

Abstract
Changes within executive function are at the root of most cognitive problems associated with Parkinson’s disease (PD). Because dopaminergic treatment does not necessarily alleviate deficits in executive function, it has been hypothesized that dysfunction of other neurotransmitters/systems besides dopamine (DA), maybe associated with this decrease in cognitive function. We have reported decreases in motor-function and dopaminergic/glutamatergic biomarkers using a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Parkinson’s mouse model. Assessment of executive function and dopaminergic/glutamatergic biomarkers within the limbic circuit has previously not been explored in our model. Our results show progressive behavioral decline in cued response task (a rodent model for frontal cortex cognitive function) with increasing weekly doses of MPTP. Although within the dorsolateral (DL) striatum mice administered MPTP showed a 63% and 83% loss of tyrosine hydroxylase (TH) and dopamine transporter (DAT) expression, respectively, there were no changes in the nucleus accumbens (NAc) or medial prefrontal cortex (mPFC). Furthermore, dopamine-1 receptor (DA-D1) and vesicular glutamate transporter-1 (VGLUT-1) expression increased in the mPFC following DA loss. There was a significant MPTP-induced decrease/increase in VGLUT-1 and vesicular glutamate transporter-2 (VGLUT-2) expression, respectively, within the DL striatum. We propose that the behavioral decline following MPTP treatment may be associated with a change in not only cortical-cortical (VGLUT-1) glutamate function, but also in striatal DA and glutamate (VGLUT-1/VGLUT-2) input.
Keywords: cued response task, Parkinson’s disease, nigrostriatal depletion, cognition, RRID:nif-0000-00313, RRID:AB_2313787, RRID:AB_2190287, RRID:AB_2193878, RRID:AB_2094980, RRID:AB_2302051, RRID:AB_2190727, RRID:AB_887876, RRID:AB_2254574, RRID:AB_476743, RRID:AB_2340875, RRID:AB_11125348, RRID:AB_11125338
Graphical abstract
1. INTRODUCTION
Parkinson’s disease (PD) is a progressive neurodegenerative disorder predominantly characterized by motor dysfunction. Aside the impairment of dopamine (DA)-stimulated motor behaviors, PD patients also experience progressive cognitive deficits. Of these cognitive deficits, the most prominent is executive function; that is, the elaboration of behavioral and cognitive responses to adverse environmental stimulation (Dubois and Pillon 1997). Unlike most motor deficits associated with PD, the non-motor deficits, including executive function, are unresponsive to dopaminergic treatment (McDowell and Chesselet 2012). Cognitive dysfunction is therefore hypothesized to be due to an imbalance of other neurotransmitters besides DA, and perhaps within other neuronal circuitry besides the basal ganglion (Darvas et al., 2014). Relative glutamate levels in PD animal models, for example, have been previously shown to change within the striatum in a time dependent manner after a unilateral nigrostriatal 6-hydroxydopamine (6-OHDA) lesion (Meshul et al., 1999) and following acute/subacute treatment with the neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Robinson et al., 2003; Holmer et al., 2005b). We have also recently reported that following progressive MPTP treatment, glutamate transporters were significantly increased (Sconce et al., 2015).
Overall there appears to be an important DA-glutamate interaction that occurs within the basal ganglia and a similar association within the limbic system that maintains cognitive function (David et al., 2005). Furthermore, previous studies investigating cognitive changes in MPTP animal models have analyzed serotonin, norepinephrine and DA levels, but glutamate’s role in executive dysfunction has yet to be determined. Characterization of dopaminergic and glutamatergic biomarkers within the limbic system needs to be elucidated in PD animal models to understand the mechanisms underlying the cognitive deficits associated with the disease.
Widely used in neurotoxin animal models of PD, MPTP selectively damages dopaminergic neurons in the basal ganglion (Blandini and Armentero 2012; Meredith et al., 2008; Tieu 2011). Many studies using animal models to investigate PD employ acute/subacute MPTP toxin paradigms where a large number of the dopaminergic neurons are lesioned in a short period of time. This method negates the progressive nature of the disease. In our progressive mouse model of PD, mice are injected with MPTP over a period of 4 weeks (5 days/week) and with increased doses for each week (8, 16, 24, and 32-mg/kg). The progressive model is valuable because it attempts to simulate the continuous neurodegeneration (Goldberg et al., 2011a) and decline in motor movement (Amende et al., 2005; Goldberg et al., 2011b) seen in clinical PD. For this study, in addition to lesioning the dopaminergic neurons by progressive administration of MPTP, cognitive deficits were behaviorally analyzed by cued response task, which requires animals to learn a stimulus-action-outcome contingency as well as the ability to exhibit behavioral flexibility depending on the stimulus present at a given time (Wilhelm and Mitchell 2008).
The purpose of this study was to examine behavioral changes via simple executive function testing and their relationship to dopaminergic and glutamatergic protein expression in the dorsolateral (DL) striatum, nucleus accumbens (NAc), and medial prefrontal cortex (mPFC) in a progressive MPTP mouse model. We hypothesize that if progressive MPTP doses elicit early cognitive decline, then we will see glutamatergic changes in the mPFC. To our knowledge, this is the first time that cognitive deficits and DA/glutamate biomarkers within the limbic system have been evaluated in a progressive mouse model of PD.
2. MATERIALS AND METHODS
2.1 Animals
25 Male C57BL/6J mice Jackson Labs, RRID:IMSR_JAX:000664, Bar Harbor, ME, USA; 9-weeks old at arrival) were housed 3–4/cage and maintained on a 12 hr light-dark cycle throughout (lights on 0600). They had ad libitum access to standard laboratory chow and water, until 2 days prior to behavioral training when mice were food restricted to approximately 90% of their normal weight until the end of the behavioral study, after which free feeding was reinstated. All procedures were approved by the Oregon Health & Science University Institutional Animal Care and Use Committee.
2.2 Cued Response Task
The cued response task was modified from a go/no-go task (apparatus and task described in Gubner et al. 2010). The task included 60 trials. Each trial began by turning on the house light for 9–24 s. Then the cue light positioned above either the left or right food well was lit; location determined randomly, with the proviso that there should be 30 trials/location. A nose poke in the cued food well during the cue period was rewarded by delivering a 20-μl sucrose solution (10% w/v) and the cue light and house light extinguished for a 10-s timeout period. The duration of the cue period was 5 s. Responses when the cue light was not lit did not earn rewards but were recorded.
All mice received some task training initially. In Phase 1, the cue period was 30 s. Mice entered Phase 2 training (cue period = 10 s) after earning 30 of the 60 possible rewards within 40 minutes on two consecutive sessions. Mice entered the experimental phase (cue period = 5 s) after earning 10 rewards on one session. During the experiment, there were 6 baseline sessions after which subjects were randomly assigned to the MPTP or saline group. Analyses of the baseline sessions indicated that all performance indices were equivalent between groups and no group x session interactions (all p > .25). The MPTP-treated group included 14 mice, injected daily i.p., 5 days/week (Monday-Friday) with doses increasing weekly [8 mg/kg, 16 mg/kg, 24 mg/kg and 32 mg/kg (expressed as free base); Sigma Aldrich, St. Louis, MO, USA] over the course of 4 weeks. The vehicle group included 11 mice, and received saline (0.1 ml/0.1 kg, i.p.). All injections were given within 1 hour after the cued response task on that day, beginning after the 6th baseline session.
After receiving 3 injections of 24 mg/kg dose (or equivalent saline session), the concentration of sucrose for the next session was increased to 25% for both groups, before being returned to 10% on subsequent sessions. This manipulation was included to examine the role of motivation in task behavior, but performance was unchanged presumably due to ceiling effects for the saline animals and floor effects preventing experience of the new contingencies in the MPTP group. Data from this session were not included in the analysis of MPTP dose effects. Following 5 sessions at the 32 mg/kg dose (or equivalent saline session), mice continued to perform the task daily for 5 additional sessions without injections being given to either group. This manipulation was included to examine MPTP recovery.
2.3 Locomotor Activity
Locomotor activity was analyzed on 2 consecutive days, beginning 9 days after the final injection of 32 mg/kg MPTP (4 days after completing the cued response task procedure) using the parallel rod activity chamber (PRAC)(Kamens and Crabbe, 2007), with photo beams added to record locomotor activity (Goldberg et al., 2011a). The values were averaged over the 2 days of testing. In addition to horizontal movement, paw slips through the parallel rods were recorded as foot faults. Mice were acclimated to the PRAC for 5 min, on each session, then activity was recorded for 5 minutes. Foot faults per total activity were expressed as a ratio of foot faults per beam breaks (FF/BB) in order to normalize the data against possible changes in locomotor (BB) activity (i.e., to evaluate whether changes in foot faults are due to changes in movement or in motor control).
2.4 Gait Dynamics
Gait was assessed using a DigiGait apparatus (Mouse Specifics, Quincy, MA), as previously reported (Goldberg et al., 2011b). Ten days following the final injection of 32 mg/kg MPTP, ventral plane videography captured the gait of each mouse through a transparent, motor-driven treadmill belt (Amende et al., 2005; Kale et al., 2004). Digital images of the paws of each mouse were taken at 150 frames/s as mice walked at a speed of 24 cm/s.
2.5 Tyrosine Hydroxylase Immunohistochemistry and DL Striatum Analysis
Ten days following the last injection of 32 mg/kg of MPTP, half the animals in each treatment group (N = 7 for the MPTP group, N = 5 for the vehicle group) were anesthetized with 1% ketamine/0.1% mg xylazine (2.0 ml/0.1 kg, i.p.), and euthanized by transcardial perfusion with 1000 U/ml of heparin in 0.1 M phosphate buffer (3 ml total) followed by fixative [2% paraformaldehyde, 1% acrolein in 0.1 M phosphate buffer; pH 7.4; ~35–50 ml]. Brains were removed and bisected coronally. The rostral half, containing the striatum, was left in 1.0% glutaraldehyde/0.5% paraformaldehyde/0.1% picric acid in 0.1M phosphate buffer for 20 hours in the cold (4 °C). The caudal half, containing the SNc and VTA, was left in 0.1M phosphate buffer at 4 °C. Both halves were then washed daily in 0.1 M phosphate buffer until sectioned. Consecutive sections through the entire SNc and VTA (beginning at Bregma - 2.9) (Paxinos and Franklin 2004) were cut at 40 μm thick and the striatum (beginning at Bregma + 1.2) cut at 60 μm thick using a vibratome (Ted Pella Inc., Redding, CA, USA). Sections were matched anatomically in each animal, verifying that the coronal sections of the SNc/VTA and striatum were similar in all treatment groups. Six sections, extending throughout the rostrocaudal portion of the SNc/VTA and striatum (up to the caudal part of the anterior commissure) were used for analyses.
The following incubations were carried out in the PELCO BioWave® Pro microwave (Ted Pella Inc., Redding, CA, USA) with the temperature limited to 35°C. Rinsing solutions were under normal pressure unless otherwise stated. Sections were incubated in 10 mM sodium citrate (pH 6) for 5 min at 550 W for antigen retrieval in a vacuum chamber that cycles the pressure down to 20 Hg and back to atmosphere repeatedly during this step (cycling vacuum), rinsed in 0.01 M phosphate buffered saline (PBS) at 150 W for 1 min., rinsed with 0.3% hydrogen peroxide (150 W, 1 min), two PBS rinses for 1 minute, and then incubated in 0.5% Triton X (550 W, 5 min, cycling vacuum). Sections were incubated with TH antibody (1:2,000 for striatum and 1:2500 for SNc/VTA; (Immunostar Cat# P22941, RRID:AB_2313787), mouse monoclonal) at 200 W for 36 min and 20 sec under continuous vacuum (20Hg, cycling the magnetron for 2 min on/3 min off/2 min on/5 min off repeating). Sections were incubated in blocking solution [10% goat serum/0.5% Triton-X 100/0.1 M phosphate buffer; pH 7.4] for two, 1 min washes at 150 W, and then exposed to biotinylated goat anti-mouse secondary antibody (1: 400; Vector, Burlingame, CA, USA) for 16 min and 20 sec under continuous vacuum (10 sec at 150 W, 4 min at 200 W, 3 min at 0 W, 4 min at 200 W, 5 min at 0 W, and 10 sec at 150 W), rinsed with PBS, then washed with imidazole working buffer [5% Imidazole buffer(0.2M), pH 9.0/16% sodium acetate (0.1M), pH7.2] (1 min at 150 W), and finally incubated with avidin-biotin complex solution (ABC, diluted according to manufacturer instruction; Vector) for 16 min and 10 sec under continuous vacuum (4 min at 200 W, 3 min at 0 W, 4 min at 200 W, 5 min at 0 W, and 10 sec at 200 W). Tissue was then rinsed with imidazole working buffer (1 min at 150 W), incubated with diaminobenzidine [0.1% DAB (Sigma D5637)+ 1.5% hydrogen peroxide in 0.1 M phosphate buffer] for 10 min 20 sec under continuous vacuum (10 sec at 200 W, 10 min at 200 W, 10 sec at 0 W), rinsed with imidazole working buffer, and finally in PBS. Tissue was mounted on gel-coated slides and dehydrated at room temperature overnight and cover-slipped using Pro-Texx® medium (Lerner, Pittsburgh, PA). Tissue from all treatment groups was processed on the same day, and all reacted with DAB for the same length of time. Optical density of the DL striatum sections, the region receiving the input from the motor cortex (McGeorge and Faull 1989), was analyzed using light microscopy, by an individual blinded to the treatment group (1.25x magnification, images analyzed using ImagePro Plus 6.3, Media Cybernetics, RRID:nif-0000-00313).
2.6 SNc/VTA TH-ir Neuron Counting
Tissue was prepared, mounted, and coverslipped as described above. TH-ir neurons only at the in-focus surface plane of immunolabeled SNc/VTA tissue were counted using light microscopy (5x magnification, images analyzed using ImagePro Plus 6.3, Media Cybernetics, RRID:nif-0000-00313) by an individual blinded to the treatment group. The number of TH-ir neurons from the left and right SNc and VTA sections were averaged for each animal for all six sections analyzed since both sides of the SNc are affected following systemic administration of this neurotoxin. Mean numbers of TH-ir neurons/section were then determined for all animals in the VTA and SNc separately in a given treatment group. Areas were identified using Paxinos and Franklin (2004) mouse stereotaxic coordinates as a guide. From these tissue section counts, the total number of labeled neurons/section was re-evaluated using the Abercrombie correction, which accounts for fragmented nuclei within each section and provides an accurate estimate when tissue thickness exceeds soma thickness by more than 50%, which is the case in this study (40 μm sections); (Clarke 1992; Smolen et al., 1983). Although this cell counting methodology may have yielded an underestimation of the total number of TH-ir neurons/section in the SNc, it is an appropriate approach for measuring the mean number of neurons/section according to recent comparisons of 2D and 3D analyses of brain tissue (Baquet et al., 2009; Benes and Lange 2001).
2.7 Western Immunoblots
Mice (N = 7 for the MPTP group, N = 6 for the vehicle group) were euthanized by cervical dislocation 10 days following the last administration of MPTP, and the mPFC, NAc, and DL striatum were dissected, and frozen at −80 °C. Using a 3 mm microdissection knife and a stereomicroscope, we bilaterally dissected the mPFC according to Paxinos and Franklin (2004), which excluded the caudal cingulate cortex. No olfactory tissue was included in the dissection. Protein was extracted from the tissue by sonication in lysis buffer [5% 1 M Tris, 2% 0.5 M EDTA, 1% Triton-X 100, 0.5% Protease Inhibitor Cocktail III (Calbiochem, USA)]. Protein concentrations of the tissue from each individual animal were measured using the BCA Protein Assay Kit (Thermo Scientific). Samples were mixed with XT Sample Buffer and XT Reducing Agent (1:10; Bio-Rad, Hercules, CA, USA) and electrophoresed on a 4–12% Bis-Tris XT Precast Gel (Bio-Rad, Hercules, CA, USA). Separated proteins were transferred to polyvinylidene difluoride membranes (Millipore, MA, USA), which were blocked in 5% non-fat dry milk in Tris–buffered saline with Tween-20 (TBST) for 60 min. Membranes were then washed three times for 5 min each in TBST, and probed with the following primary antibodies: TH at 62kDa (Immunostar Cat# P22941, RRID:AB_2313787;1:40,000, mouse monoclonal), DAT at 80kDa (Santa Cruz Biotechnology Cat# sc-14002, RRID:AB_2190287; 1:500, rabbit polyclonal), DA receptor 1 (DA-D1) at 74kDa (Santa Cruz Biotechnology Cat# sc-20822, RRID:AB_2193878; 1:200, goat polyclonal), dopamine receptor 2 (DA-D2) at 50kDa (Millipore Cat# AB5084P, RRID:AB_2094980;1:1000, rabbit polyclonal), glutamate-aspartate transporter (GLAST) at 65 kDa (Santa Cruz Biotechnology Cat# sc-15316, RRID:AB_2302051; 1:200, rabbit polyclonal), excitatory amino acid carrier 1 (EAAC1) at 57kDa (Santa Cruz Biotechnology Cat# sc-25658, RRID:AB_2190727;1:500, rabbit polyclonal), VGLUT-1 at 60kDa; (Synaptic Systems GmbH Cat# 135 303, RRID:AB_887876; 1:20,000, rabbit polyclonal), vesicular glutamate transporter 2 (VGLUT-2) at 52kDa (Synaptic Systems Cat# 135 403, RRID:AB_2254574; 1:2000, rabbit polyclonal), or β-actin at 43kDa (Sigma-Aldrich Cat# A5316, RRID:AB_476743; 1:6500, mouse monoclonal). After three 5 min washes in TBST, membranes were probed with secondary antibodies for 1 hour (ap-bovine anti-goat IgG H+L (Jackson ImmunoResearch Cat# 805-055-180, RRID:AB_2340875), ap-goat anti-mouse IgG H+L (Bio-Rad Cat# 170-6520 RRID:AB_11125348), ap-goat anti-rabbit IgG H+L (Bio-Rad Cat# 170-6518 RRID:AB_11125338) then washed again in TBST for 3 × 5 minutes. ECF substrate (GE Healthcare, Piscataway, NJ, USA) was added to the membrane prior to visualization. Visualization and quantification of the antigen-antibody binding density were performed using theTyphoon HLA7000 imaging system (GE Healthcare, Piscataway, NJ, USA) and ImagePro Plus 6.3, Media Cybernetics, RRID:nif-0000-00313. Protein densities were analyzed relative to individual β-actin densities.
2.8 Statistical Analysis
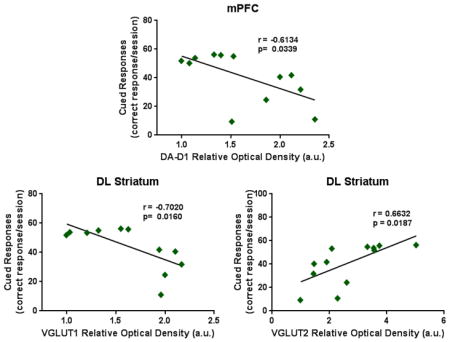
The effects of MPTP on Cued response learning task was analyzed by mixed factor one-way analysis of variance (ANOVA), with treatment as the between subjects factor and dose, or dose and session, as within subject factors where appropriate. All significant interactions of the Cued response learning task were subjected to post hoc Bonferroni-corrected t-tests, for which ‘p’ values are presented. Additional MPTP effects were examined using an ANOVA to compare vehicle and MPTP groups for histology, gait dynamics, PRAC and protein expression. All significant interactions were subjected to post hoc Tukey-Kramer HSD tests for multiple comparisons, for which ‘p’ values are presented. For gait dynamics all the measurements for the four paws were averaged for each animal. Violations of sphericity were addressed using Huynh-Feldt corrections, indicated by adjustments in the reported degrees of freedom. Pearson correlations with significant r-values were used to evaluate the correlations between cognitive behaviors and biochemical markers (Fig. 5).
Figure 5. Pearson Correlations of executive function and biochemical markers.


TH expression within the DL striatum showed a (A) significant positive correlation with cued responses and (B) a negative correlation with latency. (C) DAT levels correlated positively with cued responses within the DL striatum and (D) the DA-D1 expression levels significantly correlated negatively with cued responses within the mPFC. Also within the DL striatum, VGLUT-1 levels showed (E) a negative correlation with cued response but (F) a positive correlation with latency while VGLUT-2 levels revealed (G) a positive correlation with cued response with (H) a negative correlation with latency. (I) The expression of GLT-1 within the DL striatum correlated negatively with latency. (J) Within the NAc, EAAC1 expression levels correlated negatively with latency.
2.9 Antibody Characterization
Please see Table 1 for a list of all antibodies used.
Table 1.
2.9 Antibody Chart
| Antibody Name | Immunogen Structure | Manufacturer, Cat. #, RRID, Species, Type | Dilution |
|---|---|---|---|
| tyrosine hydroxylase | “Full length TH purified to homogeneity from PC12 cells, rat origin” | (Immunostar Cat# P22941, RRID:AB_2313787) mouse, monoclonal | 1:2000, DL striatum (IHC) 1:2500 SNc (IHC) 1:40,000 (WB) |
| dopamine transporter | AA (541–620) near the C- terminus | (Santa Cruz Biotechnology Cat# sc-14002, RRID:AB_2190287) rabbit, polyclonal | 1:500 |
| dopamine receptor 1 | (C-15) near C-terminus | (Santa Cruz Biotechnology Cat# sc-20822, RRID:AB_2193878) goat, polyclonal | 1:200 |
| dopamine receptor 2 | “28 AA peptide sequence from the human D2 receptor within cytoplasmic loop #3” | (Millipore Cat# AB5084P, RRID:AB_2094980) rabbit, polyclonal | 1:1000 |
| vesicular glutamate transporter-1 | “AA 456–560, Strep-Tag fusion protein of rat VGLUT1” | (Synaptic Systems GmbH Cat# 135 303, RRID:AB_887876) rabbit, polyclonal | 1:20,000 |
| vesicular glutamate transporter-2 | “AA 510–582, Strep-Tag fusion protein of rat VGLUT2” | (Synaptic Systems Cat# 135 403, RRID:AB_2254574) rabbit, polyclonal | 1:2000 |
| glutamate transporter 1 | AA (1–85) near the N- terminus of GLT-1 human origin | (Santa Cruz Biotechnology Cat# sc-15317, RRID:AB_2239433) rabbit, polyclonal | 1:1000 |
| excitatory amino acid carrier 1 | AA (455–524) of human origin | (Santa Cruz Biotechnology Cat# sc-25658, RRID:AB_2190727) rabbit, polyclonal | 1:500 |
| glutamate-aspartate transporter | AA (1–50) near the N- terminus of GLAST | (Santa Cruz Biotechnology Cat# sc-15316, RRID:AB_2302051) rabbit, polyclonal | 1:200 |
| beta-actin | “slightly modified β-cytoplasmic actin N- terminal peptide, Ac-Asp- Asp-Asp-Ile-Ala-Ala-Leu- Val-Ile-Asp-Asn-Gly-Ser- Gly-Lys, conjugated to KLH.” | (Sigma-Aldrich Cat# A5316, RRID:AB_476743) mouse, monoclonal | 1:6500 |
The Tyrosine hydroxylase (TH) antibody from Immunostar Inc. has well been established as a reliable marker for TH in immunohistochemistry and western immublots where mouse brain slices of the striatum and substantia nigra look like those within the manufacture’s datasheet. Additionally, western immunoblots show one band at the correct 62 kDa weight. The dopamine transporter (DAT) antibody will detect multiple bands based on the degree of glycosylation of the protein. Analysis was done on the 80kDa band or the fully glycosylated form of DAT (Li et al., 2004). The dopamine-1 receptor (DA-D1) antibody detected a single band at 74kDa and the dopamine-2 receptor (DA-D2) detected a single band at 50 kDa. The vesicular glutamate transporter 1 (VGLUT1) and 2 (VGLUT2) antibodies detected single thick bands at 60kDa and ~52kDa, respectively. The antibody for the glutamate transporter 1 (GLT-1) detected a single band at 70kDa as shown within the manufacture’s datasheet. The antibodies raised against excitatory amino acid carrier 1 (EAAC1) and glutamate aspartate transporter 1 (GLAST) detected broad single bands at 57kDa and 65kDa, respectively, as shown in the manufacture’s datasheet. Beta-Actin levels were assessed as our loading control and the antibody detects reproducibly and consistently a single band at 43kDa as shown in the manufacture’s datasheet.
3. RESULTS
3.1 Cued Response Task
There were three main dependent measures derived from the cued response task: the number of location-appropriate responses during cue period (cue responses, maximum 60/session), the latency between the cue onset and response (latency), and the rate of responding during the period prior to cue onset (precue response rate).
While performance on all of these measures was equivalent between groups at baseline, MPTP administration had profound effects on task performance. ANOVAs examining dose effects for the MPTP treatment group indicated progressive declines in cued responses across the 4 doses (Fig. 1A; F1.3,16.5 = 9.09; p=0.004), increases in latency to respond to the cue (Fig. 1B; F3,36 = 14.67; p<0.0001), and reduced responding during the precue period (Fig. 1C; F1.5,20.0 = 8.42; p=0.004). In contrast, saline treatments over the same period were associated with increases in cued responses (F1.9,20.6 = 10.57; p=0.001) and reductions in response latencies (F2.2,23.6 = 14.82; p<0.0001) as animals refined their performance. The response during the precue period was unchanged (p>0.05) in saline treated animals. These changes in performance resulted in differences between groups, as indicated by Bonferroni-corrected t-tests that emerged at different doses for different variables (Fig. 1). Comparing the saline and MPTP groups during comparable sessions at the lowest 8 mg/kg MPTP dose using Bonferroni-corrected t-tests indicated that differences in cued responses emerged after animals received 3 injections of 8 mg/kg, while differences in latency emerged after the final dose of 8 mg/kg.
Figure 1. Behavioral measures of executive function.

The performance of each mouse was measured during and following weekly administration of increasing doses of MPTP. (A) location-correct responses during the cue revealed that mice administered MPTP (n=14) failed to learn the task compared to the vehicle treated mice (n=11), while (B) latency to respond correctly following the onset of the cue increased progressively with the MPTP doses. (C) The precue period decreased significantly compared to the vehicle group following the 3rd and 4th week of MPTP administration. Values are means ± S.E.M. *p<0.01 versus vehicle group; **p<0.01 versus 32 mg/kg dose.
Ending MPTP administration did not abolish group differences but comparisons to the 32 mg/kg dose condition and the recovery sessions indicated significant improvements in performance when MPTP was discontinued (Fig. 1): cued responses increased (t(13) = −6.37, p<0.0001), latency tended to decrease (t(12) = 2.09, p=0.058), while precue responses increased (t(13) = −3.51, p=0.004; t(13) = −2.72, p=0.017). Performance by saline treated animals did not change significantly over this period.
3.2 Locomotor Activity
There were no significant differences in performance on the PRAC (beam breaks, foot faults and foot faults per total activity: all Fs < 1) (data not shown), suggesting that overall motor activity was not affected by the MPTP treatment.
3.3 Gait Dynamics
MPTP-treated mice exhibited different finer gait dynamics when compared to vehicle-treated mice for stance width variability and stance width coefficient of variation. There was a significant increase in the width between the forepaws and hind paws, as measured in centimeters, during the time the mice are in a stance position (i.e. when the paws hit the belt during running) in the MPTP group compared to animals treated with vehicle [values are means (cm) ± S.E.M: vehicle: 0.258 ± 0.03; MPTP: 0.35 ± 0.03, F1,22 = 4.6; p=0.04]. There was also an increase in the stance width coefficient of variation (variation normalized around the mean) in the MPTP compared to the vehicle treated group (values are mean % ± S.E.M: Vehicle: 13.9 ± 0.31; MPTP: 22.9 ± 2.5, F1,22 = 4.7; p=0.03).
3.4 TH-ir immunohistochemistry analysis of the dorsolateral striatum, SNc and VTA
Densitometric analysis of tyrosine hydroxylase immunoreactive (TH-ir) nerve terminals with the dorsolateral (DL) striatum showed a 63% decrease in the relative optical density labeling in the MPTP compared to the vehicle-treated group (Fig. 2Aii, C; F1,4 =14.28, p = 0.02). Cell surface counting of TH-ir neurons within the SNc and VTA 10 days following the final dose of 32 mg/kg MPTP was carried out in both treatment groups (Fig. 2Ai). The SNc of MPTP-treated animals had a 21% decrease in the number of TH-ir neurons/section in comparison to the vehicle group (Fig. 2B; F1,4 =15.32, p = 0.017). This suggests that there was a main effect of treatment as reported and seen previously with the progressive MPTP mouse model (Goldberg et al., 2011a). However, the VTA did not show significant changes in number of TH-ir neurons/section between the two treatment groups (Fig. 2B; F1,4 = 0.77, p = 0.43), suggesting that MPTP did not affect the limbic part of the mesocortical DA pathway compared to the modest lesion of the motor nigrostriatal tract.
Figure 2. TH-ir immunohistochemistry of the SNpc, VTA and DL striatum.

Changes in the mean number of TH-ir cells/section within the SNc/VTA (A. i) and DL striatum (A. ii) following the last dose of MPTP (i.e., 7 days following the 32 mg/kg dose). (B) The mean number of TH-ir cells/section within the SNc and VTA following MPTP treatment (n=7) was significantly decreased 21% in the SNc but not in the VTA compared to the vehicle group (n=5). (C) The optical density of TH in the DL striatum indicated a 63% loss in the animals treated with MPTP for 4 weeks. Values are means ± S.E.M. *p< 0.02.
3.5 Western Immunoblots
Western blot analysis for dopaminergic biomarkers of the DL striatum, NAc, and mPFC was evaluated 10 days following the last dose of MPTP. Although there were no differences in the NAc and mPFC for TH, there was a 62% decrease in TH protein expression in the DL striatum in MPTP-treated mice (Fig. 3A, D; p<0.0001), similar to what we have previously reported (Goldberg et al., 2011a, 2012). A 62% decrease in TH levels as indicated by western blot is in agreement with the 64% decrease in TH-ir optical density of the DL striatum as seen via immunohistochemistry (Fig. 2Aii). Additionally, there was a 83% decrease in DAT protein expression within the DL striatum of the MPTP treated mice (Fig. 3B, E; p<0.0002) with respect to the vehicle group. DAT expression within the NAc and mPFC did not show any changes between the MPTP and saline treated groups. DA-D1 protein analysis did not show any differences within the DL striatum or NAc, but further evaluation of the mPFC showed that the MPTP-treated mice had a significant increase of 47% in DA-D1 protein expression compared to the vehicle group (Fig. 3C, F; p=0.021). The expression of the DA-D2 receptor was also analyzed, but no differences were found between the two groups within any of the brain regions (data not shown).
Figure 3. Western blot analysis of dopaminergic biomarkers.

Western blots showing the mean relative optical density of (A) TH, (B) DAT, and the (C) DA-D1 receptor protein expression within the DL striatum, NAc and mPFC following treatment with MPTP (n=6–7) or vehicle (n=5–6). (D) TH levels significantly decreased 62% compared to vehicle within the DL striatum in mice treated with MPTP. There was a similar trend in the NAc and mPFC though not significant. (E) DAT levels were significantly reduced by 83% compared to the vehicle only within the DL striatum of mice treated with MPTP. (F) DA-D1 expression increased 47% within the mPFC compared to the vehicle in MPTP treated mice. There were no differences in DA-D1 levels in the DL striatum or NAc even though they were trending with similar increases. Values are means ± S.E.M. *p<0.05.
Western blot analysis for glutamatergic biomarkers of the DL striatum, NAc, and mPFC was additionally evaluated. Despite a lack of difference within the NAc, there was a significant increase in the protein expression of VGLUT1 in MPTP- versus vehicle-treated mice in both the DL striatum (p = 0.002) and the mPFC (Fig. 4A, E; p = 0.006). MPTP-treated mice showed a significant decrease in VGLUT2 expression compared to the vehicle group (p = 0.003) within the DL striatum, while there were no changes within the NAc, and mPFC (Fig. 4B, F). Interestingly, the levels of GLT-1 significantly decreased within the DL striatum (p = 0.007) and were trending towards a significant increase within the NAc (p = 0.077), but there were no differences within the mPFC (Fig. 4C, G). Even though no differences were observed within the DL striatum and mPFC, the expression levels of EAAC1 revealed a significant decrease within the NAc for the MPTP treated animals compared to the respective vehicle group (Fig. 4D, H; p=0.0205). Furthermore, GLAST was also analyzed, but there were no differences between the two groups within any of the brain regions (data not shown).
Figure 4. Western blot analysis of glutamatergic biomarkers.

Western blots showing the mean relative optical density of (A) VGLUT-1, (B) VGLUT-2, (C) GLT-1, and (D) EAAC1 protein expression within the DL striatum, NAc and mPFC following treatment with MPTP (n=6–7) or vehicle (n=5–6). (E) Although there were no changes in VGLUT-1 in the NAc, there was a significant 58% and 103% increase of expression compared to the vehicle within the DL striatum and mPFC, respectively, in mice treated MPTP. (F) VGLUT-2 levels significantly decreased 50% compared to the vehicle within the DL striatum of MPTP treated mice. The NAc and mPFC did not show any changes. (G) Expression of GLT-1 significantly decreased 81% compared to the vehicle within the DL striatum of the mice treated with MPTP. Though there weren’t any differences in the mPFC, there was a large increase in expression within the NAc that was trending (p=0.0791). (H) Within the NAc, EAAC1 levels significantly decreased 65% compared to the vehicle in the mice treated with MPTP. The DL striatum and mPFC did not show any differences. Values are means ±S.E.M. *p<0.05.
3.6 Correlation Analysis
Additional analysis was conducted to investigate if cognitive behaviors correlate with dopaminergic and glutamatergic biomarkers within the different brain regions. In the DL striatum TH expression correlated moderately positive with cued responses (Fig. 5A; r=0.6053; p=0.0370) and moderately negative with latency (Fig. 5B; r=−0.6239; p=0.0302). Also in the DL striatum, DAT expression correlated moderately positive with cued responses (Fig. 5C; r=0.6473; p=0.0229), while within the mPFC, DA-D1 levels correlated moderately negative with cued responses (Fig. 5D; r=−0.6134; p=0.0339). VLGUT-1 levels within the DL striatum revealed a moderate negative correlation with cued response (Fig. 5E; r=−0.7020; p=0.0160) with a highly positive correlation with latency (Fig. 5F; r=0.8524; p=0.0009). VGLUT-2 levels within the DL striatum correlated moderately positive with cued response (Fig. 5G; r=0.6632; p=0.0187) but moderately negative with latency (Fig. 5H; r=−0.5939; p=0.0417). The expression of GLT-1 with the DL striatum revealed a moderate negative correlation with latency (Fig. 5I; r=−0.6882; p=0.0404) and EAAC1 expression within the NAc correlated moderately negative with latency (Fig. 5J; r=−0.6471; p=0.0229).
4. DISCUSSION
Our progressive mouse model of PD, as reported in previous studies, consistently shows the gradual increasing loss of dopaminergic biomarkers within the nigrostriatal projections as well as motor behaviors (Goldberg et al., 2011a, b; Sconce et al., 2015a, b). In this study, we used our progressive model to investigate the cognitive deficits associated with PD via performance tests that gauge executive function. Additionally we evaluated changes in glutamatergic and dopaminergic biomarkers within the mPFC and NAc. Confirming the reproducibility of our previous studies, we found similar deficits in motor behavior in animals administered MPTP and a 63–64% decrease of TH labeling within the DL striatum by immunohistochemical and western immunoblots analysis. Further analysis of cognitive behavior revealed that mice administered MPTP had a decreased performance in the cued response task, particularly in the number of correct cued responses per session, and latency to respond to the cue. Western immunoblot analysis of the mPFC and striatum indicated a significant increase in VGLUT-1 expression in both brain regions, and a decrease in VGLUT-2 protein expression within the striatum. This suggests that alterations in cortical glutamate nerve terminals (origin of VGLUT1 protein) may play an important role in the behavioral changes observed during the progressive loss of nigrostriatal DA, besides the changes in corticostriatal (VGLUT-1) and thalamostriatal (VGLUT-2) input. Additionally, mice that were administered MPTP showed a significant increase of DA-D1 levels within the mPFC and a significant decrease of EAAC1 within the NAc. Our findings suggest that the progressive MPTP mouse model is not only advantageous for observing motor and cellular changes linked with PD within the basal ganglia, but also the model can be utilized to study cognitive and cellular changes associated with PD within the limbic circuit.
4.1 Cued response task displays cognitive deficits
Simply classified, executive function is a set of mental skills partly coordinated in the mPFC that include time/attention management, planning/organizing, integrating past with present experience, and the ability to switch focus. Of the many cognitive deficits associated with PD, a decline in executive function is very common and cued response tasks are a way of gauging executive function in rodent models. In this study, the animals treated with MPTP for 4 weeks showed sizeable decreases in cued response and increase in latency simultaneously coinciding with the increased dosing of MPTP. In contrast, saline-vehicle animals exhibited concurrent improvements in the same performance indices. This suggests that progressive administration of MPTP results in a dose-dependent progressive cognitive decline of executive function. Interestingly, following cessation of the MPTP-treatments, the MPTP mice began to show performance improvements despite the preceding behavioral data indicating that the animals treated with MPTP had failed to learn the task. We have previously reported several of the motor and gait deficits associated with progressive MPTP treatment (Goldberg et al., 2011a, b; Sconce et al., 2015a, b). However, the increasing motor impairments would most likely not have affected the overall motor behavior of the animal in order to respond to the cue. In the current study, gross motor deficits were not observed (i.e., no changes in the PRAC), only modest gait disturbances. Therefore, the underlying source of the observed learning failure is unclear. While it is possible that MPTP-treated animals may have been less motivated by the sucrose reinforcer, our observations that they rapidly consumed supplemental chow provided following the experimental session argued against this hypothesis. It’s also a possibility that peripheral MPTP toxicity could be hindering the animals’ ability to perform behavioral tests simply because the animals are sick. This could explain the recovery to vehicle levels in the precue rate after MPTP administration. However, the cued responses and the latency measures did not indicate a recovery to vehicle levels after MPTP administration even though there was some improvement. Additionally, the mice treated with MPTP did not lose any weight compared to the vehicles which would have further implicated peripheral MPTP toxicity symptomatically affecting behavioral testing. In fact MPTP treated group slightly gained more weight vs vehicle group over the 4-week toxin treatment (data not shown). Nonetheless, further studies would be required to conclusively address this supposition.
Additionally, the animals’ ability to form stimuli-response/action-outcome [SR/AO] associations (or any specific pair of associations within the SR/AO) may have been impaired, retarding response acquisition and facilitating extinction due to non-reinforcement with continued doses. This explanation for the alterations in behavior is consistent with the observation that performance began to improve (increased correct responses, decreased latency) when MPTP treatments were discontinued. Additional studies are needed to explore the precise mechanisms underlying these performance impairments, as well as studies to examine whether our performance indices (correct responses, latency, anticipatory precue response rate) differentially reflect the strength of specific pairs of the SR/AO associations.
Also intriguing in the findings of this study is that different performance indices exhibited different time courses of MPTP-associated decrement. That is, during the first week of MPTP-treatment, the number of correct cued responses declined relative to the saline-treated mice; but when these responses were made their latency did not differ between groups nor did the number of anticipatory responses. Although DA markers were not measured after the first week of MPTP in the current study, we have reported that 1 week of MPTP treatment results in a modest, but significant, loss of TH-ir nerve terminals in the striatum and TH-ir nerve cells in the SNc of about 20% (Goldberg et al., 2011a). This small change in nigrostriatal DA may be sufficient to result in a decrease in the number of cued responses but not that of the latency response or precue response rate, suggesting differential sensitivity to the loss of striatal DA compared to cued responses. Our findings suggest that with progressive nigrostriatal lesions, alterations in cognitive behavior can be easily demonstrated early on in this escalation model and that possible interventions can be tested during even the earliest phases of progressive striatal DA loss.
Using other more acute/subacute MPTP models of PD in both mice and rats, it has been reported that following toxin administration, there are alterations in working memory/object recognition (Ho et al., 2014; Castro et al., 2013), two-way active avoidance and forced swimming to evaluate the effects on cognition and depression (Barbeiro et al, 2014; Castro et al., 2013), impairment in procedural memory (Luchtman et al., 2012), and poor cognitive performance in both a passive avoidance task and in the cued version of the Morris water maze test (Kumar et al., 2009)[however, see Fifel et al (2014) for lack of effect on cognitive function following MPTP]. Although the cued response task in the current study was somewhat unique compared to the other executive/cognition tests carried out by other investigators, overall our results are in agreement with those previous findings.
In addition, the issue of DA depletion resulting in depressive behavior must also be taken into consideration in the current study, as reported by others using a more acute MPTP model of PD (Barbeiro et al, 2014; Castro et al., 2013). Although this behavior was not measured, there was no overall change in locomotor behavior, only changes in several measures of gait, following chronic/progressive treatment with MPTP compared to a significant decrease in motor behavior following acute MPTP administration (Fisher et al., 2004). It has been reported in patients that are depressed that there is an increase in glutamate levels in the mPFC (Frye et al., 2007; McEwen et al., 2012). Although glutamate levels were not measured in the current study, we find that MPTP resulted in an increase in VGLUT-1 protein expression with the mPFC. These data suggest that either there was an increase in the number of synaptic vesicles within these intra-cortical nerve terminals or that each vesicle contained more VGLUT-1 protein. Regardless, this would be consistent with the possibility that these terminals have the capacity to release more glutamate.
4.2 Gait dynamics
The observed gait changes support our previous findings (Goldberg et al., 2011b), which suggest there was a sufficient loss of nigrostriatal DA that resulted in significant gait alterations. Mice administered MPTP showed an increase in stance width as well as the stance width coefficient of variation which is essentially measuring the variability of stance. These data can be translational to clinical PD where many patients exhibit symptoms of postural instability and bradykinesia (Wu and Krishnan 2010; Schaafsma et al 2003; Hausdorff et al 1998; Blin et al 1990; Plotnik and Hausdorff 2008; Berardelli et al., 2001; Morris et al 2001, 1996; Hallett and Khoshbin, 1980), hallmarks of the disease. In our progressive model, not only can we see behavioral measures that are associated with cognition/executive function, but also, we are able to reproduce previous findings of basal ganglia associated motor deficits. Furthermore, future experiments can address both motor and cognitive decline associated amid PD within our progressive MPTP model.
4.3 Dopaminergic biomarker analysis by immunohistochemistry and western immunoblot
4.3.1 TH-ir IHC of DL striatum and SNc
TH enzyme is the limiting reaction step of DA synthesis and we have shown that TH protein levels correlate strongly with DA concentration (Goldberg et al., 2011a). Therefore in the current study, only TH was measured and not DA tissue levels. Analysis of the DL striatum showed a 63% decrease in TH relative optical density in MPTP treated mice compared to the vehicle group, which is comparable to our previous findings (Goldberg and Meshul 2011; Goldberg et al., 2011a, b). This loss of nigrostriatal DA could underlie the performance deficits in the MPTP-treated mice. Recent studies on the role of the DL striatum in terms of habit formation are of interest in the current study since it was reported that following bilateral infusion of the neurotoxin, 6-hydroxydopamine, into the rat striatum, the animals remained sensitive to reward devaluation and therefore, did not develop a habit of automatically responding to the cue (Faure et al., 2005). Following MPTP treatment a significant loss of TH and DAT was found within this same DL striatum in the current study, suggesting an association between the decrease in cued response and the loss of DA biomarkers within the DL striatum.
Interestingly, there was a relatively modest decrease in the number of TH labeled neurons/section in the SNc. It is unclear why the loss of TH-ir in the striatum was similar to what we have previously reported but the loss of TH-ir neurons in the SNc was not as robustly affected in this study as our past findings (Goldberg et al., 2011a, b). We previously reported in a subacute MPTP model (30 mg/kg/d × 7d) that caloric restriction can be neuroprotective (Holmer et al., 2005a). It’s possible that because the mice in this study were calorically restricted as part of the behavioral testing, the results indicated a modest loss of TH-ir SNc neurons despite the same MPTP dosing paradigm. Furthermore, it has been hypothesized that the loss of nigrostriatal DA begins first with the loss of DA terminals in the striatum (Hornykevitz, 1998). Over time there is a die-back phenomenon in which the terminals, then the axons, and finally the cell bodies begin to degenerate. This would be consistent with the findings of the current study in which there is a significant 64% loss of dopamine nerve terminals but only a very modest 21% loss of TH-ir DA neurons in the SNc. Nonetheless, despite the moderate loss of TH-ir SNc neurons, behavioral measures indicate sufficient DA loss, which suggests the importance of striatal versus neuronal DA depletion.
4.3.2 TH-ir VTA neurons following MPTP-administration
As previously mentioned, dopaminergic treatment does not ameliorate the cognitive deficits seen in patients with PD (McDowell and Chesselet 2012). We, therefore, hypothesized that there would be minimal loss of the DA producing neurons in the VTA, since the VTA sends dopaminergic projections to the mPFC and NAc. While there was a significant dopaminergic interaction within the nigrostriatal projection and motor behaviors, there were no significant differences in the number of TH-ir neurons/section in the VTA of MPTP-treated mice in comparison to vehicle-treated mice suggesting that the cognitive decline with MPTP administration could be due to an imbalance of neurochemicals in the brain other than DA (Pattij and Vanderschuren, 2008). However, the role of DA within either the nigrostriatal pathway or changes in DA-D1 receptors within the mPFC cannot be overlooked (Darvas et al., 2014).
One possible explanation for the unchanged VTA DA neuron population is that the VTA neurons have been reported to be less sensitive to MPTP than the SNc (Hung and Lee, 1996). It has been hypothesized that the reduced loss of VTA DA neurons either following MPTP or in patients with PD could be due to a lower density of DA transporters, DA receptors, the vesicular monoamine transporters, or the L-type calcium channels, compared to the SNc DA neurons (Reyes et al., 2013; Phani et al., 2010; Chan et al., 2007). In the current study, when comparing the SNc to the VTA DA cell loss, the SNc showed a modest but significant decrease in TH-ir/DA cells and the VTA did not, suggesting that cognitive impairment is most likely not due to VTA DA cell loss and potentially could be a downstream result of nigrostriatal cell loss (Darvas et al., 2014). When treating non-human primates with chronic exposure to low doses of MPTP, there was inconclusive cortical catecholamine and metabolite changes, but significant decreases in nigrostriatal DA. These data suggest that at least for DA, impairments in cognition could be due indirectly to depletion of this neurotransmitter within the striatum (Schneider 1990). Furthermore, because of the lack of DA neuronal loss within the VTA, imbalance of other neurotransmitters, such as glutamate, has been suggested to be associated with cognitive dysfunction (Pattij and Vanderschuren, 2008). This would also explain why treating PD patients with l-dopa alleviates motor symptoms but not necessarily executive function.
4.3.3 TH, DAT, and D1 biomarker analysis via western immunoblots of DL striatum, NAc, & mPFC
By western immunoblot analysis of the DL striatum, the expression levels of TH and DAT showed a significant decrease in MPTP-treated mice even up to 10 days following toxin treatment (62.3% and 82.6%, respectively). This is complementary with the TH-ir immunohistochemical data of the DL striatum. There were no changes in either DA biomarker within the mPFC or the NAc, which is interesting considering that both these regions receive DA input from the VTA. However, because we found no changes in the mean number of TH-ir neurons/section within the VTA following MPTP, it is not surprising.
Intriguingly, there was an increase in the protein expression of the DA-D1 receptor in the mPFC following MPTP. Although extracellular or tissue levels of cortical DA were not assessed in the current study, an increase in DA-D1 expression following MPTP would suggest a possible MPTP-induced decrease in DA release. This would be consistent with our findings of a 60% decrease in TH protein expression within the mPFC following MPTP. Although this decrease was not statistically significant, this might have resulted in a decrease in tissue levels of DA, resulting in the increase in DA-D1 receptor levels. Alterations in DA-D1 receptor levels within the mPFC have been directly associated with playing a critical role in working memory and executive function (Goldman-Rakic et al., 2004). Therefore, the cognitive dysfunction and alterations in mPFC DA-D1 receptors following MPTP treatment as reported in the current study would be consistent with the findings of Goldman-Rakic et al (2004). The DA-D1 receptor has been localized to not only DA-like nerve terminals but also post synaptically onto GABA-like interneurons within the monkey prefrontal cortex (Muley et al., 1998). In addition, it has recently been reported of an unusual presynaptic localization of the DA-D1 receptor within glutamate-like nerve terminals making an excitatory, asymmetrical synaptic contact within the primate prefrontal cortex (Paspalas and Goldman-Rakic, 2005). These authors propose that such regulation of excitation with this region of the primate brain may be involved in modulating working memory. It is also recognized that DA-D1 receptor levels need to be within a critical value to be effective in terms of mPFC function (Williams and Castner, 2006). This inverted U-shaped function suggests that in the current study, increased expression of the DA-D1 receptor within the mPFC may be too high, resulting in the reported executive dysfunction following nigrostriatal DA loss.
Also in support of possible changes in GABA function, it has been reported that PD patients have a small but significant decrease in mRNA levels in parvalbumin-positive GABA neurons, along with a decrease in glutamic acid decarboxylase mRNA levels, within the prefrontal cortex compared to non-PD controls (Lanoue et al., 2010, 2013). Presynaptic DA D-1 receptors may be capable of modulating not only GABAergic interneurons but also the release of glutamate within the prefrontal cortex. In our findings, the increase in DA D-1 receptor expression within the medial prefrontal cortex following MPTP, despite unchanged TH/DAT expression, suggests that the decrease in cognitive function (as measured by the cued response task) may also involve alterations in both GABA and glutamate release. Although future studies will focus on measuring both GABA and glutamate levels within the medial prefrontal cortex using in vivo microdialysis and quantitative immuno-gold electron microscopy, this current study analyzed glutamate transporter levels.
4.3.4 Glutamatergic biomarker analysis by western immunoblot
Novel in this bilateral lesion model, we found that animals administered progressive doses of MPTP showed an increase in VGLUT-1 and a decrease in VGLUT-2 protein expression levels within the striatum compared to the vehicle group. Our results are in agreement with Marin et al. (2011) and Massie et al. (2010), who reported an increase in VGLUT1 and a decrease in VLGUT-2 protein expression within the striatum both ipsi/contra-lateral following a nearly complete loss of nigrostriatal DA using the unilateral 6-hydroxydopamine model. In the current study, the increase in VGLUT-1 and the decrease in VGLUT-2 expression were observed nearly 7 weeks after the start of the MPTP injections. The increase in VGLUT-1 transporter levels could be due to increased activity of the corticostriatal pathway via increased turnover of synaptic vesicles and/or more VGLUT-1 protein/vesicle membrane. Interestingly, it is commonly proposed that in PD there is increased activity of the subthalamic input to the SN, which uses exclusively VGLUT-2 for glutamate uptake into the synaptic vesicle (cortical inputs use VGLUT-1).
The opposite would be hypothesized to take place for VGLUT-2 (input from the thalamus primarily) in terms of decreased protein expression in the striatum following MPTP. We have preliminary data showing that following MPTP, there is an increase in the density of nerve terminal glutamate immuno-gold labeling inside VGLUT-1 positive nerve terminals that are making an asymmetrical (i.e., excitatory) contact and this was associated with a decrease in the extracellular levels of striatal glutamate (Meshul, Moore, and Neubert, unpublished findings). However, it was reported that in the non-human primate PD model, administration of MPTP resulted in an increase in the number of VGLUT-1 labeled terminals within the striatum, with no change in the number of VGLUT-2 labeled terminals (Raju et al., 2008).
Within the medial prefrontal cortex following MPTP, there was only a change in VGLUT-1 protein expression. This cortical region projects to both the VTA and the NAc, but no changes were observed in VGLUT-1 glutamate transporter levels within the NAc. It is likely that the consequences of MPTP treatment on glutamate function within the cortex may be associated with alterations in cortical-cortical glutamate connections. Since we observed an increase in the DA D-1 receptor levels following MPTP, it is possible that these presynaptic DA receptors located on glutamate-like terminals (Paspalas and Goldman-Rakic, 2005) may be affecting the release of glutamate, resulting in a significant decline in cued response and an increase in latency as observed in the current study.
Significance Statement.
Dopamine (DA) replacement therapy for Parkinson’s disease (PD) reduce motor symptoms but does not alleviate cognitive deficits even though both brain areas utilize DA as a neurotransmitter. The long-term progressive dosing regimen in our mouse model of PD better mimics the progressive neurodegenerative nature of clinical PD. This study aims to investigate if cognitive deficits and biochemical alterations within the cognitive regions of the brain can be observed in our progressive mouse model of PD. Potential treatments can then be tested in this model to address motor and cognitive deficits.
Acknowledgments
This work was supported by the National Institutes of Health DA027580, AA10760, and DA018165 to SHM and Merit Review #1BX 001643 to CKM from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Our thanks to Cindy Moore and Rebecca Hood for technical assistance. This work was supported by the National Institutes of Health DA027580, AA10760, and DA018165 to SHM and Merit Review #1BX 001643 to CKM from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Abbreviations
- DA-D1
Dopamine receptor-1
- DA-D2
Dopamine receptor-2
- DA
Dopamine
- DAT
Dopamine transporter
- EAAC1
Excitatory amino acid carrier 1
- GABA
Gamma amino butyric acid
- GLAST
Glutamate-aspartate transporter
- mPFC
Medial prefrontal cortex
- MPTP
1-methyl-4-phenyl-1,23,6-tetrahydropyridine
- NAc
Nucleus accumbens
- PD
Parkinson’s Disease
- SNc
Substantia nigra pars compacta
- TH
Tyrosine hydroxylase
- TH-ir
Tyrosine hydroxylase immunoreactivity
- VGLUT-1
Vesicular glutamate transporter-1
- VGLUT-2
Vesicular glutamate transporter-2
- VTA
Ventral tegmental area
Footnotes
Conflict of Interest Statement:
There are no conflicts of interest involved within this research study.
Authors’ Roles:
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: LP, CKM, and SHM. Acquisition of data: LP, KAS, MDS, RLH, and VBW. Analysis and interpretation of data: LP, KAS, MDS, VBW, CKM, and SHM. Drafting of the manuscript: LP. Critical revision of the manuscript for important intellectual content: MDS, CKM, and SHM. Statistical analysis: LP, KAS, MDS, and VBW. Obtained funding: CKM and SHM. Administrative, technical, and material support: LP, KAS, MDS, VBW, CKM, and SHM. Study supervision: CKM and SHM.
References
- Amende I, Kale A, McCue S, Glazier S, Morgan JP, Hampton TG. Gait dynamics in mouse models of parkinson’s disease and huntington’s disease. J Neuroeng Rehabil. 2005;2:20. doi: 10.1186/1743-0003-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, Williams D, Brody J, Smeyne RJ. A comparison of model-based (2D) and design-based (3D) stereological methods for estimating cell number in the substantia nigra pars compacta (SNpc) of the C57BL/6J mouse. Neuroscience. 2009;161:1082–1090. doi: 10.1016/j.neuroscience.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbiero JK, Santiago R, Tonin FS, Boschen S, da Silva LM, Werner MF, da Cunha C, Lima MM, Vital MA. PPAR-alpha agonist fenofibrate protects against the damaging effects of MPTP in a rat model of Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2014;53:35–44. doi: 10.1016/j.pnpbp.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Benes FM, Lange N. Two-dimensional versus three-dimensional cell counting: A practical perspective. Trends Neurosci. 2001;24:11–17. doi: 10.1016/s0166-2236(00)01660-x. [DOI] [PubMed] [Google Scholar]
- Berardelli A, Rothwell JC, Thompson PD, Hallett M. Pathophysiology of bradykinesia in Parkinson’s disease. Brain. 2001;124(11):2131–2146. doi: 10.1093/brain/124.11.2131. [DOI] [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- Blandini F, Armentero MT. Animal models of parkinson’s disease. FEBS J. 2012;279:1156–1166. doi: 10.1111/j.1742-4658.2012.08491.x. [DOI] [PubMed] [Google Scholar]
- Blin O, Ferrandez AM, Serratrice G. Quantitative analysis of gait in Parkinson patients: increased variability of stride length. J Neurol Sci. 1990;98(1):91–97. doi: 10.1016/0022-510x(90)90184-o. [DOI] [PubMed] [Google Scholar]
- Castro AA, Wiemes BP, Matheus FC, Lapa FR, Viola GG, Santos AR, Tasca CI, Prediger RD. Atorvastatin improves cognitive, emotional and motor impairments induced by intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration in rats, an experimental model of Parkinson’s disease. Brain Res. 2013;1513:103–116. doi: 10.1016/j.brainres.2013.03.029. [DOI] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Chung EK, Chen LW, Chan YS, Yung KK. Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J Comp Neurol. 2008;511:421–437. doi: 10.1002/cne.21852. [DOI] [PubMed] [Google Scholar]
- Clarke PG. How inaccurate is the abercrombie correction factor for cell counts? Trends Neurosci. 1992;15:211–212. doi: 10.1016/0166-2236(92)90036-8. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Sagar HJ, Jordan N, Harvey NS, Sullivan EV. Cognitive impairment in early, untreated parkinson’s disease and its relationship to motor disability. Brain. 1991;114(Pt 5):2095–2122. doi: 10.1093/brain/114.5.2095. [DOI] [PubMed] [Google Scholar]
- Darvas M, Henschen CW, Pamiter RD. Contributions of signaling by dopamine neurons in dorsal striatum to cognitive behaviors corresponding to those observed in Parkinson’s disease. Neurobiol Dis. 2014;65:112–123. doi: 10.1016/j.nbd.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David HN, Ansseau M, Abraini JH. Dopamine-glutamate reciprocal modulation of release and motor responses in the rat caudate-putamen and nucleus accumbens of “intact” animals. Brain Res Rev. 2005;50:336–360. doi: 10.1016/j.brainresrev.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Dervan AG, Meshul CK, Beales M, McBean GJ, Moore C, Totterdell S, Snyder AK, Meredith GE. Astroglial plasticity and glutamate function in a chronic mouse model of parkinson’s disease. Exp Neurol. 2004;190:145–156. doi: 10.1016/j.expneurol.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Downes JJ, Roberts AC, Sahakian BJ, Evenden JL, Morris RG, Robbins TW. Impaired extra-dimensional shift performance in medicated and unmedicated parkinson’s disease: Evidence for a specific attentional dysfunction. Neuropsychologia. 1989;27:1329–1343. doi: 10.1016/0028-3932(89)90128-0. [DOI] [PubMed] [Google Scholar]
- Dubois B, Pillon B. Cognitive deficits in parkinson’s disease. J Neurol. 1997;244:2–8. doi: 10.1007/pl00007725. [DOI] [PubMed] [Google Scholar]
- Faure A, Haberland U, Conde F, Massioui NE. Lesion to the nigrostriatal dopamine system disrupts stimulus-response habit formation. J Neuroscience. 2005;25:2771–2780. doi: 10.1523/JNEUROSCI.3894-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifel K, Dkhissi-Benyahya O, Cooper HM. Lack of long-term changes in circadian, locomotor, and cognitive functions in acute and chronic MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse models of Parkinson’s disease. Chronobiol Int. 2013;30:741–755. doi: 10.3109/07420528.2012.762011. [DOI] [PubMed] [Google Scholar]
- Fisher BE, Petzinger GM, Nixon K, Hogg E, Bremmer S, Meshul CK, Jakowec MW. Exercise-induced behavioral recovery and neuroplasticity in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse basal ganglia. J Neurosci Res. 2004;77:378–390. doi: 10.1002/jnr.20162. [DOI] [PubMed] [Google Scholar]
- Flowers KA, Robertson C. The effect of parkinson’s disease on the ability to maintain a mental set. J Neurol Neurosurg Psychiatry. 1985;48:517–529. doi: 10.1136/jnnp.48.6.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman M, Oscar-Berman M. Selective delayed response deficits in parkinson’s and alzheimer’s disease. Arch Neurol. 1986;43:886–890. doi: 10.1001/archneur.1986.00520090026011. [DOI] [PubMed] [Google Scholar]
- Frye MA, Watzl J, Banakar S, O’Neill J, Mintz J, Davanzo P, Fischer J, Chirichigno JW, Ventura J, Elman S, Tsuang J, Walot I, Thomas MA. Increased anterior cingulate/medial prefrontal cortical glutamate and creatine in bipolar depression. Neuropsychopharm. 2007;32:2490–2499. doi: 10.1038/sj.npp.1301387. [DOI] [PubMed] [Google Scholar]
- Goldberg NR, Haack AK, Lim NS, Janson OK, Meshul CK. Dopaminergic and behavioral correlates of progressive lesioning of the nigrostriatal pathway with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience. 2011a;180:256–271. doi: 10.1016/j.neuroscience.2011.02.027. [DOI] [PubMed] [Google Scholar]
- Goldberg NR, Hampton T, McCue S, Kale A, Meshul CK. Profiling changes in gait dynamics resulting from progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced nigrostriatal lesioning. J Neurosci Res. 2011b;89:1698–1706. doi: 10.1002/jnr.22699. [DOI] [PubMed] [Google Scholar]
- Goldberg NR, Meshul CK. Effect of intermittent washout periods on progressive lesioning of the nigrostriatal pathway with 1-methyl-2-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Neuroscience. 2011;182:203–207. doi: 10.1016/j.neuroscience.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Goldberg NR, Fields V, Pflibsen L, Salvatore MF, Meshul CK. Social enrichment attenuates nigrostriatal lesioning and reverses motor impairment in a progressive 1-methyl-2-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. Neurobiology of Disease. 2012;45:1051–1067. doi: 10.1016/j.nbd.2011.12.024. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharm. 2004;174:3–16. doi: 10.1007/s00213-004-1793-y. [DOI] [PubMed] [Google Scholar]
- Gubner NR, Wilhelm CJ, Phillips TJ, Mitchell SH. Strain differences in behavioral inhibition in a Go/No-go Task demonstrated using 15 inbred mouse strains. Alcohol: Clin Exp Res. 2010;34(1):1–10. doi: 10.1111/j.1530-0277.2010.01219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett M, Khoshbin S. A physiological mechanism of bradykinesia. Brain. 1980;103(2):301–314. doi: 10.1093/brain/103.2.301. [DOI] [PubMed] [Google Scholar]
- Hausdorff JM, Cudkowicz ME, Firtion R, Wei JY, Goldberger AL. Gait variability and basal ganglia disorders: Stride-to-stride variations of gait cycle timing in Parkinson’s disease and Huntington’s disease. Movement Disord. 1998;13(3):428–437. doi: 10.1002/mds.870130310. [DOI] [PubMed] [Google Scholar]
- Henderson JM, Carpenter K, Cartwright H, Halliday GM. Degeneration of the centré median-parafascicular complex in Parkinson’s disease. Ann Neurol. 2000a;47:345–352. [PubMed] [Google Scholar]
- Henderson JM, Carpenter K, Cartwright H, Halliday GM. Loss of thalamic intralaminar nuclei in progressive supranuclear palsy and Parkinson’s disease: clinical and therapeutic implications. Brain. 2000b;123:1410–1421. doi: 10.1093/brain/123.7.1410. [DOI] [PubMed] [Google Scholar]
- Hietanen M, Teravainen H. Cognitive performance in early parkinson’s disease. Acta Neurol Scand. 1986;73:151–159. doi: 10.1111/j.1600-0404.1986.tb03257.x. [DOI] [PubMed] [Google Scholar]
- Holmer HK, Keyghobadi M, Moore C, Menashe RA, Meshul CK. Dietary restriction affects striatal glutamate in the MPTP-induced mouse model of nigrostriatal degeneration. Synapse. 2005a;57:100–112. doi: 10.1002/syn.20163. [DOI] [PubMed] [Google Scholar]
- Holmer HK, Keyghobadi M, Moore C, Meshul CK. L-dopa-induced reversal in striatal glutamate following partial depletion of nigrostriatal dopamine with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neuroscience. 2005b;136:333–341. doi: 10.1016/j.neuroscience.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O. Biochemical aspects of Parkinson’s disease. Neurology. 1998;51:S2–9. doi: 10.1212/wnl.51.2_suppl_2.s2. [DOI] [PubMed] [Google Scholar]
- Hung HC, Lee EH. The mesolimbic dopaminergic pathway is more resistant than the nigrostriatal dopaminergic pathway to MPTP and MPP+ toxicity: role of BDNF gene expression. Brain Res Mol Brain Res. 1996;41:14–26. doi: 10.1016/0169-328x(96)00062-9. [DOI] [PubMed] [Google Scholar]
- Ho SC, Hsu CC, Pawlak CR, Tikhonova MA, Lai TJ, Amstislavskaya TG, Ho YJ. Effects of ceftriaxone on the behavioral and neuronal changes in an MPTP-induced Parkinson’s disease rat model. Behav Brain Res. 2014;268:177–184. doi: 10.1016/j.bbr.2014.04.022. [DOI] [PubMed] [Google Scholar]
- Kale A, Amende I, Meyer GP, Crabbe JC, Hampton TG. Ethanol’s effects on gait dynamics in mice investigated by ventral plane videography. Alcohol Clin Exp Res. 2004;28:1839–1848. doi: 10.1097/01.alc.0000148103.09378.81. [DOI] [PubMed] [Google Scholar]
- Kamens HM, Crabbe JC. The parallel rod floor test: A measure of ataxia in mice. Nat Protoc. 2007;2:277–281. doi: 10.1038/nprot.2007.19. [DOI] [PubMed] [Google Scholar]
- Karachi C, Grabli D, Bernard FA, Tande D, Wattiez N, Belaid H, Bardinet E, Prigent A, Nothacker H-P, Hunot S, Hartmann A, Lehericy S, Hirsch EC, Francois C. Cholinergic mesencephalic neurons are involved in gait and postural disorders in Parkinson disease. J Clin Investigation. 2010;120(8):2745–2754. doi: 10.1172/JCI42642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Kaundal RK, More S, Sharma SS. Beneficial effects of pioglitazone on cognitive impairment in MPTP model of Parkinson’s disease. Behav Brain Res. 2009;197:398–403. doi: 10.1016/j.bbr.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Lanoue AC, Blatt GJ, Soghomonian JJ. Decreased parvalbumin mRNA expression in DL prefrontal cortex in Parkinson’s disease. Brain Research. 2013 doi: 10.1016/j.brainres.2013.07.025. pii:S0006–8993 (13)01012–3. doi:10.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoue AC, Dumitriu A, Myers RH, Soghomonian JJ. Decreased glutamic acid decarboxylase mRNA expression in prefontal cortex in Parkinson’s disease. Exp Neurol. 2010;226:207–217. doi: 10.1016/j.expneurol.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin BE, Llabre MM, Weiner WJ. Cognitive impairments associated with early parkinson’s disease. Neurology. 1989;39:557–561. doi: 10.1212/wnl.39.4.557. [DOI] [PubMed] [Google Scholar]
- Li LB, Nianhang C, Sammanda R, Limen C, Xiao-Nan C, Wang Lijuan C, Reith Maarten EA. The role of N-glycosylation in function and surface trafficking of the human dopamine transporter. J Biol Chem. 2004;279(20):21012–21020. doi: 10.1074/jbc.M311972200. [DOI] [PubMed] [Google Scholar]
- Luchtman DW1, Meng Q, Song C. Ethyl-eicosapentaenoate (E-EPA) attenuates motor impairments and inflammation in the MPTP-probenecid mouse model of Parkinson’s disease. Behav Brain Res. 2012;226:386–396. doi: 10.1016/j.bbr.2011.09.033. [DOI] [PubMed] [Google Scholar]
- Marin C, Bonastre M, Aguilar E, Jiménez A. The metabotropic glutamate receptor antagonist 2-methyl-6-(phenylethynyl) pyridine decreases striatal VGlut2 expression in association with an attenuation of L-DOPA-induced dyskinesias. Synapse. 2011;65:1080–1086. doi: 10.1002/syn.20941. [DOI] [PubMed] [Google Scholar]
- Massie A, Schallier A, Vermoesen K, Arckens L, Michotte Y. Biphasic and bilateral changes in striatal VGLUT1 and 2 protein expression in hemi-Parkinson rats. Neurchemistry International. 2010;57:111–118. doi: 10.1016/j.neuint.2010.04.019. [DOI] [PubMed] [Google Scholar]
- McDowell K, Chesselet MF. Animal models of the non-motor features of parkinson’s disease. Neurobiol Dis. 2012;46:597–606. doi: 10.1016/j.nbd.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen AM1, Burgess DT, Hanstock CC, Seres P, Khalili P, Newman SC, Baker GB, Mitchell ND, Khudabux-Der J, Allen PS, LeMelledo JM. Increased glutamate levels in the medial prefrontal cortex in patients with postpartum depression. Neuropsychopharm. 2012;37:2428–2435. doi: 10.1038/npp.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeorge AJ, Faull RL. The organization of the projection from the cerebral cortex to the striatum in the rat. Neuroscience. 1989;29:503–537. doi: 10.1016/0306-4522(89)90128-0. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Kang UJ. Behavioral models of Parkinson’s disease: A new look at an old problem. Mov Disord. 2006;21(10):1595–1606. doi: 10.1002/mds.21010. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S, Beales M, Meshul CK. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of parkinson’s disease. Exp Neurol. 2009;219:334–340. doi: 10.1016/j.expneurol.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith GE, Sonsalla PK, Chesselet MF. Animal models of parkinson’s disease progression. Acta Neuropathol. 2008;115:385–398. doi: 10.1007/s00401-008-0350-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshul CK, Emre N, Nakamura CM, Allen C, Donohue MK, Buckman JF. Time-dependent changes in striatal glutamate synapses following a 6-hydroxydopamine lesion. Neuroscience. 1999;88:1–16. doi: 10.1016/s0306-4522(98)00189-4. [DOI] [PubMed] [Google Scholar]
- Morris ME, Huxham F, McGinley J, Dodd K, Iansek R. The biomechanics and motor control of gait in Parkinson’s disease. Clin Biomechan. 2001;16(6):459–470. doi: 10.1016/s0268-0033(01)00035-3. [DOI] [PubMed] [Google Scholar]
- Morris ME, Matyas TA, Iansek R, Summers JJ. Temporal stability of gait in Parkinson’s disease. Physical Therapy. 1996;76(7):763–777. doi: 10.1093/ptj/76.7.763. [DOI] [PubMed] [Google Scholar]
- Muly EC, 3rd, Szigeti O, Goldman-Rakic PS. D1 receptor in interneurons of macaque prefrontal cortex: distribution and subcellular localization. J Neurosci. 1998;18:10553–10565. doi: 10.1523/JNEUROSCI.18-24-10553.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen AM, James M, Leigh PN, Summers BA, Marsden CD, Quinn NP, Lange KW, Robbins TW. Fronto-striatal cognitive deficits at different stages of parkinson’s disease. Brain. 1992;115(Pt 6):1727–1751. doi: 10.1093/brain/115.6.1727. [DOI] [PubMed] [Google Scholar]
- Paspalas CD, Goldman-Rakic PS. Presynaptic D1 dopamine receptors in primate prefrontal cortex: target-specific expression in the glutamatergic synapse. J Neurosci. 2005;25:1260–1267. doi: 10.1523/JNEUROSCI.3436-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattij T, Vanderschuren LJ. The neuropharmacology of impulsive behaviour. Trends Pharmacol Sci. 2008;29:192–199. doi: 10.1016/j.tips.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Gulf Professional Publishing; 2004. [Google Scholar]
- Phani S, Gonye G, Lacovitte L. VTA neurons show a potentially protective transcriptional response to MPTP. 2010. Brain Res. 2010;1343:1–13. doi: 10.1016/j.brainres.2010.04.061. [DOI] [PubMed] [Google Scholar]
- Plotnik M, Hausdorff JM. The role of rhythmicity and bilateral coordination of stepping in the pathophysiology of freezing of gait in Parkinson’s disease. Movement Disord. 2008;23(2):444–450. doi: 10.1002/mds.21984. [DOI] [PubMed] [Google Scholar]
- Raju DV, Ahern TH, Shah DJ, Wright TM, Standaert DG, Hall RA, Smith Y. Differential synaptic plasticity of the corticostriatal and thalamostriatal systems in an MPTP-treated monkey model of parkinsonism. Eur J Neurosci. 2008;27:1647–1658. doi: 10.1111/j.1460-9568.2008.06136.x. [DOI] [PubMed] [Google Scholar]
- Reyes S, Cottam V, Kirik D, Double KL, Halliday GM. Variability in neuronal expression of dopamine receptors and transporters in the substantia nigra. Movement Disorder. 2013 doi: 10.1002/mds.25493. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Riederer P, Wuketich S. Time course of nigrostriatal degeneration in Parkinson’s disease. J Neural Transm. 1976;38:277–301. doi: 10.1007/BF01249445. [DOI] [PubMed] [Google Scholar]
- Robinson S, Freeman P, Moore C, Touchon JC, Krentz L, Meshul CK. Acute and subchronic MPTP administration differentially affects striatal glutamate synaptic function. Exp Neurol. 2003;180:74–87. doi: 10.1016/s0014-4886(02)00050-x. [DOI] [PubMed] [Google Scholar]
- Schaafsma JD, Giladi N, Balash Y, Bartels AL, Urevich GT, Hausdorff JM. Gait dynamics in Parkinson’s disease: relationship to Parkinsonian features, falls and response to levodopa. J Neurol Sci. 2003;212(1–2):47–53. doi: 10.1016/s0022-510x(03)00104-7. [DOI] [PubMed] [Google Scholar]
- Schneider JS. Chronic exposure to low doses of MPTP. II. neurochemical and pathological consequences in cognitively-impaired, motor asymptomatic monkeys. Brain Res. 1990;534:25–36. doi: 10.1016/0006-8993(90)90108-n. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Kovelowski CJ., 2nd Chronic exposure to low doses of MPTP. I. cognitive deficits in motor asymptomatic monkeys. Brain Res. 1990;519:122–128. doi: 10.1016/0006-8993(90)90069-n. [DOI] [PubMed] [Google Scholar]
- Sconce MD, Churchill MJ, Moore C, Meshul CK. Intervention with 7, 8-dihydroxyflavone blocks further striatal terminal loss and restores motor deficits in a progressive mouse model of Parkinson’s disease. Neuroscience. 2015;290:454–471. doi: 10.1016/j.neuroscience.2014.12.080. [DOI] [PubMed] [Google Scholar]
- Sconce MD, Churchill MJ, Greene RE, Meshul CK. Intervention with exercise restores motor deficits but not nigrostriatal loss in a progressive MPTP mouse model of Parkinson’s disease. Neuroscience. 2015;299:156–174. doi: 10.1016/j.neuroscience.2015.04.069. [DOI] [PubMed] [Google Scholar]
- Sharpe MH. Auditory attention in early parkinson’s disease: An impairment in focused attention. Neuropsychologia. 1992;30:101–106. doi: 10.1016/0028-3932(92)90019-i. [DOI] [PubMed] [Google Scholar]
- Sharpe MH. Distractibility in early parkinson’s disease. Cortex. 1990;26:239–246. doi: 10.1016/s0010-9452(13)80353-x. [DOI] [PubMed] [Google Scholar]
- Smolen AJ, Wright LL, Cunningham TJ. Neuron numbers in the superior cervical sympathetic ganglion of the rat: A critical comparison of methods for cell counting. J Neurocytol. 1983;12:739–750. doi: 10.1007/BF01258148. [DOI] [PubMed] [Google Scholar]
- Tieu K. A guide to neurotoxic animal models of parkinson’s disease. Cold Spring Harb Perspect Med. 2011;1:a009316. doi: 10.1101/cshperspect.a009316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuckovic MG, Wood RI, Holschneider DP, Abernathy A, Togasaki DM, Smith A, Petzinger GM, Jakowec MW. Memory, mood, dopamine, and serotonin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse model of basal ganglia injury. Neurobiol Dis. 2008;32:319–327. doi: 10.1016/j.nbd.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm CJ, Mitchell SH. Rats bred for high alcohol drinking are more sensitive to delayed and probabilistic outcomes. Genes Brain Behav. 2008;7:705–713. doi: 10.1111/j.1601-183X.2008.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GV, Castner SA. Under the curve: critical issues for elucidating D1 receptor function in working memory. Neurosci. 2006;139:263–276. doi: 10.1016/j.neuroscience.2005.09.028. [DOI] [PubMed] [Google Scholar]
- Wu Y, Krishnan S. Statistical analysis of gait rhythm in patients with Parkinson’s disease. IEEE Transactions on Neural Systems and Rehabilitation Engineering. 2010;18(2):150–158. doi: 10.1109/TNSRE.2009.2033062. [DOI] [PubMed] [Google Scholar]