

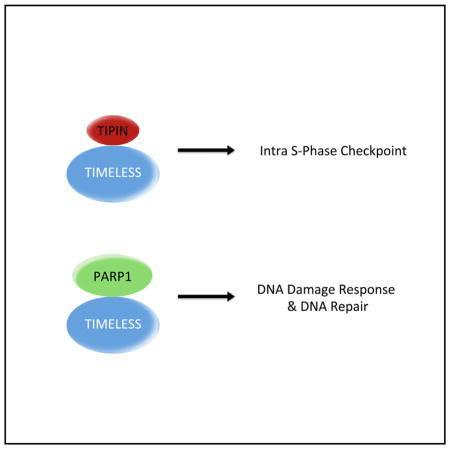
SUMMARY
PARP1 is the main sensor of single- and double-strand breaks in DNA and, in building chains of poly(ADP-ribose), promotes the recruitment of many downstream signaling and effector proteins involved in the DNA damage response (DDR). We show a robust physical interaction between PARP1 and the replication fork protein TIMELESS, distinct from the known TIMELESS-TIPIN complex, which activates the intra-S phase checkpoint. TIMELESS recruitment to laser-induced sites of DNA damage is dependent on its binding to PARP1, but not PARP1 activity. We also find that the PARP1-TIMELESS complex contains a number of established PARP1 substrates, and TIMELESS mutants unable to bind PARP1 are impaired in their ability to bind PARP1 substrates. Further, PARP1 binding to certain substrates and their recruitment to DNA damage lesions is impaired by TIMELESS knockdown, and TIMELESS silencing significantly impairs DNA double-strand break repair. We hypothesize that TIMELESS cooperates in the PARP1-mediated DDR.
Graphical abstract
INTRODUCTION
TIMELESS (tim1) is a protein originally characterized in Drosophila melanogaster as a core component of the circadian clock that regulates daily rhythms. Orthologs in many species, including mammals, have been identified, but mammalian TIMELESS shares greater similarity to a Drosophila paralog of tim1, called timeout or tim2 (Gotter, 2006). While tim2 retains a residual role for light entrainment, suggesting an evolutionary link to tim1, it is mainly an essential gene required for DNA replication and chromosome stability (Benna et al., 2010). Accordingly, although mammalian TIMELESS has a circadian function in the superchiasmatic nucleus of the mouse (Barnes et al., 2003; McFarlane et al., 2010), it is best characterized for its role in regulating the response to DNA replication stress (Leman and Noguchi, 2012; McFarlane et al., 2010). At least partially because of this fundamental function, knockout of murine TIMELESS confers early embryonic lethality, usually during the pre- or peri-implantation period (embryonic day 5.5 [E5.5] to E7.5) (Gotter et al., 2000).
In response to the stalling of DNA replication forks, single-stranded DNA (ssDNA) regions are generated and rapidly coated by the replication protein A (RPA) complex. TIPIN (TIMELESS interacting protein) recognizes and binds RPA-coated DNA in a conserved complex with TIMELESS (Gotter, 2003). Subsequent interaction with CLASPIN promotes ATR-mediated phosphorylation and activation of CHK1, resulting in the inhibition of CDK1 and, with it, mitotic events (Ciccia and Elledge, 2010). In C. elegans and mammals, the TIMELESS-TIPIN complex has been demonstrated to interact with members of the cohesin complex, and these studies have suggested a role in establishing and maintaining sister chromatid cohesion during and after DNA replication (Leman and Noguchi, 2012). Significantly, until now, TIPIN has been the major known binding partner of TIMELESS in both mammals and yeast.
In a genome-wide small interfering RNA (siRNA) screen, TIMELESS was identified as a gene involved in maintaining genome stability, as measured by spontaneous formation of γ-H2AX foci when its expression is silenced (Paulsen et al., 2009). Other effects of TIMELESS depletion include greater genomic instability (more frequent breaks and abnormal chromosomes upon metaphase spread), enhanced formation of double-strand breaks (DSBs) in S phase cells, and increased RAD51 and RAD52 foci (Leman and Noguchi, 2012). Moreover, after TIMELESS knockdown in serum-released fibroblasts, sister chromatid exchange (SCE) substantially increased, suggesting that TIMELESS may have a role in preventing recombination events during unperturbed DNA replication (Urtishak et al., 2009).
In summary, TIMELESS has an established role in the intra-S phase checkpoint, which requires its association with TIPIN. However, TIMELESS appears to play additional less characterized functions, including in circadian clock regulation. Because of our interest in both the response to genotoxic stress (Bassermann et al., 2008; Busino et al., 2003; D’Angiolella et al., 2012; Peschiaroli et al., 2006; Skaar et al., 2009) and circadian clock regulation (Busino et al., 2007; Xing et al., 2013), we decided to investigate further the cellular functions of TIMELESS and found that TIMELESS robustly binds PARP1 (also called ADP-ribosyltransferase 1 or ARTD1).
PARP family proteins polymerize poly(ADP-ribose) (PAR) onto acceptor proteins using the metabolite NAD+ as a substrate; indeed, they are the primary consumers of cellular NAD+ (Barkauskaite et al., 2015; Thomas and Tulin, 2013). PARylation, the process of adding branched PAR chains to proteins, has been implicated in numerous cellular and developmental functions, from chromatin remodeling and transcriptional control to DNA damage recognition and repair to stem cell differentiation, apoptosis, and glycolysis (Bai, 2015). PARylation of proteins occurs mainly on Lys, Glu, or Asp residues and can be formed by branched or elongated chains. The human PARP protein family is composed of 17 PARPs, of which 3 (PARP1, PARP2, and PARP3) are known to possess DNA binding activity. PARP1 is the main sensor of single-strand breaks (SSBs) and DSBs in DNA, and its localization is restricted to the nucleus, unless cleaved just prior to apoptosis when DNA repair becomes futile and the cellular pool of NAD+ and ATP should be preserved.
PARylated chains can grow to over 200 U of ADP-ribose, serving as a large, negatively charged platform for other proteins. In the presence of nicks and breaks, PARP1 polymerizes extensive amounts of PAR chains onto histone and other proteins, including itself, and is, in essence, its own best target. PARylation of histones proximal to DNA damage results in an alteration in the net charge of histones and the unwinding of the nucleosome-DNA complex, providing access to DNA lesions for repair. Auto- and substrate-PARylation by PARP1 establishes and amplifies the DNA damage signal, providing a cellular flare for recruitment of necessary repair factors and activation of effector proteins involved in the DNA damage response (DDR), including the master regulators of the DDR: ATM, ATR, and DNA-PK (Ciccia and Elledge, 2010).
There appears to be some ability for PARP2 to compensate for the absence of PARP1, since PARP1−/− mice are viable whereas PARP1−/−;PARP2−/− double-knockout mice are embryonic lethal (Bai, 2015). PARP1 knockout mice, however, do display a number of phenotypes consistent with the known functions of PARP1. Lack of PARP1 activity leads to slow cell-cycle progression and sensitization to genotoxic stress. PARP1−/− cells exhibit radiosensitivity and genomic instability when challenged with genotoxic agents. Further, PARP1 knockout and knockdown and chemical inhibition of PARP1 increase the formation of γ-H2AX and RAD51 foci, suggesting a dependence on homologous recombination (HR) repair in cells lacking functional PARP1. In fact, a small-molecule PARP1 inhibitor has been recently approved for clinical use in breast cancer patients with gene mutations conferring a loss of HR (e.g., BRCA1, BRCA2) (Feng et al., 2015).
While traditionally associated with two different pathways of maintaining genome integrity, we identified a robust, physical interaction between TIMELESS and PARP1. In the studies presented herein, we investigated the functional importance of this interaction and suggest that TIMELESS contributes to the DDR functions of PARP1.
RESULTS
TIMELESS Forms a Stable Complex with PARP1
We noticed that when we immunoprecipitated tagged TIMELESS after its expression in human cells, by staining with either Ponceau S or silver staining dyes, we consistently detected a second, faster migrating band that co-precipitated at an apparent, near-stoichiometric ratio with TIMELESS (Figures 1A and S1A). Inserting a FLAG-tag on either the N terminus or the C terminus of TIMELESS co-precipitated the identical band, suggesting that the band is not a cleavage or degradation product of the bait protein. To uncover the identity of this putative binding partner, we coupled affinity purification with mass spectrometry. STREP-FLAG-tagged TIMELESS or an empty vector construct were transfected into HEK293T cells. Protein complexes were purified and subjected to SDS-PAGE (Figure S1A). The co-precipitating band was then excised, subjected to mass spectrometry analysis, and identified as PARP1, a protein of the molecular weight corresponding to our observed band.
Figure 1. TIMELESS Physically Interacts with PARP1.

(A) HEK293T cells were transfected with the indicated FLAG-tagged proteins. 24 hr post-transfection, cells were harvested and lysed. Cell extracts were subjected to immunoprecipitation with α-FLAG resin and stained with Ponceau S. MWM, molecular weight markers.
(B) The experiment was performed as in (A), except that whole-cell extracts (WCEs) were subjected to immunoprecipitation (IP) with α-FLAG resin and immunoblotted as indicated. EV, empty vector. Asterisks denote FLAG-tagged proteins.
(C) The experiment was performed as in (B).
(D) Lysates of U2OS cells were immunoprecipitated with either an antibody against TIMELESS or a rabbit IgG and immunoblotted as indicated.
(E) Super-resolution images of TIMELESS and PARP1 in U2OS cells selected from regions of the nucleus shown in Figure S1D. Scale bar, 400 nm.
(F) Super-resolution images of TUNEL, TIMELESS, and PARP1 in neocarzinostatin treated U2OS cells showing that TIMELESS and PARP1 are found at DNA damage sites.
To confirm TIMELESS-PARP1 interaction and evaluate its specificity, we immunoprecipitated a panel of N-terminally FLAG-tagged circadian clock proteins with an anti-FLAG resin. We found that TIMELESS was the only protein able to bind endogenous PARP1, despite the fact that the levels of immunoprecipitated TIMELESS were much lower than that of any other immunoprecipitated protein (Figure 1B). We confirmed the reciprocal interaction by immunoprecipitating FLAG-tagged PARP1 and demonstrating its binding to endogenous TIMELESS (Figure 1C). In contrast, FLAG-tagged NAMPT (the enzyme providing NAD+ to PARPs), FLAG-tagged PARG (the major de-PARylating enzyme), and FLAG-tagged IDUNA (an ubiquitin ligase that binds PARylated substrates) did not co-precipitate TIMELESS (Figures 1C and S1B). Notably, interaction between endogenous PARP1 and endogenous TIMELESS was also observed (Figure 1D).
Size exclusion chromatography showed that much of the endogenous pool of PARP1 resides in rather low-molecular-weight complexes in whole-cell extracts (peaking in fraction 15), estimated to reflect monomeric PARP1 (Figure S1C). However, the subpopulation of endogenous PARP1 bound to FLAG-tagged TIMELESS shifted to much higher molecular weights, peaking in fractions 6–8, suggesting that TIMELESS and PARP1 form a complex containing other proteins.
The interaction between TIMELESS and PARP1 appeared to be direct and not mediated by DNA, since it was stable in the presence of nucleases (TurboNuclease or Benzonase) and after sonication. Accordingly, when we employed super-resolution microscopy, we found that in U2OS cells TIMELESS and PARP1 foci showed a close association and often overlapped (Figures 1E, 1F, and S1D), supporting the hypothesis of a direct physical binding.
Mapping of the PARP1 Binding Motif in TIMELESS
Next, we systematically mapped the binding site in TIMELESS that is responsible for its interaction with PARP1. Using the COILS software, we performed a coiled-coil structure prediction analysis of the protein in order to avoid disruption of highly ordered regions of the protein when generating truncation mutants (Figure S2A). Based on this analysis, we created N-terminally FLAG-tagged truncated TIMELESS mutants, whose N terminus also contains a canonical SV40 nuclear localization signal, in order to avoid mischaracterization of the binding capacity of a mutant that cannot properly localize to the nucleus. The first set of truncation mutants indicated the importance of the far C terminus of TIMELESS, between residues 1,089 and 1,100 of the 1,208-amino-acid protein, for PARP binding (Figures 2A and S1C). This result was confirmed and extended by generating 5-amino-acid deletion mutants within the 1,070–1,109 region (summarized in Figure 2B). Next, we undertook triple Ala scanning mutagenesis of this region as well as mutation (both to Ala and Asp) of the phosphorylable residues present in this region and its surroundings (Figures 2A–2C). Briefly, the results of the Ala scanning mutagenesis showed that residues 1,089–1,092 (TQLR) and 1,097–1,099 (SLS) are crucial for the interaction between TIMELESS and PARP1. Interestingly, of 12 mutants of phosphorylable residues only one, T1078D (a TIMELESS mutant in which Thr1078 was mutated to a phosphomimicking Asp residue), was entirely unable to bind PARP1 (Figure 2A). The results of this extensive mutational analysis are summarized in Figure 2B, and they suggest that seven residues (1,089–1092 and 1,097–1,099) of TIMELESS are implicated in its interaction with PARP1 and that phosphorylation of Thr1078 may inhibit this binding. We compared this region of human TIMELESS to that of other species and found that the region is largely conserved in vertebrates (Figure S2B).
Figure 2. Identification of the PARP1 Binding Site in TIMELESS.

(A) HEK293T cells were transfected with either an empty vector (EV), FLAG-tagged TIMELESS, or FLAG-tagged TIMELESS mutants as indicated. Asterisks indicate the insertion of a STOP codon after the numerically specified codon. 24 hr post-transfection, WCEs were subjected to IP with α-FLAG resin and immunoblotted as indicated.
(B) Schematic representation of TIMELESS mutants tested for binding to PARP1. TIMELESS mutants that interacted with endogenous PARP1 are designated with the symbol (+); those unable to co-precipitate PARP1 are designated with the symbol (−).
(C) The experiment was performed as in (A), except that different TIMELESS mutants were used.
TIMELESS Is Recruited to DNA Damage Sites in a PARP1-Dependent but PARP1-Activity-Independent Manner
Having found that TIMELESS stably binds to PARP1, we hypothesized that TIMELESS may have an unexplored role in DNA repair, distinct from its previously characterized role in signaling DNA replication stress. We thus generated cells stably expressing fluorescently tagged versions of both TIMELESS and PARP1 under the control of a weak retroviral promoter from which exogenous proteins are expressed at near-physiological levels (Figure S3A). Using these tools, we investigated the recruitment of TIMELESS and PARP1 to laser-induced sites of DNA damage (Mortusewicz et al., 2007). We observed that both proteins are recruited to sites of DNA damage “spots” with identical kinetics (Figure 3A), within seconds (the limit of detection) of the introduction of DNA damage.
Figure 3. TIMELESS Requires PARP1, but Not Its Activity, to Be Recruited to DNA Damage Sites.

(A) TIMELESS-Tomato and PARP1-GFP were stably co-transfected in U2OS cells and their recruitment to sites of DNA damage was captured by live cell imaging following laser microirradiation. Times are indicated in seconds.
(B) U2OS cells stabling expressing GFP-TIMELESS were depleted of PARP1 for three days using siRNA oligo #1, and the kinetics of TIMELESS recruitment to DNA damage sites were assessed by live cell imaging of laser-induced lesions. For each condition, n ≥ 28. SE is shown for each time point. Panels below show efficiency of knockdowns using western blot.
(C) U2OS cells stabling expressing GFP-TIMELESS were treated for 1 hr with inhibitors to PARP1, ATM, DNA-PK, and CDK1 prior to laser microirradiation. Next, kinetics of TIMELESS recruitment to DNA damage sites were assessed by live-cell imaging of laser-induced lesions. PARP1 knockdown (using oligo #2) was used for comparison. For each condition, n ≥ 20. SE is shown for each time point.
(D) U2OS cells stabling expressing either wild-type GFP-TIMELESS or two GFP-TIMELESS mutants that do not bind PARP1 were subjected to laser micro-irradiation. Next, kinetics of recruitment of wild-type TIMELESS and TIMELESS mutants to DNA damage sites were assessed by live-cell imaging of laser-induced lesions. For each condition, n ≥ 15. SE is shown for each time point.
In subsequent experiments, we generated kinetic plots using the intensity data derived from the live-cell images. In order to evaluate recruitment per se, and to subtract the contribution of protein diffusion, i.e., a fluorescence recovery after photobleaching (FRAP) effect, we developed an analysis method to compare the damage spot to the nearby bleached area. A region “A” at the center of the lesion was compared to a region “B” in the area immediately adjacent, where bleaching also occurred. We subtracted the background intensity from a distant, dark “C” region of the slide from both A and B, such that we defined a relative fluorescence unit (RFU) as (A − C)/(B − C) (Figure S3B). This value was calculated for each frame of each image, and we set a stringency that the starting image should have a value with no greater than 10% deviation (0.9 < (A − C)/(B − C) > 1.1). We then averaged these values over the course of many live-cell imaging time courses for a given sample (n ranging between 15 to 50 per experimental group) and computed the SE for each time point. Using this method, we characterized the dependence of TIMELESS on PARP1 for recruitment to these sites of damage. GFP-TIMELESS peaked at DNA damage sites at ~8 s after irradiation, after which the signal decreased steadily (Figures 3B–3D). Approximately 50% of GFP-TIMELESS dissociated after 45 s, and complete dissociation occurred within 2 min (not shown). When PARP1 protein levels were reduced via siRNA (using two different oligos, individually), TIMELESS recruitment was greatly reduced and nearly abolished (Figures 3B, 3C, and S3E). However, when cells were challenged with the PARP1 enzymatic inhibitor PJ34, TIMELESS recruitment was not reduced and, interestingly, was significantly enhanced (Figures 3C, S3C, and S3E). This distinction is important, because PARP1 enzymatic inhibition completely abolishes the recruitment of PARP1 substrates and many early DNA damage effectors to sites of DNA damage (Izhar et al., 2015; Mortusewicz et al., 2007). We concluded that it is the physical interaction with PARP1 protein, and not PARP1 enzymatic function, that influences the recruitment of TIMELESS to sites of DNA damage.
We further validated this hypothesis by evaluating the ability of TIMELESS mutants that do not bind PARP1 to be recruited to sites of laser-induced DNA damage. We investigated the kinetics of recruitment of these TIMELESS mutants and found that the T1078D point mutation, consistently and with little variation, completely abolished the recruitment of TIMELESS to DNA damage sites (Figures 3D, S3D and S3E), in agreement with its ability to inhibit PARP1 binding. Similarly, the SLS/AAA mutant was also robustly and significantly impaired in its ability to move to DNA damage sites. However, the TQL/AAA mutant, which by immunoprecipitation does not appear to bind PARP1 (Figure 2C), showed a more modest impairment (although still significant), owing to a loosely bimodal distribution of data (Figure S3D), which could be explained by a residual binding between the two proteins in intact cells. We therefore concluded that TIMELESS recruitment to sites of DNA damage is dependent upon its ability to interact with PARP1.
We further characterized the recruitment of TIMELESS to laser-induced sites of damage using candidate inhibitors for proteins whose enzymatic activity often allows the recruitment of DNA repair factors at damage sites. We inhibited ATM (KU60019), ATR (AZ20), DNA-PK (NU7441), and CDK1 (RO3306) and found that none of these impaired TIMELESS recruitment in response to laser ablation (Figures 3C and S3C).
The TIMELESS-PARP1 Complex Binds Many PARP1 Substrates Involved in the DNA Damage Response
The fraction of PARP1 that immunoprecipitates with TIMELESS resides in high-molecular-weight protein complexes (Figure S1B). In order to understand the context of the interaction between TIMELESS and PARP1 and the other proteins involved in these larger complexes, we again employed a proteomic approach. We expressed both FLAG-tagged TIMELESS and hemagglutinin (HA)-tagged PARP1 in HEK293T cells and then performed a sequential immunopurification of the two proteins. First, we pulled down TIMELESS using an anti-FLAG antibody and eluted by competition with an excess of FLAG peptide. 10% of the eluate of the first immunoprecipitation was set aside, and the remaining 90% was subjected to a second immunoprecipitation using HA resin and elution using 1% SDS. The two eluates were evaluated by silver staining (Figure S4A) and mass spectrometry analysis (Figure S4B). We uncovered that many proteins previously described as definitive or putative PARP1 substrates (i.e., mostly proteins involved in the early phases of the DDR) were present in both the first and second eluate, suggesting that they are present in the same complex with TIMELESS and PARP1. In contrast, TIPIN, whose binding to TIMELESS is relevant in the context of the response to DNA replication stress, was found to elute with TIMELESS in the first immunoprecipitation, but not in the TIMELESS-PARP1 complex analyzed after the second immunoprecipitation. Similarly, DNA-PK, KU70, and KU80 were eluted only in the first immunoprecipitation. Therefore, we cannot exclude that TIMELESS and PARP1 bind DNA-PK, KU70, and KU80 independently of each other.
We next confirmed the binding of these interactors by immunoprecipitation followed by western blotting. We were able to not only confirm the binding of PARP1 to known interactors, but also demonstrate their interaction with TIMELESS (Figure 4A). We also confirmed that TIPIN only bound TIMELESS, but not PARP1. Interestingly, DNA-PK was found to stably co-precipitate only with TIMELESS, but its binding to PARP1 was observed as only slightly above background. The binding data across many experiments are summarized in Figure S4C. Finally, we immunoprecipitated the TIMELESS mutants that cannot bind PARP1 and found that these mutants were significantly impaired in their binding to the PARP1 substrates (Figure 4B).
Figure 4. TIMELESS Stabilizes PARP1 Interaction with Its Substrates upon DNA Damage.

(A) HEK293T cells were transfected with either an EV or the indicated FLAG-tagged proteins. 24 hr post-transfection, WCEs were subjected to IP with α-FLAG resin and immunoblotted as indicated. Ex. Prot., exogenous proteins.
(B) The experiment was performed as in (A). Gels in the bottom panels were loaded as indicated in the upper panels, except that MWMs were omitted. Asterisks denote nonspecific bands.
(C) U2OS cells were transfected overnight with either an siRNA to TIMELESS or a control non-targeting siRNA (siCTRL). 24 hr later cells were then transfected with either an EV or FLAG-tagged PARP1. 24 hr post-transfection, WCEs were subjected to IP with α-FLAG resin and immunoblotted as indicated. White asterisks denote non-specific band. The experiment was repeated three times with identical results.
(D) PARP1 recruitment is impaired after laser microirradiation following knockdown of TIMELESS. U2OS cells stabling expressing PARP1-GFP were depleted of TIMELESS for three days using siRNA, and the kinetics of PARP1recruitment to DNA damage sites were assessed by live-cell imaging of laser-induced lesions. For each condition, n ≥ 15. SE is shown for each time point.
(E) KU80 recruitment is impaired after laser microirradiation following knockdown of TIMELESS. U2OS cells stabling expressing KU80-GFP were depleted of TIMELESS for 3 days using siRNA, and the kinetics of KU80 recruitment to DNA damage sites were assessed by live-cell imaging of laser-induced lesions. For each condition, n ≥ 20. SE is shown for each time point.
(F) Quantification of NHEJ measured as fold change in frequency of repair of EJ5-GFP, resulting in GFP-positive cells after expression of I-SceI in cells treated with the indicated siRNA oligos or drugs. Bar graphs represent the mean of eight independent experiments ± SD. p < 0.0001 (unpaired t test, using GraphPad software). Panel below shows efficiency of knockdown using western blot.
(G) Quantification of HR measured as fold change in frequency of repair of DR-GFP, resulting in GFP-positive cells after expression of I-SceI in cells treated with the indicated siRNA oligos or drugs. Bar graphs represent the mean of eight independent experiments ± SD. p < 0.0001 (unpaired t test, using GraphPad software). Panels below show efficiency of knockdowns using western blot.
To further characterize the relationship between TIMELESS and PARP1, we performed a number of experiments in which TIMELESS levels were reduced using siRNA oligos. We first investigated whether the absence of TIMELESS would affect PARP1 ability to interact with its substrates. We found that TIMELESS depletion led to a reduction of PARP1 binding to substrates exclusively in cells in which DNA damage was induced by treating cells for 15 min with neocarzinostatin (Figure 4C). This effect cannot be secondary to changes in the cell-cycle profile, since TIMELESS depletion does not affect the cell-cycle distribution (Unsal-Kaçmaz et al., 2005; Yang et al., 2010).
We then asked whether TIMELESS has any role in the recruitment of PARP1. When TIMELESS expression was silenced (using two different oligos, individually), PARP1 recruitment to laser-induced sites of DNA damage was reduced, but not abolished (Figure 4D). TIMELESS depletion also robustly and significantly impaired the recruitment of the DSB effector KU80 to DNA lesions (Figure 4E). Representative knockdown of TIMELESS is shown in Figures 4F and 4G.
Finally, to understand the functional consequences of TIMELESS silencing, we used I-SceI-based reporter assays that generate a flow-cytometric readout to assess the contribution of TIMELESS to repair of DSBs. The DR-GFP reporter assays measures the efficiency of repair by HR, and the EJ5-GFP reporter assay measures repair by non-homologous end-joining (NHEJ). Cells depleted of TIMELESS exhibited highly significant diminished repair in both systems (Figures 4F and 4G). Interestingly, TIMELESS silencing did not synergize with DNA-PK inhibition in impairing NHEJ (Figure 4F), suggesting that they cooperate in the same mechanism of repair. Together, these data suggest that TIMELESS plays a role in regulating DNA DSB repair.
DISCUSSION
Both TIMELESS and PARP1 have established roles in the response to genotoxic stress: TIMELESS (together with its known interactor, TIPIN) as a mediator of the response to DNA replication stress, enabling the activation of the CHK1 signaling cascade, which in turn inhibits mitosis; and PARP1 as a key initiator of the response to SSBs and DSBs, leading to activation of the CHK2 signaling cascade, which in turn amplifies the DDR for efficient DNA repair (Ciccia and Elledge, 2010). While these two pathways have been shown to be capable of significant redundancy, especially in downstream steps of their activation, TIMELESS and PARP1, as very early mediators of very different types of stress, have not previously been shown to interact or contribute to the same types of DNA repair.
In this study, we show that TIMELESS forms a stable and, apparently, near-stoichiometric complex with PARP1. In fact, co-immunoprecipitates display similar intensity of bands with both Ponceau S and silver staining dyes (Figures 1A, S1A, and S4A) and a comparable number of PARP1 and TIMELESS peptides by mass spectrometry (Figure S4B). These data, together with the overlapping signal of TIMELESS and PARP1 foci detected by super-resolution microscopy (Figures 1E and 1F), suggest that the binding between TIMELESS and PARP1 may be direct. Accordingly, the crystal structure of two proteins binding to each other has been recently resolved at the atomic level (Xie et al., 2015).
The complex that TIMELESS forms with TIPIN is distinct from the TIMELESS-PARP1 complex, since TIPIN does not co-immunoprecipitate with PARP1 (as detected by either immunoblot or mass spectrometry), and certain TIMELESS mutants, which are unable to interact with PARP1, still bind TIPIN (Figures 4A–4C, S4B, and S4C).
We found that seven single amino acids in TIMELESS are involved in its binding with PARP1 (Figure 2). Interestingly, Thr1089 and Ser1099, which are in the PARP1 binding domain of TIMELESS, are also part of a TQ and SQ motif, respectively. However, neither the Ala nor Asp mutant of these two residues is sufficient to abrogate binding of TIMELESS to PARP1 (Figure 2), suggesting that their phosphorylation may not play a role in regulating their interaction. Accordingly, ATM and ATR inhibitors have no effect on the TIMELESS-PARP1 complex (data not shown). In contrast, mutation of Thr1078 to Asp completely inhibits TIMELESS-PARP1 interaction, leading to the speculation that phosphorylation of Thr1078 by a yet to be identified kinase may be a prerequisite to dissociate the two proteins, likely by inhibiting the contact between Gln1076, Phe1079, and Arg1081 in TIMELESS with Ile879, Phe851, and Asp993 in PARP1, respectively (Xie et al., 2015).
Using laser microirradiation coupled with live-cell imaging, we have also established that TIMELESS is rapidly recruited to sites of DNA damage and this recruitment is dependent on PARP1 binding and independent of PARP1’s enzymatic activity, which in fact increases the speed, intensity, and residence of TIMELESS recruitment (Figures 3B, 3C, and S3B). Interestingly, TIMELESS behaves in a manner opposite to that of known PARP1 substrates, which are unable to accumulate at the laser damage site when PARylation is enzymatically inhibited (Bai, 2015; Thomas and Tulin, 2013).
Significantly, upon DNA damage, TIMELSSS silencing decreases the interaction of PARP1 with its substrates and impairs PARP1 and KU80 recruitment to laser-induced sites of DNA damage (Figures 4C–4E). However, recruitment of LIG3 and XRCC1 is not significantly affected (not shown). These findings suggest that TIMELESS stabilizes complex formation of PARP1 with its substrates at the level of DNA lesions, at least in the earliest moments of the response to DNA damage and for certain substrates. Accordingly, TIMELESS knockdown also significantly affects DNA DSB repair (Figures 4F and 4G). Rescue experiments with wild-type TIMELESS and various mutants were attempted. Unfortunately, they were technically challenging because persistent TIMELESS knockdown induced cell death, which was difficult to control using exogenous proteins. Thus, the demonstration that TIMELESS is involved in DNA repair by virtue of its interaction with PARP1 will require clustered regularly interspaced short palindromic repeat (CRISPR) knockin cell lines, which will enable manipulation of the system with greater precision.
In sum, our work suggests that TIMELESS and PARP1 operate in a complex to mediate DNA repair. Thus, TIMELESS plays a role distinct from its established, TIPIN-dependent function in the intra-S phase checkpoint. Significantly, PARP1 has many functions, including cytoplasmic and nuclear (Daniels et al., 2015), yet how PARP1 works in response to different stimuli and recognizes different substrates remains unknown. Our findings suggest that TIMELESS is a cofactor for the DDR functions of PARP1, and impairment of this critical axis confers significant deficits in the early response to DNA DSBs as well as to resolution of DSBs via canonical repair pathways.
EXPERIMENTAL PROCEDURES
Laser-Induced DNA Damage and Live-Cell Imaging
Cells were plated at a density of 75,000 per well on a four-well Lab-Tek II chambered number 1.5 borosilicate coverglass and incubated overnight before live-cell imaging. RNA knockdown experiments were performed 2 or 3 days prior to microscopy for TIMELESS and PARP1 knockdowns, respectively. Cell culture medium was exchanged to DMEM lacking phenol red and supplemented with sodium pyruvate and HEPES buffer on the day of data collection. Imaging was performed using a DeltaVision Elite inverted microscope system (Applied Precision), using a PlanApo 60× oil 1.42 numerical aperture objective from Olympus. Excitation was achieved with a 7 Color Combined Insight solid state illumination system and was equipped with a polychroic beam splitter and filter sets to support the following wavelengths pertinent to these studies: GFP (525/48) and mCherry (625/45). The system is equipped with a CoolSNAP HQ2 camera and SoftWorx imaging software version 5.0. DNA lesions were introduced using a 405-nm, 50-mW laser at 100% power for 0.5 or 1 s as indicated. Three pre-laser images were recorded in all cases; the number and interval of post-laser images is indicated for each experiment and varied by the protein studied in each experiment. Recruitment intensity was analyzed using a macro written for ImageJ that calculated the ratio of intensity of a circumscribed laser spot A to the adjacent area B such that an RFU for each data collection point was calculated by the equation RFU = (A − C)/(B − C), where C is the background intensity of an unpopulated area of the image. In cases in which recruitment was not detectable, A was determined by use of laser coordinates recorded in the data log file.
Antibodies
The following antibodies were used: anti-TIMELESS (Bethyl Laboratories, A300-961A and A300-960A, the latter used for IP), anti-PARP1 (Invitrogen, 436400), anti-PARP1 (Cell Signaling Technology, 9542 and 46D11, the latter used for IF), anti-FLAG M2 (Sigma, F3165), anti-p-Chk1 (Cell Signaling, 2344), anti-p-Chk2 (Cell Signaling, 2661), anti-p-ATM (Cell Signaling, 4526), anti-LIG3 (Bethyl, A301-637A), anti-HLTF (Bethyl, A300-229A), anti-KU70 (Santa Cruz Biotechnology, sc-9033), anti-KU80 (Neomarkers, MS-285-P1; Cell Signaling, 2180), anti-XRCC1 (Cell Signaling, 2735), anti-DNA-PK (Santa Cruz, sc-5282), anti-DNA-PK (Cell Signaling, 12311), anti-TIPIN (Bethyl, A301-474A-1), anti-RPA1 (Santa Cruz, sc46504), anti-RPA2 (Millipore, 04-1481), anti-SSRP1 (Abcam, ab26212), anti-SPT16 (Cell Signaling, 12191), anti-SKP1 (Santa Cruz, sc-5281), and anti-phospho-H2AX (Millipore, 05-636).
Supplementary Material
Highlights.
TIMELESS forms a complex with PARP1 distinct from the TIMELESS-TIPIN complex
TIMELESS is recruited to laser-induced DNA damage sites
TIMELESS recruitment is dependent on PARP1 binding, but not PARP1 activity
TIMELESS binds PARP1 substrates involved in the DNA damage response
Acknowledgments
The authors thank D.M. Ofri and Y. Yin for their contribution to this study; M. Cammer, Y. Shiloh, and P. Voigt for experimental advice and support; and M. Jasin, H. Leonhardt, D. Reinberg, and J. Stark for reagents. M.P. is grateful to T.M. Thor and T.B. Balduur for continuous support. This work was funded by grants from the NIH (R37-CA076584), New York State Health Department (NYSTEM-N11G-255), and Shifrin-Meyer Breast Cancer Discover Fund (SMBCDF) to M.P.; the NIH (GM057691, CA187612, GM110385, and GM108119), SMBCDF, and the Ralph S. French Charitable Foundation Trust to E.R.; NIH 5T32CA009161 to L.M.Y.; NIH T32GM088118 to D.A.R., and an NIH/NIA F30AG038215 fellowship to L.M.Y. The Proteomics Resource Center directed by B.U. is partially supported by the NCI CCSG grant P30CA016087. M.P. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Supplemental information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.09.017.
AUTHOR CONTRIBUTIONS
Conceptualization, L.M.Y. and M.P.; Methodology, L.M.Y. and M.P.; Investigation, L.M.Y., A.M., P.P.-D., and D.A.R.; Data Curation, L.M.Y., D.N.M., and B.U.; Writing, L.M.Y. and M.P.; Funding Acquisition, L.M.Y., E.R., and M.P.; Visualization, L.M.Y.; Supervision, L.M.Y., D.A.R., A.S., and M.P.
References
- Bai P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol Cell. 2015;58:947–958. doi: 10.1016/j.molcel.2015.01.034. [DOI] [PubMed] [Google Scholar]
- Barkauskaite E, Jankevicius G, Ahel I. Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol Cell. 2015;58:935–946. doi: 10.1016/j.molcel.2015.05.007. [DOI] [PubMed] [Google Scholar]
- Barnes JW, Tischkau SA, Barnes JA, Mitchell JW, Burgoon PW, Hickok JR, Gillette MU. Requirement of mammalian Timeless for circadian rhythmicity. Science. 2003;302:439–442. doi: 10.1126/science.1086593. [DOI] [PubMed] [Google Scholar]
- Bassermann F, Frescas D, Guardavaccaro D, Busino L, Peschiaroli A, Pagano M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell. 2008;134:256–267. doi: 10.1016/j.cell.2008.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benna C, Bonaccorsi S, Wülbeck C, Helfrich-Förster C, Gatti M, Kyriacou CP, Costa R, Sandrelli F. Drosophila timeless2 is required for chromosome stability and circadian photoreception. Curr Biol. 2010;20:346–352. doi: 10.1016/j.cub.2009.12.048. [DOI] [PubMed] [Google Scholar]
- Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426:87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, Draetta GF, Pagano M. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316:900–904. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, Pagano M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149:1023–1034. doi: 10.1016/j.cell.2012.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels CM, Ong SE, Leung AK. The Promise of Proteomics for the Study of ADP-Ribosylation. Mol Cell. 2015;58:911–924. doi: 10.1016/j.molcel.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng FY, de Bono JS, Rubin MA, Knudsen KE. Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Mol Cell. 2015;58:925–934. doi: 10.1016/j.molcel.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter AL. Tipin, a novel timeless-interacting protein, is developmentally co-expressed with timeless and disrupts its self-association. J Mol Biol. 2003;331:167–176. doi: 10.1016/s0022-2836(03)00633-8. [DOI] [PubMed] [Google Scholar]
- Gotter AL. A Timeless debate: resolving TIM’s noncircadian roles with possible clock function. Neuroreport. 2006;17:1229–1233. doi: 10.1097/01.wnr.0000233092.90160.92. [DOI] [PubMed] [Google Scholar]
- Gotter AL, Manganaro T, Weaver DR, Kolakowski LF, Jr, Possidente B, Sriram S, MacLaughlin DT, Reppert SM. A time-less function for mouse timeless. Nat Neurosci. 2000;3:755–756. doi: 10.1038/77653. [DOI] [PubMed] [Google Scholar]
- Izhar L, Adamson B, Ciccia A, Lewis J, Pontano-Vaites L, Leng Y, Liang AC, Westbrook TF, Harper JW, Elledge SJ. A systematic analysis of factors localized to damaged chromatin reveals PARP-dependent recruitment of transcription factors. Cell Rep. 2015;11:1486–1500. doi: 10.1016/j.celrep.2015.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leman AR, Noguchi E. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle. 2012;11:3945–3955. doi: 10.4161/cc.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane RJ, Mian S, Dalgaard JZ. The many facets of the Tim-Tipin protein families’ roles in chromosome biology. Cell Cycle. 2010;9:700–705. doi: 10.4161/cc.9.4.10676. [DOI] [PubMed] [Google Scholar]
- Mortusewicz O, Amé JC, Schreiber V, Leonhardt H. Feedback-regulated poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007;35:7665–7675. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell. 2009;35:228–239. doi: 10.1016/j.molcel.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschiaroli A, Dorrello NV, Guardavaccaro D, Venere M, Halazonetis T, Sherman NE, Pagano M. SCFbetaTrCP-mediated degradation of Claspin regulates recovery from the DNA replication checkpoint response. Mol Cell. 2006;23:319–329. doi: 10.1016/j.molcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Skaar JR, Richard DJ, Saraf A, Toschi A, Bolderson E, Florens L, Washburn MP, Khanna KK, Pagano M. INTS3 controls the hSSB1-mediated DNA damage response. J Cell Biol. 2009;187:25–32. doi: 10.1083/jcb.200907026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Tulin AV. Poly-ADP-ribose polymerase: machinery for nuclear processes. Mol Aspects Med. 2013;34:1124–1137. doi: 10.1016/j.mam.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unsal-Kaçmaz K, Mullen TE, Kaufmann WK, Sancar A. Coupling of human circadian and cell cycles by the timeless protein. Mol Cell Biol. 2005;25:3109–3116. doi: 10.1128/MCB.25.8.3109-3116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urtishak KA, Smith KD, Chanoux RA, Greenberg RA, Johnson FB, Brown EJ. Timeless Maintains Genomic Stability and Suppresses Sister Chromatid Exchange during Unperturbed DNA Replication. J Biol Chem. 2009;284:8777–8785. doi: 10.1074/jbc.M806103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie S, Mortusewicz O, Ma HT, Herr P, Helleday T, Qian C. TIMELESS interacts with PARP-1 to promote homologous recombination repair. Mol Cell. 2015 doi: 10.1016/j.molcel.2015.07.031. Published online September 2, 2015 http://dx.doi.org/10.1016/j.molcel.2015.07.031. [DOI] [PubMed]
- Xing W, Busino L, Hinds TR, Marionni ST, Saifee NH, Bush MF, Pagano M, Zheng N. SCF(FBXL3) ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature. 2013;496:64–68. doi: 10.1038/nature11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wood PA, Hrushesky WJ. Mammalian TIMELESS is required for ATM-dependent CHK2 activation and G2/M checkpoint control. J Biol Chem. 2010;285:3030–3034. doi: 10.1074/jbc.M109.050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.