

Abstract
It has been shown that vitamin C (VC) is transported at synaptic boutons, but how this occurs has not been elucidated. In this study, we investigated the role of the sodium-dependent vitamin C transporter-2 (SVCT2) in transporting VC at the cortical nerve terminal. Immunostaining of cultured mouse superior cervical ganglion cells showed the SVCT2 to be expressed in presynaptic boutons, co-localizing with the vesicular monoamine transporter-2 and the norepinephrine transporter. Immunoblotting of enriched cortical synaptosomes demonstrated that the SVCT2 was enriched in presynaptic fractions, confirming a predominant presynaptic location. In crude synaptosomes, known inhibitors of SVCT2 inhibited uptake of VC. Further, the kinetic features of VC uptake were consistent with SVCT2-mediated function. VC was also found to efflux from synaptosomes by a mechanism not involving the SVCT2. Indeed, VC efflux was substantially offset by re-uptake of VC on the SVCT2. The presence and function of the SVCT2 at the presynaptic nerve terminal suggest that it is the transporter responsible for recovery of VC released into the synaptic cleft.
Keywords: Synaptosome, Antioxidant, Ganglia, Cortical neurons, Membrane transport
Graphical abstract
The vitamin C (VC) transporter, SVCT2, co-localizes with the vesicular monoamine transporter-2 and the norepinephrine transporter in the presynaptic boutons of cultured mouse superior cervical ganglion cells. The SVCT2 is also functional in cortical synaptosomes suggesting that it is responsible for the recovery of VC released into the synaptic cleft.
Introduction
Vitamin C (VC), an antioxidant not synthesized in humans, is an essential nutrient in the central nervous system (CNS) and in neurons in particular (Wilson 1997; Hediger 2002). A balance of transport into and out of the cells determines the neuronal content of VC. Neuronal VC uptake appears to be mediated exclusively by the Sodium-dependent Vitamin C Transporter-type 2 (SVCT2) (Harrison and May 2009), consistent with the observation that mice lacking the SVCT2 have very low levels of VC in brain tissue despite the ability to synthesize the vitamin on their own (Sotiriou et al. 2002). The transporter is sodium- and energy-dependent, accumulating VC against its chemical gradient by co-transporting sodium cations into cells down an electrochemical gradient established by the membrane bound Na+/K+-ATPase (Godoy et al. 2006). This transporter can concentrate VC many-fold. For example, whereas cerebrospinal fluid VC levels are 200-300 μM, brain tissue levels are in the low millimolar range (Harrison and May 2009).
Despite its hydrophilicity and negative charge at physiologic pH, VC can efflux from astrocytes and neurons, especially in response to glutamate uptake (May et al. 2006). Mechanisms for VC efflux in the brain are unclear, but are mediated in part by channel proteins (Rice 2000; Wilson 2005). For example, glutamate uptake causes cell swelling that in turn triggers volume-regulated anion channels (VRACs) to release osmolytes to prevent swelling-induced cell lysis. VC appears to be one of the substances released on VRACs in astrocytes (Wilson et al. 2000) and neurons (May et al. 2006).
Whether and how VC is transported in specialized organelles within neurons is unknown and has received less attention. Studies in primary cultures of mouse hippocampal neurons suggested that the SVCT2 is localized to the plasma membrane of the cell body and punctate structures in axons (Qiu et al. 2007). These structures might contain mitochondria (Azzolini et al. 2013; Munoz-Montesino et al. 2014) or even synaptic vesicles headed for synapses. Indeed, VC uptake and efflux has been described in synaptosomes (Grunewald and Fillenz 1984), but whether either phase involves the SVCT2 is unknown. Therefore, we performed the studies described in this paper to investigate SVCT2 expression and function in VC uptake in synaptosomes.
Materials and Methods
Animal Mice, Wild type (Gulo+/+; SVCT2 +/+)
Mice denoted as “wild-type mice” are from C57BL/6J background. These mice were originally obtained from Jackson laboratories (stock #000664, RRID: IMSR_JAX:000664) and a colony was maintained in-house. All animals were housed in tub cages in a temperature- and humidity-controlled vivarium on a 12:12-h light:dark cycle with lights on at 6 AM. All procedures conformed to Institutional Animal Use Committee guidelines. Mice had free access to food and water for the duration of experiment and were 4-6 months old.
Superior cervical ganglion (SCG) neuronal preparation and culture
SCGs were cultured as previously described (Matthies et al. 2009). Briefly, superior cervical ganglia from 1–3 day old wild type pups (C57Bl/6J) were dissected and incubated for 30 min at 37°C in collagenase (3 mg/ml; Sigma) and trypsin (0.5 mg/mL/Gibco). The digestion was terminated by treating for 2 h at 37°C with 10% fetal bovine serum (FBS, Atlanta Biologicals) in UltraCulture medium (BioWhittaker). Enrichment of neurons was accomplished by differentially plating for 2 hours on dishes in UltraCulture supplemented with 3% FBS, nerve growth factor (NGF, 20 ng/mL; Harlan) and 2 mM L-glutamine. Neurons were collected, centrifuged for 5 min, suspended in supplemented UltraCulture medium and transferred onto poly-D-lysine and collagen-coated glass-bottomed MatTek dishes. After 24 h of incubation at 37°C, cultures were treated with 1 μM 5-fluoro-5-deoxyuridine (FdU; Sigma) and grown for 14–28 days in supplemented UltraCulture medium prior to examination (Matthies et al. 2009).
Immunostaining and microscopy
Cultured superior cervical ganglion (SCG) neurons were immunostained as previously described (Matthies et al, 2009). In short, cultured SCG neurons were serum starved for 1 h in DMEM:F12 and subsequently fixed with PBS containing 0.9 mM CaCl2 and 0.049 mM MgCl2 (Ca2+/Mg2+) and 4% paraformaldehyde, washed three times with PBS with Ca2+/Mg2+, permeabilized, and blocked with PBS/4% bovine serum albumin (BSA)/0.15% Tween-20. Cultures were immunostained using the following antibodies, goat polyclonal anti-SVCT2 (Santa Cruz Biotechnology, SC-9926, RRID: AB_661205), rabbit polyclonal anti-vesicular monoamine transporter-2 (Santa Cruz Biotechnology, SC-15314, RRID: AB_2187867), mouse monoclonal anti-norepinephrine transporter (MAb Technologies, NET05-2) dissolved in PBS plus 4% BSA and 0.05% Tween-20. Both anti-SVCT2 and anti-NET immunoreactivities have been verified using the respective mouse knockouts (Qiu et al. 2007;Matthies et al. 2009). Primary antibodies were visualized with the appropriate covalently Alexa-labeled secondary antibody from Invitrogen. Immunofluorescence was imaged using a PerkinElmer UltraView confocal with a Nikon Eclipse 2000-U microscope equipped with a 60× lens with a numerical aperture of 1.49. Z-series were taken and shown are sections at the widest point of boutons (center of the bouton). Image processing was performed using ImageJ (RRID: nif-0000-30467) and Adobe Photoshop (RRID: SciRes_000161).
Synaptosome preparations and functional assessments
Before obtaining brain tissue for synaptosome preparation, mice were deeply anesthetized with isoflurane and killed quickly by decapitation.
Crude Synaptosome Preparation
Crude synaptosomes were prepared as described by Shirey-Rice, et al. (Shirey-Rice et al. 2013). The whole brain cortex was removed and added to 9 ml of homogenization buffer (0.32 M sucrose/4.3 mM HEPES solution, 1.54 μM aprotinin, 10.7 μM leupeptin, 0.948 μM pepstatin, and 200 μM phenylmethylsulfonyl fluoride, pH 7.4) in a 10 mL glass homogenizer tube. The mixture was homogenized with a hand-held Teflon pestle (approximately 7 strokes). The homogenate was then transferred to a 15 mL plastic tube for centrifugation at 1000 g for 5 min at 4°C. The supernatant (S1) was then transferred to a 15 mL plastic tube and then centrifuged at 12,000 ×g for 15 min at 4°C. The pellet was suspended in assay buffer and then transferred to an Eppendorf tube for assays.
Enriched Synaptosome Preparation
Enriched synaptosomes were prepared as described by Betke, et al. (Betke et al. 2014) with modifications. Crude synaptosomes were obtained similar to protocol above with the following additions for enrichment. Supernatant (S1) was transferred to 50 ml a conical tube containing S1a. The supernatant was brought to 20 ml with 0.32 M sucrose solution and centrifuged at 10,000 ×g at 4°C for 20 min to produce P2 pellet (contains crude synaptosomes, mitochondria, and microsomes). P2 pellets were gently suspended in 3.4 ml of 0.32 M sucrose solution and aliquot was taken for Western blotting. The remaining suspension of the P2 pellet was applied to a discontinuous sucrose gradient. This was prepared in Beckman 15 ml polycarbonate tubes (Cat. #342080) in which 3 ml of 0.8 M, 1.0 M and 1.2 M sucrose solutions were slowly layered in that order. Three milliliters of crude synaptosomes were layered on top of gradient and balanced with 0.32 M sucrose solution. Samples were centrifuged at 100,000 ×g in a Beckman Optima LE-80K ultracentrifuge using an SW 40 Ti rotor at 4°C for 2 hours. Enriched synaptosomes were collected from the interface of the 1.0 M and 1.2 M solutions and transferred to clean 15 ml conical tubes. Fractions were diluted with 3 ml of 0.32 M sucrose solution and centrifuged in same rotor at 100,000 ×g for 30 min at 4°C.
Synaptosome Fractionation
Pre- and post-synaptic fractions of the enriched synaptosome preparation were obtained by the protocol of Betke, et al. (Betke et al. 2014) with modifications. The P2 pellet from an enriched or crude synaptosome preparation was suspended in 4 ml hypotonic lysis buffer (Same as above with the addition of 0.1 mM CaCl2 and 1 mM MgCl2, pH 7.4). This was incubated on ice for 20 min to lyse membranes and transferred to 5 ml ultracentrifuge tubes (Beckman Ultra Clear, #344057). Samples were centrifuged at 100,000 ×g in a SW 55 Ti rotor in a Beckman centrifuge (Optima LE-80K) for 120 min at 4°C. The supernatant (S3), which contains cytosolic and perisynaptic proteins from both pre- and post-synaptic terminals, was transferred to another tube and placed on ice. Pellet P3 was suspended in 1 ml of Tris buffer (20 mM Tris (pH 8.0), 1% Triton X-100, 1.54 μM aprotinin, 10.7 μM leupeptin, 0.948 μM pepstatin, and 200 μM PMSF). This was transferred to plastic 2 ml Eppendorf tubes and incubated on ice for 20 min. The samples were centrifuged at 10,000 ×g for 30 min at 4°C, and the supernatant (S4) containing the presynaptic fraction was transferred to Eppendorf tubes. The pellet (P4) was suspended in 200 μl of 1× PBS/1% SDS and centrifuged at 10,000 ×g for 30 min to obtain the supernatant (S5) containing the postsynaptic fraction as described. To ensure adequate amounts of cytosolic/perisynaptic protein were studied, samples were concentrated using 3K concentrator tubes (Amincon Ultra, Millipore) that were centrifuged at 3400 rpm at 4°C until volumes reached ∼250 μl, whereupon membranes were washed with homogenization buffer, and transferred to new Eppendorf tubes for assays.
Transport assays
All pharmacologic inhibitors including sulfinpyrazone (S9509), phloretin (P7912), and 3-O-methylglucose (M4879) were purchased from Sigma-Aldrich. VC (L-Ascorbic acid, Sigma-Aldrich A4403) was added to samples when appropriate. All uptake and efflux assays with synaptosomes were performed in KRH assay buffer (10 mM HEPES, 128 mM, 5.2 mM KCl, 1 mM NaH2PO4, 2.2 mM MgSO4 1.2 mM CaCl2, 10 mM glucose, pH 7.4) at 4°C.
Radioactive VC Assays
Crude synaptosomes were transferred to glass reaction tubes and preincubated for 10 min at 37°C. Transport was started by adding indicated concentrations of 14C-VC (L-[1-14C] ascorbic acid, 4 μCi/mmol, Perkin Elmer, NEC146050UC). Transport was stopped at the indicated times by filtrations of synaptosomes over 0.3% polyethylenimine-coated glass fiber filters (Whatman GF/B; Whatman) using a cell harvester (Brandel Inc., Gaithersburg, MD) and ice-cold wash buffer (assay buffer without glucose). Radioactive VC contents accumulated on filters were determined by liquid scintillation counting (Perkin-Elmer, Tri-Carb 2910 TR). Mean values for specific uptake (pmol/mg protein ± SEM) were determined from at least three experiments. Background 14C-VC signal was determined by assessing the signal detected after lysing synaptosomes. This amount was then subtracted from all of the readings.
Unlabeled VC Assays
Assay was started by transferring crude synaptosomes to 1.5 mL Eppendorf tubes and placing these in a water bath at 37°C for allotted times. Transport was initiated by adding the indicated concentration of unlabeled VC. Transport was stopped at the indicated times with 1 mL of ice-cold wash buffer (assay buffer without glucose). Synaptosomes were then pelleted by centrifugation at 16,200 ×g for 2 min. VC was measured as ascorbic acid in metaphosphoric acid extracts of tissues as described (Harrison et al. 2008) using ion-pair HPLC with electrochemical detection.
Fura Red-AM Assay
Crude synaptosome preparations were pelleted and suspended in 1.3 mL of Fura Red-AM™, (Invitrogen, F3021) solution, containing 5 mM glucose and 4 μM Fura Red-AM™ dye. The latter is a visible light–excitable Fura-2 analog that can cross cell membranes but is trapped within cells or sealed synaptosomes (Yates et al. 1992). The mixture was then incubated in a 37°C bath for 30 min, pelleted and suspended in 1.3 mL of the assay buffer. Following this, 50 μL of the preparation was treated as noted in text at 37°C. Reaction was stopped by addition of 1 mL of ice-cold assay buffer (no glucose), centrifuged and lysed with 60% methanol in water. 100 μL aliquots of the supernatant were transferred to a clear plate in a microtiter plate reader and fluorescence was read at excitation 420/emission 610 nm.
Western blot analysis
Immunoblotting was performed as previously described in (Qiao and May 2008). Solubilized protein (5-20 μg) was subjected to SDS-polyacrylamide gel electrophoresis (7.5% gel) according to Laemmli's method (Laemmli and Favre 1973). Primary antibodies used for immunoblotting were obtained from several sources. Santa Cruz Biotechnology provided antibodies to SVCT2 (SC-9926, 1:200), vesicular monoamine transporter-2 (VMAT2, SC-15314, RRID: AB_2187867, 1:200), actin (SC-1616 RRID: AB_10160631, 1:400), and G-protein β subunit (GβP, SC-378, RRID: AB_631542, 1:10000). Other antibodies included glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Millipore, MAB-374, 1:20000), N-methyl-D-aspartate-1 (NMDAR1, BD Biosciences, 556308, RRID: AB_396353, 1:2000), and postsynaptic density protein-95 (PSD-95, Neuromab, 75-028, RRID: AB_2307331 1:20000). Secondary antibodies (1:10000-20000) that were used for immunodetection are the following: anti-rabbit IgG-conjugated horseradish peroxidase (Santa Cruz Biotechnology, SC-2030, RRID: AB_631747), anti-goat IgG-conjugated horseradish peroxidase (Santa Cruz Biotechnology, SC-2020, RRID: AB_631728), goat anti-rabbit IgG-conjugated horseradish peroxidase (PerkinElmer, NEF812001EA) and goat anti-mouse (PerkinElmer, NEF822E001EA). Antibody detection was performed with Western Lightening Plus-ECL Enhanced Chemiluminescence Substrate Kit (Perkin Elmer) and Carestream Kodak BioMax MR Film (Kodak, Rochester, NY). Band intensity from X-ray film detection was analyzed by densitometry using ImageJ software from the National Institutes of Health. Antibodies were used as described in Table 1.
Table 1. Antibodies Used in Methods.
| Antibody Name | Immunogen Structure | Manufacturer, Catalog #, RRID, Species, Type (mono- poly- clonal) | Concentration Used |
|---|---|---|---|
| Primary | |||
| SVCT2 | Epitope mapping near the N-terminus of SVCT2 of rat origin | Santa Cruz Biotechnology, SC-9926, RRID: AB_661205, goat polyclonal | 1:200 |
| VMAT2 | Epitope corresponding to amino acids 44-133 mapping near the N-terminus of synaptic vesicle monoamine transporter 2 (VMAT 2) of human origin | Santa Cruz Biotechnology, SC-15314, RRID: AB_2187867, rabbit polyclonal | 1:200-400 |
| NET | Raised against a peptide (amino acids 05-17) coupled to KLH by the addition of an C-terminal cysteine | MAb Technologies, NET05, mouse monoclonal | |
| Actin | Epitope mapping at the C-terminus of Actin of human origin | Santa Cruz Biotechnology, SC-1616, RRID: AB_10160631, goat polyclonal | 1:400 |
| GβP | Epitope mapping at the C-terminus of Gβ of mouse origin | Santa Cruz Biotechnology, SC- 378, RRID: AB_631542, rabbit polyclonal | 1:10000 |
| GAPDH | Antibody clone number 6C5, Glyceraldehyde-3-phosphate dehydrogenase from rabbit muscle. | Milllipore, MAB-374, mouse monoclonal | 1:20000 |
| NMDAR1 | Rat NMDAR1 aa. 660-811 Recombinant Protein | BD Biosciences, 556308, RRID: AB_396353, Mouse monoclonal | 1:2000 |
| PSD-95 | Fusion protein amino acids 77-299 (PDZ domains 1 and 2) of human PSD-95 (also known as Postsynaptic density protein 95, Disks large homolog 4, Synapse-associated protein 90, DLG4, Dlgh4 and SAP-90, accession number P78352). Clone K28/43 | Neuromab, 75-028, RRID: AB_2307331, mouse monoclonal | 1:20000 |
| Secondary | |||
| Anti-rabbit IgG-conjugated horseradish peroxidase | Cruz Marker™ compatible secondary antibodies recognize an epitope common to each of the Cruz Marker™ molecular weight standards | Santa Cruz Biotechnology, SC-2030, RRID: AB_631747, Goat polyclonal | 1:20000-30000 |
| Anti-goat IgG-conjugated horseradish peroxidase | Cruz Marker™ compatible secondary antibodies recognize an epitope common to each of the Cruz Marker™ molecular weight standards | Santa Cruz Biotechnology, SC-2020, RRID: AB_631728, donkey polyclonal | 1:5000-10000 |
| Anti-Rabbit IgG-conjugated horseradish peroxidase | Rabbit IgG | PerkinElmer, NEF812001EA, goat polyclonal | 1:20000 |
| Anti-mouse IgG- | Mouse-IgG | PerkinElmer, NEF822E001EA, goat | 1:10000-20000 |
| Conjugated horseradish peroxidase | polyclonal | ||
Statistics
Graph Pad Prism 6 software, (La Jolla, CA) was used to determine statistical significance (by one-way ANOVA or unpaired t-testing, as indicated), apparent Km and Vmax values (by hyperbolic Michaelis-Menten analysis), and for least-squares fitting of monoexponential decay. For the latter, data was logarithmically transformed and fitted using linear regression; with line slopes compared using the extra sum-of-squares test.
Results
Neuronal SVCT2 localization
To determine if the SVCT2 is found in presynaptic compartments in neurons we utilized cultured mouse SCG, which have large presynaptic boutons (∼5 μm in diameter). This allows determination of presynaptic localization. Using confocal microscopy, it is possible to take a section through the center of the bouton that forms the presynaptic side of the synaptic cleft. Since it shows proteins located in the presynaptic bouton membrane and in vesicles just below it, this approach can be used to determine whether proteins localize to this important region. Therefore, SCGs were triple-immunostained with antibodies to the norepinephrine transporter (NET) (Figure 1A), to the SVCT2 (Figure 1B), and to the VMAT2 (Figure 1C). NET and VMAT2 are found in the presynaptic boutons in these cultured SCGs (Figure 1A, C; (Matthies et al. 2009). Co-localization of the SVCT2 with either of these two transporters would help to demonstrate that SVCT2 can localize to a presynaptic compartment. The SVCT2 antibody stained axons where NET was also found and presynaptic structures including varicosities and boutons that corresponded to the predominant localization of VMAT (Figure 1D). SVCT2 localizes to the boutons (Figure 1D and 1E, insets), overlapping the distribution of both NET and VMAT. This indicates that the SVCT2 can be expressed at presynaptic sites most likely at the plasma membrane with NET and in internal organelles.
Figure 1. Immunostaining of the superior cervical ganglion presynaptic bouton.
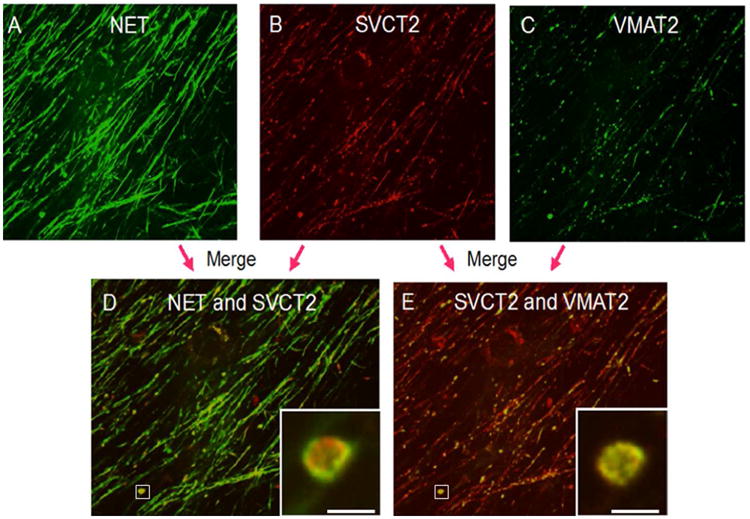
Top panel: Immunostaining for [A] norepinephrine transporter, [B] sodium-dependent vitamin C transporter-2 and [C] vesicular monoamine transporter-2. Bottom panel: Co-localization of transporters, merging of immunostaining images of [D] NET and SVCT2 and [E] SVCT2 and VMAT2. Scale bar is 5 microns. Inset comes from lower magnification area denoted in the white box.
NET, norepinephrine transporter; SVCT2, sodium-dependent vitamin C transporter-2; VMAT2, vesicular monoamine transporter-2
SVCT2 expression at the cortical presynaptic terminal
To assess the location of SVCT2 protein within the nerve terminal, we prepared synaptosomes from cortical tissue of male wild-type mice 4-6 months of age. Enriched synaptosome preparations were separated into three fractions: (1) cytosolic, which contains mostly detergent-soluble cytosolic proteins as well as synaptic vesicles and perisynaptic membrane fragments, (2) presynaptic, and (3) postsynaptic. The relative enrichment of synaptic proteins was compared to whole synaptosomes (unfractionated preparation) and assessed by Western blotting (Figure 2A and B). The cytosolic marker glyceraldehyde 3-phosphate dehydrogenase, (GAPDH, Figure 2C), was enriched in the cytosolic fraction. VMAT2 (Figure 2D), was also found in the cytosolic fraction, but was more prominent in the presynaptic fraction. Two proteins located in the postsynaptic terminal, NMDAR1 (Figure 2E) and PSD-95 (Figure 2F) were enriched in the postsynaptic fraction. These markers confirmed that the fractionation procedure enriches sub-cellular proteins found in these parts of the synapse. Under these conditions, The SVCT2, although it migrated for unknown reasons at different locations in the various fractions, was predominantly enriched in the presynaptic fraction (Figure 2G), suggesting that SVCT2 is indeed localized at the synapse, at the presynaptic terminal.
Figure 2. SVCT2 is localized in presynaptic synaptosomes.
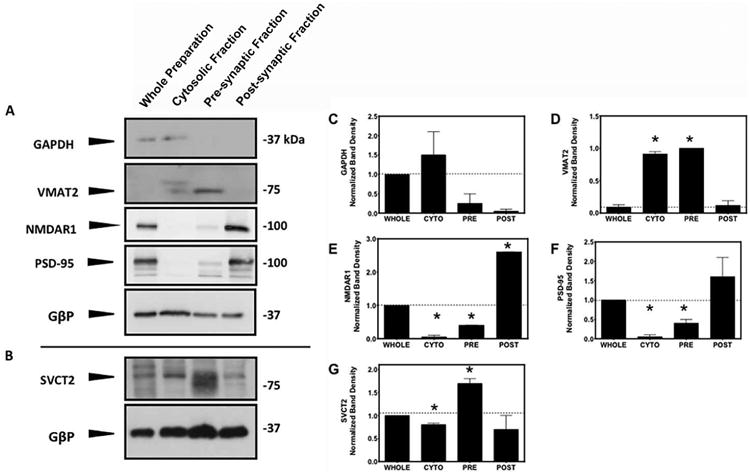
[A] Western immunoblots of cortical synaptosome preparations and fractionated samples. Samples containing 5 μg of protein were subjected to SDS-PAGE and immunoblotting. Samples were probed for the cytosolic protein, glyceraldehyde 3-phosphate dehydrogenase; for the presynaptic protein, vesicular monoamine transporter-2; and for two postsynaptic proteins, N-methyl-D-aspartate-1 and postsynaptic density protein-95. G-protein β subunit served as a loading control.[B] Samples containing 30 μg of protein were subjected to SDS-PAGE and immunoblotting under same fractionation conditions and probed for SVCT2. Bands in each lane [A and B] were normalized by GβP expression in the same blot and compared to the initial crude synaptosome preparation (first lane). Quantification of [C] GAPDH, [D] VMAT2, [E] NMDAR1, [F] PSD-95 and [G] SVCT2 are represented as the mean ± SEM, N=2. *P<0.05 where fractions are compared to control group (WHOLE) using two-tailed, unpaired T-test.
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; VMAT2, vesicular monoamine transporter-2; N-methyl-D-aspartate-1, NMDAR1, postsynaptic density protein-95, PSD-95; G-protein β subunit, GβP
SVCT2 function in crude cortical synaptosomes
Net VC uptake
To determine if the SVCT2 functions at the nerve terminal and in which direction, we performed experiments to characterize net VC uptake. We found that crude synaptosomes contained endogenous VC at ∼80 nmol/g (Figure 3A). Upon incubation for 30 min with 250 μM unlabeled VC, a two-fold increase in VC content was observed (Figure 3A). Two known SVCT2 inhibitors, sulfinpyrazone and phloretin prevented this uptake (Figure 3A). Similar results were observed in studies using 0.05 μCi radiolabeled 14C-VC, which also showed net uptake of 14C-VC uptake at ∼80-100 nmol/g when incubated for 30 min (Figure 3B). Radiolabeled VC uptake was also inhibited by the pre-incubation with sulfinpyrazone, phloretin, or unlabeled VC, as well as by replacement of buffer sodium with choline. It has been shown that the oxidized form of VC, dehydroascorbic acid (DHA) can be transported via the GLUT3 in neurons (Duarte et al. 2005; Rumsey et al. 1997). However, the GLUT transporter competitive inhibitor, 3-O-methylglucose at a concentration that will substantially compete for DHA did not affect 14C-VC uptake. Furthermore, in these experiments 0.5 mM glutathione was added to ensure that VC was not oxidized. This indicated that 14C-VC uptake was independent of GLUT function.
Figure 3. Net VC uptake in cortical crude synaptosomes.
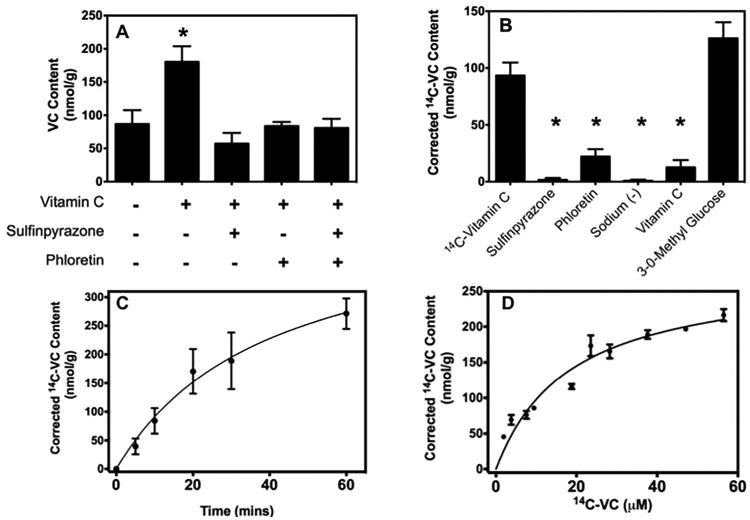
[A] Crude synaptosomes were incubated for 30 min without (control) and with 250 μM VC only or 250 μM VC and 10 min pre-incubation with 1 mM sulfinpyrazone and/or 0.1 mM phloretin. [B] Crude synaptosomes were pre-incubated for 10 min with 1 mM SPZ, 0.1 mM PHL, sodium-free KRH, 1 mM unlabeled VC, or 40 mM 3-O-methylglucose before the addition of 14C-VC (0.05 μCi, 7 μM) and further incubated for 30 min. [C] Crude synaptosomes were incubated with 14C-VC (0.05μCi, 7μM) for different periods of time or [D] for 10 min with increasing amounts of unlabeled VC. The calculated Km and Vmax were 18.3 ± 4.7 μM and 277 ± 28.1 nmol/g/30 min, respectively. The results represent the mean ± SEM (n=3). *P<0.05, compared control bar (far left) by one-way ANOVA with post-hoc testing.
SPZ, sulfinpyrazone; PHL, phloretin; VC, Vitamin C
Kinetic characterization of VC uptake
To determine if the kinetic properties of VC uptake in synaptosomes were similar to those expected if transport occurred via the SVCT2, time and concentration curves were performed using 0.05 μCi of radiolabeled VC. The uptake of 14C-VC plateaued with time (Figure 3C). Using an incubation time in the linear phase of uptake (10 min), uptake was saturable with an apparent Km of 18.31 ± 4.7 μM and a Vmax of 277 ± 28.15 nmol/g/30 min (Figure 3D). These data suggest that the VC uptake in synaptosomes is consistent with SVCT2 function.
VC efflux from cortical synaptosomes
VC transport is a balance between uptake and efflux (May and Qu 2009). In order to assess the contribution of VC movement out of synaptosomes to net uptake, a series of efflux studies were performed to observe the release of endogenous VC from non-loaded synaptosomes over time. Spontaneous endogenous VC efflux was measured, since net uptake would depend on efflux occurring at 37°C.
Temperature dependence and role of the SVCT2 in spontaneous VC efflux
No VC efflux was observed at 4°C, whereas at 37°C, the endogenous VC concentration steadily decreased over time and after 30 min had decreased to ∼80–100 nmol/g, comparable to the uptake experiments in the absence of added VC over the same time frame (Figure 4A and B). Similarly, when synaptosomes were loaded with Fura Red™ dye for 30 min at 37°C, centrifuged, and suspended in buffer, there was insignificant efflux of the trapped dye over 30 min at 37°C (results not shown). Together, these results suggest that the synaptosomes were not damaged or leaky under the conditions of our experiment. To determine the contribution of SVCT2 function to VC efflux, synaptosomes were incubated with the SVCT2 inhibitors sulfinpyrazone and phloretin, for 30 min at 37°C. The presence of these inhibitors did not inhibit efflux but rather enhanced loss of VC as a consequence of inhibition of reuptake. This resulted in a decrease of VC content of 50% and 25%, respectively (Figure 4C and E). This is likely due to the decrease in VC re-uptake caused by the inhibition of the SVCT2, resulting in an apparent increase of net efflux. This confirms the unidirectional transport of VC by SVCT2 and that the transporter in its native orientation is not responsible for VC efflux, but that influx does play a role in VC homeostasis in the synaptosome.
Figure 4. Time-dependent spontaneous VC Efflux.
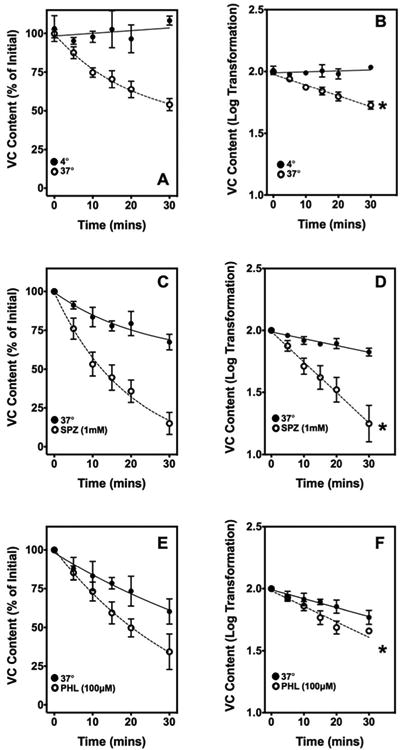
Time course of spontaneous VC efflux. [A] Relative VC content over 30 min at 4°C (closed circles) and 37°C (open circles). The initial synaptosomal VC concentration was ∼250-300 nmol/g (n=6). [B] Logarithmic transformation of data in panel A. [C] Relative VC content over 30 min of efflux at 37°C with (open circles) and without (closed circles) 1 mM sulfinpyrazone (n=4). [D] Logarithmic transformation of data in panel C. [E] Relative VC content over 30 mins of efflux at 37°C with (open circles) and without (closed circles) 100 μM phloretin (n=4). [F] Logarithmic transformation of data in panel C. In all the log-transformed panels, an asterisk (*) denotes a significant difference between the slopes of the two lines (p<0.05).
SPZ, sulfinpyrazone; PHL, phloretin; VC, Vitamin C
Discussion
In this work we found that the major neuronal transporter of VC, the SVCT2, is present in mouse brain synaptosomes and can localize to the presynaptic bouton. Kinetic and inhibitor experiments suggested that the SVCT2 mediates VC uptake into synaptosomes and opposes its spontaneous efflux.
The location of the SVCT2 in neurons will reflect areas or organelles with high VC content. Data from knock-out mice (Sotiriou et al. 2002) and kinetic studies in neuronal cells (Qiu et al. 2007) clearly show that the SVCT2 mediates global VC uptake in neurons. As we have shown in this study, it also is present in nerve fibers and synaptic boutons of SCG neurons in the peripheral nervous system, where it co-localized with both the NET and VMAT2 in the presynaptic bouton. Both NET and VMAT are known to be present in presynaptic areas. Additionally, our studies confirmed previous results that NET is found on the plasma membrane and in internal organelles, whereas VMAT is also found in internal organelles lacking NET (Matthies et al. 2009).
Additional evidence that the SVCT2 is expressed in nerve terminals was obtained in immunoblots of enriched brain cortical synaptosomes. As predicted by its presence in the terminal bouton, it was not surprising to find the SVCT2 was localized to the presynaptic synaptosomal fraction, along with the VMAT2, as suggested by the immunocytochemistry experiment. The SVCT2 thus likely accounted for endogenous VC found in freshly prepared synaptosomes. The content of VC in mouse brain cortical synaptosomes measured in this study (80-100 nmol per gram of measured protein) was similar to that of 90 nmol/g protein observed for rat brain synaptosomes (Kuo et al. 1978). When considered relative to protein content, the synaptosome content of VC is 3-4-fold higher than that in whole brain (Kuo et al. 1978). Considering that some VC will be lost during homogenization, this supports previous results showing a high neuronal content of VC relative to other cell types, such as astrocytes, which do not contain the SVCT2 in vivo (Berger and Hediger 2000; Berger et al. 2003).
The SVCT2 was also expressed in the cytosolic/perisynaptic fraction of synaptosomes. This may reflect SVCT2 localization in distinct organelles or vesicles, such as neurosecretory vesicles and mitochondria. As noted above, there may be co-localization of the SVCT2 with the VMAT2 in secretory vesicles. It has also recently been shown that mitochondria from HEK-293 (Munoz-Montesino et al. 2014) and U937 cells (Azzolini et al. 2013) express a functional SVCT2. Such localization might contribute to the punctate distribution of the SVCT2 in axons of primary culture hippocampal neurons (Qiu et al. 2007).
As expected from the presence of the SVCT2 in presynaptic membranes of brain cortical neurons, our studies in synaptosomes showed that VC was taken up against a concentration gradient with kinetic and inhibitor properties that match those of the plasma membrane SVCT2 in other cell types. For example, VC uptake in synaptosomes was saturable, with an apparent Km of 18 μM which fits with a Km of 20-30 μM determined for this protein when the human isoform was over-expressed in cultured cells (Daruwala et al. 1999; Takanaga et al. 2004). Further, transport of radiolabeled VC was inhibited by VC itself, as well as by phloretin, sulfinpyrazone, and removal of sodium from the medium, which are known to impair SVCT2 function (Wilson 2005). Significantly, VC transport was not inhibited by 40 mM 3-O-methylglucose, which at this concentration will substantially inhibit the GLUT-type transporters. This indicates that radiolabeled VC uptake occurred on the SVCT2 and not as radiolabeled DHA on a GLUT-type transporter with subsequent reduction and retention in the synaptosomes.
VC also effluxed from synaptosomes. Spontaneous VC efflux occurred at 37°C, but not at 4°C. This, as well as the finding that Fura-Red AM, which will be trapped in cells after initial loading and de-esterification (Yates et al. 1992), did not leak out of synaptosomes indicates that efflux occurred on a transporter or channel and was not due to diffusion or release from damaged synaptosomes. This conclusion agrees with that of previous studies of VC efflux from rat synaptosomes (Grunewald and Fillenz 1984). As to the mechanism of VC efflux, consistent with the literature, VC efflux mediated by SVCT2 was ruled out by the finding that known SVCT2 inhibitors actually increased net VC efflux, which would be expected from decreased VC re-uptake on the SVCT2. Thus the main contribution of the SVCT2 to synaptosomal VC homeostasis was to retain it via its uptake.
The proteins involved in the mechanism of spontaneous VC efflux, which has been observed in astrocytes (Siushansian et al. 1996) and neurons (May et al. 2006), remain unknown. However, there are a few postulated protein-mediated pathways that include hetero-exchange mechanisms and several types of anion channels such as VRACs (Corti et al. 2010).
In regard to the latter, lack of molecular identification of VRACs is a significant drawback in the assessment of their role in VC efflux. Furthermore, as the putative VRAC contribution accounts for less than 50% of the VC efflux by many of the cells tested, including hepatocyte-like Hep-G2 cells (Upston et al. 1999), SH-SY5Y neuroblastoma cells (May et al. 2006), and K562 cells (Lane and Lawen 2008) other release pathways are probably significant contributors. Further studies will have to be performed in order to characterize proteins involved in VC efflux. Understanding transport mechanisms for VC are crucial to understanding VC's role at the synapse.
Efflux and re-uptake of VC likely contributes to its homeostasis in the synapse. It is known that the extracellular VC concentration in the CNS is maintained homeostatically (Schenk et al. 1982). For example, Meile and Fillenz (Miele and Fillenz 1996) reported using voltammetry that extracellular VC levels spontaneously recovered within a few minutes after induced perturbations in either direction in awake, behaving rats. Further, the level of behavioral activation in rodents has been correlated with the level of striatal VC release (O'Neill et al. 1984), the latter mediated by the striatal glutamatergic system (Rebec et al. 2005). Homeostatic regulation of extracellular VC coupled with behavioral responses suggest that the extracellular compartment of brain tissue might be an important site of action for VC (Rice 2000). By analogy, spontaneous or induced efflux of VC into the synaptic cleft could contribute to this homeostasis and provide an anti-oxidative environment needed to protect certain catecholaminergic neurotransmitters from destruction.
The present results show that mouse brain synaptosomes contain the VC transporter SVCT2 and that this likely mediates their net concentrative uptake of VC. It follows that the SVCT2 may function to take up VC that has been released into the synaptic cleft. Glutamate has been shown to increase VC release from rat brain synaptosomes, which we also found in our studies of mouse brain cortex preparations (Pierce 2014). In addition to maintaining basal VC levels in the presynaptic nerve terminal, it also may be important to clear VC that has been released into the synaptic cleft in response to glutamate stimulation.
Significance Statement.
The vitamin C (VC) transporter, SVCT2, is present and functional at cortical nerve terminals. These data suggests that SVCT2 is responsible for the recovery of VC released into the synaptic cleft. Understanding how VC is transported at important regions of the neuron helps to clarify the bigger picture of brain health and function. This is crucial for the advancement of therapeutic options that can combat specific forms of oxidative stress that are implicated in neurodegeneration.
Acknowledgments
We would like to thank William Parker for his help in collecting VC efflux data.
Grant Information: Research was supported by National Institutes of Health grants NS 057674 and 5 T32 AR59039.
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
Author's roles: All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: MP, JM. Data collection: MP, AR, KB, HM. Analysis and interpretation: MP, AR, KB, HM. Writing the article: MP, JM. Critical revision of the article: MP, HM, JM. Final approval of the article: MP, AR, KB, HM, JM. Statistical analysis: MP. Obtained funding: JM. Overall responsibility: MP.
References
- Azzolini C, Fiorani M, Cerioni L, Guidarelli A, Cantoni O. Sodium-dependent transport of ascorbic acid in U937 cell mitochondria. IUBMB Life. 2013;65:149–153. doi: 10.1002/iub.1124. [DOI] [PubMed] [Google Scholar]
- Berger UV, Hediger MA. The vitamin C transporter SVCT2 is expressed by astrocytes in culture but not in situ. Neuroreport. 2000;11:1395–1399. doi: 10.1097/00001756-200005150-00009. [DOI] [PubMed] [Google Scholar]
- Berger UV, Lu XC, Liu W, Tang Z, Slusher BS, Hediger MA. Effect of middle cerebral artery occlusion on mRNA expression for the sodium-coupled vitamin C transporter SVCT2 in rat brain. J Neurochem. 2003;86:896–906. doi: 10.1046/j.1471-4159.2003.01891.x. [DOI] [PubMed] [Google Scholar]
- Betke KM, Rose KL, Friedman DB, Baucum AJ, Hyde K, Schey KL, Hamm HE. Differential localization of G protein betagamma subunits. Biochemistry. 2014;53:2329–2343. doi: 10.1021/bi500091p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti A, Casini AF, Pompella A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch Biochem Biophys. 2010;500:107–115. doi: 10.1016/j.abb.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Daruwala R, Song J, Koh WS, Rumsey SC, Levine M. Cloning and functional characterization of the human sodium-dependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999;460:480–484. doi: 10.1016/s0014-5793(99)01393-9. [DOI] [PubMed] [Google Scholar]
- Duarte AI, Santos MS, Oliveira CR, Rego AC. Insulin neuroprotection against oxidative stress in cortical neurons--involvement of uric acid and glutathione antioxidant defenses. Free Radic Biol Med. 2005;39:876–889. doi: 10.1016/j.freeradbiomed.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Godoy A, Ormazabal V, Moraga-Cid G, Zuniga FA, Sotomayor P, Barra V, Vasquez O, Montecinos V, Mardones L, Guzman C, Villagran M, Aguayo L, Onate S, Reyes AM, Carcamo JG, Rivas CI, Vera JC. Mechanistic insights and functional determinants of the transport cycle of the ascorbic acid transporter SVCT2. Activation by sodium and absolute dependence on bivalent cations. J Biol Chem. 2006;282:615–624. doi: 10.1074/jbc.M608300200. [DOI] [PubMed] [Google Scholar]
- Grunewald RA, Fillenz M. Release of ascorbate from a synaptosomal fraction of rat brain. Neurochem Int. 1984;6:491–500. doi: 10.1016/0197-0186(84)90120-7. [DOI] [PubMed] [Google Scholar]
- Harrison FE, May JM. Vitamin C function in the brain: Vital role of the ascorbate transporter (SVCT2) Free Radic Biol Med. 2009;45:719–730. doi: 10.1016/j.freeradbiomed.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison FE, Yu SS, Van Den Bossche KL, Li L, May JM, McDonald MP. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J Neurochem. 2008;106:1198–1208. doi: 10.1111/j.1471-4159.2008.05469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hediger MA. New view at C. Nat Med. 2002;8:445–446. doi: 10.1038/nm0502-445. [DOI] [PubMed] [Google Scholar]
- Kuo CH, Yonehara N, Hata F, Yoshida H. Subcellular distribution of ascorbic acid in rat brain. Jpn J Pharmacol. 1978;28:789–791. doi: 10.1254/jjp.28.789. [DOI] [PubMed] [Google Scholar]
- Laemmli UK, Favre M. Maturation of the head of bacteriophage T4. J Mol Biol. 1973;80:575–599. doi: 10.1016/0022-2836(73)90198-8. [DOI] [PubMed] [Google Scholar]
- Lane DJ, Lawen A. Non-transferrin iron reduction and uptake are regulated by transmembrane ascorbate cycling in K562 cells. J Biol Chem. 2008;283:12701–12708. doi: 10.1074/jbc.M800713200. [DOI] [PubMed] [Google Scholar]
- Matthies HJ, Han Q, Shields A, Wright J, Moore JL, Winder DG, Galli A, Blakely RD. Subcellular localization of the antidepressant-sensitive norepinephrine transporter. BMC Neurosci. 2009;10:65. doi: 10.1186/1471-2202-10-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May JM, Li L, Hayslett K, Qu ZC. Ascorbate transport and recycling by SH-SY5Y neuroblastoma cells: Response to glutamate toxicity. Neurochem Res. 2006;31:785–794. doi: 10.1007/s11064-006-9077-z. [DOI] [PubMed] [Google Scholar]
- May JM, Qu ZC. Ascorbic acid efflux and re-uptake in endothelial cells: maintenance of intracellular ascorbate. Mol Cell Biochem. 2009;325:79–88. doi: 10.1007/s11010-008-0022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele M, Fillenz M. In vivo determination of extracellular brain ascorbate. J Neurosci Methods. 1996;70:15–19. doi: 10.1016/S0165-0270(96)00094-5. [DOI] [PubMed] [Google Scholar]
- Munoz-Montesino C, Roa FJ, Pena E, Gonzalez M, Sotomayor K, Inostroza E, Munoz CA, Gonzalez I, Maldonado M, Soliz C, Reyes AM, Vera JC, Rivas CI. Mitochondrial ascorbic acid transport is mediated by a low-affinity form of the sodium-coupled ascorbic acid transporter-2. Free Radic Biol Med. 2014;70:241–254. doi: 10.1016/j.freeradbiomed.2014.02.021. [DOI] [PubMed] [Google Scholar]
- O'Neill RD, Fillenz M, Sundstrom L, Rawlins JN. Voltammetrically monitored brain ascorbate as an index of excitatory amino acid release in the unrestrained rat. Neurosci Lett. 1984;52:227–233. doi: 10.1016/0304-3940(84)90166-6. [DOI] [PubMed] [Google Scholar]
- Qiao H, May JM. Development of ascorbate transport in brain capillary endothelial cells in culture. Brain Res. 2008;1208:79–86. doi: 10.1016/j.brainres.2008.02.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu S, Li L, Weeber EJ, May JM. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J Neurosci Res. 2007;85:1046–1056. doi: 10.1002/jnr.21204. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Witowski SR, Sandstrom MI, Rostand RD, Kennedy RT. Extracellular ascorbate modulates cortically evoked glutamate dynamics in rat striatum. Neurosci Lett. 2005;378:166–170. doi: 10.1016/j.neulet.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Rice ME. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- Rumsey SC, Kwon O, Xu GW, Burant CF, Simpson I, Levine M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J Biol Chem. 1997;272:18982–18989. doi: 10.1074/jbc.272.30.18982. [DOI] [PubMed] [Google Scholar]
- Schenk JO, Miller E, Gaddis R, Adams RN. Homeostatic control of ascorbate concentration in CNS extracellular fluid. Brain Res. 1982;253:353–356. doi: 10.1016/0006-8993(82)90709-0. [DOI] [PubMed] [Google Scholar]
- Shirey-Rice JK, Klar R, Fentress HM, Redmon SN, Sabb TR, Krueger JJ, Wallace NM, Appalsamy M, Finney C, Lonce S, Diedrich A, Hahn MK. Norepinephrine transporter variant A457P knock-in mice display key features of human postural orthostatic tachycardia syndrome. Dis Model Mech. 2013;6:1001–1011. doi: 10.1242/dmm.012203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siushansian R, Dixon SJ, Wilson JX. Osmotic swelling stimulates ascorbate efflux from cerebral astrocytes. J Neurochem. 1996;66:1227–1233. doi: 10.1046/j.1471-4159.1996.66031227.x. [DOI] [PubMed] [Google Scholar]
- Sotiriou S, Gispert S, Cheng J, Wang YH, Chen A, Hoogstraten-Miller S, Miller GF, Kwon O, Levine M, Guttentag SH, Nussbaum RL. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nature Med. 2002;8:514–517. doi: 10.1038/0502-514. [DOI] [PubMed] [Google Scholar]
- Takanaga H, Mackenzie B, Hediger MA. Sodium-dependent ascorbic acid transporter family SLC23. Pflugers Arch. 2004;447:677–682. doi: 10.1007/s00424-003-1104-1. [DOI] [PubMed] [Google Scholar]
- Upston JM, Karjalainen A, Bygrave FL, Stocker R. Efflux of hepatic ascorbate: a potential contributor to the maintenance of plasma vitamin C. Biochem J. 1999;342:49–56. [PMC free article] [PubMed] [Google Scholar]
- Wilson JX. Antioxidant defense of the brain: a role for astrocytes. Can J Physiol Pharmacol. 1997;75:1149–1163. [PubMed] [Google Scholar]
- Wilson JX. Regulation of vitamin C transport. Annu Rev Nutr. 2005;25:105–125. doi: 10.1146/annurev.nutr.25.050304.092647. [DOI] [PubMed] [Google Scholar]
- Wilson JX, Peters CE, Sitar SM, Daoust P, Gelb AW. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000;858:61–66. doi: 10.1016/s0006-8993(99)02433-6. [DOI] [PubMed] [Google Scholar]
- Yates SL, Fluhler EN, Lippiello PM. Advances in the use of the fluorescent probe fura-2 for the estimation of intrasynaptosomal calcium. J Neurosci Res. 1992;32:255–260. doi: 10.1002/jnr.490320215. [DOI] [PubMed] [Google Scholar]