

Abstract
Alcohol engages signaling pathways in the brain. Midkine (MDK) is a neurotrophic factor that is overexpressed in the prefrontal cortex of alcoholics. MDK and one of its receptors, anaplastic lymphoma kinase (ALK), also regulate behavioral responses to ethanol in mice. The goal of this study was to determine whether MDK and ALK expression and signaling are activated by ethanol. We found that ethanol treatment of neuroblastoma cells increased MDK and ALK expression. We also assessed activation of ALK by ethanol in cells and found that ALK and ALK-dependent extracellular signal-regulated kinase (ERK) and signal transducer and activator of transcription 3 (STAT3) phosphorylation increased rapidly with ethanol exposure. Similarly, treatment of cells with recombinant MDK protein increased ALK, ERK and STAT3 phosphorylation, suggesting that ethanol may utilize MDK to activate ALK signaling. In support of this, transfection of cells with MDK siRNAs attenuated ALK signaling in response to ethanol. Ethanol also activates ERK signaling in the brain. We found that inhibition of ALK or knockout of MDK attenuated ethanol-induced ERK phosphorylation in mouse amygdala. These results demonstrate that ethanol engages MDK and ALK signaling, which has important consequences for alcohol-induced neurotoxicity and the regulation of behaviors related to alcohol abuse.
Keywords: Addiction, Alcohol, Midkine, ALK, ERK, STAT3
Introduction
Midkine (MDK) is a growth factor that regulates many biological processes including development, inflammation, cancer and tissue repair after injury (Kadomatsu et al. 2013). In the nervous system, MDK is expressed in the brain and spinal cord during development, promotes neurite outgrowth and acts as a neurotrophic factor (Maruta et al. 1993, Muramatsu et al. 1993, Kikuchi et al. 1993, Michikawa et al. 1993). MDK protects neurons from injury and is upregulated after cerebral and retinal ischemia (Yoshida et al. 1995, Miyashiro et al. 1998, Mochizuki et al. 1998, Wada et al. 2002) and spinal cord injury (Muramoto et al. 2013). MDK expression in the brain is also responsive to toxic insult, for example after chronic exposure to drugs of abuse. Mdk expression increases in the hippocampus and ventral tegmental area of rats after chronic morphine exposure (Ezquerra et al. 2007, Garcia-Perez et al. 2014) and Mdk knockout (MdkKO) mice display more activated astrocytes in the striatum compared to wild-type mice after amphetamine exposure (Gramage et al. 2011). It has been hypothesized that MDK may protect against neuron damage and neurodegeneration caused by exposure to neurotoxic agents (Herradon & Perez-Garcia 2014). In support of this, MDK expression is higher in the prefrontal cortex of human alcoholics compared to non-alcoholic control subjects (Flatscher-Bader et al. 2005, Flatscher-Bader & Wilce 2008, Flatscher-Bader & Wilce 2006), suggesting that MDK might be an ethanol-responsive gene. However, MDK expression is also elevated in the brains of ethanol-naïve mice genetically predisposed to consume high amounts of alcohol (Mulligan et al. 2006) and MdkKO mice exhibit altered sensitivity to ethanol-induced ataxia and the rewarding properties of ethanol (Vicente-Rodriguez et al. 2014). It is currently not known whether MDK modulates behavioral responses to ethanol due solely to a genetic predisposition or if MDK gene expression and MDK-dependent signaling pathways are regulated by ethanol exposure.
ALK is a tyrosine kinase receptor for MDK that also mediates behavioral responses to drugs of abuse (Lasek et al. 2011a, Lasek et al. 2011b). Alk knockout mice are slower to recover from sedating doses of ethanol and drink more ethanol in a binge drinking test compared to wild-type mice (Lasek et al. 2011b). Regulation of Alk gene expression and signaling in response to ethanol has not been examined. Given that Mdk and Alk knockout mice show altered behavioral responses to ethanol, it is feasible that ALK and MDK coordinately regulate ethanol sensitivity. In neuronal cells, MDK activates the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) and phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) pathways to promote cell survival and proliferation (Lorente et al. 2011, Owada et al. 1999a, Owada et al. 1999b, Stoica et al. 2002). ALK also activates the MAPK/ERK (Gouzi et al. 2005, Kuo et al. 2007, Moog-Lutz et al. 2005, Motegi et al. 2004, Souttou et al. 2001), PI3K/AKT (Galkin et al. 2007, Gouzi et al. 2005, Stoica et al. 2002) and signal transducer and activator of transcription 3 (STAT3) signaling pathways (Sattu et al. 2013, Moog-Lutz et al. 2005). Activation of ALK by MDK is important for the proliferation of sympathetic neurons (Reiff et al. 2011) and resistance of glioma cells to cell death by cannabinoids (Lorente et al. 2011).
Ethanol also activates MAPK/ERK (Ibba et al. 2009, Sanna et al. 2002, Spanos et al. 2012, Thorsell et al. 2013, Zhu et al. 2013), PI3K/AKT (Neasta et al. 2011) and STAT3 (Bachtell et al. 2002) signaling in the mammalian brain. Activation of these signaling pathways alters behavioral responses to ethanol. For instance, several members of the MAPK/ERK pathway are overexpressed in high ethanol-preferring mice and inhibition of MAPK signaling alters ethanol self-administration and binge-like drinking in mice (Faccidomo et al. 2009, Agoglia et al. 2015). Knockout of Rasgrf2, which regulates Ca2+-dependent activation of the MAPK/ERK pathway, decreases ethanol consumption in mice (Mulligan et al. 2011, Stacey et al. 2012). We hypothesize that ethanol-induced activation of the MAPK/ERK, PI3K/AKT or STAT3 pathways involves MDK and ALK. Activation of MDK and ALK signaling might be an adaptive neuroprotective response and/or a mechanism through which ethanol elicits its behavioral effects.
Here, we present evidence that MDK and ALK are ethanol-responsive. We employed the neuroblastoma cell lines SH-SY5Y and IMR-32. SH-SY5Y cells express ALK and MDK and differentiate into dopamine-like neurons when treated with retinoic acid (Korecka et al. 2013, Nakagawara et al. 1995, Stoica et al. 2002). IMR-32 cells express full-length, wild-type ALK and thus provide a useful model for examining ALK signaling (Janoueix-Lerosey et al. 2008). To extend these studies to an in vivo system, we examined phosphorylation of ERK in response to ethanol in the mouse amygdala in the presence and absence of the ALK inhibitor TAE684 (Galkin et al. 2007) and in MdkKO mice. Our results provide the first demonstration that ethanol alters ALK and MDK signaling pathways.
Materials and methods
Materials
Antibodies to phosphorylated ALK (pALK, Tyr 1278, #6941), ERK1/2 (#9102), phosphorylated ERK1/2 (pERK, Thr 202/Tyr 204, #4370), STAT3 (#12640), and phosphorylated STAT3 (pSTAT3, Tyr 705, #9145) were purchased from Cell Signaling Technology (Danvers, MA, USA). The polyclonal rabbit antibody to ALK was purchased from Life Technologies (Carlsbad, CA, USA, #51-3900). β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO, USA, clone AC-15, #A5441). Antibodies to AKT (sc-8312) and phosphorylated AKT (pAKT, sc-7985-R) were from Santa Cruz Biotechnology (Dallas, TX, USA). MDK antibody was purchased from Abcam (ab36038, Cambridge, UK). Horseradish peroxidase (HRP)-conjugated anti-rabbit and mouse secondary antibodies were purchased from Bio-Rad (Des Plaines, IL, USA). TAE684 was purchased from Selleck Chemicals (Houston, TX, USA) and recombinant MDK protein was purchased from R&D systems (Minneapolis, MN, USA). siGENOME MDK siRNAs (D003677-02 and D003677-03) and a control siRNA (D-001206-13-05) were purchased from GE Healthcare (Dharmacon RNAi, Lafayette, CO, USA).
Cell culture
The human neuroblastoma cell lines SH-SY5Y and IMR-32 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and were incubated at 37°C in 5% CO2. SH-SY5Y cells were cultured in a 1:1 mixture of Eagle’s Minimum Essential Medium (EMEM) and F12 medium containing 10% FBS. SH-SY5Y cells were differentiated into dopamine-like neurons using retinoic acid by culturing in Neurobasal medium containing B27 supplement and GlutaMAX (Life Technologies) and treating for 3 days with 10 μM all-trans retinoic acid (Sigma-Aldrich). Just before ethanol treatment, medium was changed to 1:1 mixture of EMEM and F12 medium. IMR-32 cells were cultured in EMEM containing 10% FBS. For treatment of IMR-32 cells with ethanol and TAE684, cells were cultured to 90% confluence and starved in serum-free medium for 3 h. Cells were treated with 100 nM TAE684 for 30 min prior to 100 mM ethanol treatment. The IC50 of TAE684 on cell lines expressing oncogenic ALK is 4 nM and concentrations of TAE684 > 50 nM have been shown to completely inhibit ALK-dependent ERK and STAT3 phosphorylation in tumor cell lines (Galkin et al. 2007). For treatment of IMR-32 cells with recombinant MDK protein and TAE684, cells were cultured to 90% confluence and starved in serum free EMEM medium for 3 h. TAE684 (100 nM) was added 30 min prior to addition of 100 ng/mL recombinant MDK.
RNA isolation and quantitative real-time PCR (qPCR)
Total RNA was isolated from IMR-32 and SH-SY5Y cells using the GeneJet RNA Purification Kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. RNA was reverse transcribed using the Maxima First Strand cDNA Synthesis Kit for RT-PCR (Thermo Scientific) and qPCR performed using Maxima qPCR Master Mix. Sequences of primers were as follows: ALK forward primer: 5′-GTGCCATGCTGCCAGTTAAG-3′, ALK reverse primer: 5′-TGGTTGCTTTTGCTGGGGTA-3′; MDK forward primer: 5′-AAGGAGTTTGGAGCCGACTG-3, MDK reverse primer: 5-CATTGTAGCGCGCCTTCTTC-3′. Amplification of GAPDH was used as a normalization control for total RNA input. Relative expression of ALK and MDK were determined using the dCq method.
Western blots
IMR-32 cells were lysed in 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 1 μg/ml aprotinin, and EDTA-free Complete Protease Inhibitor Cocktail tablets (Roche Diagnostics, Indianapolis, IN, USA). Lysate protein concentration was determined using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA) and equal amounts of protein (20 μg) were subjected to SDS-PAGE and transferred to PVDF membranes. Primary antibodies were diluted in 5% BSA in TBST (25 mM Tris-HCl, pH 7.4, 137 mM NaCl, and 0.1% Tween 20). Secondary antibodies were HRP-conjugated goat anti-mouse or anti-rabbit IgG. The membranes were developed with ECL detection reagents (Pierce). Films were scanned and densitometry performed using ImageJ software.
MDK siRNA transfections
IMR-32 cells grown to 60% confluence in 6-well plates were transfected using Lipofectamine 2000 (Life Technologies) with 50 nM of control siRNA or MDK siRNA in Opti-MEM medium. Five h after transfection, 2 ml of fresh complete MEM medium containing 10% FBS was added. The cells were cultured for additional 72 h and analyzed for protein expression by Western blotting. Ethanol (100 mM) was added to media 5 min before processing lysates.
Mice
Male C57BL/6J mice (8 weeks old, 20-30 grams) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). MdkKO mice have been described previously (Nakamura et al. 1998). MdkKO mice were re-derived from frozen embryos at the University of Illinois at Chicago (UIC) Transgenic Production Service and heterozygotes were bred in-house for experiments. Male and female MdkKO mice were group-housed with same sex littermates. All mice were kept in a temperature- and humidity-controlled room on a 14 hour light/dark cycle with lights on at 6 a.m. and off at 8 p.m. Food and water were available ad libitum. All procedures with mice were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the UIC Animal Care and Use Committee. All efforts were taken to minimize pain and discomfort.
Ethanol administration and immunohistochemistry
Naïve mice were randomized and treated with 3 g/kg ethanol or saline (10 ml/kg) intraperitoneally in their home cages. For TAE684 experiments, mice were treated 5 hours prior to ethanol or saline administration by oral gavage with vehicle (90% PEG 300, 10% 1-methyl-2-pyrrolidinone) or 10 mg/kg TAE684 in vehicle in a volume of approximately 0.1 mL. This dose of TAE684 is effective in reducing ALK-dependent lymphomas in mice without causing toxicity (Galkin et al. 2007). Mice were euthanized 30 min after ethanol or saline administration using pentobarbital followed by transcardial perfusion with PBS and 4% paraformaldehyde. Brains fixed overnight in 4% paraformaldehyde. After incubation overnight in 30% sucrose in PBS, 40 μm coronal sections were cut and treated with 0.25% Triton X-100 for 30 min, blocked in 3% BSA and then incubated with pERK antibody. Sections were incubated with biotin-conjugated goat anti-rabbit secondary antibody and developed using the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA). Images were acquired using a Zeiss AxioScope.A1 microscope fitted with a 5 megapixel AxioCam ERc 5s color camera using a 10x objective lens and ZenLite image acquisition and archiving software (Carl Zeiss, Thornwood, NY, USA). Images from 2-3 sections from the amygdala of each mouse were acquired for quantification and 3-6 mice were treated in each group (4 groups per experiment). ImageJ software was used for analysis of staining density in the amygdala.
Statistical analysis
For all experiments, analysis was done using GraphPad Prism software (San Diego, CA, USA) and based on the combined data from 3 independent experiments done in triplicate. qPCR data was analyzed using a 1 way ANOVA followed by Bonferroni’s multiple comparisons test. Western blotting data and pERK intensity in the central nucleus of the amygdala (CeA) were analyzed using 2 way repeated measures (RM) or regular 2 way ANOVA followed by Holm-Sidak’s multiple comparisons test.
Results
Ethanol increases MDK expression in neuroblastoma cells
To determine if MDK gene expression is altered by ethanol, SH-SY5Y and IMR-32 cells were treated with 20, 50 or 100 mM ethanol for 4 h and analyzed for MDK expression by qPCR. Fig. 1a shows that MDK expression increased by 14% after 20 and 50 mM ethanol and by 28% after 100 mM ethanol treatment in SH-SY5Y cells (1 way ANOVA: F(3,32) = 9.92, p < 0.0001). Bonferroni’s multiple comparisons test indicated that the increase in MDK expression was significant at all three concentrations of ethanol when compared to the untreated control (20 mM, p = 0.041; 50 mM, p = 0.035; 100 mM, p < 0.0001). In IMR-32 cells, MDK expression also increased with ethanol treatment (Fig. 1e, 1 way ANOVA: F(3,20) = 6.07, p = 0.004), but the effect was only significant at 100 mM ethanol (a 21% increase, p = 0.002). MDK expression was also examined at various time points (1-6 h) after 100 mM ethanol treatment. MDK expression significantly increased after ethanol treatment in SH-SY5Y (Fig. 1b, 1 way ANOVA: F(4,10) = 6.06, p = 0.0096) and IMR-32 cells (Fig. 1f, 1 way ANOVA: F(3,8) = 11.7, p = 0.0027). In SH-SY5Y cells, MDK expression increased by 33% at 4 and 6 h compared to the 0 h time point (4 h, p = 0.009; 6 h, p = 0.011) and in IMR-32 cells, MDK expression increased by 12%, 16%, and 22% at 2, 4 and 6 h, respectively, compared to the 0 h time point (2 h, p = 0.036; 4 h, p = 0.008; 6 h, p = 0.0014). We also found that the change in MDK gene expression corresponds with an increase in MDK protein levels. SH-SY5Y and IMR-32 cells were treated with 100 mM ethanol for 1-6 h and MDK protein was analyzed by Western blotting (Fig. 1c, g). Quantification of the Western blots indicated that MDK protein levels were significantly elevated after ethanol treatment in SH-SY5Y (Fig. 1d, 1 way ANOVA: F(4,10) = 6.3, p = 0.0085) and IMR-32 cells (Fig. 1h, 1 way ANOVA: F(3,8) = 11.54, p = 0.0028). In SH-SY5Y cells, ethanol treatment increased MDK protein levels by 46% and 58% at 2 and 4 h, respectively (2 h, p = 0.025; 4 h, p = 0.0058). By 6 h, MDK protein levels were still elevated by 18% above the 0 h time point, but this increase was not significant. In IMR-32 cells, ethanol treatment led to a significant increase in MDK protein of 142% at 4 h and 92% at 6h (4 h, p = 0.0012; 6 h, p = 0.017). Together, these data indicate that MDK expression increases with ethanol treatment in 2 neuroblastoma cell lines.
Fig. 1.

Ethanol increases MDK expression in SH-SY5Y and IMR-32 cells. SH-SY5Y (a–d) or IMR-32 (eh) cells were treated with ethanol and MDK mRNA or protein expression was analyzed by qPCR or Western blotting, respectively. (a, e) MDK mRNA expression 4 h after treatment with 20, 50 or 100 mM ethanol. (b, f) Time course showing MDK mRNA expression after 100 mM ethanol treatment. (c, g) Representative Western blot showing an increase in MDK protein expression at various time points after 100 mM ethanol treatment. (d, h) Quantification of Western blotting results using ImageJ. Data are presented as the mean ± S.D. *p < 0.05, **p < 0.01, ****p < 0.0001.
Ethanol increases ALK expression in neuroblastoma cells
We next tested if ALK gene and protein expression are altered by ethanol treatment. In SH-SY5Y and IMR-32 cells, 4 h of ethanol treatment at 20, 50 and 100 mM increased ALK mRNA expression as measured by qPCR (SH-SY5Y, Fig. 2a, 1 way ANOVA: F(3,32) = 10.24, p < 0.0001; IMR-32, Fig. 2e, 1 way ANOVA: F(3,20) = 8.08, p = 0.001). Ethanol significantly increased ALK expression in SH-SY5Y cells by 19% at 20 mM, 20% at 50 mM and 32% at 100 mM compared to the untreated control (20 mM, p = 0.011; 50 mM, p = 0.0054; 100 mM, p < 0.0001). In IMR-32 cells, 100 mM ethanol significantly increased ALK transcription by 23% (p = 0.0015). Figs. 2b, f show that 100 mM ethanol treatment over time resulted in significantly elevated ALK mRNA levels in both SH-SY5Y and IMR-32 cells (SH-SY5Y, 1 way ANOVA: F(4,10) = 6.92, p = 0.0062; IMR-32, 1 way ANOVA: F(3,8) = 8.79, p = 0.0065). Ethanol increased ALK mRNA by approximately 35% after 4 h and 30% after 6 h in both SH-SY5Y and IMR-32 cells (SH-SY5Y, 4 h, p = 0.0048, 6 h, p = 0.014; IMR-32, 4 h, p = 0.0043, 6 h, p = 0.025). Similar to the increase in ALK gene expression, we also found that ALK protein levels were elevated after ethanol treatment in SH-SY5Y and IMR-32 cells (Figs. 2c and g). The increase in ALK protein was observed for the full-length ALK protein (molecular weight, 220 kDa) and for truncated 140 kDa ALK (Moog-Lutz et al. 2005). Quantification of ALK Western blots from ethanol-treated SH-SY5Y and IMR-32 cells are shown in Figs. 2d, h. A 2 way RM ANOVA comparing expression of each ALK variant over time showed a significant effect of time, but no difference between the two ALK variants or a variant by time interaction in SH-SY5Y cells (effect of time, F(3,6) = 12.01, p = 0.0060). There was a significant increase in ALK 220 at 4 h (33% increase, p = 0.041). Similar results were obtained in IMR-32 cells treated with ethanol (Fig. 2h, effect of time, F(3,12) = 16.04, p = 0.0002). There were significant increases in ALK 220 and 140 at 2 h (ALK 220, 33% increase, p = 0.012; ALK 140, 36% increase, p = 0.01) and 4 h (ALK 220, 50% increase, p = 0.0006; ALK 140, 28% increase, p = 0.042). These results indicate that transcription and subsequent ALK protein expression are increased by ethanol treatment in SH-SY5Y and IMR-32 neuroblastoma cells.
Fig. 2.

Ethanol increases ALK expression in SH-SY5Y and IMR-32 cells. SH-SY5Y (a–d) or IMR-32 (e–h) cells were treated with ethanol and ALK mRNA or protein expression was analyzed by qPCR or Western blotting, respectively. (a, e) ALK expression increases 4 h after treatment with 20, 50 or 100 mM ethanol. (b, f) Time course showing ALK expression after 100 mM ethanol treatment. (c, g) Representative Western blots showing an increase in the 220 and 140 kDa isoforms of ALK at various time points after 100 mM ethanol treatment. (d, h) Quantification of Western blotting results using ImageJ. Data are presented as the mean ± S.D. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Ethanol activates ALK signaling in IMR-32 cells
To determine if ethanol rapidly activates ALK signaling, we treated IMR-32 cells with 100 mM ethanol for 5-30 min and examined pALK, pERK, pSTAT3 and pAKT in the presence and absence of the ALK inhibitor, TAE684. The Western blots shown in Fig. 3 demonstrate that treating IMR-32 cells with ethanol caused a rapid increase in the phosphorylation of two isoforms of ALK (the 220 and 140 kDa proteins (Moog-Lutz et al. 2005)), ERK and STAT3. Increased phosphorylation was evident after 5 and 15 min of ethanol exposure and returned to baseline levels after 30 min. Enhanced phosphorylation of all 3 proteins was blocked by pre-treatment with 100 nM TAE684. pAKT did not change with ethanol treatment (Figs. 3a and e). Quantification of the Western blots indicates that pALK 220 and 140 were significantly elevated after ethanol treatment (Fig. 3b, 2 way RM ANOVA: effect of time, F(7,28) = 30.3, p < 0.0001; effect of ALK variant, F(1,4) = 79.3, p = 0.0009; time by variant interaction, F(7,28) = 7.37, p < 0.0001). pALK 220 increased by 41% after 5 min (p = 0.05) of ethanol treatment, and pALK 140 increased by 51% after 5 min (p = 0.003) and 44% after 15 min (p = 0.0098) of ethanol treatment. In addition, TAE684 pre-treatment completely blocked the increase in pALK 220 and 140 when compared to the DMSO control within each time point (5 min: pALK 220, p = 0.0032; pALK 140, p < 0.0001; 15 min: pALK 140, p < 0.0001). We also found that baseline pALK 140, but not 220, was significantly reduced by 56% after pre-treatment with TAE684 in the absence of ethanol (p = 0.001).
Fig. 3.
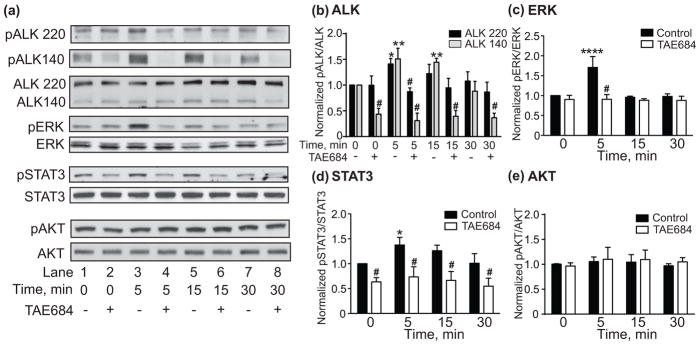
Ethanol activates ALK signaling in IMR-32 cells. (a) Representative Western blots showing increased pALK 220 and 140, pERK, and pSTAT3, but not pAKT within 5 min of 100 mM ethanol treatment. Total ALK, ERK, STAT3, and AKT Western blots are shown below each phosphorylated protein blot for comparison. The effect of ethanol on phosphorylation was blocked by pre-treatment with the ALK inhibitor TAE684 (+, lanes 2, 4, 6 and 8) compared to the DMSO control (-, lanes 1, 3, 5 and 7). (b–e) Quantification of Western blots using ImageJ. Significant increases in pALK (b), pERK (c), and pSTAT3 (d), but not pAKT (e) were observed within 5 min of ethanol treatment, effects that were blocked by TAE684. Data are presented as mean ± S.D. *p < 0.05, **p < 0.01 compared to 0 minute time point within DMSO controls. #Significant difference when comparing DMSO control vs. TAE684 treatment within each time point.
pERK was significantly increased by ethanol treatment. This effect was blocked by pre-treatment with TAE684 (Fig. 3c, 2 way RM ANOVA: effect of time, F(3,12) = 14.41, p = 0.0003; effect of TAE684, F(1,4) = 28.34, p = 0.006; time by TAE684 interaction, F(3,1) = 13.02, p = 0.0004). Post hoc tests indicated that ethanol significantly increased ALK-dependent pERK by 71% after 5 min (p < 0.0001). Ethanol also significantly enhanced ALK-dependent pSTAT3 (Fig. 3d, 2 way RM ANOVA: effect of time, F(3,12) = 7.096, p = 0.0053; effect of TAE684, F(1,4) = 32.9, p = 0.0046; time by TAE684 interaction, F(3,12) = 1.71, p = 0.218). Post hoc Holm-Sidak’s multiple comparisons test indicated that ethanol significantly increased pSTAT3 by 38% at 5 min (p = 0.0125). Interestingly, TAE684 pre-treatment reduced baseline pSTAT3 by 37% in the absence of ethanol (p = 0.0088). Together, these results indicate that ethanol rapidly activates ALK signaling through the ERK and STAT3 pathways in a neuronal cell line.
MDK activates ALK signaling in IMR-32 cells
To determine if MDK activates ALK signaling pathways in IMR-32 cells, recombinant MDK protein was added to IMR-32 culture medium in the presence and absence of TAE684. pALK, pERK, pSTAT3 and pAKT were analyzed by Western blotting. MDK treatment of IMR-32 cells increased pALK, pERK and pSTAT3, but not pAKT, within 5 min (Fig. 4a). The effect of MDK on IMR-32 signaling was blocked by TAE684, suggesting that MDK acts through ALK to activate the ERK and STAT3 pathways. The graphs in Figs. 4b–e are results from the quantification of the Western blots. There was a significant effect of MDK on pALK 220 and 140 (Fig. 4b, 2 way RM ANOVA: effect of time, F(5,20) = 54.3, p < 0.0001; effect of ALK variant, F(1,4) = 77.92, p = 0.0009; time by variant interaction, F(5,20) = 13.58, p < 0.0001). pALK 220 significantly increased by 44% after 5 min (p = 0.0079) and by 60% after 15 min (p = 0.0004) of MDK treatment. pALK 140 significantly increased by 39% after 15 min of MDK treatment (p = 0.017). As in Fig. 3b, we observed a significant reduction in baseline pALK 140 after pre-treatment with TAE684 (Fig. 4b, 90% reduction, p < 0.0001).
Fig. 4.
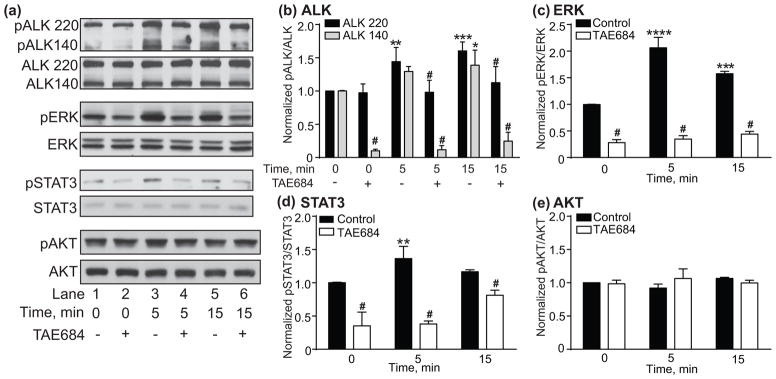
MDK activates ALK signaling in IMR-32 cells. (a) Representative Western blots showing increased pALK 220 and 140, pERK and pSTAT3, but not pAKT within min of treatment with recombinant MDK protein. Total ALK, ERK, STAT3, and AKT Western blots are shown below each phosphorylated protein blot for comparison. The effect of MDK was blocked by pre-treatment with the ALK inhibitor TAE684 (lanes 2, 4 and 6) compared to the DMSO control (lanes 1, 3 and 5). (b–e) Quantification of Western blots using ImageJ showing significant increases in pALK (b), pERK (c), and pSTAT3 (d) but not pAKT (e) within 5 min of ethanol treatment, effects that were blocked by TAE684. Data are presented as mean ± S.D. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 compared to 0 minute time point within DMSO controls. #Significant difference when comparing DMSO control vs. TAE684 treatment within each time point.
Quantification of the ERK Western blots indicated that pERK was significantly increased by MDK treatment and blocked by pre-treatment with TAE684 (Fig. 4c, 2 way RM ANOVA: effect of time: F(2,8) = 58.7, p < 0.0001; effect of TAE684, F(1,4) = 782.4, p < 0.0001; time by TAE684 interaction, F(2,8) = 44.7, p < 0.0001). pERK significantly increased by 106% after 5 min (p < 0.0001) and by 58% after 15 min (p = 0.0001) of exposure to MDK. In addition, TAE684 reduced baseline pERK by approximately 70% (p < 0.0001). pSTAT3 was also significantly increased by MDK treatment in an ALK-dependent manner (Fig. 4d, 2 way RM ANOVA: effect of time: F(2,8) = 13.5, p = 0.0027; effect of TAE684, F(1,4) = 97.32, p = 0.0006; time by TAE684 interaction: F(2,8) = 13.54, p = 0.0027). MDK treatment significantly increased pSTAT3 by 36% after 5 min (p = 0.0089). Similar to what we observed in Fig. 3d, baseline pSTAT3 was significantly reduced by TAE684 pre-treatment (Fig. 4d, 65% reduction, p < 0.0001). These results indicate that MDK activates ERK and STAT3 signaling through ALK in IMR-32 cells.
Activation of ALK signaling by ethanol is dependent on MDK expression
To determine if activation of ALK signaling in response to ethanol is dependent on MDK expression, we transfected MDK siRNAs into IMR-32 cells and examined the effect of ethanol on pALK, pERK, pSTAT3 and pAKT by Western blotting. As shown in Fig. 5a, two independent MDK siRNAs were effective in reducing MDK protein expression. Ethanol increased pALK 220 and 140, pERK, and pSTAT3, and this increase was reduced in cells that were transfected with MDK siRNAs. As we observed previously, there was no effect of ethanol on pAKT (Fig. 5a and f). Quantification of the Western blots is shown in Fig. 5b-f. Both of the MDK siRNAs (siMDK2 and siMDK3) significantly reduced MDK expression by approximately 50% (Fig. 5b, 2 way ANOVA: effect of siRNA, F(2,6) = 126.0, p < 0.0001; effect of ethanol, F(1,6) = 2.49, p = 0.166; siRNA by ethanol interaction, F(2,6) = 11.87, p = 0.0082). For siMDK2, there was no effect of ethanol on MDK expression. However, ethanol significantly reduced MDK expression in siMDK3-transfected cells by 23% (p = 0.010).
Fig. 5.

Ethanol activates ALK signaling through MDK. (a) Representative Western blots showing that increased pALK 220 and 140, pERK and pSTAT3 in response to 100 mM ethanol is attenuated by MDK siRNAs (siMDK2 and siMDK3) compared to a control siRNA (siCTL). Total ALK, ERK, STAT3, and AKT Western blots are shown below each phosphorylated protein blot for comparison. MDK blot shows the extent of knockdown by siMDK2 and siMDK3 and Actin blot is shown to indicate that equal protein amounts were loaded onto the gel. Ethanol treatment (Lanes 2, 5 and 6) is indicated by a + sign below the blots. (b–f) Quantification of Western blots using ImageJ showing significant decreases in MDK protein with siMDK2 and siMDK3transfection (b) and increases in pALK (c), pERK (d), and pSTAT3 (e) but not pAKT (f) within 5 min of ethanol treatment, effects that were attenuated by siMDK2 and siMDK3. Data are presented as mean ± S.D. ***p < 0.001, ****p < 0.0001 when comparing ethanol-treated sample to control. ~ Significant siRNA by ethanol interaction. #Significant difference between siCTL and siMDK in ethanol-treated cells.
Ethanol significantly increased pALK220 and 140 (Fig. 5c, 2 way ANOVA: effect of ethanol, F(5,24) = 27.35, p < 0.0001; effect of ALK variant, F(1,24) = 2.28, p = 0.144; ethanol by ALK variant interaction, F(5,24) = 0.587, p = 0.710). In cells transfected with control siRNA (siCTL), pALK 220 increased by 59% (p < 0.0001) and pALK 140 increased by 76% (p < 0.0001) after ethanol treatment. Increased pALK in response to ethanol was abolished in cells transfected with either siMDK2 or siMDK3. Ethanol significantly increased pERK in IMR-32 cells transfected with a control siRNA and this effect was reduced in cells transfected with siMDK2 and siMDK3 (Fig. 5d, 2 way ANOVA: effect of ethanol, F(1,12) = 51.87, p < 0.0001; effect of siRNA, F(2,12) = 4.30, p = 0.039; ethanol by siRNA interaction, F(2,12) = 6.50, p = 0.012). IMR-32 cells transfected with the control siRNA showed a 59% increase in pERK in response to ethanol (p < 0.0001), and this increase was reduced by half in cells transfected with siMDK2 (p = 0.017) and siMDK3 (p = 0.035). Finally, ethanol increased pSTAT3 in IMR-32 cells transfected with the control siRNA (Fig. 5e, 2 way ANOVA: effect of ethanol, F(1,12) = 46.16, p < 0.0001; effect of siRNA, F(2,12) = 4.18, p = 0.042; ethanol by siRNA interaction, F(1,12) = 6.79, p = 0.011). pSTAT3 increased by 67% in cells transfected with siCTL (p < 0.0001) and this increase was reduced by half in cells expressing siMDK2 (p = 0.025) and siMDK3 (p = 0.015). Together, these results indicate that the ethanol-induced activation of ALK, ERK and STAT3 depends on MDK expression in IMR-32 cells.
ALK inhibition attenuates the ethanol-stimulated increase in pERK in the amygdala
Administration of ethanol to mice increases pERK in the CeA (Spanos et al. 2012). To determine if ALK regulates ERK activation in response to ethanol in the amygdala, we treated male mice with vehicle or the ALK inhibitor TAE684. We found that ethanol increased pERK in the CeA of vehicle-treated mice and that this increase was attenuated in mice treated with TAE684 (Fig. 6a-d). Quantification of the intensity of pERK antibody staining in the CeA indicated that pERK increased by 160% in mice treated with ethanol when compared to mice treated with saline. Mice treated with TAE684 prior to ethanol showed a blunted ethanol response of 68%, representing a 35% reduction in pERK in response to ethanol (Fig. 6e). A 2 way ANOVA indicated significant main effects of ethanol and TAE684 and a trend towards an interaction between ethanol and TAE684 (effect of ethanol, F(1,68) = 54.4, p < 0.0001; effect of TAE684, F(1, 68) = 11.93, p = 0.001; ethanol by TAE684 interaction, F(1,68) = 3.34, p = 0.072). Similar results were obtained with a 2 g/kg ethanol injection, although changes were not significant. pERK increased by 72% in mice treated with 2 g/kg ethanol compared to saline (Fig. 6f, p = 0.058) and pERK decreased by 25% in mice treated with TAE684 compared to vehicle-treated mice (Fig. 6g, p = 0.16). These results suggest that ALK activity contributes to increased pERK in the CeA in response to ethanol.
Fig. 6.
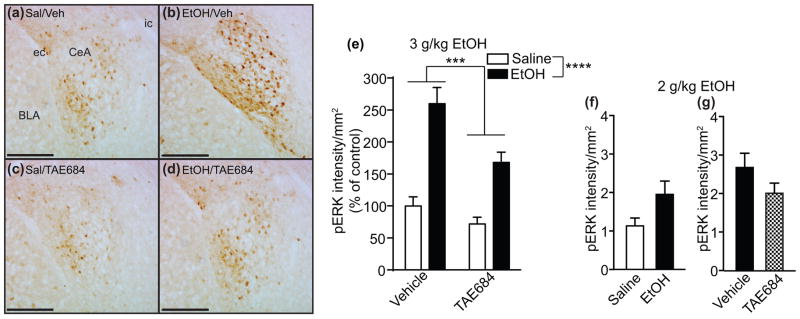
ALK inhibition attenuates ethanol-stimulated increase in pERK in the amygdala. (a–d) Images showing pERK staining in the mouse CeA after saline (a, c) or 3 g/kg ethanol (b, d) treatment and vehicle (a, b) or TAE684 treatment (c, d). (e) Quantification of pERK intensity using Image J. There were significant effects of ethanol and TAE684 treatments. (f) pERK intensity after injection of 2 g/kg ethanol or saline. (g) pERK staining in mice treated with 2 g/kg ethanol and TAE684 or vehicle. Data are presented as mean ± S.E.M. ***p <0.001, ****p < 0.0001. Abbreviations: Sal, saline; Veh, vehicle; EtOH, ethanol; CeA, central nucleus of the amygdala; BLA, basolateral amygdala; ec, external capsule; ic, internal capsule. Scale bar, 200 μm.
Reduced pERK in the amygdala of MdkKO mice treated with ethanol
We next tested if MDK is involved in the increase in pERK in response to ethanol using male and female MdkKO mice. As shown in Fig. 7a–d, pERK increased in the CeA of wild-type female mice treated with ethanol. The increase in pERK staining in response to ethanol was reduced in female MdkKO mice. Quantification of pERK immunostaining (Fig. 7e) demonstrated a 286% increase in pERK in female wild-type mice treated with ethanol compared to saline. In female MdkKO mice, ethanol treatment resulted in a 108% increase in pERK, representing a 47% reduction in ethanol response when compared to wild-type mice. A 2 way ANOVA indicated a significant main effect of ethanol treatment, a trend towards a main effect of genotype, and a significant ethanol by genotype interaction (effect of ethanol, F(1, 41) = 16.93, p = 0.0002; effect of genotype, F(1,41) = 3.15, p = 0.084; ethanol by genotype interaction, F(1,41) = 4.28, p = 0.045). Interestingly, the attenuation of pERK in response to ethanol was only observed in female, but not male (data not shown), MdkKO mice. These results suggest that MDK contributes to the activation of ERK in the CeA in response to ethanol in female mice.
Fig. 7.
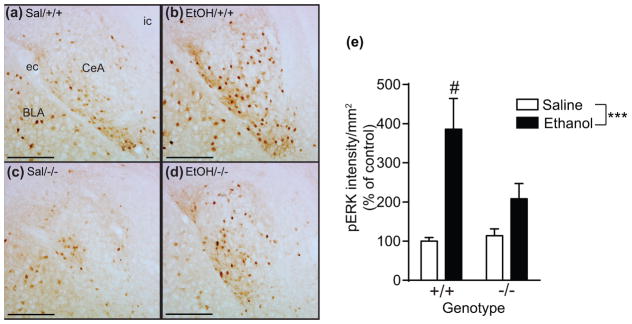
Reduced pERK in the amygdala of MdkKO mice treated with ethanol. (a–d) Images showing pERK staining in the CeA of female wild-type (a, b; +/+) and MdkKO (c, d; −/−) mice 30 min after saline (a, c) or 3 g/kg ethanol (b, d) treatment. (e) Quantification of pERK staining using Image J. There was a significant effect of ethanol treatment and a treatment by genotype interaction. Data are presented as mean ± S.E.M. ***p < 0.001; #p = 0.045, treatment by genotype interaction. Abbreviations: Sal, saline; EtOH, ethanol; CeA, central nucleus of the amygdala; BLA, basolateral amygdala; ec, external capsule; ic, internal capsule. Scale bar, 200 μm.
Discussion
We provide the first direct evidence that ethanol alters the expression of MDK and ALK and rapidly activates MDK and ALK signaling pathways in cultured neuroblastoma cells and in the brain. Treating SH-SY5Y and IMR-32 neuroblastoma cells with ethanol increased the transcription of MDK and ALK within 4 h. The effect of ethanol on ALK and MDK expression was observed in dopamine-like, differentiated SH-SY5Y (Figs. 1 and 2) and in undifferentiated SH-SY5Y cells (data not shown). We have also observed that ethanol exposure increases the expression of mouse Mdk in Neuro-2a cells and in primary striatal neurons (D.H. and A.W.L., unpublished results). Thus, at least in the case of MDK, ethanol exposure increases the expression of this gene in several neuronal cell types. The doses of ethanol used in our studies represent blood alcohol concentrations ranging from 0.09% (20 mM, intoxicating) to 0.46% (100 mM, sedating). We found that SH-SY5Y cells appear to be more sensitive to the effects of ethanol compared to IMR-32 cells. Low to moderate concentrations of ethanol (20 and 50 mM) were able to increase MDK and ALK transcription in dopamine-like SH-SY5Y cells, whereas 100 mM ethanol was required to increase the expression of these genes in IMR-32 cells. These results suggest that certain types of neurons, such as dopamine neurons, may be more sensitive to the effects of ethanol in activating MDK and ALK transcription. The mechanisms by which ethanol alters the transcription of MDK and ALK are currently unknown and remain an important area for investigation. It would also be beneficial to know if repeated or chronic exposure to ethanol causes MDK and ALK protein levels to be persistently elevated.
MDK mRNA and protein levels were previously found to be higher in the prefrontal cortex of alcoholics compared to non-alcoholic control subjects (Flatscher-Bader et al. 2005, Flatscher-Bader & Wilce 2006, Flatscher-Bader & Wilce 2008). In those studies it was not feasible to determine if levels of MDK are higher in alcoholics due to an inherent genetic predisposition or as a result of chronic alcohol consumption, since they were performed in post-mortem human brain. Gene expression microarray data from ethanol-naïve mice selectively bred to consume high amounts of alcohol indicate that Mdk expression is higher in the brains of these mice (Mulligan et al. 2006). This suggests that increased MDK levels in the brain might predispose individuals to consume excessive amounts of alcohol. Our data also provide evidence that MDK expression increases with ethanol treatment, indicating that MDK may be both a predisposing gene for alcohol abuse and responsive to alcohol exposure. Because MDK is upregulated following tissue injury, it is not entirely surprising that alcohol also increases MDK expression. Here, we found that MDK expression is induced by ethanol exposure in neuronal cells. This is in contrast to what was observed in the prefrontal cortex of alcoholics, where MDK expression was induced in astrocytes (Flatscher-Bader & Wilce 2008). In addition, MDK is highly expressed in astrocytes after ischemia in human and rat brain (Mochizuki et al. 1998, Wada et al. 2002). The reason for this discrepancy is unknown. One possibility is that the increase in MDK expression in neuroblastoma cultures is due to the partially differentiated state of the cells. Another possibility is that acute versus chronic ethanol exposure may differentially affect neurons vs astrocytes. Acute ethanol may have an immediate and transient impact on MDK expression in neurons, whereas chronic alcohol exposure may induce MDK in astrocytes in response to brain injury.
In addition to finding that ethanol increases MDK and ALK expression, we discovered that ethanol has a rapid effect on ALK signaling, which is likely independent of the transcriptional effect. Ethanol activated ALK signaling in IMR-32 cells, which express wild-type ALK. pALK (220 and 140 kDa variants) increased after 5 min of ethanol treatment, remained elevated at 15 min and normalized by 30 min. The increase in pALK was blocked by pre-treatment with the ALK inhibitor, TAE684. TAE684 decreased baseline pALK 140 but not 220, raising the intriguing possibility that ALK 140 might be a constitutively active form of ALK. Moog-Lutz et al demonstrated that the 140 kDa form of ALK results from cleavage of the full-length 220 kDa ALK protein in the extracellular domain, resulting in a truncated version of ALK that expresses part of the extracellular domain and the full transmembrane and kinase domains (Moog-Lutz et al. 2005). To our knowledge, our results provide the first evidence that ALK 140 may be a constitutively active kinase. In concert with increased pALK in response to ethanol treatment, we also observed increased pERK and pSTAT3, which were blocked by pre-treatment with TAE684, indicating that ethanol activates ERK and STAT3 signaling through ALK. Wild-type ALK has been previously shown to activate ERK and STAT3 signaling in response to agonist antibody treatment with similar kinetics to what we observed upon ethanol treatment (Moog-Lutz et al. 2005, Sattu et al. 2013). ALK-dependent neurite outgrowth in PC12 and SK-N-SH neuroblastoma cells requires ERK signaling (Souttou et al. 2001, Motegi et al. 2004) and activation of STAT3 by ALK appears to be important for increasing MYCN expression and neuroblastoma proliferation (Sattu et al. 2013). In contrast to ERK and STAT3, we did not observe any changes in pAKT upon ethanol treatment, indicating specificity in the activation of ALK signaling by ethanol in IMR-32 cells. Although ALK activates AKT, it does so as an oncogenic protein, when artificially localized to the cytoplasm and when overexpressed, suggesting that activation of AKT may not occur through wild-type ALK in neurons (Gouzi et al. 2005, Palmer et al. 2009, Kuo et al. 2007). The mechanisms by which ethanol activates ALK signaling are not known. Ethanol might directly activate ALK through an effect on cellular membranes and dimerization of the receptor. However, our results suggest that MDK may act as an intermediary in the process.
We found that ethanol increases pERK in IMR-32 cells. Others have found that ethanol treatment can either increase or decrease pERK in vitro (Kalluri & Ticku 2003, Roivainen et al. 1995, Seiler et al. 2001). For instance, acute ethanol treatment decreases pERK in cultured cortical neurons and in SH-SY5Y cells (Kalluri & Ticku 2003, Seiler et al. 2001), whereas chronic ethanol treatment increases pERK in cortical neurons and PC-12 cells (Kalluri & Ticku 2003, Roivainen et al. 1995). These data indicate that acute vs. chronic ethanol treatment differentially affects signaling in these cells. However, acute ethanol treatment in vivo can also increase or decrease pERK, depending on the brain region (Spanos et al. 2012, Zhu et al. 2013). We propose that the response to ethanol with regard to activation of ERK signaling is dependent on both cell type and treatment regimen.
We found that MDK activates ALK signaling in IMR-32 cells in a manner similar to ethanol. Treatment of IMR-32 cells with recombinant MDK protein resulted in increased pALK, pERK and pSTAT3 within 5 min. Activation of these signaling pathways was ALK-dependent, since effects were blocked by pre-treatment with TAE684. We also found that TAE684 reduced baseline pERK and pSTAT3, suggesting that there is some level of ALK-depending signaling that occurs under quiescent conditions. This may be due to constitutive signaling by ALK 140, since baseline phosphorylation of this ALK variant was also reduced by TAE684 treatment. Cleavage of ALK to the 140 kDa form releases an 80 kDa extracellular fragment and is predicted to eliminate MDK binding from ALK 140 (Mazot et al. 2011, Moog-Lutz et al. 2005), since the MDK binding site on ALK has been mapped to amino acids 391-401 (Stoica et al. 2002, Stylianou et al. 2009). However, we found that pALK 140 was enhanced by MDK treatment. MDK also binds to the receptor protein tyrosine phosphatase β/ζ (RPTPβ/ζ) (Maeda et al. 1999). RPTPβ/ζ negatively regulates ALK signaling by dephosphorylating the receptor (Perez-Pinera et al. 2007). MDK binding to RPTPβ/ζ is hypothesized to inactivate the phosphatase, thereby activating ALK signaling. This would occur in a manner similar to PTN binding to RPTPβ/ζ, another putative ALK ligand (Perez-Pinera et al. 2007, Muramatsu 2014). It is possible that the ability of MDK to activate ALK signaling in IMR-32 cells is indirect, through MDK binding to and inactivating RPTPβ/ζ.
Evidence that MDK is important for the ability of ethanol to activate ALK signal transduction is provided by our experiments in which we reduced MDK expression using siRNAs. Transfection of IMR-32 cells with MDK siRNAs decreased MDK protein levels by approximately 50%. Reducing MDK levels abolished the ethanol-stimulated increase in pALK and attenuated the ethanol-stimulated increase in pERK and pSTAT3. We also performed experiments in which we treated IMR-32 cells with the MDK inhibitor, iMDK (Hao et al. 2013), and observed similar attenuation of the ethanol-stimulated increase in pALK, pERK and pSTAT3 (A.W.L and D.H., unpublished results). These results indicate that MDK contributes to the activation of ALK signaling in response to ethanol. How ethanol might rapidly activate MDK/ALK signaling is not known, but it is possible that ethanol increases MDK secretion from cells, thus activating ALK. We hypothesize that this is a separate event from the ethanol-induced increase in MDK transcription, since increased MDK expression occurs within hours and the activation of ALK signaling by ethanol happens within minutes. MDK protein levels in IMR-32 cells and mouse amygdala must be sufficient to promote ALK activation in response to ethanol without an increase in de novo protein synthesis.
We extended our studies to an in vivo model of ethanol-stimulated signaling. Ethanol rapidly increases pERK in the CeA, which is an important brain region that controls alcohol consumption (Ibba et al. 2009, Spanos et al. 2012, Thorsell et al. 2013, Roberto et al. 2012). We found that inhibition of ALK by TAE684 attenuated the increase in pERK in response to ethanol, indicating that ALK in the CeA contributes to ethanol-stimulated ERK signaling. We also found that MDK contributes to ethanol-stimulated ERK signaling in the CeA, since MdkKO mice had decreased pERK in response to ethanol. We found a sex difference in this response. The attenuation in ethanol-stimulated pERK was only seen in female, but not male, MdkKO mice. MDK is an estrogen-regulated gene, so it is possible that higher levels of MDK are present in female mouse brain (Zhao et al. 2012). As a result, the effect of knocking out Mdk in female mice might have a more pronounced effect on signaling in response to ethanol. Although we found a role for MDK and ALK signaling in ethanol-stimulated pERK in the CeA, signaling through MDK and ALK is not the only mechanism of ERK activation in response to ethanol. Others have found that the neuropeptide S receptor and the dopamine D1 receptor also contribute to increased pERK after ethanol treatment (Thorsell et al. 2013, Ibba et al. 2009). Ethanol activation of ERK signaling in the CeA probably results from the activation of several receptors.
What are the implications for the ability of ethanol to activate ERK and STAT3 via the MDK/ALK axis? MDK and ALK are clearly important for regulating behavioral responses to ethanol (Lasek et al. 2011b, Vicente-Rodriguez et al. 2014). ERK signaling also regulates behavioral responses to ethanol and other drugs of abuse (Faccidomo et al. 2009, Girault et al. 2007, Peana et al. 2013, Jeanblanc et al. 2013, Agoglia et al. 2015). This suggests that the behavioral alterations in Mdk and Alk knockout mice could be due to altered activation of the ERK signaling pathway. Targeting this pathway may provide a viable therapeutic strategy to curb excessive alcohol consumption. Less is known about the role of STAT3 signaling in response to ethanol and ethanol-related behaviors, although pSTAT3 in the Edinger-Westphal nucleus is elevated after ethanol exposure (Bachtell et al. 2002). Interestingly, STAT3 plays a role in alcoholic liver disease and may protect against alcoholic liver injury and inflammation (Gao 2012). Since MDK plays a role in inflammation (Weckbach et al. 2011), activation of the STAT3 pathway may be a mechanism through which inflammatory processes in the brain are engaged with chronic alcohol consumption. Our data provide a starting point for determining whether ethanol-stimulation of MDK and ALK signaling contributes to alcohol abuse.
Acknowledgments
This study was supported by the National Institutes of Health, National Institute on Alcohol Abuse and Alcoholism (AA020912 and AA016654). We would like to thank Roberta Franks of the UIC Transgenic Production Service for assistance in re-deriving the MDKKO mice from frozen embryos.
Abbreviations
- MDK
midkine
- ALK
anaplastic lymphoma kinase
- MAPK/ERK
mitogen-activated protein kinase/extracellular signal-regulated kinase
- PI3K/AKT
phosphoinositide 3-kinase/protein kinase B
- STAT3
signal transducer and activator of transcription 3
- MdkKO
midkine knockout
- CeA
central nucleus of the amygdala
Footnotes
Laboratory of origin: Dr. Amy Lasek, University of Illinois at Chicago
conflict of interest disclosure
The authors declare no conflicts of interest.
References
- Agoglia AE, Sharko AC, Psilos KE, Holstein SE, Reid GT, Hodge CW. Alcohol alters the activation of ERK1/2, a functional regulator of binge alcohol drinking in adult C57BL/6J mice. Alcoholism, clinical and experimental research. 2015;39:463–475. doi: 10.1111/acer.12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtell RK, Tsivkovskaia NO, Ryabinin AE. Alcohol-induced c-Fos expression in the Edinger-Westphal nucleus: pharmacological and signal transduction mechanisms. The Journal of pharmacology and experimental therapeutics. 2002;302:516–524. doi: 10.1124/jpet.102.036046. [DOI] [PubMed] [Google Scholar]
- Ezquerra L, Perez-Garcia C, Garrido E, Diez-Fernandez C, Deuel TF, Alguacil LF, Herradon G. Morphine and yohimbine regulate midkine gene expression in the rat hippocampus. European journal of pharmacology. 2007;557:147–150. doi: 10.1016/j.ejphar.2006.11.024. [DOI] [PubMed] [Google Scholar]
- Faccidomo S, Besheer J, Stanford PC, Hodge CW. Increased operant responding for ethanol in male C57BL/6J mice: specific regulation by the ERK1/2, but not JNK, MAP kinase pathway. Psychopharmacology. 2009;204:135–147. doi: 10.1007/s00213-008-1444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatscher-Bader T, van der Brug M, Hwang JW, Gochee PA, Matsumoto I, Niwa S, Wilce PA. Alcohol-responsive genes in the frontal cortex and nucleus accumbens of human alcoholics. Journal of neurochemistry. 2005;93:359–370. doi: 10.1111/j.1471-4159.2004.03021.x. [DOI] [PubMed] [Google Scholar]
- Flatscher-Bader T, Wilce PA. Chronic smoking and alcoholism change expression of selective genes in the human prefrontal cortex. Alcoholism, clinical and experimental research. 2006;30:908–915. doi: 10.1111/j.1530-0277.2006.00106.x. [DOI] [PubMed] [Google Scholar]
- Flatscher-Bader T, Wilce PA. Impact of alcohol abuse on protein expression of midkine and excitatory amino acid transporter 1 in the human prefrontal cortex. Alcoholism, clinical and experimental research. 2008;32:1849–1858. doi: 10.1111/j.1530-0277.2008.00754.x. [DOI] [PubMed] [Google Scholar]
- Galkin AV, Melnick JS, Kim S, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:270–275. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B. Hepatoprotective and anti-inflammatory cytokines in alcoholic liver disease. Journal of gastroenterology and hepatology. 2012;27(Suppl 2):89–93. doi: 10.1111/j.1440-1746.2011.07003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Perez D, Luisa Laorden M, Nunez C, Victoria Milanes M. Glial activation and midkine and pleiotrophin transcription in the ventral tegmental area are modulated by morphine administration. Journal of neuroimmunology. 2014;274:244–248. doi: 10.1016/j.jneuroim.2014.07.017. [DOI] [PubMed] [Google Scholar]
- Girault JA, Valjent E, Caboche J, Herve D. ERK2: a logical AND gate critical for drug-induced plasticity? Current opinion in pharmacology. 2007;7:77–85. doi: 10.1016/j.coph.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Gouzi JY, Moog-Lutz C, Vigny M, Brunet-de Carvalho N. Role of the subcellular localization of ALK tyrosine kinase domain in neuronal differentiation of PC12 cells. Journal of cell science. 2005;118:5811–5823. doi: 10.1242/jcs.02695. [DOI] [PubMed] [Google Scholar]
- Gramage E, Martin YB, Ramanah P, Perez-Garcia C, Herradon G. Midkine regulates amphetamine-induced astrocytosis in striatum but has no effects on amphetamine-induced striatal dopaminergic denervation and addictive effects: functional differences between pleiotrophin and midkine. Neuroscience. 2011;190:307–317. doi: 10.1016/j.neuroscience.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Hao H, Maeda Y, Fukazawa T, et al. Inhibition of the growth factor MDK/midkine by a novel small molecule compound to treat non-small cell lung cancer. PloS one. 2013;8:e71093. doi: 10.1371/journal.pone.0071093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herradon G, Perez-Garcia C. Targeting midkine and pleiotrophin signalling pathways in addiction and neurodegenerative disorders: recent progress and perspectives. British journal of pharmacology. 2014;171:837–848. doi: 10.1111/bph.12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba F, Vinci S, Spiga S, Peana AT, Assaretti AR, Spina L, Longoni R, Acquas E. Ethanol-induced extracellular signal regulated kinase: role of dopamine D1 receptors. Alcoholism, clinical and experimental research. 2009;33:858–867. doi: 10.1111/j.1530-0277.2009.00907.x. [DOI] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugieres L, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- Jeanblanc J, Logrip ML, Janak PH, Ron D. BDNF-mediated regulation of ethanol consumption requires the activation of the MAP kinase pathway and protein synthesis. The European journal of neuroscience. 2013;37:607–612. doi: 10.1111/ejn.12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadomatsu K, Kishida S, Tsubota S. The heparin-binding growth factor midkine: the biological activities and candidate receptors. Journal of biochemistry. 2013;153:511–521. doi: 10.1093/jb/mvt035. [DOI] [PubMed] [Google Scholar]
- Kalluri HS, Ticku MK. Regulation of ERK phosphorylation by ethanol in fetal cortical neurons. Neurochemical research. 2003;28:765–769. doi: 10.1023/a:1022822119560. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Muramatsu H, Muramatsu T, Kim SU. Midkine, a novel neurotrophic factor, promotes survival of mesencephalic neurons in culture. Neuroscience letters. 1993;160:9–12. doi: 10.1016/0304-3940(93)90904-y. [DOI] [PubMed] [Google Scholar]
- Korecka JA, van Kesteren RE, Blaas E, Spitzer SO, Kamstra JH, Smit AB, Swaab DF, Verhaagen J, Bossers K. Phenotypic characterization of retinoic acid differentiated SH-SY5Y cells by transcriptional profiling. PloS one. 2013;8:e63862. doi: 10.1371/journal.pone.0063862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AH, Stoica GE, Riegel AT, Wellstein A. Recruitment of insulin receptor substrate-1 and activation of NF-kappaB essential for midkine growth signaling through anaplastic lymphoma kinase. Oncogene. 2007;26:859–869. doi: 10.1038/sj.onc.1209840. [DOI] [PubMed] [Google Scholar]
- Lasek AW, Gesch J, Giorgetti F, Kharazia V, Heberlein U. Alk is a transcriptional target of LMO4 and ERalpha that promotes cocaine sensitization and reward. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011a;31:14134–14141. doi: 10.1523/JNEUROSCI.3415-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek AW, Lim J, Kliethermes CL, et al. An evolutionary conserved role for anaplastic lymphoma kinase in behavioral responses to ethanol. PloS one. 2011b;6:e22636. doi: 10.1371/journal.pone.0022636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente M, Torres S, Salazar M, et al. Stimulation of the midkine/ALK axis renders glioma cells resistant to cannabinoid antitumoral action. Cell death and differentiation. 2011;18:959–973. doi: 10.1038/cdd.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N, Ichihara-Tanaka K, Kimura T, Kadomatsu K, Muramatsu T, Noda M. A receptor-like protein-tyrosine phosphatase PTPzeta/RPTPbeta binds a heparin-binding growth factor midkine. Involvement of arginine 78 of midkine in the high affinity binding to PTPzeta. The Journal of biological chemistry. 1999;274:12474–12479. doi: 10.1074/jbc.274.18.12474. [DOI] [PubMed] [Google Scholar]
- Maruta H, Bartlett PF, Nurcombe V, et al. Midkine (MK), a retinoic acid (RA)-inducible gene product, produced in E. coli acts on neuronal and HL60 leukemia cells. Growth factors. 1993;8:119–134. doi: 10.3109/08977199309046932. [DOI] [PubMed] [Google Scholar]
- Mazot P, Cazes A, Boutterin MC, et al. The constitutive activity of the ALK mutated at positions F1174 or R1275 impairs receptor trafficking. Oncogene. 2011;30:2017–2025. doi: 10.1038/onc.2010.595. [DOI] [PubMed] [Google Scholar]
- Michikawa M, Kikuchi S, Muramatsu H, Muramatsu T, Kim SU. Retinoic acid responsive gene product, midkine, has neurotrophic functions for mouse spinal cord and dorsal root ganglion neurons in culture. Journal of neuroscience research. 1993;35:530–539. doi: 10.1002/jnr.490350509. [DOI] [PubMed] [Google Scholar]
- Miyashiro M, Kadomatsu K, Ogata N, Yamamoto C, Takahashi K, Uyama M, Muramatsu H, Muramatsu T. Midkine expression in transient retinal ischemia in the rat. Current eye research. 1998;17:9–13. doi: 10.1076/ceyr.17.1.9.5257. [DOI] [PubMed] [Google Scholar]
- Mochizuki R, Takeda A, Sato N, Kimpara T, Onodera H, Itoyama Y, Muramatsu T. Induction of midkine expression in reactive astrocytes following rat transient forebrain ischemia. Experimental neurology. 1998;149:73–78. doi: 10.1006/exnr.1997.6687. [DOI] [PubMed] [Google Scholar]
- Moog-Lutz C, Degoutin J, Gouzi JY, Frobert Y, Brunet-de Carvalho N, Bureau J, Creminon C, Vigny M. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. The Journal of biological chemistry. 2005;280:26039–26048. doi: 10.1074/jbc.M501972200. [DOI] [PubMed] [Google Scholar]
- Motegi A, Fujimoto J, Kotani M, Sakuraba H, Yamamoto T. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. Journal of cell science. 2004;117:3319–3329. doi: 10.1242/jcs.01183. [DOI] [PubMed] [Google Scholar]
- Mulligan MK, Ponomarev I, Hitzemann RJ, et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:6368–6373. doi: 10.1073/pnas.0510188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Rhodes JS, Crabbe JC, Mayfield RD, Harris RA, Ponomarev I. Molecular profiles of drinking alcohol to intoxication in C57BL/6J mice. Alcoholism, clinical and experimental research. 2011;35:659–670. doi: 10.1111/j.1530-0277.2010.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu H, Shirahama H, Yonezawa S, Maruta H, Muramatsu T. Midkine, a retinoic acid-inducible growth/differentiation factor: immunochemical evidence for the function and distribution. Developmental biology. 1993;159:392–402. doi: 10.1006/dbio.1993.1250. [DOI] [PubMed] [Google Scholar]
- Muramatsu T. Structure and function of midkine as the basis of its pharmacological effects. British journal of pharmacology. 2014;171:814–826. doi: 10.1111/bph.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramoto A, Imagama S, Natori T, et al. Midkine overcomes neurite outgrowth inhibition of chondroitin sulfate proteoglycan without glial activation and promotes functional recovery after spinal cord injury. Neuroscience letters. 2013;550:150–155. doi: 10.1016/j.neulet.2013.06.025. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Milbrandt J, Muramatsu T, Deuel TF, Zhao H, Cnaan A, Brodeur GM. Differential expression of pleiotrophin and midkine in advanced neuroblastomas. Cancer research. 1995;55:1792–1797. [PubMed] [Google Scholar]
- Nakamura E, Kadomatsu K, Yuasa S, et al. Disruption of the midkine gene (Mdk) resulted in altered expression of a calcium binding protein in the hippocampus of infant mice and their abnormal behaviour. Genes to cells : devoted to molecular & cellular mechanisms. 1998;3:811–822. doi: 10.1046/j.1365-2443.1998.00231.x. [DOI] [PubMed] [Google Scholar]
- Neasta J, Ben Hamida S, Yowell QV, Carnicella S, Ron D. AKT signaling pathway in the nucleus accumbens mediates excessive alcohol drinking behaviors. Biological psychiatry. 2011;70:575–582. doi: 10.1016/j.biopsych.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owada K, Sanjo N, Kobayashi T, Mizusawa H, Muramatsu H, Muramatsu T, Michikawa M. Midkine inhibits caspase-dependent apoptosis via the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase in cultured neurons. Journal of neurochemistry. 1999a;73:2084–2092. [PubMed] [Google Scholar]
- Owada K, Sanjyo N, Kobayashi T, Kamata T, Mizusawa H, Muramatsu H, Muramatsu T, Michikawa M. Midkine inhibits apoptosis via extracellular signal regulated kinase (ERK) activation in PC12 cells. Journal of medical and dental sciences. 1999b;46:45–51. [PubMed] [Google Scholar]
- Palmer RH, Vernersson E, Grabbe C, Hallberg B. Anaplastic lymphoma kinase: signalling in development and disease. The Biochemical journal. 2009;420:345–361. doi: 10.1042/BJ20090387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peana AT, Giugliano V, Rosas M, Sabariego M, Acquas E. Effects of L-cysteine on reinstatement of ethanol-seeking behavior and on reinstatement-elicited extracellular signal-regulated kinase phosphorylation in the rat nucleus accumbens shell. Alcoholism, clinical and experimental research. 2013;37(Suppl 1):E329–337. doi: 10.1111/j.1530-0277.2012.01877.x. [DOI] [PubMed] [Google Scholar]
- Perez-Pinera P, Zhang W, Chang Y, Vega JA, Deuel TF. Anaplastic lymphoma kinase is activated through the pleiotrophin/receptor protein-tyrosine phosphatase beta/zeta signaling pathway: an alternative mechanism of receptor tyrosine kinase activation. The Journal of biological chemistry. 2007;282:28683–28690. doi: 10.1074/jbc.M704505200. [DOI] [PubMed] [Google Scholar]
- Reiff T, Huber L, Kramer M, Delattre O, Janoueix-Lerosey I, Rohrer H. Midkine and Alk signaling in sympathetic neuron proliferation and neuroblastoma predisposition. Development. 2011;138:4699–4708. doi: 10.1242/dev.072157. [DOI] [PubMed] [Google Scholar]
- Roberto M, Gilpin NW, Siggins GR. The central amygdala and alcohol: role of gamma-aminobutyric acid, glutamate, and neuropeptides. Cold Spring Harbor perspectives in medicine. 2012;2:a012195. doi: 10.1101/cshperspect.a012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roivainen R, Hundle B, Messing RO. Ethanol enhances growth factor activation of mitogen-activated protein kinases by a protein kinase C-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:1891–1895. doi: 10.1073/pnas.92.6.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna PP, Simpson C, Lutjens R, Koob G. ERK regulation in chronic ethanol exposure and withdrawal. Brain research. 2002;948:186–191. doi: 10.1016/s0006-8993(02)03191-8. [DOI] [PubMed] [Google Scholar]
- Sattu K, Hochgrafe F, Wu J, et al. Phosphoproteomic analysis of anaplastic lymphoma kinase (ALK) downstream signaling pathways identifies signal transducer and activator of transcription 3 as a functional target of activated ALK in neuroblastoma cells. The FEBS journal. 2013;280:5269–5282. doi: 10.1111/febs.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler AE, Ross BN, Rubin R. Inhibition of insulin-like growth factor-1 receptor and IRS-2 signaling by ethanol in SH-SY5Y neuroblastoma cells. Journal of neurochemistry. 2001;76:573–581. doi: 10.1046/j.1471-4159.2001.00025.x. [DOI] [PubMed] [Google Scholar]
- Souttou B, Carvalho NB, Raulais D, Vigny M. Activation of anaplastic lymphoma kinase receptor tyrosine kinase induces neuronal differentiation through the mitogen-activated protein kinase pathway. The Journal of biological chemistry. 2001;276:9526–9531. doi: 10.1074/jbc.M007333200. [DOI] [PubMed] [Google Scholar]
- Spanos M, Besheer J, Hodge CW. Increased sensitivity to alcohol induced changes in ERK Map kinase phosphorylation and memory disruption in adolescent as compared to adult C57BL/6J mice. Behavioural brain research. 2012;230:158–166. doi: 10.1016/j.bbr.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey D, Bilbao A, Maroteaux M, et al. RASGRF2 regulates alcohol-induced reinforcement by influencing mesolimbic dopamine neuron activity and dopamine release. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:21128–21133. doi: 10.1073/pnas.1211844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica GE, Kuo A, Powers C, Bowden ET, Sale EB, Riegel AT, Wellstein A. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. The Journal of biological chemistry. 2002;277:35990–35998. doi: 10.1074/jbc.M205749200. [DOI] [PubMed] [Google Scholar]
- Stylianou DC, Auf der Maur A, Kodack DP, Henke RT, Hohn S, Toretsky JA, Riegel AT, Wellstein A. Effect of single-chain antibody targeting of the ligand-binding domain in the anaplastic lymphoma kinase receptor. Oncogene. 2009;28:3296–3306. doi: 10.1038/onc.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsell A, Tapocik JD, Liu K, et al. A novel brain penetrant NPS receptor antagonist, NCGC00185684, blocks alcohol-induced ERK-phosphorylation in the central amygdala and decreases operant alcohol self-administration in rats. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:10132–10142. doi: 10.1523/JNEUROSCI.4742-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Rodriguez M, Perez-Garcia C, Haro M, Ramos MP, Herradon G. Genetic inactivation of midkine modulates behavioural responses to ethanol possibly by enhancing GABA(A) receptor sensitivity to GABA(A) acting drugs. Behavioural brain research. 2014;274:258–263. doi: 10.1016/j.bbr.2014.08.023. [DOI] [PubMed] [Google Scholar]
- Wada M, Kamata M, Aizu Y, Morita T, Hu J, Oyanagi K. Alteration of midkine expression in the ischemic brain of humans. Journal of the neurological sciences. 2002;200:67–73. doi: 10.1016/s0022-510x(02)00134-x. [DOI] [PubMed] [Google Scholar]
- Weckbach LT, Muramatsu T, Walzog B. Midkine in inflammation. The Scientific World Journal. 2011;11:2491–2505. doi: 10.1100/2011/517152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Goto M, Tsutsui J, Ozawa M, Sato E, Osame M, Muramatsu T. Midkine is present in the early stage of cerebral infarct. Brain research. Developmental brain research. 1995;85:25–30. doi: 10.1016/0165-3806(94)00183-z. [DOI] [PubMed] [Google Scholar]
- Zhao G, Nie Y, Lv M, He L, Wang T, Hou Y. ERbeta-mediated estradiol enhances epithelial mesenchymal transition of lung adenocarcinoma through increasing transcription of midkine. Molecular endocrinology. 2012;26:1304–1315. doi: 10.1210/me.2012-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Wang Y, Zhao B, Wei S, Xu M, Liu E, Lai J. Differential phosphorylation of GluN1-MAPKs in rat brain reward circuits following long-term alcohol exposure. PloS one. 2013;8:e54930. doi: 10.1371/journal.pone.0054930. [DOI] [PMC free article] [PubMed] [Google Scholar]