggered activity was captured in five rat hearts. In the example shown in Fig. 2, two consecutive runs of EAD-mediated triggered activity arose from the LV base of an isolated rat heart, causing focal VT at mean CL of 85 ms [CI: 71-99 ms]. Four isolated rat hearts exposed to hypokalemia for 90 min with 10 μmol/l nifedipine present remained in sinus rhythm throughout, but developed VT/VF after nifedipine was removed (Online Supplement, Fig. S1). Similarly, when extracellular [Ca] was reduced from 1.8 to 0.9 mmol/l to attenuate intracellular Ca loading, four rat hearts exposed to hypokalemia for 90 minutes remained in sinus rhythm, but subsequently developed VT/VF when [Ca] was raised to 1.8 mmol/l (Online Supplement, Fig. S2).
Figure 1.

Hypokalemia-induced VF in adult isolated Langendorff rabbit (A) and rat (B) hearts exposed to 2.7 mmol/l [K]o. Left panel: sinus rhythm during normokalemia, as recorded with a pseudo-ECG, bipolar electrograms from the left atrium (LA) and right ventricle (RV) and an epicardial intracellular microelectrode (ME). Middle and right panel: onset of EAD-mediated triggered activity (*) from sinus rhythm during hypokalemia. Arrows in B indicate DADs.
Figure 2.

Optical action potential recordings and activation maps during EAD-mediated triggered activity in an isolated-perfused rat heart exposed to hypokalemia (2.7 mmol/l [K]). A. Optical voltage fluorescence (FV) traces from 4 adjacent sites labeled 1-4 in panel B, illustrating two runs of EAD-mediated triggered activity (b1-b13) arising spontaneous from sinus rhythm (beats S1 and S6). B. Optical voltage fluorescence snapshots corresponding to the labeled beats in A. C. Isochronal activation maps of a sinus beat (S6) and the subsequent triggered beat (b7) indicating the different sites of origin.
Dofetilide sensitizes isolated rabbit hearts to hypokalemia-induced VT/VF
We next tested the level of hypokalemia required to induce VT/VF in isolated rabbit hearts. Fig. 3 shows that as the extracellular [K] was lowered from 5.4 to 4.0, 3.5, 2.7, 2.0 or 1.0 mmol/l, the incidence of VT/VF within 90 minutes progressively increased to 100% at 2.0 and 1.0 mmol/l. Fit to a Hill equation, the estimated [K]o concentration at which 50% of hearts developed VT/VF was 2.7 mmol/l. The time to onset of VT/VF also decreased as hypokalemia became more severe, from 55 min in one heart at 3.5 mmol/l [K]o, to 47 min (range: 28-80 min), 15.8 min (range: 10-22 min) and 9.6 min (range: 8-11 min) at 2.7, 2.0 and 1.0 mmol/l [K]o, respectively (Fig. 3B).
Figure 3.
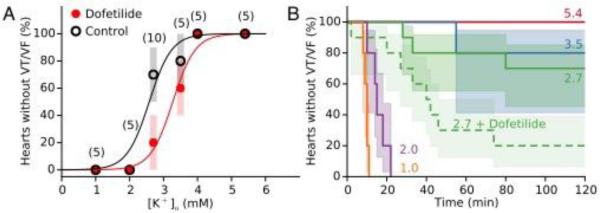
K dependence of hypokalemia-induced VT/VF in isolated rabbit hearts, in the absence and presence of dofetilide. A. When extracellular K concentration was lowered from the control value of 5.4 mmol/l to 4.0, 3.5, 2.7, 2.0 or 1.0 mmol/l, the incidence of VT/VF within 90 minutes progressively increased to 100% at 2.0 and 1.0 mmol/l. Data show median and 83% CI for the number of hearts indicated in parentheses, in the absence (black circles) or presence (red circles) of 1 μmol/l dofetilide. The significantly increased by dofetilide (P=0.03). Curves are fits to a Hill equation in the absence (black curve) and presence of dofetilide (red curve). B. Kaplan-Meier survival curves comparing time to onset of VT/VF, with 83% CI indicated by shading (solid lines=without dofetilide, dashed line=with dofetilide). Rate of VT/VF onset was significantly faster at 1.0 and 2.0 mmol/l [K] than at higher [K] (P=0.0002-0.002), and significantly faster at 2.7 mmol/l [K] in the presence of dofetilide than in its absence (P=0.02).
Since hypokalemia is known to potentiate arrhythmogenic effects of Class III antiarrhythmic drugs, we also studied the effects of progressive hypokalemia on VT/VF with 1 μM dofetilide present (Fig. 3). At 5.4 mmol/l [K]o, dofetilide prolonged APD from 206 ms [CI: 202-210 ms] to 219 ms [CI: 213-225 ms] (n=5, P=0.03), but failed to induce VT/VF, even after [K]o was lowered to 4.0 mmol/l (n=5 in each group). However, at 2.7 mmol/l [K]o, dofetilide accelerated (Fig. 3B) and significantly increased the percentage of hearts developing VT/VF from 30% (n=10) to 80% (n=10) (P=0.03) (Fig. 3A). The fitted Hill curve predicted that dofetilide shifted the [K]o threshold at which 50% of hearts developed VT/VF within 90 min from 2.7 to 3.3 mmol/l (Fig. 3A). In the Online Supplement, Fig. S3 shows an example of a heart which failed to develop VT/VF after 90 min of moderate hypokalemia (2.7 mmol/l), and subsequently developed VT/VF after adding dofetilide. Dofetilide prolonged the APD at 2.7 mmol/l from 263 ms [CI: 258-268 ms] to 281ms [CI: 277-286 ms] (n=10, P=0.005).
The CaMKII inhibitor KN-93 and the late INa blocker GS-967 prevent hypokalemia-induced VT/VF
Since hypokalemia and dofetilide affect K currents immediately, the delay between the onset of hypokalemia and EAD-mediated VT/VF (from 9.6 to 55 min depending on the degree of hypokalemia) implies that additional factors are at play. Based on previous reports that CaMKII inhibition suppressed EADs induced by clofilium in rabbits7, 8, we investigated whether CaMKII inhibition prevented arrhythmias induced by hypokalemia alone. We used 2.0 mmol/l [K]o (severe hypokalemia) since this concentration induced VT/VF in 100% of rabbit hearts within 15 min. Pre-treatment with the CaMKII inhibitor KN-93 (1 μmol/l, Fig. 4B), but not KN-92 at the same concentration (Fig. 4C), prevented VT/VF during a 30 min exposure to 2.0 mmol/l [K]o in all three hearts tested, even after dofetilide was added. Similar findings were also obtained in 3 out of 4 isolated rat hearts exposed to 2.7 mmol/l [K]o (Online Supplement, Fig. S4), consistent with findings in mouse hearts12.
Figure 4.

Suppression of hypokalemia-induced VF by CaMKII inhibition or late INa blockade.Representative ECG and RV electrograms from isolated rabbit ventricles at the times indicated after reducing [K]o from 5.4 to 2.0 mmol/l. A. No drug, illustrating the spontaneous transition from sinus rhythm to VT/VF after 10 min. B. KN-93 (1 μmol/l), illustrating maintenance of sinus rhythm after 30 min. C. Same heart as in B, 3 min after replacing KN-93 with KN-92 (1 μmol/l), illustrating spontaneous onset of VT/VF. D. GS-967 (1 μmol/l), illustrating maintenance of sinus rhythm after 30 min. E. Same heart as in D, 38 min after washing out GS-967, illustrating onset of spontaneous VT/VF.
Since an important CaMKII target implicated in EAD formation is late INa21, we exposed 4 isolated rabbit hearts to severe hypokalemia (2.0 mmol/l) in the presence of the selective late INa blocker GS-967 (1 μmol/l)17. In all 4 hearts studied, pre-treatment with GS-967 prevented VT/VF during a 30 min exposure to 2.0 mmol/l [K]o (Fig. 4D-E).
To determine directly whether moderate hypokalemia increased basal (autophosphorylated) CaMKII activity, we measured tissue CaMKII activity levels in isolated rabbit ventricles. During baseline arterial perfusion with 5.4 mmol/l [K]o, basal tissue CaMKII activity in 3 hearts averaged 47% [CI: 44-49] of maximal CaMKII activity determined by adding saturating Ca and CaM to the same tissue sample. Thirty minutes after lowering [K]o to 2.7 mmol/l in a separate group of 5 hearts, CaMKII activity increased to 71% [CI: 50-92] of maximal (P=0.025) (Fig. 5). Although 3 of these 5 hearts developed transient (1-2 min in 2 hearts) or persistent VT/VF (1 heart) during the hypokalemic period, the elevation in CaMKII activity was similar to two hearts that remained in sinus rhythm throughout (open circles, Fig. 5).
Figure 5.

Basal (autophosphorylated) tissue CaMKII activity level, expressed as percent of maximal Ca/CaM-stimulated CaMKII activity, in isolated rabbit ventricular tissue arteriallyperfused with 5.4 mmol/l or 2.7 mmol [K]o for 30 mins. Individual data points, median, 95% confidence intervals and p value are indicated. For 2.7 mmol/l data, open circles indicate hearts in which no VT/VF occurred during hypokalemia (2 hearts), and open squares indicate hearts in which transient (1-2 min episodes in 2 hearts) or sustained VT/VF developed (1 heart) during hypokalemia.
CaMKII inhibitors and the late INa blocker GS-967 suppress hypokalemia-induced EADs in isolated ventricular myocytes
Moderate hypokalemia (2.7 mmol/l) readily induces bradycardia-dependent EADs in isolated rabbit and rat ventricular myocytes16, 22, 23. Fig. 6A shows that in isolated rabbit myocytes, hypokalemia-induced EADs (2nd row) were unaffected by KN-92 (3rd row), but completely suppressed by KN-93 at the equivalent concentration of 1 M (4th row). EADs recurred when KN-93 was washed out (not shown). Similar results were obtained in 4 other myocytes.
Figure 6.

Suppression of hypokalemia-induced EADs by CaMKII inhibition and late INa blockade in isolated patch-clamped rabbit ventricular myocytes. Membrane voltage (Vm) traces during pacing at PCL 6 s. Top to bottom traces: A. [K]o = 5.4 mmol/l; 2.7 mmol/l; 2.7 mmol/l + 1 μmol/l KN-92; 2.7 mmol/l + 1 μmol/l KN-92. B. [K]o = 5.4 mmol/l + AIP; 2.7 mmol/l + AIP (dialyzed for 30 min). C. [K]o = 5.4 mmol/l; 2.7 mmol/l; 2.7 mmol/l; 2.7 mmol/l + 1 μmol/l GS-967 (bottom). Right panels show superimposed action potentials under the various conditions for A-C.
The more selective CaMKII inhibitor peptide AIP also suppressed EADs induced by 2.7 mmol/l [K]o (Fig. 6B). In these experiments, the myocyte was maintained in normal Tyrode's solution with 5.4 mmol/l [K]o for 20-30 min following patch rupture to allow the peptide to diffuse into the cytoplasm. After 20 min of dialysis, reducing [K]o from 5.4 to 2.7 mmol/l caused EADs to appear transiently and then disappear within 10 min (n=4). After 30 min of dialysis, no EADs occurred when [K]o was lowered (n=4). Without AIP, hypokalemia-induced EADs were always persistent and never spontaneously resolved (n>50). Similar to KN-93 and AIP, the selective late INa blocker GS-967 (1 mol/l)17 also suppressed EADs in 4 isolated rabbit ventricular myocytes exposed to 2.7 mmol/l [K]o (Fig. 6C).
Together with the intact heart experiments, these findings indicate that CaMKII activation is required for hypokalemia to induce EAD-mediated VT/VF, with CaMKII-induced potentiation of late INa a required component.
Computer simulations reveal CaMKII activation as a key positive feedback element in hypokalemia-induced EADs
Hypokalemia can increase intracellular Ca loading both by prolonging APD and by inhibiting the Na-K pump10. To dissect the relative importance of these factors in activating CaMKII during moderate hypokalemia (2.7 mmol/l), we performed computer simulations using a rabbit ventricular AP and Ca cycling model18 incorporating CaMKII signaling24. The reported effects of CaMKII activation on ICa,L and late INa21, 25, 26,27, both of which are known to potentiate EADs27, were simulated (see Methods). During pacing at a CL of 1 s, reducing [K]o from 5.4 to 2.7 mmol/l caused both intracellular [Na] and [Ca] to rise, progressively activating CaMKII. By enhancing late INa and ICa,L, the increased CaMKII activity further enhanced Na and Ca loading, leading to further CaMKII activation in a positive feedback loop (Fig. 7A & B). Both intracellular Na and Ca continued to rise, APD prolonged markedly and after a delay of 10.9 min, EADs appeared. In contrast, if CaMKII activation was disabled to simulate the effects of KN-93 or AIP, intracellular [Na] and [Ca] still increased over the same time period to values of 15.5 mmol/l for [Na] and 0.59 μmol/l for mean [Ca], respectively, but eventually leveled off at 21.2 mmol/l. During an 83 min-long simulation, no EADs occurred and APD stabilized at 217 ms. In the model with intact CaMKII signaling, the time to onset of EADs progressively shortened as [K]o was reduced (Fig. 7C, black trace), similar to the experimental findings (Fig. 3B). Complete blockade of IKr to simulate the effects of dofetilide accelerated the time to onset of EADs at lower [K]o (Fig. 7C, red trace).
Figure 7.

Effects of hypokalemia on membrane voltage (Vm), [Ca]i, and [Na]i in the rabbit ventricular AP model paced at a CL of 1s. A. Vm at various times corresponding to panel B, for [K]o = 5.4 mmol/l (blue traces); [K]o = 2.7 mmol/l (red traces); and [K]o = 2.7 mmol/l with CaMKII signaling disabled (green traces). B-C. Corresponding time course of changes in [Ca]i, (B) and [Na]i (C). [Ca]i and [Na]i accelerate rapidly with the onset of EADs, and systolic [Ca]i then transiently decreases when EADs transition to repolarization failure. D. Time to onset of the first EAD versus level of [K]o, with normal IKr (black) or IKr blocked (red) to simulate the effects of dofetilide.
To analyze which factors (APD prolongation, Na-K pump inhibition, CaMKII activation) were essential for EADs to develop, we performed additional simulations (Table 1). If intracellular [Na] was clamped to its control value of 10.9 mmol/l to prevent any increase during hypokalemia, APD prolonged modestly, but Ca accumulation and CaMKII activation was attenuated, such that no EADs developed (column d), even when APD was further prolonged by blocking IKr to simulate the effects of a Class III antiarrhythmic drug like dofetilide (column e). These findings indicate that APD prolongation alone, due to inhibition of K currents by hypokalemia and/or a Class III antiarrhythmic drug, does not elevate intracellular Ca sufficiently to cause EADs in this model unless hypokalemia-induced Na-K pump inhibition is also present. Conversely, if the suppressant effects of hypokalemia on IKr and IK1 were selectively eliminated by making both conductances insensitive to [K]o, while preserving hypokalemia-induced Na-K pump inhibition, intracellular Na and Ca accumulation still increased sufficiently to activate CaMKII and caused EADs to appear after only a slight delay from 10.9 to 11.1 min (column f). If IKr was blocked to simulate a Class III antiarrhythmic agent under the same conditions, the time to EAD onset modestly accelerated to 9.8 min (column g). These findings indicate that in this model, hypokalemia-induced Na-K pump inhibition resulting in CaMKII activation is sufficient to cause EADs, whether or not K conductances are concomitantly reduced.
Table 1.
Effects of simulated hypokalemia on [Na]i, mean [Ca]i (averaged throughout the cardiac cycle), late INa (as a surrogate index of CaMKII activity), APD (ms) and EAD development in the rabbit ventricular AP model under various conditions (a)-(g). Note that unless otherwise indicated below, CaMKII signaling in the model is intact. APD values in (b), (f) and (g) are immediately prior to the onset of EADs (10.9 min, 11.1 min and 9.8 min, respectively). For the other conditions, values are after 12 min to permit comparison, although EADs did not develop in any of these simulations up to 83 min.
| a | b | c | d | e | f | g | |
|---|---|---|---|---|---|---|---|
| [Na]i (mmol/l) | 10.9 | 15.7 | 15.5 | 10.9 | 10.9 | 16.0 | 15.3 |
| Mean[Ca]i (μmol/l) | 0.31 | 1.05 | 0.59 | 0.31 | 0.32 | 1.11 | 0.98 |
| Late INa (% of peak INa) | 0.14 | 0.64 | 0.11 | 0.14 | 0.14 | 0.66 | 0.57 |
| APD (ms) | 215 | 443 | 217 | 243 | 265 | 453 | 458 |
| EADs | No | Yes | No | No | No | Yes | Yes |
(a)[K]o= 5.4 mmol/l (control)
(b)[K]o= 2.7 mmol/l
(c)[K]o= 2.7 mmol/l with CaMKII signaling disabled
(d)[K]o= 2.7 mmol/l with [Na]i clamped at 10.9 mmol/l
(e)same as (d) + 100% IKr block;
(f)[K]o= 2.7 mmol/l with K conductances insensitive to [K]o
(g)same as (f) + 100% IKr block
Discussion
In this study, we have documented in two species with different underlying AP characteristics that moderate hypokalemia, at levels commonly observed clinically, is a potent inducer of EAD-mediated arrhythmias, even in the absence of QT prolonging drugs. The most novel contribution is our demonstration that the mechanism depends not only on K current suppression by hypokalemia, but also on hypokalemia-induced Na-K pump inhibition leading to Na and Ca overload and CaMKII activation. CaMKII activation initiates a positive feedback cascade, further exacerbating Na and Ca accumulation by inducing late INa and increasing ICa,L, which progressively impair repolarization reserve (Fig. 8). Thus, our findings provide insight into why most experimental models in which EAD-mediated arrhythmias were induced by K channel blocking drugs in normal heart tissue also included hypokalemia (and often hypomagnesemia) as a co-factor. This is reasonable, given clinical observations that hypokalemia often accompanies TdP induced by Class III antiarrhythmic drugs28, 29, but also emphasizes the critical independent role that hypokalemia plays via Na-K pump inhibition with consequent CaMKII activation. In our isolated rabbit heart preparation, dofetilide, despite significantly prolonging APD, was ineffective at inducing EAD-mediated arrhythmias when extracellular [K] was normal. Rather dofetilide sensitized the heart to hypokalemia, such that VT/VF occurred at a higher extracellular [K] when dofetilide was present (Fig. 3).
Figure 8.

Schema of positive feedback loops (red and blue arrows) promoting intracellular Na and Ca overload, CaMKII activation and EADs during hypokalemia. The potentiation of the blue positive feedback loop by Class III antiarrhythmic (AA) drugs such as dofetilide is also shown. INaK=Na-K pump current.
We do not mean to imply that QT prolonging drugs such as dofetilide pose no arrhythmic risk in the absence of hypokalemia. In settings in which CaMKII activity is already elevated by other factors, such as heart disease, exercise or other drugs, additional CaMKII activation via hypokalemia-induced Na-K pump inhibition may not be required to induce the positive feedback cascade in Fig. 8. In this context, the positive feedback loop between CaMKII activation and Na and Ca loading has been recently emphasized as a key factor driving maladaptive excitation-contraction coupling remodeling and arrhythmogenesis in heart failure13, 21. Our findings predict that this paradigm is significantly potentiated by chronic hypokalemia, a common complication of diuretic therapy in heart failure. Combined with other factors reducing repolarization reserve in heart disease, this may explain the greater pro-arrhythmic risk of Class III antiarrhythmic drugs in diseased hearts, and also why it is easier to identify studies of drug-induced TdP during normokalemia in diseased heart models30, in which CaMKII activity is already elevated. The traditional explanation for the pro-arrhythmic synergy between K channel blocking drugs and hypokalemia is that hypokalemia further reduces repolarization reserve by suppressing IKr, IK1 and Ito, whose conductances are allosterically regulated by extracellular [K]5, 6. In addition, the sensitivity of some K channels to block by Class III antiarrhythmic drugs is increased by hypokalemia31. Recently, however, Aronsen et al10 documented significant Na-K pump inhibition by moderate hypokalemia (2.7 mmol/l) in isolated rat ventricular myocytes, sufficient to increase intracellular Na and Ca to a level inducing spontaneous diastolic Ca waves. They did not directly record triggered arrhythmias, nor did they investigate the role of CaMKII activation in their study. Nevertheless, their direct measurements of hypokalemia-induced intracellular Na and Ca accumulation are compatible with our experimental data that the same level of moderate hypokalemia increased tissue CaMKII activity (Fig. 5).
As analyzed quantitatively in the computer model, two factors contribute to hypokalemia-induced Na-K pump inhibition. Given a K0.5 of the Na-K pump for [K]o of 1.9 mmol/l32, reducing [K]o from 5.4 to 2.7 mmol/l is predicted to reduce pump activity by only 21% (i.e. if the membrane potential is clamped to prevent hyperpolarization and other ion concentrations are held constant). However, the Na-K pump is also electrogenic and is inhibited by membrane hyperpolarization. Thus, when hypokalemia shifts the K equilibrium potential to more negative values, the resting membrane potential also becomes hyperpolarized (from −82 to −100 mV in isolated rabbit ventricular myocytes), which is predicted to reduce pump activity by 28% if all ion concentrations are held constant. The combined effects of both reduced extracellular [K] and hyperpolarization acutely reduced Na-K pump activity by 43% in the model, before any other ion concentrations besides [K]o changed. These findings agree quantitatively with the direct measurements of Na-K pump current in isolated rat myocytes by Aronsen et al10, which decreased by approximately 50% at 2.7 mmol/l [K]33. Together, these factors contribute to a slow rise in intracellular [Na], which inhibits the ability of the Na-Ca exchanger to remove Ca from the cell (Fig. 8).
However, Na-K pump inhibition and APD prolongation by themselves were not sufficient to induce either dramatic increases in intracellular Ca or EADs, unless the effects of CaMKII activation on the ICa,L and late INa current were also simulated. When these effects of CaMKII are added, positive feedback loops are introduced (Fig. 8) – as CaMKII is activated, the resulting phosphorylation of L-type Ca channels augments their peak current25 which further increases Ca loading during the action potential. CaMKII-dependent phosphorylation of Na channels also increases late INa, exacerbating Na loading 21, 26. As shown in Fig. 7, these effects accelerate intracellular Na and Ca accumulation during hypokalemia, in addition to further compromising repolarization reserve due to hypokalemia-induced K conductance suppression. Thus, in the model, the reduction of repolarization reserve by hypokalemia was not, by itself, sufficient to induce EADs, even if IKr was also blocked to simulate a Class III antiarrhythmic drug such as dofetilide (Table 1). Na-K pump inhibition leading to CaMKII activation with ICa,L and INa modification was required for EADs to develop.
Limitations
In the intact heart experiments, we could not directly measure increases in intracellular Ca expected to activate CaMKII, although we documented increased tissue CaMKII activity during moderate hypokalemia (2.7 mmol/l). Microelectrode and optical mapping recordings were confined to the epicardium, and we cannot exclude the possibility that arrhythmias due to EADs, DADs or other mechanisms arose from deeper levels. Nevertheless, we did directly record EADs and triggered activity from epicardial myocytes (e.g. Figs. 1 & 2). In previous studies14, 34, we also showed that endocardial cryoablation did not prevent EADs and EAD-mediated arrhythmias induced by a variety of stressors.
It has been reported that hypokalemia induces removal of Kr channels from the surface membrane35. However, the time course (hours) is too slow to explain the approximately 10-55 min delay before the onset of VT/VF in our isolated rabbit and rat heart models. Moreover, IKr is not a major repolarizing current in the rat ventricle, so that its removal from the surface membrane should be of minor consequence36. Similarly, our experiments with dofetilide were too short-term to account for late INa activation augmentation due to phosphoinositide 3-kinase inhibition, since this effect required more than 30 min and probably several hours9.
Regarding the computer modeling, we simulated only the reported effects of CaMKII on late INa and ICa,L amplitude relevant to EAD formation. However, CaMKII also affects ICa,L kinetics, as well as additional targets involved in excitation-contraction coupling, Ca cycling and repolarization, such as ryanodine receptors, IK1 and Ito, that may also play important roles in EAD formation, as well as promoting Ca cycling abnormalities predisposing to Ca waves and DADs13. We did observe DADs accompanying EADs in some hearts (Fig. 1B), although they did not appear to play an important role in initiating VT/VF. The CaMKII-mediated effects on ICa,L and late INa were sufficient to explain the experimental observations on EADs without incorporating these additional aspects, and block of late INa alone using GS-967 was sufficient to prevent EADs and EAD-mediated arrhythmias in both isolated myocytes and intact hearts.
We assumed a single K0.5 value (1.9 mmol/l) for the [K]o dependence of the Na-K pump corresponding to the dominant α1 isoform in heart32, 37. However, the α2 isoform comprises about 12-25% in rodents37, 38, and has a higher K0.5 (2.9 mmol/l)32, which, overall, would exacerbate Na-K pump inhibition by hypokalemia. The K0.5 of both isoforms are also modulated dynamically by PKC/PKA-mediated phosphorylation of phospholemman32, which we did not simulate.
Clinical Implications
Hypokalemia, defined as a serum level <3.5 mmol/l, is a common electrolyte abnormality in clinical practice. Serum K values <3.6 mmol/l were seen in over 20% of hospitalized patients39 and in as many as 10-40% of patients on thiazide diuretics1. Hypokalemia is known to promote ventricular arrhythmias and sudden cardiac death2-4, was present in 49% of patients resuscitated from out-of-hospital VF40, has been implicated in electrical storm41 and is also commonly associated with drug-induced TdP42. Our findings in rabbit and rat hearts provide insight into the molecular basis for these pro-arrhythmic effects of hypokalemia, demonstrating that clinically-relevant levels (2.0-3.5 mmol/l) have the potential to produce life-threating arrhythmias even in the absence of K channel blocking drugs or frank heart disease. In settings in which repolarization reserve is already compromised, such as heart failure and congenital or acquired long QT syndromes, moderate hypokalemia is likely to be even more arrhythmogenic and should be rigorously avoided during diuretic therapy. Heart failure patients, in which CaMKII activity is already elevated and repolarization reserve is reduced due to electrical remodeling, may be especially vulnerable. In addition, because CaMKII is already activated in heart failure, Class III antiarrhythmic drugs may be arrhythmogenic even when [K]o is normal. Finally, our findings with GS-967 suggest that the pro-arrhythmic effects of hypokalemia and Class III antiarrhythmic drugs can be abrogated by blocking late INa, a key target of CaMKII which, by increasing intracellular Na and Ca loading, fuels the positive feedback loop that further accelerates CaMKII activation (Fig. 8), enhancing the onset of EAD-mediated arrhythmias inhibition.
Supplementary Material
Acknowledgements
We thank Dr. Luiz Belardinelli (Gilead Sciences Inc.) for generously supplying GS-967, and Hayk Stepanyan and Dr. Guillaume Calmettes for technical assistance.
Funding Sources: This work was supported by NIH/NHLBI grant P01 HL079831 and the Laubisch and Kawata Endowments (JNW).
Footnotes
Disclosures: Drs. Karagueuzian and Qu receive research funding from Gilead Sciences, Inc.
References
- 1.Schulman M, Narins RG. Hypokalemia and cardiovascular disease. Am J Cardiol. 1990;65:4E–9E. doi: 10.1016/0002-9149(90)90244-u. discussion 22E-23E. [DOI] [PubMed] [Google Scholar]
- 2.Macdonald JE, Struthers AD. What is the optimal serum potassium level in cardiovascular patients? J Am Coll Cardiol. 2004;43:155–61. doi: 10.1016/j.jacc.2003.06.021. [DOI] [PubMed] [Google Scholar]
- 3.Cooper HA, Dries DL, Davis CE, Shen YL, Domanski MJ. Diuretics and risk of arrhythmic death in patients with left ventricular dysfunction. Circulation. 1999;100:1311–5. doi: 10.1161/01.cir.100.12.1311. [DOI] [PubMed] [Google Scholar]
- 4.Siscovick DS, Raghunathan TE, Psaty BM, Koepsell TD, Wicklund KG, Lin X, Cobb L, Rautaharju PM, Copass MK, Wagner EH. Diuretic therapy for hypertension and the risk of primary cardiac arrest. N Engl J Med. 1994;330:1852–7. doi: 10.1056/NEJM199406303302603. [DOI] [PubMed] [Google Scholar]
- 5.Sanguinetti MC, Jurkiewicz NK. Role of external Ca and K in gating of cardiac delayed rectifier K currents. Pflugers Arch. 1992;420:180–6. doi: 10.1007/BF00374988. [DOI] [PubMed] [Google Scholar]
- 6.Osadchii OE. Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fundam Clin Pharmacol. 2010;24:547–59. doi: 10.1111/j.1472-8206.2010.00835.x. [DOI] [PubMed] [Google Scholar]
- 7.Anderson ME, Braun AP, Wu Y, Lu T, Wu Y, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- 8.Mazur A, Roden DM, Anderson ME. Systemic administration of calmodulin antagonist W-7 or protein kinase A inhibitor H-8 prevents torsade de pointes in rabbits. Circulation. 1999;100:2437–42. doi: 10.1161/01.cir.100.24.2437. [DOI] [PubMed] [Google Scholar]
- 9.Yang T, Chun YW, Stroud DM, Mosley JD, Knollmann BC, Hong C, Roden DM. Screening for acute IKr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation. 2014;130:224–34. doi: 10.1161/CIRCULATIONAHA.113.007765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aronsen JM, Skogestad J, Lewalle A, Louch WE, Hougen K, Stokke MK, Swift F, Niederer S, Smith NP, Sejersted OM, Sjaastad I. Hypokalaemia induces Ca overload and Ca waves in ventricular myocytes by reducing Na,K -ATPase alpha2 activity. J Physiol. 2015;593:1509–21. doi: 10.1113/jphysiol.2014.279893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 2010;48:112–21. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Killeen MJ, Gurung IS, Thomas G, Stokoe KS, Grace AA, Huang CL. Separation of early afterdepolarizations from arrhythmogenic substrate in the isolated perfused hypokalaemic murine heart through modifiers of calcium homeostasis. Acta Physiol (Oxf) 2007;191:43–58. doi: 10.1111/j.1748-1716.2007.01715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swaminathan PD, Purohit A, Hund TJ, Anderson ME. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res. 2012;110:1661–77. doi: 10.1161/CIRCRESAHA.111.243956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morita N, Sovari AA, Xie Y, Fishbein MC, Mandel WJ, Garfinkel A, Lin S-F, Chen P-S, Xie L-H, Chen F, Qu Z, Weiss JN, Karagueuzian HS. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am J Physiol Heart Circ Physiol. 2009;297:H1594–1605. doi: 10.1152/ajpheart.00579.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morita N, Lee JH, Xie Y, Sovari A, Qu Z, Weiss JN, Karagueuzian HS. Suppression of re entrant and multifocal ventricular fibrillation by the late sodium current blocker ranolazine. J Am Coll Cardiol. 2011;57:366–375. doi: 10.1016/j.jacc.2010.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen TP, Xie Y, Garfinkel A, Qu Z, Weiss JN. Arrhythmogenic consequences of myofibroblast-myocyte coupling. Cardiovasc Res. 2012;93:242–51. doi: 10.1093/cvr/cvr292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belardinelli L, Liu G, Smith-Maxwell C, Wang WQ, El-Bizri N, Hirakawa R, Karpinski S, Li CH, Hu L, Li XJ, Crumb W, Wu L, Koltun D, Zablocki J, Yao L, Dhalla AK, Rajamani S, Shryock JC. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J Pharmacol Exp Ther. 2013;344:23–32. doi: 10.1124/jpet.112.198887. [DOI] [PubMed] [Google Scholar]
- 18.Restrepo JG, Weiss JN, Karma A. Calsequestrin-mediated mechanism for cellular calcium transient alternans. Biophys J. 2008;95:3767–89. doi: 10.1529/biophysj.108.130419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calmettes G, Drummond GB, Vowler SL. Making do with what we have: use your bootstraps. J Physiol. 2012;590:3403–6. doi: 10.1113/jphysiol.2012.239376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010;7:1891–9. doi: 10.1016/j.hrthm.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species-activated Ca/calmodulin kinase IIdelta is required for late INa augmentation leading to cellular Na and Ca overload. Circ Res. 2011;108:555–65. doi: 10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madhvani RV, Xie Y, Pantazis A, Garfinkel A, Qu Z, Weiss JN, Olcese R. Shaping a new Ca conductance to suppress early afterdepolarizations in cardiac myocytes. J Physiol. 2011;589:6081–92. doi: 10.1113/jphysiol.2011.219600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bapat A, Nguyen TP, Lee JH, Sovari AA, Fishbein MC, Weiss JN, Karagueuzian HS. Enhanced sensitivity of aged fibrotic hearts to angiotensin II- and hypokalemia-induced early afterdepolarization-mediated ventricular arrhythmias. Am J Physiol Heart Circ Physiol. 2012;302:H2331–40. doi: 10.1152/ajpheart.00094.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hund TJ, Rudy Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation. 2004;110:3168–3174. doi: 10.1161/01.CIR.0000147231.69595.D3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca/calmodulin-dependent protein kinase mediates Ca-induced enhancement of the L-type Ca current in rabbit ventricular myocytes. Circ Res. 1994;75:854–61. doi: 10.1161/01.res.75.5.854. [DOI] [PubMed] [Google Scholar]
- 26.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca/calmodulin-dependent protein kinase II regulates cardiac Na channels. J Clin Invest. 2006;116:3127–38. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qu Z, Xie LH, Olcese R, Karagueuzian HS, Chen PS, Garfinkel A, Weiss JN. Early afterdepolarizations in cardiac myocytes: beyond reduced repolarization reserve. Cardiovasc Res. 2013;99:6–15. doi: 10.1093/cvr/cvt104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zehender M, Hohnloser S, Just H. QT-interval prolonging drugs: mechanisms and clinical relevance of their arrhythmogenic hazards. Cardiovasc Drugs Ther. 1991;5:515–30. doi: 10.1007/BF03029779. [DOI] [PubMed] [Google Scholar]
- 29.Wenzel-Seifert K, Wittmann M, Haen E. QTc prolongation by psychotropic drugs and the risk of Torsade de Pointes. Dtsch Arztebl Int. 2011;108:687–93. doi: 10.3238/arztebl.2011.0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Volders PG, Sipido KR, Vos MA, Kulcsar A, Verduyn SC, Wellens HJ. Cellular basis of biventricular hypertrophy and arrhythmogenesis in dogs with chronic complete atrioventricular block and acquired torsade de pointes. Circulation. 1998;98:1136–47. doi: 10.1161/01.cir.98.11.1136. [DOI] [PubMed] [Google Scholar]
- 31.Yang T, Roden DM. Extracellular potassium modulation of drug block of IKr. Implications for torsade de pointes and reverse use-dependence. Circulation. 1996;93:407–11. doi: 10.1161/01.cir.93.3.407. [DOI] [PubMed] [Google Scholar]
- 32.Han F, Tucker AL, Lingrel JB, Despa S, Bers DM. Extracellular potassium dependence of the Na,K-ATPase in cardiac myocytes: isoform specificity and effect of phospholemman. Am J Physiol Cell Physiol. 2009;297:C699–705. doi: 10.1152/ajpcell.00063.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weiss JN. Palpitations, potassium and the pump. J Physiol. 2015;593:1387–8. doi: 10.1113/jphysiol.2014.285924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morita N, Lee JH, Bapat A, Fishbein MC, Mandel WJ, Chen PS, Weiss JN, Karagueuzian HS. Glycolytic inhibition causes spontaneous ventricular fibrillation in aged hearts. Am J Physiol Heart Circ Physiol. 2011;301:H180–H191. doi: 10.1152/ajpheart.00128.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N, Zhang S. Extracellular K concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest. 2009;119:2745–57. doi: 10.1172/JCI39027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouchard R, Clark RB, Juhasz AE, Giles WR. Changes in extracellular K concentration modulate contractility of rat and rabbit cardiac myocytes via the inward rectifier K current IK1. J Physiol. 2004;556:773–90. doi: 10.1113/jphysiol.2003.058248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonough AA, Zhang Y, Shin V, Frank JS. Subcellular distribution of sodium pump isoform subunits in mammalian cardiac myocytes. Am J Physiol. 1996;270:C1221–7. doi: 10.1152/ajpcell.1996.270.4.C1221. [DOI] [PubMed] [Google Scholar]
- 38.Berry RG, Despa S, Fuller W, Bers DM, Shattock MJ. Differential distribution and regulation of mouse cardiac Na,K-ATPase alpha1 and alpha2 subunits in T-tubule and surface sarcolemmal membranes. Cardiovasc Res. 2007;73:92–100. doi: 10.1016/j.cardiores.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 39.Paice BJ, Paterson KR, Onyanga-Omara F, Donnelly T, Gray JM, Lawson DH. Record linkage study of hypokalaemia in hospitalized patients. Postgrad Med J. 1986;62:187–91. doi: 10.1136/pgmj.62.725.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thompson RG, Cobb LA. Hypokalemia after resuscitation from out-of-hospital ventricular fibrillation. J Am Med Assoc. 1982;248:2860–3. [PubMed] [Google Scholar]
- 41.Israel CW, Barold SS. Electrical storm in patients with an implanted defibrillator: a matter of definition. Ann Noninvasive Electrocardiol. 2007;12:375–82. doi: 10.1111/j.1542-474X.2007.00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Astrom-Lilja C, Odeberg JM, Ekman E, Hagg S. Drug-induced torsades de pointes: a review of the Swedish pharmacovigilance database. Pharmacoepidemiol Drug Saf. 2008;17:587–92. doi: 10.1002/pds.1607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.