

Abstract
Oligomeric states of the amyloid β-protein (Aβ) appear to be causally related to Alzheimer’s disease (AD). Recently, two familial mutations in the amyloid precursor protein gene have been described, both resulting in amino acid substitutions at Ala2 (A2) within Aβ. An A2V mutation causes autosomal recessive early onset AD. Interestingly, heterozygotes enjoy some protection against development of the disease. An A2T substitution protects against AD and age-related cognitive decline in non-AD patients. Here, we use ion mobility-mass spectrometry (IM-MS) to examine the effects of these mutations on Aβ assembly. These studies reveal different assembly pathways for early oligomer formation for each peptide. A2T Aβ42 formed dimers, tetramers, and hexamers, but dodecamer formation was inhibited. In contrast, no significant effects on Aβ40 assembly were observed. A2V Aβ42 also formed dimers, tetramers, and hexamers, but no dodecamers. However, A2V Aβ42 formed trimers, unlike A2T or wild type (wt) Aβ42. In addition, the A2V substitution caused Aβ40 to oligomerize similar to wt Aβ42, as evidenced by the formation of dimers, tetramers, hexamers, and dodecamers. In contrast, wt Aβ40 formed only dimers and tetramers. These results provide a basis for understanding how these two mutations lead to, or protect against, AD. They also suggest that the Aβ N-terminus, in addition to the oft discussed central hydrophobic cluster and C-terminus, can play a key role in controlling disease susceptibility.
Keywords: Amyloid β-Protein, Familial Alzheimer’s Disease, A2T, A2V, Oligomerization, Ion Mobility Spectrometry, Mass Spectrometry
Introduction
The amyloid β-protein (Aβ) plays an important role in Alzheimer’s disease (AD) pathogenesis1-2. Aβ is produced from the amyloid precursor protein (APP) by successive endoproteolytic cleavages by β- and γ-secretase. Aβ exists in the body primarily in two forms, 40- (Aβ40) or 42-residues (Aβ42) in length. Although Aβ40 is present in the body at a concentration ≈10-fold that of Aβ42, the latter peptide is more toxic and is the primary component of amyloid plaques3. Although pathognomonic for AD, fibril-containing plaques appear not be the primary pathologic agents in AD: The primacy of Aβ oligomers has been suggested instead4-5. In solution, Aβ40 and Aβ42 monomers are both intrinsically disordered, yet they display distinct aggregation pathways on the way to fibril formation. Aβ40 initially forms small oligomers including dimers and tetramers, whereas Aβ42 forms larger aggregates, including dimers, tetramers, hexamers, and dodecamers. These oligomer states appear to be on-pathway for fibril formation6-7. Of these oligomers, the 56-kDa dodecamer has been shown to be a proximate toxic agent for AD pathology8-9.
Although most AD cases occur sporadically, ~5% of AD cases are caused by mutations in the APP10-11, presenilin 1 (PS1)12-13, or presenilin 2 (PS2)14 genes. These familial AD (FAD) cases, often lead to early onset of disease (<60 years of age). Numerous FAD-related mutations in the APP gene have been identified and many of them are near β- or γ-secretase cleavage sites. This results most commonly in overproduction of Aβ or relative increases in the amount of Aβ42 that is produced relative to Aβ4015. However, as many mutations occur within the Aβ region, it is very likely that these substitutions would alter the structural and aggregation properties of the resultant Aβ42 and Aβ40 peptides. Notably, many mutations in the APP gene result in amino acid substitutions within the central region of Aβ, as, for example, Flemish (A21G)16, Arctic (E22G)17, Dutch (E22Q)18, Osaka (E22Δ)19, Italian (E22K)20 and D23N (Iowa)21 mutations. The resulting peptides exhibit distinct aggregation propensity and toxicity. The central region of Aβ has been shown to be crucial for the initial nucleation of Aβ folding and assembly22. Mutations near this region may disrupt the Aβ conformation, resulting in increased aggregation propensity and formation of toxic oligomers23. On the other hand, the role of the N-terminus in aggregation, toxicity and pathology has been less thoroughly studied due to the fact that this region appears disordered in the fibril state24-25_ENREF_22. However, as with the central region of Aβ, a number of APP mutations result in amino acid substitutions at the N-terminus, and these substitutions alter Aβ assembly. These include the English (H6R)26-27, Tottori (D7N)26-29, and Taiwanese (D7H)30 mutations. The importance of the Aβ N-terminus in disease causation thus is clear. Most recently, two new APP mutations have been described that result in the substitutions A2T and A2V can be important in Aβ structure and assembly31-32. In the work presented here, we elucidate the effects on early Aβ assembly of these two recently discovered mutations.
The A2T substitution substantially decreases AD risk, as well as protecting against age-related cognitive decline in the elderly without AD31. It is thought to be the first example of a sequence variant that protects against AD. The A2T substitution occurs immediately adjacent to the β-secretase site, and indeed, the mutation has been found to reduce Aβ production ~20% in heterozygous carriers. Such a reduction may be responsible for its protective function in AD pathology31. However, as the mutation is within the Aβ sequence, it is possible that the A2T mutation also changes the aggregation properties of Aβ proteins, thus contributing to its protective effect, a possibility we investigate here.
The mutation causing the A2V substitution results in early onset AD in the homozygotes, whereas some protection against AD is observed in heterozygotes32. In contrast to the A2T substitution, A2V increases Aβ production. Interestingly, co-incubation of A2V Aβ42 and wt Aβ42 produced slower aggregation rates than exhibited by either peptide alone, as well as decreased toxicity32. The A2V substitution accelerates Aβ42 oligomerization and also leads to the production of annular structures with a higher hydrophobicity than wt Aβ4233.
A consensus regarding the effects of the A2T and A2V substitutions on Aβ assembly has not been reached. Two recent studies of A2T and A2V peptides reported different aggregation kinetics by thioflavin T (ThT) fluorescence studies. Benilova et al. showed that the A2T substitution has little effect on Aβ42 aggregation, but did affect its solubility34. Maloney et al., in contrast, showed that the A2T mutant had a lower aggregation propensity compared to the A2V mutant or to wt Aβ4235. For Aβ40, the A2T mutant was shown to aggregate similarly to wt, whereas the A2V mutant exhibited faster aggregation and a shorter lag phase, making this peptide behave more Aβ42-like34-35_ENREF_23.
To improve our understanding of the A2T and A2V substitutions, we used ion mobility coupled to mass spectrometry (IM-MS) to examine the early assembly and subsequent aggregation of these mutant peptides. IM-MS can separate species with the same mass-to-charge (z/n) ratio but different shapes or sizes36. As a consequence, it has successfully revealed the structures of Aβ oligomers and the effects of small molecule inhibitors of Aβ assembly7,29,37-42. We examine here the early oligomer distributions of A2T- and A2V-containing Aβ40 and Aβ42 to understand how each assembles and whether the early assembly pathways are identical or different. We also examine the early oligomer distributions of mixtures of wt and mutant peptides to understand how each affects the other’s assembly. This provides the means to model in vitro the homozygous and heterozygous states that exist in humans. These studies provide mechanistic insights into the aetiology of FAD, mechanisms of protection from FAD, and potential targets for therapeutic agents.
Results
Different oligomer distributions of wt and mutant Aβ42
Mass spectra of wt Aβ42, A2T, and A2V were recorded individually and are shown in Figure 1a-c. Four common peaks were observed for each peptide, corresponding to z/n ratios of −4, −3, −5/2 and −2, where z is charge and n is oligomer size. The mass spectrum of A2V Aβ42 was interesting because in addition to the four peaks, another peak was observed between z/n = −3 and −5/2 in the spectrum, corresponding to z/n = −8/3. This indicates the A2V mutant forms a trimer, which is not observed for wt or A2T Aβ42. Moreover, there is another peak between z/n = −4 and −3 for A2V, denoted by *, which is assigned as fragment peak or impurity (see supporting information, Figure S3, for detailed discussion of this peak assignment).
Figure 1.

a-c) Mass spectra of A2T, A2V and wt Aβ42. The charge state of each species is noted with z/n, where z is the charge and n is oligomer number. The peak marked with * in panel b is assigned as a fragment peak or impurity (see discussion in the SI). d-f) ATDs of z/n = −5/2 peaks for A2T, A2V and wt Aβ42. The oligomer order (n) is noted for each feature. The dashed lines represent the peak shape for a single conformation. Injection energy studies of the z/n = −5/2 A2T and A2V Aβ42 peaks are provided in Figuer S1. The injection energy in panels d, e, and f is 40 eV.
The arrival time distributions (ATDs) of the z/n = −5/2 peaks for all three Aβ42 alloforms are shown in Figure 1d-e. The ATD of wt Aβ42 shows four features, with arrival times at ~710, 670, 610 and 540 μs, which were previously assigned as Aβ42 dimer, tetramer, hexamer, and dodecamer, respectively, based on their calculated collision cross sections (See reference 7 for detailed discussion of these assignments). However, the ATD of A2T or A2V Aβ42 (Figure 1d or e) shows only three features, with arrival times at ~710, 670, 610 μs which were assigned as dimer, tetramer, and hexamer, respectively, based on their calculated cross sections. There is no feature at lower arrival time observed in either of the ATD for mutants, indicating no other oligomers larger than hexamers are formed. These results suggest the formation of Aβ42 dodecamer is inhibited by both A2T and A2V mutations.
To assign the peaks in the ATDs unambiguously, and to better understand the oligomer distributions of the Aβ42 mutants, the −5/2 ATDs for Aβ42 mutants were measured at different injection energies. At low injection energy, the ions are rapidly thermalized by cooling collisions with the helium gas in the drift cell and therefore large complexes can be preserved through the process. At high injection energy, the ions are given sufficient energy to lead to internal excitation which can cause isomerization into low energy structure or dissociation of large noncovalent complexes into smaller species. As shown in Figure S1, the ATDs measured at intermediate injection energy (40 eV) are the same ones shown in Figure 1d and 1e. When the injection energy is lowered to 25 eV (Figure S1 top panel), the hexamer peak becomes especially prominent, whereas the tetramer and dimer features decrease. However, there are still no peaks with lower arrival times observed, suggesting that oligomers of size dodecamer or larger are not formed in solution. At high injection energy (100 eV, Figure S1 bottom panel), the hexamer peak disappears whereas the tetramer and dimer peaks dominate the spectrum. This suggests hexamer dissociation into smaller oligomers. These injection energy studies are fully consistent with the assignment of the three peaks in the ATDs as dimer, tetramer, and hexamer.
Ion mobility study of z/n = −2 and −8/3 peaks: A2V Aβ42 forms trimers
The z/n = −2 Aβ42 is a relative low charge state of the Aβ42 alloforms and possibly consists of high order oligomers, making its ATD of interest. The signal of the z/n = −2 peak for wt Aβ42 is too low to obtain a reliable ATD, therefore no data is shown. However, we were able to record ATDs for the −2 peaks of A2T and A2V Aβ42 (Figure 2).
Figure 2.
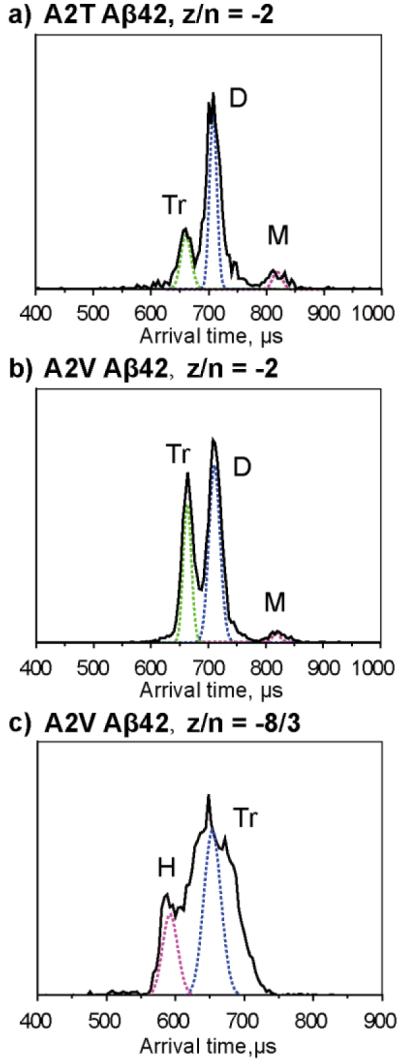
a) and b), ATDs of the z/n = −2 peaks for A2T and A2V Aβ42, respectively. c) ATD of the z/n = −8/3 peak for A2V. The dashed lines represent the peak shape for a single conformation. The oligomer order is noted for each feature where M represents monomer, D represents dimer, Tr represents trimer and H represents hexamer. The injection energy is 40 eV. Summary of ATDs for each peak and their cross sections for Aβ42 alloforms is given in Figure S7.
The ATD of −2 A2T Aβ42 shows three features, with arrival times at ~820, 720, 670 μs, which can be assigned as monomer, dimer, and trimer, respectively. Similarly, the ATD of the z/n = −2 A2V Aβ42 shows three features, corresponding to monomer, dimer and trimer. However, the relative intensity of the A2T trimer is much lower than that of its dimer while the relative intensity of the A2V trimer is comparable to that of its dimer, indicating the formation of trimer is more favored for A2V Aβ42. Injection energy results (Figure S2b) support this trend. At low injection energies the trimer of A2V Aβ42 is dominant while the trimer of A2T Aβ42 remains minor, indicating trimer in the A2V mutant is significant in solution.
The ATD of z/n = −8/3 A2V Aβ42 shows two features, with arrival times at ~660 and 590 μs observed, which correspond to an A2V trimer and hexamer, respectively (Figure 3c). The breadth of the trimer feature indicates there is a family of trimer structures existing in the solution. The injection energy study of −8/3 A2V Aβ42 (Figure S2c) indicates the hexamer feature increases at lowest energies and the trimer peak gets sharper. At high energy (100 eV), the hexamer feature disappears and the broad trimer feature becomes the dominant peak.
Figure 3.

Ion mobility study of an equimolar mixture of wt and A2T Aβ42. a) A full mass spectrum of wt/A2T Aβ42 mixture and a zoom-in spectrum of z/n = −5/2 peaks which contains three species which correspond to wt Aβ42 homo-oligomers, wt/A2T Aβ42 hetero-oligomers and A2T Aβ42 homo-oligomers. b-d) ATDs of the three −5/2 oligomer peaks. The oligomer order (n) is noted for each feature. The dashed lines represent the peak shape for a single conformation. The injection energy in panels b, c, and d is 40 eV.
Taken together, these ion mobility results reveal that the oligomerization pattern is different for each of the alloforms. wt Aβ42 forms dimer, tetramer, hexamer, and dodecamer. A2T and A2V Aβ42 form dimer, tetramer, and hexamer, without the formation of dodecamer, but A2V forms a significant trimer which is only very minor in A2T and may not be present in wt Aβ42 at all.
Transmission electron microscopy (TEM) images were recorded for the same Aβ42 samples after 5 days’ incubation in the room temperature and the results are shown in Figure S9. The wt Aβ42 forms long fibrils after 5 days’ incubation, while the A2T and A2V Aβ42 form some short fibrils or protofibrils along with some long fibrils. These results are consistent with previous study34 and suggest the oligomers we detected are on-pathway.
Mixtures of wt and mutant Aβ42: The effects on wt Aβ42 oligomerization
The A2T mutation has been shown to protect carriers from AD or normal age-related cognitive decline31. To model the effects of this peptide in heterozygotes, we created an equimolar mixture of A2T and wt Aβ42 and then performed MS (Figure 3). Four sets of peaks were observed, corresponding to z/n = −4, −3, −5/2 and −2 charge states. A zoom-in spectrum of the −5/2 region using the QTOF-MS was obtained and shows that there are three peaks with charge state of −5/2, which correspond to −5/2 wt Aβ42 homo-oligomers, wt/A2T hetero-oligomers (1:1 ratio), and A2T homo-oligomers. The ATDs of these three peaks (Figure 3b, c, d) display a similar oligomer distribution with three features with arrival times of ~710, 670 and 600 μs. We assign these features as dimers, tetramers, and hexamers, respectively. Note that no feature at shorter arrival time was observed, indicating there is no homo-/hetero-dodecamer or higher oligomer formation. These results indicate that the A2T mutant forms small hetero-oligomers (up to hetero-hexamers) with wt Aβ42 and inhibits the formation of wt Aβ42 dodecamer or higher oligomers.
Previous studies showed that A2V is a recessive mutation that causes early-onset of AD in homozygotes but appears protective in heterozygotes32. To provide insight into this these observation, we performed ion mobility studies on an equimolar mixture of wt and A2V Aβ42 (Figure 4). Similar to the A2T/wt mixture, the A2V/wt mixture shows three −5/2 peaks, corresponding to wt Aβ42 homo-oligomers, wt/A2V hetero-oligomers (1:1 ratio) and A2V homo-oligomers. The ATDs of these −5/2 peaks all show three features that can be assigned as dimer, tetramer, and hexamer, respectively. The data show that A2V Aβ42 forms small hetero-oligomers (only up to hexamers) with wt Aβ42 and prevents the formation of larger oligomers.
Figure 4.

Ion mobility study of an equimolar mixture of wt Aβ42 and A2V mutant. a) A full mass spectrum of A2V/wt Aβ42 mixture and a zoom-in spectrum of z/n = −5/2 peaks which contains three species which correspond to wt Aβ42 homo-oligomers, wt /A2V Aβ42 hetero-oligomers and A2V Aβ42 homo-oligomers. b-d) ATDs of the three −5/2 oligomer peaks. The oligomer order (n) is noted for each feature. The dashed lines represent the peak shape for a single conformation. The injection energy in panels b, c, and d is 40 eV.
There is no −8/3 trimer peak observed in the equimolar mixture of wt and A2V Aβ42. Moreover, the ATD of the z/n = −2 peak for the wt/A2V mixture (Figure S4b) shows a dominant dimer peak and only a minor trimer peak, unlike A2V alone (Figure 2b). These results indicate that A2V trimer formation is inhibited by wt Aβ42.
Ion mobility study of Aβ40 mutants: A2V Aβ40 forms hexamer and dodecamer
We next examined the effects of the A2T and A2V substitutions on Aβ40 assembly (see Figure 5). The mass spectra for the A2T and A2V mutants (Figure 5b and c) showed four peaks, corresponding to z/n = −4, −3, −5/2 and −2, which is similar to that of the wt Aβ40 (Figure 5a).
Figure 5.

a-c) Mass spectra of Aβ40 wt , A2T and A2V. The charge state of each species is noted with z/n, where z is the charge and n is oligomer number. d-f) ATDs of z/n = −5/2 peaks for Aβ40 wt, A2T and A2V. g-i) ATDs of z/n = −2 peaks for Aβ40 wt, A2T and A2V. The oligomer order (n) is noted for each feature. The dashed lines represent the peak shape for a single conformation. The injection energy in panels d, e, f, g, h, and i is 40 eV. Summary of ATDs for each peak and their cross sections for Aβ40 alloforms is given in Figure S8.
The ATD of z/n = −5/2 wt Aβ40 (Figure 5d) displays two features, with arrival times at ~710 and 670 μs, which were previously assigned as Aβ40 dimer and tetramer, respectively7. The ATD of the z/n = −5/2 A2T Aβ40 (Figure 5e) again shows two features, with arrival times at ~710 and 670 μs, corresponding to dimer and tetramer, respectively. This ATD was similar to that of wt Aβ40. However, the ATD of z/n = −5/2 A2V Aβ40 (Figure 5f) shows four features with arrival times of ~710, 670, 620 and 550 μs, which can be assigned as dimer, tetramer, hexamer and dodecamer, respectively. Hence, A2V Aβ40 forms hexamers and dodecamers, something not observed for wt or A2T Aβ40. This is consistent with previous ThT studies showing A2V Aβ40 displays a shorter lag phase during aggregation, which is similar to that of wt Aβ4234-35.
The ATDs of z/n = −2 Aβ40 alloforms were recorded and are shown in Figure 5g-i. The ATD of wt Aβ40 shows a dominant dimer peak at ~720 μs and a small monomer peak at ~840 μs. The dimer peak is slightly broad at the bottom, which indicates there might be a small amount of trimer formed. The ATDs of A2T and A2V Aβ40 (Figure 5h and i) show one additional peak, with a shorter arrival time at ~680 μs, which is assigned as trimer. The relative intensity of the A2V trimer is greater than that of the A2T trimer or wt Aβ40 trimer. This is consistent with the results of z/n = −5/2 peaks, which suggests that A2V Aβ40 is aggregating into larger oligomers than is A2T and wt Aβ40.
In summary, the A2T mutation does not significantly change Aβ40 oligomerization. The A2V mutation, in contrast, promotes Aβ40 oligomerization and causes it to undergo a more "Aβ42-like" aggregation process. Although the relative intensity of the A2V dodecamer is smaller than observed for wt Aβ42 (Figure 1f), the Aβ40 isoform is ten times more abundant than Aβ42 in vivo. Hence this is a significant result and is fully consistent with the fact homozygous carriers of the A2V mutation develop early-onset of AD.
The TEM results of Aβ40 samples after 5 days’ incubation (Figure S9) showed that they all formed short fibrils along with some annuli-like aggregates. Interestingly, the Aβ40 A2V fibrils are thinner and tend to clump together to form plaques, indicating the aggregation of Aβ40 A2V is faster than wt or A2T Aβ40.
The results of co-incubation experiments using wt and mutant Aβ40 are shown in the Figure S5 and S6. The A2T/wt Aβ40 mixture shows formation of homo-/hetero-dimer and tetramer, which is similar to that of wt Aβ40, indicating no enhancement of aggregation by A2T. Similarly, the A2V/wt mixture shows only homo- and hetero-dimer and tetramer. This is important because it indicates wt Aβ40 inhibits formation of A2V hexamer or dodecamer. Hence, heterozygous A2V carriers are protected from dodecamer formation while homozygous A2V carriers are not.
Discussion
Our results show that amino acid substitutions at Ala2 of Aβ affect Aβ42 oligomerization (summarized in Figure 6). The Iceland mutation A2T was observed to prevent the formation of Aβ42 dodecamer, which was previously identified as an important neurotoxin in AD8-9. These results are consistent with previous studies demonstrating that A2T is a protective mutation31. However, our ion mobility studies show that the A2T mutation does not have a significant effect on oligomerization of the less toxic Aβ40 isoform.
Figure 6.
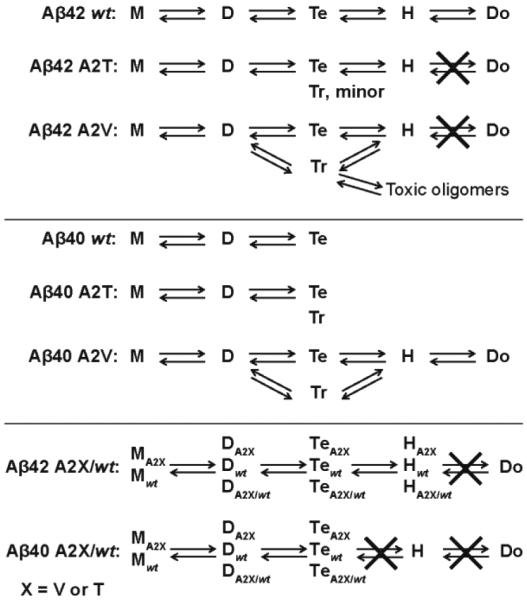
Different oligomerization patterns for wt Aβ, A2T, A2V alloforms and mixtures. M, D, Tr, Te, H, Do represent monomer, dimer, trimer, tetramer, hexamer and dodecamer, respectively.
The A2V mutation was observed to inhibit the formation of Aβ42 dodecamer as well. However, the A2V mutation leads to a much greater fraction of Aβ42 trimer formation and observation of a unique z/n = −8/3 trimer peak that contains a significant fraction of hexamer not formed in A2T or wt Aβ42. This result implies A2V Aβ42 may adopt another early assembly pathway through trimer that leads to toxic oligomers before going on to form fibrils. Even more interestingly, the A2V mutation shows significant effects on Aβ40 assembly, resulting in the formation of Aβ40 hexamer and dodecamer, which are not observed for A2T or wt Aβ40. Hence, the A2V mutation changes the Aβ40 aggregation pathway into a Aβ42-like pathway, which is consistent with a previous ThT fluorescence study showing A2V has a shorter aggregation lag phase than wt Aβ4034. Although the relative intensity of dodecamer in A2V Aβ40 is smaller than for wt Aβ42 (Figure 1f), this peptide is ten times more abundant than Aβ42 in vivo and hence the fact it produces potentially toxic oligomer states will be strongly amplified in vivo. This result, while not proof, is entirely consistent with the fact the A2V mutant results in early onset AD in homozygous carriers32.
The effects of the A2T and A2V substitutions on wt Aβ42 oligomerization were evaluated by co-incubating equimolar mixtures of the mutant and wt Aβ42 proteins. Both mutants formed small hetero-oligomers with wt Aβ42, including dimers, tetramers, and hexamers. However, no hetero- or homo-dodecamers were observed, indicating the formation of Aβ42 dodecamer is inhibited by the mutants. Co-incubation of A2T and wt Aβ40 shows formation of dimers and tetramers, which is similar to that of wt Aβ40, indicating no enhancement of aggregation by A2T (Figure S5). However, co-incubation of A2V and wt Aβ40 shows only homo- and hetero-dimer and tetramer formation, indicating thathexamer and dodecamer formation is inhibited (Figure S6). This indicates that rapid A2V aggregation is inhibited by wt Aβ40. These results are consistent with previous studies suggesting that A2T protects against AD and A2V heterozygous carriers are not affected by this mutation31-32.
The N-terminus of Aβ is relatively hydrophilic and appears to exist in a disordered state. It thus has been argued that it plays only a modest (or no) role in controlling Aβ assembly compared to the central hydrophobic cluster region or the C-terminus 24. However, we find here that single A2T and A2V amino acid substitutions do affect Aβ oligomerization quite significantly, offering a mechanistic explanation for the phenotypes of humans expressing the cognate genes.
Threonine (T) and valine (V) have similar sizes but different hydrophobicity. The substitution of the neutral alanine (A) with a nucleophilic threonine or a hydrophobic valine will change the hydrophobicity of the N-terminus region and perhaps change the conformation of Aβ. A recent simulation study of A2T and A2V Aβ42 showed significantly different conformational landscapes of the Aβ42 monomer43. The A2T Aβ42 mutant makes the N-terminus more polar, which displays unusual long-range electrostatic interactions with residues such as Lys16 and Glu2243. Through such electrostatic interactions, the hairpin structure in the central hydrophobic region is disrupted, resulting in a population of unique conformations with only a C-terminal hairpin. In contrast, the A2V Aβ42 shows an enhanced double-hairpin population due to hydrophobic interactions between the N-terminus and distant hydrophobic regions (Central hydrophobic core and C-terminus hydrophobic region)43. A previous simulation showed that the A2V mutation reduced the intrinsic disorder and increased the hairpin population in the Aβ(1-28) monomer44. In addition, a previous MD simulation study showed that the N-terminus of Aβ40 displayed a β-strand structure at Ala-2-Phe-4 which was not present in Aβ4245. The hydrophilic N-termini of Aβ proteins are on the surface of the oligomers, thus the presence of an N-terminal β-strand in Aβ40 might prevent the hydrophobic core of the oligomers from adding additional Aβ40 molecules to form larger oligomers, which explains why Aβ40 aggregates slower and forms smaller oligomers than Aβ42. Therefore the substitution of Ala with a hydrophobic Val may disrupt the formation of a hydrophilic N-terminal β-strand and make the hydrophobic core accessible for other Aβ40 molecules, shifting the A2V Aβ40 oligomerization toward those of Aβ42. Our ion mobility studies reveal different oligomerization for Aβ proteins with a single mutation in the N-terminus region and imply the importance of the N-terminus region for Aβ assembly, results consistent with these previous studies43-45.
In this work, we have demonstrated that IMS-MS is becoming a powerful tool to carry out studies that lead to understanding of AD familial mutations. This is of significance as single mutations have been implied to be important in disease aetiology. For instance, recently the G127V mutation in a prion variant has been shown to completely protect transgenic mice from prion disease46. Hence understanding the mechanism of these positive substitutions becomes important for future therapeutic development. Thus IMS-MS can be used as a new tool to study other systems of this kind and provide an insight into their structure-disease relationship.
Conclusions
The A2T mutation prevents formation of Aβ42 dodecamer both in homo- and heterozygotes. The dodecamer has been implicated as a proximate toxic agent in AD.
The A2V mutation in homozygotes also prevents dodecamer formation in Aβ42 but promotes trimer formation which may initiate a new pathway for early oligomer formation in Aβ42.
The A2V mutation in homozygotes promotes hexamer and dodecamer formation in Aβ40, whereas wt Aβ40 assembly terminates at the tetramer. Since Aβ40 is 10 times more prevalent than Aβ42 in vivo, facilitation of Aβ40 hexamer and dodecamer formation may well explain why the A2V mutation causes early onset of AD in homozygotes.
Both the A2T and A2V mutations eliminate dodecamer formation in heterotypic mixtures with wt Aβ40 and Aβ42, consistent with the protective effects of these substitutions in heterozygotes.
Ion mobility methods are emerging as an important new tool in developing an understanding of the effect of familial mutations on Aβ assembly in AD and the assembly of other mutated protein systems.
Methods
Peptide and Sample preparation
Full-length Aβ and mutants were synthesized by N-9-fluorenylmethoxycarbonyl (FMOC) chemistry47. The peptides were purified by reverse-phase HPLC and their quality validated by mass spectrometry and amino acid analysis. Samples were prepared in 10 mM ammonium acetate buffer, pH 7.4, at a final peptide concentration of 10 μM. Equimolar mixtures of wt and mutant Aβ were prepared at a total peptide concentration of 10 μM (5 μM of each peptide).
Mass Spectrometry and Ion Mobility spectrometry Analysis
Most data were recorded on a home-built ion mobility spectrometry-mass spectrometer48 or a Micromass QTOF2 quadrupole/time-of-flight tandem mass spectrometer. The home-built instrument is composed of a nano-electrospray ionization (nano-ESI) source, an ion funnel, a temperature-controlled drift cell and a quadrupole mass filter followed by an electron multiplier for ion detection.
Briefly, for ion-mobility measurements, ions are generated continuously by a nano-ESI source, focused and stored in the ion funnel. A pulse of ions is injected into a temperature-controlled drift cell filled with 3-5 torr helium gas, where they gently pass through under the influence of a weak electric field. The injection energy can be varied from ~20 to ~150 eV. At low injection energy, the ions are rapidly thermalized by cooling collisions with the helium gas in the drift cell. At high injection energy, the ions are given energy that can lead to internal excitation before reaching thermal equilibrium. Such internal excitation can cause isomerization into low energy structure or dissociation of large noncovalent complexes into small species37. Usually the injection energy is kept as low as possible to minimize thermal heating of the ions during the injection process (The injection energy studies are provided in the Supporting information, Figure S1 and S2). The ions exiting the drift cell are mass analyzed with a quadrupole mass filter, detected by a conversion dynode and channel electron multiplier allowing a mass spectrum to be obtained.
The pulse of ions into the drift cell starts a clock at t = 0 and ends at t = tA when the ions reach the detector. This allows an arrival time distribution (ATD) to be obtained. The ATD can be related to the time the ions spend in the drift cell which is directly related to the ion mobility and collision cross section of the analyte ion49. The width of the ATD can be compared to the width calculated for a single analyte ion structure49, which gives information on the structural distribution favored in the ATD.
Transmission Electron Microscopy (TEM)
Microscopic analysis was performed using a FEI T-20 transmission electron microscope operating at 200 kV. The Aβ samples were prepared using the same procedure as for the mass spectrometry analysis. The samples were incubated at room temperature for 5 days. For TEM measurements, 10 μL aliquots of samples were spotted on glow-discharged, carbon-coated copper grids (Ted Pella, Inc). The samples were stained with 10 mM sodium metatungstate for 10 min and gently rinsed twice with deionized water. The sample grids were then dried at room temperature before TEM analysis.
Supplementary Material
Acknowledgements
We thank Margaret Condron at UCLA for synthesizing and purifying the Aβ proteins used in this work and the mass spectrometry facility in the Department of Chemistry and Biochemistry at UCSB.
Funding: The work has been supported by the National Institutes of Health grant AG047116 (to M.T.B.), AG041295 (to D. B. T.). The Material Research Laboratory Shared Experimental Facilities are supported by the MRSEC Program of the NSF under Award No. DMR 1121053 (a member of the NSF-funded Materials Research Facilities Network, www.mrfn.org).
Abbreviations
- FAD
Familial Alzheimer’s disease
- Aβ
amyloid β-protein
- IMS-MS
ion mobility spectrometry-mass spectrometry
- ATD
arrival time distribution
Footnotes
Supporting Information:
Additional IMS-MS data of injection energy studies for A2T and A2V proteins, discussion of peak assignment for possible pentamer for A2V Aβ42, ATDs of z/n = −2 for the A2T/wt and A2V/wt Aβ42 mixtures, IMS-MS data of mixtures of A2T or A2V and wt Aβ0, summary of ATDs and cross sections for A2T and A2V Aβ proteins, TEM images for Aβ40 and Aβ42 proteins. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes: The authors declare no competing financial interest.
References
- 1.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Alzheimer's disease: Genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 3.Jakob-Roetne R, Jacobsen H. Alzheimer's disease: From pathology to therapeutic approaches. Angew. Chem., Int. Ed. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 4.Teplow D. On the subject of rigor in the study of amyloid β-protein assembly. Alzheimer's Res. Ther. 2013;5:39. doi: 10.1186/alzrt203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden E, Teplow D. Amyloid β-protein oligomers and Alzheimer's disease. Alzheimer's Res. Ther. 2013;5:60. doi: 10.1186/alzrt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid β-protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. U. S. A. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernstein SL, Dupuis NF, Lazo ND, Wyttenbach T, Condron MM, Bitan G, Teplow DB, Shea J-E, Ruotolo BT, Robinson CV, et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat. Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 9.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl. Acad. Sci. U. S. A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang J, Lemaire H-G, Unterbeck A, Salbaum JM, Masters CL, Grzeschik K-H, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 11.O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annu. Rev. Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 13.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada C-M, Kim G, Seekins S, Yager D, et al. Familial Alzheimer's disease–linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 14.Levy-Lahad E, Wasco W, Poorkaj P, Romano D, Oshima J, Pettingell W, Yu C, Jondro P, Schmidt S, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 15.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Hendriks L, van Duijn CM, Cras P, Cruts M, Van Hul W, van Harskamp F, Warren A, McInnis MG, Antonarakis SE, Martin J-J, et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the β-amyloid precursor protein gene. Nat. Genet. 1992;1:218–221. doi: 10.1038/ng0692-218. [DOI] [PubMed] [Google Scholar]
- 17.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, et al. The 'Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 18.Levy E, Carman M, Fernandez-Madrid I, Power M, Lieberburg I, van Duinen S, Bots G, Luyendijk W, Frangione B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- 19.Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, et al. A new amyloid β variant favoring oligomerization in Alzheimer's-type dementia. Ann. Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- 20.Miravalle L, Tokuda T, Chiarle R, Giaccone G, Bugiani O, Tagliavini F, Frangione B, Ghiso J. Substitutions at codon 22 of Alzheimer's Aβ peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J. Biol. Chem. 2000;275:27110–27116. doi: 10.1074/jbc.M003154200. [DOI] [PubMed] [Google Scholar]
- 21.Grabowski TJ, Cho HS, Vonsattel JPG, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- 22.Baumketner A, Bernstein SL, Wyttenbach T, Lazo ND, Teplow DB, Bowers MT, Shea J-E. Structure of the 21–30 fragment of amyloid β-protein. Protein Sci. 2006;15:1239–1247. doi: 10.1110/ps.062076806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krone MG, Baumketner A, Bernstein SL, Wyttenbach T, Lazo ND, Teplow DB, Bowers MT, Shea J-E. Effects of familial Alzheimer’s disease mutations on the folding nucleation of the amyloid β-protein. J. Mol. Biol. 2008;381:221–228. doi: 10.1016/j.jmb.2008.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE. The Alzheimer’s peptides Aβ40 and 42 adopt distinct conformations in water: A combined MD / NMR study. J. Mol. Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takeda T, Klimov DK. Probing the effect of amino-terminal truncation for Aβ1−40 peptides. J. Phys. Chem. B. 2009;113:6692–6702. doi: 10.1021/jp9016773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hori Y, Hashimoto T, Wakutani Y, Urakami K, Nakashima K, Condron MM, Tsubuki S, Saido TC, Teplow DB, Iwatsubo T. The tottori (D7N) and english (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 2007;282:4916–4923. doi: 10.1074/jbc.M608220200. [DOI] [PubMed] [Google Scholar]
- 27.Ono K, Condron MM, Teplow DB. Effects of the english (H6R) and tottori (D7N) familial Alzheimer disease mutations on amyloid β-protein assembly and toxicity. J. Biol. Chem. 2010;285:23186–23197. doi: 10.1074/jbc.M109.086496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wakutani Y, Watanabe K, Adachi Y, Wada-Isoe K, Urakami K, Ninomiya H, Saido TC, Hashimoto T, Iwatsubo T, Nakashima K. Novel amyloid precursor protein gene missense mutation (D678N) in probable familial Alzheimer’s disease. J. Neurol., Neurosurg. Psychiatry. 2004;75:1039–1042. doi: 10.1136/jnnp.2003.010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gessel MM, Bernstein S, Kemper M, Teplow DB, Bowers MT. Familial Alzheimer’s disease mutations differentially alter amyloid β-protein oligomerization. ACS Chem. Neurosci. 2012;3:909–918. doi: 10.1021/cn300050d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen W-T, Hong C-J, Lin Y-T, Chang W-H, Huang H-T, Liao J-Y, Chang Y-J, Hsieh Y-F, Cheng C-Y, Liu H-C, et al. Amyloid-Beta (Aβ) D7H mutation increases oligomeric Aβ42 and alters properties of Aβ-zinc/copper assemblies. PLoS ONE. 2012;7:e35807. doi: 10.1371/journal.pone.0035807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, et al. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 32.Di Fede G, Catania M, Morbin M, Rossi G, Suardi S, Mazzoleni G, Merlin M, Giovagnoli AR, Prioni S, Erbetta A, et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science. 2009;323:1473–1477. doi: 10.1126/science.1168979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Messa M, Colombo L, del Favero E, Cantù L, Stoilova T, Cagnotto A, Rossi A, Morbin M, Di Fede G, Tagliavini F, et al. The peculiar role of the A2V mutation in amyloid-β (Aβ) 1–42 molecular assembly. J. Biol. Chem. 2014;289:24143–24152. doi: 10.1074/jbc.M114.576256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benilova I, Gallardo R, Ungureanu A-A, Castillo Cano V, Snellinx A, Ramakers M, Bartic C, Rousseau F, Schymkowitz J, De Strooper B. The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of amyloid-β (Aβ) aggregation. J. Biol. Chem. 2014;289:30977–30989. doi: 10.1074/jbc.M114.599027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, van der Brug M, Liu Y, Ernst JA, Watts RJ, et al. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J. Biol. Chem. 2014;289:30990–31000. doi: 10.1074/jbc.M114.589069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wyttenbach T, Bowers MT. Gas-phase conformations: The ion mobility/ion chromatography method. In: Schalley C, editor. Modern Mass Spectrometry. Springer; Berlin Heidelberg: 2003. pp. 207–232. [Google Scholar]
- 37.Bernstein SL, Wyttenbach T, Baumketner A, Shea J-E, Bitan G, Teplow DB, Bowers MT. Amyloid β-protein: Monomer structure and early aggregation states of Aβ42 and its Pro19 alloform. J. Am. Chem. Soc. 2005;127:2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 38.Gessel MM, Wu C, Li H, Bitan G, Shea J-E, Bowers MT. Aβ(39–42) modulates Aβ oligomerization but not fibril formation. Biochemistry. 2011;51:108–117. doi: 10.1021/bi201520b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee S, Zheng X, Krishnamoorthy J, Savelieff MG, Park HM, Brender JR, Kim JH, Derrick JS, Kochi A, Lee HJ, et al. Rational design of a structural framework with potential use to develop chemical reagents that target and modulate multiple facets of Alzheimer’s disease. J. Am. Chem. Soc. 2014;136:299–310. doi: 10.1021/ja409801p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng X, Gessel MM, Wisniewski ML, Viswanathan K, Wright DL, Bahr BA, Bowers MT. Z-Phe-Ala-diazomethylketone (PADK) disrupts and remodels early oligomer states of the alzheimer disease Aβ42 protein. J. Biol. Chem. 2012;287:6084–6088. doi: 10.1074/jbc.C111.328575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roychaudhuri R, Lomakin A, Bernstein S, Zheng X, Condron MM, Benedek GB, Bowers M, Teplow DB. Gly25-Ser26 amyloid β-protein structural isomorphs produce distinct Aβ42 conformational dynamics and assembly characteristics. J. Mol. Biol. 2014;426:2422–2441. doi: 10.1016/j.jmb.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng X, Liu D, Klärner F-G, Schrader T, Bitan G, Bowers MT. Amyloid β-protein assembly: The effect of molecular tweezers CLR01 and CLR03. J. Phys. Chem. B. 2015;119:4831–4841. doi: 10.1021/acs.jpcb.5b00692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das P, Murray B, Belfort G. Alzheimer’s protective A2T mutation changes the conformational landscape of the Aβ1–42 monomer differently than does the A2V mutation. Biophys. J. 2015;108:738–747. doi: 10.1016/j.bpj.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen PH, Tarus B, Derreumaux P. Familial alzheimer A2V mutation reduces the intrinsic disorder and completely changes the free energy landscape of the Aβ1–28 monomer. J. Phys. Chem. B. 2014;118:501–510. doi: 10.1021/jp4115404. [DOI] [PubMed] [Google Scholar]
- 45.Urbanc B, Cruz L, Yun S, Buldyrev SV, Bitan G, Teplow DB, Stanley HE. In silico study of amyloid β-protein folding and oligomerization. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17345–17350. doi: 10.1073/pnas.0408153101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asante EA, Smidak M, Grimshaw A, Houghton R, Tomlinson A, Jeelani A, Jakubcova T, Hamdan S, Richard-Londt A, Linehan JM, et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature. 2015 doi: 10.1038/nature14510. Published online Jun 10, 2015 DOI: 10.1038/nature14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lomakin A, Chung DS, Benedek GB, Kirschner DA, Teplow DB. On the nucleation and growth of amyloid β-protein fibrils: Detection of nuclei and quantitation of rate constants. Proc. Natl. Acad. Sci. U. S. A. 1996;93:1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wyttenbach T, Kemper PR, Bowers MT. Design of a new electrospray ion mobility mass spectrometer. Int. J. Mass Spectrom. 2001;212:13–23. [Google Scholar]
- 49.Mason EA, McDaniel EW. Transport Properties of Ions in Gases. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, FRG: 2005. Kinetic theory of mobility and diffusion: Sections 5.1 – 5.2; pp. 137–193. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.