

Summary
Mitochondria need to be juxtaposted to phagosomes to synergistically produce ample reactive oxygen species (ROS) in phagocytes for pathogens killing. However, how phagosomes transmit signal to recruit mitochondria remains unclear. Here, we report that the kinases Mst1 and Mst2 function to control ROS production by regulating mitochondrial trafficking and mitochondrion-phagosome juxtaposition. Mst1 and Mst2 activate Rac GTPase to promote Toll-like receptor (TLR)-triggered assembly of the TRAF6-ECSIT complex that is required for mitochondrial recruitment to phagosomes. Inactive forms of Rac, including the human Rac2D57N mutant, disrupt the TRAF6-ECSIT complex by sequestering TRAF6, and severely dampen ROS production and greatly increase susceptibility to bacterial infection. These findings demonstrate the TLR-Mst1-Mst2-Rac signalling axis to be critical for effective phagosome-mitochondrion function and bactericidal activity.
Phagocytes are specialized immune cells that engulf harmful microorganisms and destroy them in phagosomes1, 2. The destruction process mainly depends on the production of large amounts of reactive oxygen species (ROS), long thought to be generated entirely via the phagosomal membrane NADPH ox idase (NOX) machinery3, 4. The GTP-charged active form of the Rho family GTPase Rac1 and Rac2 is required for phagosomal NOX activation5. In unstimulated phagocytes, GDP-charged inactive Rac1 and Rac2 form a complex with a Rho-GDP dissociation inhibitor (Rho-GDI) protein. Rac1 and Rac2 activation are initiated by their release from Rho-GDI, resulting in part from the phosphorylation of Rho-GDI by the kinases PAK or PKCα6, 7. The importance of Rac-GTP in phagocyte function is illustrated by a human immunodeficiency syndrome characterized by severe bacterial infections arising from a mutation in Rac2 (Rac2D57N) that results in constitutive GDP binding accompanied by impaired ROS production in phagocytes8-10.
In addition to phagosomal NOX activity, maximal phagocytic ROS generation and bactericidal activity require mitochondrial ROS (mROS) production11-15. The engagement of a subset of macrophage Toll-like receptors (TLR1, TLR2 and TLR4) leads to the translocation of mitochondria to phagosomes, mediated by the assembly of a complex between the ubiquitin ligase TNF receptor-associated factor 6 (TRAF6), and the mitochondrial protein evolutionarily conserved signalling intermediate in Toll pathways (ECSIT), resulting in the augmentation of mROS production and bactericidal activity11. Furthermore, the heightened innate immune response and increased inflammatory cytokine production by macrophages from patients with TNF receptor-associated periodic syndrome (TRAPS), results from high mitochondrial, rather than NOX-mediated, ROS production16. Thus, ROS production by macrophage mitochondria is required for optimal bactericidal activity and innate immune responses. However, how cells achieve the precise temporal and spatial coordination of phagosomal and mitochondrial ROS induction is still incompletely understood.
The kinases Mst1 and Mst2 are the closest mammalian homologues of the Drosophila melanogaster kinase Hippo, which inhibits cell proliferation and promotes apoptosis during development by inhibiting Yap and Taz through a kinase cascade formed by the scaffolding proteins WW45 and Mob1, and the kinases Lats1 and Lats217-30. However, it is less appreciated that human Mst1 deficiency results in a complex combined immunodeficiency syndrome with recurrent bacterial and viral infections, lymphopenia and variable neutropenia31, 32. In mice, Mst1 and Mst2 are important regulators of T cell adhesion, migration, proliferation and apoptosis33-41. However, the role of Mst1 and Mst2 in innate immunity is as yet largely unexplored.
In the present study, we found that kinases Mst1 and Mst2 are important for optimal ROS production and bactericidal activity of phagocytes by promoting the activation of the small GTPase Rac and mitochondrial trafficking and juxtaposition to the phagosome through assembly of a TRAF6-ECSIT complex.
Results
Higher susceptibility of sepsis in Mst1 and Mst2 null mice
We used a previously described hematopoietic cell-specific knockout of Mst1 and Mst2 (Mst1-/-Mst2fl/flVav-Cre) mice to study the role of the kinases Mst1 and Mst2 in the immune system40. The great majority of Mst1−/−Mst2fl/flVav-Cre mice exhibited multiple or recurrent infections such as pneumonia (Fig. 1a) and lung abscesses, as well as unprovoked inflammation in multiple organs, manifesting as colitis and/or glomerulonephritis (Supplementary Fig. 1a). We also observed that some animals showed massive extramedullary haematopoiesis and granulopoiesis in the spleen (Supplementary Fig. 1b, c). The median survival time for Mst1−/−Mst2fl/flVav-Cre mice was approximately 6 months.
Figure 1. Losses of Mst1 and Mst2 increase susceptibility to bacterial sepsis.

(a) Representative lung tissues and H&E staining of lung sections from wild type and Mst1−/−Mst2fl/flVav-Cre mice. Scale bar, 100 μm.
(b-e) Mst1fl/flMst2fl/fl (WT) or Mst1fl/flMst2fl/flLyz2-Cre (cDKO) mice (n=10 mice per group per experiment) subjected to sublethal CLP; mortality (Mantel–Cox test)(b), serum cytokines measured by ELISAs (c), inflammatory cell infiltration in the lungs as shown by H&E staining (d) and the bacterial loads (colony forming units, CFU) measured in the lung, liver, spleen, kidney and peritoneal fluid (e) after CLP induction. Scale bar, 50 μm.
(f) Immunoblot analysis of P-p38, P-Jnk, IκBα and P-Mob1 in WT BMDMs stimulated with indicated TLR agonists for different durations.
(g) Immunoblot analysis of P-Mob1 and MyD88 in WT or MyD88 KO RAW246.7 cells stimulated with the indicated agonists for different durations.
(h) WT or cDKO BMDMs were stimulated with LPS for the indicated times, followed by immunoblot analysis with the indicated antibodies.
(i) WT and cDKO BMDMs or neutrophils were treated with LPS (100 ng/ml) for the indicated times. Cytokine (IL-6 and TNF) production was measured by ELISA.
Data were assessed with Student's t-test and are presented as mean ± s.d. ns, no significant, * p<0.05, ** p<0.01, *** p<0.001 compared with respective controls of biological replicates in c (n=3), e (n=5) and i (n=3). Data are one experiment representative of two (e) or three (a-d, f-i) independent experiments with similar results.
The high incidence of bacterial infections in the Mst1 and Mst2 double-deficient mice prompted us to investigate the role of Mst1 and Mst2 in the innate immune system, which provides immediate defence against infection by activating phagocytes. We ablated Mst1 and Mst2 in myeloid cells by crossing Mst1fl/flMst2fl/fl mice with mice expressing the lysozyme promoter-driven Cre recombinase gene (Lyz2-Cre). The conditional Mst1fl/flMst2fl/flLyz2-Cre knockout mice (hereafter referred to as cDKO) were born at a normal ratio and exhibited no significant differences compared to Mst1fl/flMst2fl/fl wild type (WT) littermate controls in circulating lymphocyte, monocyte and granulocyte numbers according to peripheral blood counts (Supplementary Fig. 1d). Flow cytometric analysis indicated that, compared to WT littermates, the percentages of Gr-1+CD11b+ neutrophils and F480+CD11b+ macrophages were significantly decreased in the bone marrow, spleen and blood from cDKO mice, whereas the composition and activation status of T cells and B cells in the spleen, lymph nodes or blood were comparable between WT and cDKO mice (Supplementary Fig. 1e, f).
In contrast to the Mst1-/-Mst2fl/flVav-Cre mice, cDKO mice did not show spontaneous inflammation or infections over the first 7 months of life. However, in a model of septic peritonitis, cecal ligation and puncture (CLP), the bacterial peritonitis killed ∼80% of cDKO mice but only ∼40% of WT littermates (Fig. 1b). In response to infection, the rapid mobilization of neutrophils and emergency myelopoiesis were comparable in blood of both WT and cDKO mice (Supplemental Fig. 1g). Compared with WT control animals, cDKO mice exhibited significantly higher serum concentrations of the inflammatory cytokines IL-6, TNF and IL-1β at 6 or 12 h after CLP induction (Fig. 1c) and more severe inflammation in lung and kidney tissues at 24 h (Fig. 1d and Supplementary Fig. 1h). In addition, bacterial invasion of the lung, liver, spleen, kidney and peritoneal fluid after CLP induction was significantly higher in cDKO mice (Fig. 1e). These results indicate that the loss of both the Mst1 and Mst2 kinases in myeloid cells caused increased susceptibility to bacterial infection and an enhanced inflammatory response.
TLR induces Mst1 and Mst2 activation for bacterial clearance
Phagocyte TLRs act as primary sentinels for pathogen detection through their binding of microbial-associated molecular patterns (MAMPs), which serve to initiate pro-inflammatory responses1, 2, 42, 43. To examine whether Mst1 and Mst2 participate in TLR-mediated innate host responses to bacterial infections, we stimulated bone marrow-derived macrophages (BMDMs) with an array of TLR ligands, including lipopolysaccharide (LPS, a TLR4 agonist), Pam3CSK4 (TLR1 and TLR2 agonist), lipoteichoic acid (LTA, TLR2 agonist), poly(I:C) (TLR3 agonist), R848 (TLR7 and TLR8 agonist) and CpG DNA (TLR9 agonist), and examined the phosphorylation of Mob1, a physiological substrate of the kinases Mst1 and Mst2. We observed that stimulation of the cell-surface TLRs (TLR1, TLR2 and TLR4) significantly enhanced the phosphorylation of Mob1 (P-Mob1), whereas the endosomal TLR ligands (R848 or CpG DNA) did not change the P-Mob1, except for a moderate increase in P-Mob1 induced by poly(I:C) (Fig. 1f and supplementary Fig. 1i). Using a MyD88-knockout macrophage-like Raw264.7 cell line, we further demonstrated that LPS, Pam3CKS4 and LTA, predominately derived from bacteria, activated kinases Mst1 and Mst2 via a MyD88-dependent pathway (Fig. 1g). We therefore employed LPS and Pam3CKS4 in subsequent studies as representative stimuli for the activation of Mst1 and Mst2 in phagocytes.
The deletion of Mst1 and Mst2 did not alter the TLR-induced activation of the mitogen-activated protein kinases p38, Jnk or Erk (Fig. 1h) but resulted in an increased induction of pro-inflammatory cytokines, such as IL-6 and TNF, in BMDMs upon LPS stimulation (Fig. 1i). These findings suggest that the higher susceptibility to CLP-induced bacterial sepsis in cDKO mice is not due to a lack of pro-inflammatory response to bacterial infections. We then examined bacterial phagocytosis by co-incubating FITC-labelled Escherichia Coli (E. coli) or Listeria Monocytogenes (L. Monocytogenes) with BMDMs or neutrophils isolated from WT or cDKO animals. Flow cytometric analysis showed that cDKO cells exhibit a decrease in phagocytosis of both E. coli and L. monocytogenes compared to WT cells (Fig. 2a). However, compared with WT cells, the number of live intracellular bacteria was significantly higher in cDKO BMDMs or neutrophils measured at later time points after bacterial infection, indicating that in addition to a modest bacterial uptake, cDKO phagocytes are significantly defective in the intracellular killing of bacteria (Fig. 2b). Quantification of immunofluorescence micrographs of macrophages incubated with E. coli stably expressing GFP (GFP-E. coli) confirmed that Mst1 and Mst2 deficiency caused both impaired bacterial engulfment and defective clearance (Fig. 2c, d). These results indicate that the TLR-mediated Mst1 and Mst2 activation is required for phagocytosis and efficient bacterial clearance.
Figure 2. Mst1 and Mst2 deficient myeloid cells are defective in bacterial killing and ROS induction.

(a) Mst1fl/flMst2fl/fl (WT) and Mst1fl/flMst2fl/flLyz2-Cre (cDKO) BMDMs left uninfected or infected for 10 min at 37°C or 4°C with FITC-labelled E. coli (E. coli-FITC) at a multiplicity of infection (MOI) of 20 or L. monocytogenes (Lm-FITC) at MOI of 10, as analysed by flow cytometry.
(b) WT and cDKO BMDMs or neutrophils were infected with L. monocytogenes or E. coli (MOI, 10). CFUs were quantified at the indicated time points.
(c, d) WT and cDKO BMDMs infected with GFP-expressing E. coli (GFP-E. coli) for the indicated times were washed, fixed and stained with DAPI. The GFP-E. coli (green) were visualized (c) and quantified (d) by fluorescence microscopy. Scale bar, 20 μm.
(e) WT and cDKO BMDMs or neutrophils were stained with MitoSOX (mROS) or CM-H2DCFDA (cellular ROS) for 30 min and analysed by flow cytometry.
(f) WT (top panels) or cDKO BMDMs (lower panels) were labeled with CFSE, and then mixed with unlabeled cDKO or WT cells and the CellROX and DAPI coupled E. coli. The phagosomal ROS was visualized by fluorescence microscopy. WT and cDKO cells indicated with stars and arrows respectively. Scale bar, 20 μm.
(g) WT and cDKO BMDMs or neutrophils were stimulated with the indicated TLR agonists for 6 h, and mROS production was measured with MitoSOX using flow cytometry. LPS, Lipopolysaccharide; Pam3, Pam3CSK4; LTA, Lipoteichoic acid.
(h) mROS and cellular ROS in WT and cDKO BMDMs or neutrophils were measured as described in (e) after E. coli or L. monocytogenes infection. Data were assessed with Student's t test and are represented as mean ± s.d. n=5, biological replicates. * p<0.05, ** p<0.01, *** p<0.001 compared with respective controls or as indicated (b, d). Data are from one experiment representative of two (f) or three (a-e, g, h) independent experiments with similar results.
Mst1 and Mst2 deletion results in impaired mROS induction
ROS plays an essential role in bacterial killing by phagocytes4, 12. In addition to phagosomal NADPH-oxidase, mROS are an essential source of ROS in this process. Using ROS-sensitive dyes (mitoSOX to measure the mROS superoxide and CM-H2DCFDA or CellROX to measure total cellular H2O2) and flow cytometry, we observed that unstimulated WT and cDKO BMDMs exhibit the same amounts of mROS, whereas the basal amount of total cellular ROS is moderately increased in cDKO cells (Fig. 2e). To measure phagosomal ROS, E. coli were coupled with CellROX dye as well as the DAPI for visualization. CFSE-labelled WT BMDMs were mixed with unlabeled cDKO BMDMs or vice verse, and then mixed cells were infected with CellROX-DAPI-E. coli. Our data clearly showed that the induction of phagosomal ROS is greatly impaired in cDKO BMDMs upon the bacteria infection (Fig. 2f).
As shown above, engagement of the cell surface TLRs (TLR1, TLR2 and TLR4), but not the endosomal TLRs (TLR3, TLR7, TLR8 and TLR9), augmented mROS and total cellular ROS in BMDMs and neutrophils (Fig. 2g and Supplementary Fig. 2a). Notably, ligand stimulation of TLR1, TLR2 or TLR4 on cDKO BMDMs or neutrophils caused little or no increase in mROS and total cellular ROS compared with the WT cells (Fig. 2g and Supplementary Fig. 2a), and whole bacterial infection also yielded substantially reduced concentrations of total ROS in cDKO cells (Fig. 2h and Supplementary Fig. 2b). We therefore questioned whether the greatly diminished induction of mROS in cDKO cells was due to a reduced ability of their mitochondria to generate ROS. In contrast, rotenone, oligomycin or antimycin A, which are electron transport inhibitors known to increase mitochondrial superoxide generation, augmented mROS production to a similar extent in both WT and cDKO BMDMs (Supplementary Fig. 2c). In addition, the number of mitochondria, the major electron transport chain components and the mitochondrial membrane potential were also comparable between WT and cDKO cells (Supplementary Fig. 2d, e, f). Thus, a lack of Mst1 and Mst2 does not diminish the capacity of mitochondria for ROS production and some other factors are responsible for the impaired phagosomal mROS production.
Mst1 and Mst2 control mitochondrion-phagosome juxtaposition
We next examined how Mst1 and Mst2 deficiency affects mitochondrial ROS induction in phagocytes after TLR1, TLR2 or TLR4 activation. Because mitochondria are recruited to phagosomes containing intracellular pathogens11, we investigated whether this process is impaired in cDKO phagocytes. Latex beads coated with MAMPs were phagocytosed by BMDMs and enabled visualization of the juxtaposition of phagosomes to mitochondria. In contrast to WT BMDMs, cDKO BMDMs displayed markedly less mitochondrial cupping around the phagocytosed Pam3CSK4- and LPS-coated beads (Fig. 3a, upper two rows). In addition, many more bacteria co-localized with the mitochondria in WT BMDMs than in cDKO cells during GFP-E. coli infection (Fig. 3b, upper two rows).
Figure 3. Mst1 and Mst2 regulate the recruitment of mitochondria to phagosomes via the small GTPase Rac.

(a) Mst1fl/flMst2fl/fl (WT), Mst1fl/flMst2fl/flLyz2-Cre (cDKO) and cDKO-Rac1G12V BMDMs were incubated with uncoated, LPS-coated or Pam3CSK4-coated latex beads, and mitochondrial networks (Mito) were immunostained with HSP60 antibodies. Confocal images were acquired. Colocalization of beads (green) and mitochondria (red) are indicated by short arrows. Scale bar, 20 μm.
(b) The distribution of mitochondrial networks (HSP60, red) in WT, cDKO, cDKO-Rac1G12V BMDMs and WT BMDMs treated with cytochalasin D or Rac inhibitor NSC23766 after infection with GFP-E. coli (green). Colocalization of E. coli (green) and mitochondria (red) is indicated by arrows. Scale bar, 20 μm.
(c) WT, Rac1 G12V, cDKO and cDKO-Rac1G12V BMDMs were treated with DMSO or cytochalasin D and immunostained with F-actin antibodies (green). Scale bar, 20 μm.
(d) WT BMDMs and neutrophils were pretreated with DMSO or NSC23766 for 30 min followed by LPS stimulation for 3 h. mROS and cellular ROS were measured by MitoSox red dye and CM-H2DCFDA, respectively, using flow cytometry.
(e) Active (GTP-bound) Rac1 levels in WT, cDKO or cDKO-Rac1G12V BMDMs, or in WT and cDKO neutrophils, were detected in GST-PAK70-106 (GST-PAK) with a pull-down assay. GST-PAK protein was stained by Coomassie blue (CoBlue).
(f) mROS and cellular ROS in WT, cDKO and cDKO-Rac1G12V BMDMs stimulated with LPS for 3 h were measured by MitoSox and CellRox, respectively, using flow cytometry.
Images shown are representative of approximately 100 cells (a, b and c). Data are from one experiment representative of three independent experiments with similar results (a-f).
Mst1 deficiency in T cells causes cytoskeletal disorganization39, 40. Similarly, cDKO BMDMs exhibit disrupted F-actin organization compared to WT cells (Fig. 3c, upper-left two panels). To determine whether the cytoskeletal reorganization is required for the juxtaposition of phagosomes and mitochondria in macrophages, WT BMDMs were treated with cytochalasin D. As expected, this F-actin inhibitor disrupted the cytoskeleton in a manner similar to that observed in cDKO BMDMs (Fig. 3c, lower left two panels, and Supplementary Fig. 3a) and significantly reduced the co-localization of GFP-E. coli with mitochondria (Fig. 3b, third row from top), as well as the induction of mROS upon LPS stimulation (Supplementary Fig. 3b). Mst1 and Mst2 regulate chemokine-stimulated F-actin reorganization in thymocytes by promoting the activation of the small GTPase Rac40. Treatment of BMDMs with the Rac inhibitor (NSC23766) decreased the co-localization of mitochondria with GFP-E. coli (Fig. 3b, compare top row with fourth row from top) and blocked the LPS-stimulated increase of both mROS and total cellular ROS in BMDMs and neutrophils (Fig. 3d).
Although the overall abundance of Rac in cDKO BMDMs and neutrophils was similar to that in WT, the amount of Rac-GTP was greatly reduced, as estimated by pull-down with GST-tagged PAK70-106 (Fig. 3e). To determine whether the impaired mitochondrial recruitment in cDKO BMDMs is caused by defective Rac activation, we crossed cDKO mice with R26StopFLRACDA mice, which carry an inducible, constitutively active RacG12V (hereafter cDKO-RacG12V). These mice express the constitutively active RacG12V in cDKO myeloid lineages (Fig. 3e). BMDMs of the cDKO-RacG12V mice, displayed normal F-actin organization (Fig. 3c, upper right panel and Supplementary Fig. 3a), mitochondrion-phagosome juxtaposition (Fig. 3a, b, compare bottom row with the two top rows) and mROS and cellular ROS production upon LPS stimulation (Fig. 3f). These results indicate that the activation of Rac by Mst1 and Mst2 is critical for innate host defence by positively regulating the recruitment of mitochondria to phagosomes for optimal mROS induction.
Mst1 and Mst2 activate PKCα to disrupt the LyGDI-Rac complex
We next investigated the mechanism by which Mst1 and Mst2 regulate Rac activity in phagocytes. The pull-down of Flag-tagged Mst2 transiently expressed in Raw264.7 cells retrieved several endogenous proteins selectively, as identified by mass spectrometry, including PKCs and LyGDI proteins (Fig. 4a). LyGDI belongs to a family of three Rho GDP dissociation inhibitors that bind Rac-GDP in a cytosolic complex. The PKCα-mediated phosphorylation of LyGDI at Ser31 disrupts the interaction between LyGDI and Rac, releasing Rac-GDP, which translocates to the membrane and undergoes guanyl nucleotide exchange6. Although neither Mst1 nor Mst2 bound directly to LyGDI, PKCα did bind strongly to LyGDI (Fig. 4b). Transiently expressed HA-tagged Mst2 (HA-Mst2) bound to multiple co-expressed Flag-PKC isoforms (PKCα, β, γ, δ, ε, η, θ, ι and ζ; Supplementary Fig. 4a); however, only PKCα exhibited an up-shift band, as visualized using Phos-tag SDS-PAGE, when co-expressed with WT but not kinase-dead (KD) Mst2, indicating that PKCα is a substrate of Mst2 kinase (Fig. 4c). Mst2-mediated PKCα phosphorylation, as well as the Mst2 binding site, is located on the N-terminal regulatory domain of PKCα (Fig. 4d and supplementary Fig. 4b). Mass spectrometric analysis and site-directed mutagenesis further revealed that PKCα is phosphorylated by Mst2 kinase at Ser226 and Thr228 (Fig. 4e and Supplementary Fig. 4c). The LPS-stimulated increase in PKCα (Thr638), an autophosphorylation site reflective of PKCα activation, was greatly reduced in cDKO BMDMs compared with WT (Fig. 4f). Thus, the phosphorylation of PKCα at Ser226 and Thr228 by Mst1 and Mst2 is required for the optimal activation of PKCα. Consistent with this notion, co-expression with Mst2 kinase enhanced the interaction between PKCα and LyGDI (Fig. 4g) and this in turn promotes the phosphorylation of LyGDI by PKCα (Fig. 4h). LyGDI phosphorylation led to its dissociation from the LyGDI-Rac1 complex (Fig. 4i) that augmented Rac GTP charging (Fig. 4j). To confirm the results observed with transfection, the ability of LPS to diminish the interaction between LyGDI and Rac1 was greatly reduced in cDKO BMDMs compared with the WT (Fig. 4k). These results establish that the kinases Mst1 and Mst2, via the activation of PKCα, disrupt the LyGDI-Rac interaction to promote Rac activation.
Figure 4. Mst1 and Mst2 regulate the activation of Rac through PKC-LyGDI.

(a) LyGDI and PKCα were identified by mass spectrometry in a Flag-tagged Mst2 pull-down assay using Raw264.7 cell lysates.
(b) Co-immunoprecipitation (Co-IP) assay of 293T cells expressing HA-LyGDI and Flag-Mst1, Flag-Mst2 or Flag-PKCα as indicated. Cell lysates were immunoprecipitated with anti-Flag antibody and analysed by immunoblotting with antibodies as indicated.
(c) Flag-tagged PKC α, β, γ, δ, ε, η, θ, ι, orζ was co-expressed with HA-Mst2 or HA-Mst2 kinase dead form (KD) in 293T cells. Protein extracts were immunoprecipitated with Flag antibody-conjugated beads and analysed using Phostag SDS-PAGE after treatment with or without calf-intestinal alkaline phosphatase (CIP).
(d) Co-IP and Phos-tag analysis (as in c) of 293T cells expressing HA-Mst2 or HA-Mst2 (KD) and Flag-PKCα N-terminal (NT) or Flag-PKCα C-terminal (CT) as indicated.
(e) Phos-tag analysis (as in c) in 293T cells expressing Flag-PKCα, Flag-PKCα, (S226A), Flag-PKCα (T228A) or Flag-PKCα (S226A-T228A) and HA-Mst2 as indicated.
(f) Immunoblot analysis of P-PKCα (T638) and PKCα in lysates of Mst1fl/flMst2fl/fl (WT) and Mst1fl/flMst2fl/flLyz2-Cre (cDKO) BMDMs stimulated with LPS for the indicated times.
(g) Co-IP assay (as in b) of 293T cells expressing HA-LyGDI, Flag-PKCα and Myc-Mst2 as indicated.
(h) Co-IP and Phos-tag analysis (as in c) of 293T cells expressing HA-LyGDI, Flag-PKCα, Myc-Mst2 and Myc-Mst2 (KD) as indicated.
(i) Co-IP assay (as in b) of 293T cells expressing HA-Rac1, Myc-LyGDI, Flag-PKCα, Flag-Mst2 and Flag-Mst2 (KD) as indicated. Cell lysates were immunoprecipitated with anti-Myc antibody.
(j) Endogenous active (GTP-bound) Rac levels in 293T cells expressing Flag-PKCα, Flag-Mst2 and/or Flag-Mst2 (KD) as indicated was detected using an anti-Rac1 antibody in a GST-PAK70-106 (GST-PAK) pull-down assay.
(k) Immunoblot analysis of indicated proteins from lysates and immunoprecipitates of WT and cDKO BMDMs treated with LPS for 15 min.
Data are from one experiment representative of three independent experiments with similar results (b-k).
TLR signalling induces the TRAF6-mediated Rac ubiquitination
As shown above (Fig. 3b), the infection of WT BMDMs with E. coli promotes the co-localization of phagocytosed bacteria with mitochondria, a response that is lost in cDKO BMDMs but restored by the expression of constitutively active Rac1G12V. Moreover, infection of WT BMDMs with E. coli increases the co-localization of Rac with phagosomes, and this response is greatly reduced in cDKO BMDMs or in WT BMDMs treated with the Rac inhibitor NSC23766 (Supplementary Fig. 5a). To identify candidate Rac effectors of signalling upon bacterial infection, we incubated purified Flag-tagged Rac1G12V or inactive Rac1T17N with extracts from BMDMs and identified, by mass spectroscopy, the endogenous proteins co-purified with the Flag immunoprecipitation (IP). Among the proteins identified was the E3 ubiquitin ligase TRAF6, a key intermediate in TLR signalling, that specifically co-precipitated with Flag-RacT17N but not with Flag-Rac1G12V (Supplementary Fig. 5b). The selective interaction of TRAF6 with inactive Rac1T17N, but not with active Rac1G12V, was confirmed by the cotransfection of Flag-TRAF6 with HA-Rac1WT, HA-Rac1T17N (i.e., Rac-GDP) or HA-Rac1G12V (i.e., Rac-GTP) in 293T cells followed by Flag IP (Fig. 5a).
Figure 5. TRAF6 positively regulates the Lys63-linked ubiquitination of Rac1 in vitro and in vivo.

(a) Strong association of inactive Rac1 with TRAF6. Co-IP assay of 293T cells expressing HA-Rac1WT, HA-Rac1G12V, or HA-Rac1T17N and Flag-TRAF6 as indicated. Cell lysates were immunoprecipitated with anti-Flag antibody and analysed by immunoblotting with antibodies as indicated.
(b) In vitro ubiquitylation assay performed with recombinant Rac1, WT TRAF6, or catalytically inactive mutant TRAF6C70A, together with the indicated components.
(c) Ubiquitination and Co-IP assay (as in a) of 293T cells expressing Flag-Rac1, Myc-TRAF6 and HA-ubiquitin (Ub) in different combinations as indicated. Cells were treated with DMSO or the proteasome inhibitor MG132. Immunoblotting with anti-HA antibody shows the level of Rac ubiquitylation.
(d) Cell lysates of 293T cells expressing Flag-Rac1WT or Flag-Rac1K16R and Myc-TRAF6 as indicated combinations were incubated with GST-PAK70-106 (GST-PAK). The association of active (GTP-bound) Rac1 with GST-PAK70-106 was revealed by Rac1 immunoblotting. GST-PAK70-106 protein was stained by Coomassie blue (CoBlue).
(e) Immunoblot analysis of K63-linked polyubiquitin of Rac1 from immunoprecipitates and lysates of WT or TRAF6 knockdown BMDMs treated with LPS for 15 min.
(f) TRAF6 mediates the K63-linked polyubiquitin of Rac1 on lysine 16 residues. Immunoblotting assays show Rac1 ubiquitination in 293T cells expressing Flag-Rac1WT, Flag-Rac1K16R or Flag-Rac1K147R and Myc-TRAF6 as indicated. Data are from one experiment representative of three independent experiments with similar results (a-f).
TRAF6 functions with the ubiquitin-conjugating (E2) complex (consisting of UEV1A and UBE2N) to catalyse the synthesis of K63-linked polyubiquitin chains on target proteins, including TRAF6 itself44. The incubation of Rac1 with TRAF6 and standard components required for in vitro ubiquitination demonstrated the ability of TRAF6 to catalyse the ubiquitination of Rac1 (Fig. 5b). The mutation of a cysteine residue (Cys-70) in the RING-domain of TRAF6 required for its E3 ligase activity abolished the ubiquitination of Rac1 (Fig. 5b). Furthermore, the co-transfection of TRAF6 and Rac1 with wild-type HA-ubiquitin or HA-ubiquitin variants that permit only K63 or K48 linkage established TRAF6-catalysed Rac1 polyubiquitination as exclusively K63 linked (Supplementary Fig. 5c). Consistent with the lack of K48-linked ubiquitination, the proteasome inhibitor MG132 did not increase the amount of ubiquitinated Rac (Fig. 5c). TRAF6 catalysed Rac1 ubiquitination and enhanced the activation state of Rac1 (Fig. 5d). The downregulation of endogenous TRAF6 protein in BMDMs and Raw264.7 cells using shRNA (shTRAF6-1) (Supplementary Fig. 5d) was accompanied by a marked reduction of total and K63-linked Rac ubiquitination and activation in resting and LPS-stimulated BMDMs (Fig. 5e).
To map the ubiquitin acceptor site(s) in Rac1, we individually substituted all 17 lysines in the Rac1 sequence with arginine and measured the ubiquitination of these Rac1 variants in 293T cells (Supplementary Fig. 5e). Only the Rac1K16R mutant showed a strong decrease in polyubiquitination compared to the Rac1WT, whereas several mutants, e.g., Rac1K147R, exhibited a relatively small reduction (Supplementary Fig. 5c). The co-transfection of Flag-tagged Rac1K16R or Rac1K147R with Myc-tagged TRAF6 in 293T cells demonstrated that lysine-16 is the ubiquitin acceptor site (Fig. 5f) and is required for TRAF6 induced Rac1 activation (Fig. 5d). These results establish that the TRAF6 directly binds to Rac and catalyses K63-linked polyubiquitination of Rac lysine 16 in vitro and in vivo.
TRAF6-mediated ubiquitination is crucial for Rac activation
We observed above that endogenous TRAF6 bound selectively to Rac1T17N as compared with RacWT or RacG12V (Fig. 5a) and co-localizes strongly with Rac1T17N, but not with Rac1G12V, at the cell periphery (Supplementary Fig. 6a). In contrast, GST-TRAF6 binds Rac1 in vitro to a similar extent, whether Rac1 is charged with GDP or GTP (Fig. 6a). Conversely, unlike WT TRAF6, the association of catalytically inactive TRAF6C70A with Rac1WT, Rac1T17N or Rac1G12V in 293T cells was comparable (Fig. 6b). We therefore sought to define the effect of Rac1 guanyl nucleotide charging on its ability to undergo ubiquitination by TRAF6. Myc-TRAF6 expressed in 293T cells ubiquitinated co-expressed Rac1WT and, to a greater extent, Rac1G12V, but was unable to ubiquitinate Rac1T17N (Fig. 6c). In addition, the Rac1 inhibitor (NSC23766) diminished the ubiquitination of Rac1 by coexpressed TRAF6 (Supplementary Fig. 6b). In vitro, loading of Rac1 with GTPγS (a nonhydrolyzable GTP) was required for TRAF6-catalysed ubiquitination, whereas Mg2+-free and inactive Rac1-GDP were both resistant to ubiquitination (Fig. 6d). The impaired activation of endogenous Rac observed in cDKO BMDMs (Fig. 3e) was paralleled by a diminished Rac ubiquitination in LPS-stimulated cDKO BMDMs compared with the WT (Fig. 6e). Thus, TRAF6 preferentially ubiquitinates the GTP-charged active form of Rac.
Figure 6. TRAF6 maintains a Rac1 activation state via Lys63-linked ubiquitination.

(a) Direct association of TRAF6 and Rac1 in vitro (pull-down). Beads coupled to GST or GST-TRAF6 were incubated with recombinant purified His-Rac1, the Mg2+-free form of Rac1 (EDTA), or Rac1 loaded with GDP or GTPγS, as indicated. The HECT E3 ubiquitin ligase (HACE1)-associated Rac1 (top: Rac1) and input Rac1 (bottom) were determined by Rac1 immunoblotting.
(b) The association of TRAF6C70A with Rac1WT, Rac1T17N or Rac1G12V. Co-IP assay of 293T cells expressing HA-Rac1WT, HA-Rac1G12V, or HA-Rac1T17N and Flag-TRAF6 or Flag-TRAF6C70A as indicated. Cell lysates were immunoprecipitated with anti-Flag antibody and analysed by immunoblotting with antibodies as indicated.
(c) The preferential ubiquitination of active Rac1 by TRAF6 in 293T cells. Ubiquitination and co-IP assay (as in b) of 293T cells expressing Flag-Rac1WT, Flag-Rac1G12V, or Flag-Rac1T17N and/or Myc-TRAF6 as indicated.
(d) GTP-dependent ubiquitination of Rac1 by TRAF6 in vitro. Recombinant Rac1 loaded with GTPγS, GDP or the Mg2+-free form (EDTA) was used in the ubiquitination reaction.
(e) Immunoblot analysis of Rac1 ubiquitination in the lysates and immunoprecipitates of Mst1fl/flMst2fl/fl (WT) and Mst1fl/flMst2fl/flLyz2-Cre (cDKO) BMDMs upon LPS treatment.
(f, g) LPS treatment enhanced the activity of Rac1 and decreased the association of TRAF6 and Rac1 in Raw264.7 cells (e) and BMDMs (f). The Rac1 inhibitor (NSC23766) (e) or Mst1/2 deletion (f) recovered the association of TRAF6 and Rac1 upon LPS treatment.
(h) The association of Rac1, TRAF6 and LyGDI was decreased in WT BMDMs but enhanced in cDKO BMDMs upon LPS stimulation.
Data are from one experiment representative of three independent experiments with similar results (a-h).
LPS stimulation of RAW264.7 cells increased Rac GTP charging and was accompanied by a decrease in the association of Rac with TRAF6. The inhibition of Rac GTP charging with NSC23766, although diminishing Rac ubiquitination (Supplementary Fig. 6b), prevented the LPS-induced dissociation of TRAF6 from Rac1 (Fig. 6f). The LPS stimulation of BMDMs also reduced the association of Rac1 with TRAF6 (Fig. 6g) and with LyGDI (Fig. 6h), which do not occur in LPS-stimulated cDKO BMDMs (Fig. 6g, h). These results indicated that the LPS induced the release of Rac-GDP from LyGDI by initiating Rac1 GTP charging, enabling the TRAF6-catalysed K63 linked polyubiquitination of Rac-GTP at Lys16 followed by the dissociation of TRAF6 from Rac-GTP. Rac ubiquitination, although catalysed subsequent to Rac GTP charging, clearly augments and presumably stabilizes Rac GTP charging. These results established that TRAF6-mediated Lys 63-linked ubiquitination further enhance Rac GTP charging and activation.
Active Rac promotes the assembly of the TRAF6-ECSIT complex
It was previously demonstrated that the recruitment of mitochondria to phagosomes and increased mROS production are mediated by the assembly of a complex of TRAF6 with ECSIT in the mitochondria11. We found that upon E. coli infection of BMDMs, the colocalization of TRAF6 and ECSIT with the bacteria that occurs in the WT does not occur in cDKO cells, and this defect is rescued in cDKO-Rac1G12V BMDMs (Fig. 7a). To investigate how Rac1-GTP promotes the assembly of the TRAF6-ECSIT complex, we performed pull-down assays to determine the relationships of these three molecules. ECSIT interacts with the C-terminal MATH domain (AA351-522) of TRAF6 (Supplementary Fig. 7a), which is the same region of TRAF6 that binds to Rac1 (Supplementary Fig. 7b, c). The co-expression of TRAF6 and ECSIT with Rac1 variants showed that Rac1WT only modestly displaced ECSIT from TRAF6 but inactive RacT17N did so strongly. Furthermore Rac1G12V had little or no effect on the TRAF6-ECSIT complex (Fig. 7b). Immunofluorescence staining further confirmed that active, but not inactive, Rac1 greatly enhanced the co-localization of ECSIT and TRAF6 in HeLa cells (Supplementary Fig. 7d). These data indicate that the tight interaction of TRAF6 and inactive Rac1 is likely due to inefficient ubiquitination of inactive Rac1 by TRAF6. Thus inactive Rac acts in a dominant negative fashion to block the association of TRAF6-ECSIT through the competitive binding of ECIST and inactive Rac to TRAF6.
Figure 7. Rac modulates the assembly of the TRAF6-ECSIT complex for the juxtaposition of mitochondria and phagosomes.

(a) The immunofluorescence staining shows the co-localization of TRAF6 (red) and ECSIT (green) with the bacteria in Mst1fl/flMst2fl/fl (WT) and Mst1fl/flMst2fl/flLyz2-Cre-Rac1G12V (cDKO-Rac1G12V) BMDMs but not in cDKO BMDMs when infected with E. coli. Scale bar, 20 μm.
(b) Co-IP assay of 293T cells expressing Myc-ECSIT, Flag-TRAF6 and HA-Rac1WT, HA-Rac1G12V, or HA-Rac1T17N as indicated. Cell lysates were immunoprecipitated with anti-Flag antibody and analysed by immunoblotting with antibodies as indicated.
(c) Cell lysates of 293T cells expressing Flag-Rac2WT or Flag-Rac2D57N and Myc-TRAF6 as indicated were incubated with GST-PAK70-106 (GST-PAK). The association of active (GTP-bound) Rac2 with GST-PAK70-106 was revealed by Rac2 immunoblotting. GST-PAK70-106 protein was stained by Coomassie blue (CoBlue).
(d) Association of inactive Rac2D57N with TRAF6. Co-IP assay (as in b) of 293T cells expressing HA-Rac2WT, HA-Rac2D57N and Flag-TRAF6 as indicated.
(e) Competitive binding of ECIST and Rac2D57N with TRAF6. Co-IP assay (as in b) of 293T cells expressing Myc-ECSIT, Flag-TRAF6 and HA-Rac2WT or HA-Rac2D57N as indicated.
(f) WT and cDKO BMDMs were infected with adenoviruses expressing control vector, Rac2WT or Rac2D57N, then treated with LPS for 3 h. mROS and cellular ROS were measured by MitoSOX (mROS) and CellROX (cellular ROS), respectively, using flow cytometry.
(g, h) WT BMDMs infected with adenoviruses expressing Rac2WT or Rac2D57N were incubated with E. coli. Immunofluorescence staining shows the co-localization of ECSIT (purple) and TRAF6 (red) and with the bacteria (blue) (g), as well as the mitochondrial networks (Mito) (HSP60, red) with the bacteria (blue)
(h), in Rac2WT- but not Rac257N-infected BMDMs, as indicated by arrows. Scale bar, 20 μm.
Images shown are representative of approximately 100 cells (a, g, h). Data are from one experiment representative of three independent experiments with similar results (a-h).
An inactive mutation in Rac2 (Rac2D57N) causes a human immunodeficiency syndrome characterized by a significant reduction in ROS production in phagocytes8-10. We found that as with inactive Rac1T17N, TRAF6 failed to ubiquitinate the inactive Rac2D57N (Supplementary Fig. 7e). Rac2D57N binds TRAF6 better than Rac2WT (Fig. 7d), and Rac2D57N also efficiently dissociates the TRAF6-ECSIT complexes (Fig. 7e). Furthermore, the induction of mROS and cellular ROS upon LPS stimulation was blocked in WT BMDMs infected with adenoviruses expressing Rac2 D57N (Ad-Rac2 D57N), whereas cDKO BMDMs infected with Ad-Rac2WT displayed normal mROS and cellular ROS production upon LPS stimulation (Fig. 7f). In addition, Ad-Rac2 D57N also blocked the formation of ECSIT and TRAF6 complex (Fig. 7g) and mitochondrion-phagosome juxtaposition (Fig. 7h) during bacterial infections. These results indicate that the GTP charging and ubiquitination of Rac during the bacterial infection of BMDMs is critical for dissociating Rac from the TRAF6 MATH domain to enable the assembly of a TRAF6-ECSIT complex, this in turn mediates mitochondrion-phagosome juxtaposition and an increase in mitochondrial ROS production.
Rac1G12V knock-in rescues the Mst1 and Mst2 null phenotype
cDKO-RacG12V BMDMs displayed normal F-actin organization, mitochondrion-phagosome association and mROS production upon LPS stimulation or bacterial infections (Fig. 3a-c, e, f and Supplementary Fig. 3a). Furthermore, a constitutively active knock-in of Rac1G12V fully rescued the Mst1 and Mst2-deficient phenotype, as shown by the restoration of phagocyte bactericidal activity and normal resistance to bacteraemia during CLP-induced bacterial sepsis (Fig. 8a, b). These results demonstrate that the mediation of Rac activation by Mst1 and Mst2 is a critical event for phagocytes defending against infection. Taken together, our findings reveal TLR-Mst1-Mst2-PKC-Rac-TRAF6-ECSIT signalling in phagocytes as an important bactericidal mechanism (Supplementary Fig. 8a).
Figure 8. Rac1G12V knock-in fully rescues the Mst1/2-deficient phenotype.
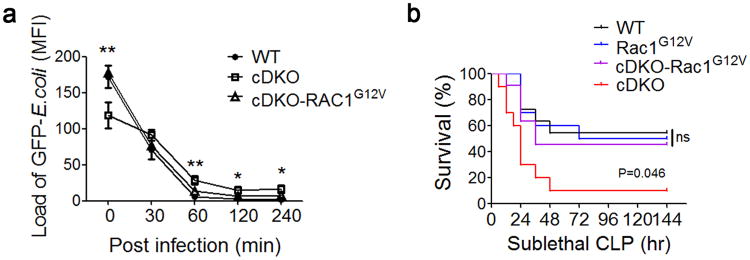
(a) Mst1fl/flMst2fl/fl (WT), Mst1fl/flMst2fl/flLyz2-Cre (cDKO) and cDKO-Rac1G12V BMDMs were infected with GFP-E. coli for the indicated times and the GFP-E. coli was quantified by fluorescence microscopy. Data were assessed with Student's t test and are represented as mean ± s.d. n=5. * p<0.05, ** p<0.01 in a comparison of cDKO and cDKO-Rac1G12V groups.
(b) Mortality of WT (n=11), Rac1G12V (n=10), cDKO (n=10) or cDKO-Rac1G12V (n=11) mice subjected to sublethal CLP. The Mantel–Cox test, p=0.046 compared between cDKO and cDKO-Rac1G12V groups.
Data are from one experiment representative of three independent experiments with similar results (a, b).
Discussion
High amounts of ROS are generated in phagocytes upon stimulation and phagocytosis, and this is necessary for killing ingested pathogens. How phagocytes generate these large amounts of ROS remains incompletely understood. Herein, we identify a new aspect of TLR signalling involved in host defence: i.e., the role of Mst1 and Mst2 in the activation of Rac and the recruitment of mitochondria to phagosomes for optimal ROS production. Stimulation of cell surface TLRs activates Mst1 and Mst2 to phosphorylate and stimulate PKCα, which in turn phosphorylates LyGDI, causing the release of inactive GDP-bound Rac. Once GTP charged, Rac undergoes K63-linked ubiquitination by TRAF6, which strongly augments further Rac-GTP charging. The importance of Mst1 and Mst2's contribution to Rac-GTP charging is shown by the ability of myeloid cell-specific expression of constitutively active RacG12V to restore the defective mitochondrial-phagosomal association and eliminate the increased susceptibility to bacterial infection in mice lacking myeloid Mst1 and Mst2 expression. Thus, Mst1 and Mst2 kinases are critical components of TLR1, TLR2 and TLR4 signalling that support host defences against invading pathogens.
Macrophages generate mROS through TLR signalling in response to bacteria, and they do so by coupling this signalling to mitochondrial complex I by promoting the association of TRAF6 with the mitochondrial complex 1 assembly factor ECSIT11. Nevertheless, the mechanisms by which TLR signalling engages components from both mitochondria and phagosomes to form the active oxidase machinery for ROS production remains incompletely defined. Here, we show that TRAF6 mediates the ubiquitination of Rac and maintains its active GTP-bound state for the full activation of ROS-generating machinery during an antimicrobial response. An inactivating mutation in human Rac2 (Rac2D57N) results in a significant reduction in ROS production by phagocytes and leads to severe bacterial infections, a phenotype similar to that of Mst1 and Mst2-double deficient mice8, 9. We found that Mst1 and Mst2 signalling controls mitochondrial trafficking and the mitochondrion-phagosome association by stimulating Rac activation. Rac-GTP, in addition to its key role in the activation of NADPH oxidase, mediates the actin reorganization that is required for mitochondrial trafficking to phagosomes. Rac2D57N binds GDP but not GTP, and Rac-GDP binds competitively to TRAF6 with ECSIT, thereby interdicting the formation of a TRAF6-ECSIT complex and preventing the association of mitochondria with phagosomes. Thus, Rac is a key downstream regulator of mROS and phagosomal ROS and the Mst1 and Mst2 stimulation of Rac activation is critical for optimal ROS production during pathogen phagocytosis.
In summary, this work demonstrates that kinases Mst1 and Mst2 are key regulators of microbe-elicited ROS production and that they act through a previously unrecognized signalling cascade: i.e., TLR-Mst1-Mst2-PKC-Rac-TRAF6-ECIST. The downstream effectors of the TLR-MyD88 complex for the activation of the kinases Mst1 and Mst2 remain to be identified. Recently, the kinase Mst4 was shown to limit inflammatory responses through the direct phosphorylation of TRAF645. It will be interesting to determine whether the kinases Mst1 and Mst2 can also phosphorylate TRAF6 and, if so, whether this affects the TRAF6-mediated regulation of Rac activation. Previous studies showed that the phosphorylation of FoxO transcription factors by Mst1 protects against neuronal cell death induced by oxidative stress46-48. However, the Mst1-FoxO pathway still remains controversial in immune cells33, 35, 40. Thus, additional studies are needed to identify in full the mechanisms through which TLR-Mst1-Mst2 signalling regulates microbe-elicited ROS production during an antimicrobial response, thereby regulating the killing of the pathogens and the self-antioxidant defence needed for cell survival. Such studies will provide molecular insight that will aid in the design of therapeutic agents for antimicrobial infection.
Methods
Animals
The Mst1 and Mst2 gene conditional knockouts were previously described. Wild type C57BL/6, Vav-Cre mice (stock number 008610), Lyz2-Cre mice (stock number 004781) and C57BL/6-Gt(ROSA)26Sortm9(Rac1*,EGFP)Rsky/J mice (stock number 012361) were originally purchased from the Jackson Laboratory. All mice were maintained under specific pathogen-free conditions at the Xiamen University Laboratory Animal Center (XMULAC). This mouse experiment was approved by the Institutional Animal Care and Use Committee and was in strict accordance with good animal practice as defined by the XMULAC.
Chemicals and reagents
The Pam3csk4, LTA, CpG, Poly(I:C) and R848 were obtained from invivoGen (San Diego, CA, USA). The LPS, rotenone, antimycin A, cytochalasin D, MG132 (C2211), GDP (G7127) and GTPγS (G8634) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The NSC23766 (1177865-17-6) was purchased from Millipore (Billerica, MA, USA). The Mitochondrial Membrane Potential Assay Kit with JC-1 was purchased from Beyotime Biotech (Shanghai, China). Oligo nucleotides were synthesized by Sangon Biotech (Shanghai, China) (Supplementary Table 1). The Phos-tag TM Acrylamide AAL-107 was purchased from the NARD Institute Ltd (Japan).
Flow cytometry assays
Single cells isolated from the bone marrow, spleen, lymph nodes or blood were stained with the indicated fluorescence-conjugated antibodies for 20 min, washed, and resuspended with 1% BSA/PBS flow cytometry assays (FACS) staining buffer containing DAPI (Invitrogen, Carlsbad, CA, USA). Stained cells were analysed with a BD LSRFortessa™ flow cytometer (BD Biosciences, Bedford, USA). Flow cytometry data were plotted and quantified using median fluorescence intensity (MFI) using FlowJo software (Treestar, Ashland, US). The fluorescence-conjugated antibodies, anti-CD3ε (145-2C11), anti-CD62L (MEL14), anti-CD11b (M1/70), anti-B220 (RA3-6B2), anti-CD44 (IM7), anti-F4/80 (BM8) and anti-Gr-1 (RB6-8C5) antibodies, were from Biolegend (San Diego, CA, USA) or BD Biosciences.
ROS and mROS measurements
For FACS-based measurements, cells (RAW246.7 cells, BMDMs or neutrophils) were plated in non-tissue-culture-treated dishes and treated with various TLR agonists or infected with bacteria as indicated. The concentrations of stimulants were as follows: 1 μg/ml LPS, 1μg/ml Pam3CSK4, 2 μg/ml LTA, 10 μg/ml poly(I:C), 1 μM CpG, 1μg/ml R848, 500 nM rotenone, 1mg/ml Oligomycin, or 5 μM Antimycin A. Culture medium was removed, cells were washed with PBS, and then incubated with MitoSOX (to measure the mROS superoxide; Invitrogen) and/or CM-H2DCFDA or CellROX (to measure total cellular H2O2; Invitrogen) at 2.5 μM final concentration in serum-free DMEM (Invitrogen) for 30 min at 37 °C. The cells were washed with warmed PBS, removed from the plates with cold PBS containing 1 mM EDTA by pipetting, pelleted at 1,500 r.p.m. for 3 min, immediately re-suspended in cold PBS containing 1% FBS and subjected to flow cytometry analysis.
For luminol-based ROS measurements, a total of 1 × 105 BMDMs or neutrophils were seeded in each well of a luminometer plate in DMEM with 10% (vol/vol) FBS. Before the assay, cells were washed once with warm Hank's balanced-salt solution (HBSS) without phenol red. Then, 200 μl HBSS containing 100 μM luminol and 5 units of horseradish peroxidase (Sigma) was added to each well. Cells were incubated for 10 min at 37°C, followed by stimulation with L. monocytogenes (MOI, 10) or E. coli (MOI, 20). ROS production was monitored every minute for 2 h with a luminometer (an MRX Revelation 96-well multiscanner, Dynex Technologies, Chantilly, VA, USA).
Cell culture
The 293T, HeLa and RAW264.7 cell lines were obtained from the ATCC and tested negative for mycoplasma contamination and cultured in DMEM supplemented with 10% FBS and 1× penicillin/streptomycin (Invitrogen).
For BMDMs, the femur and tibia were collected from mice of each genotype, and bone marrow cells were flushed with complete DMEM containing 50 mg/ml streptomycin and 10% FBS. Erythrocytes were removed via red blood cell lysis buffer treatment, and the cell suspension was filtered through a 40 μm cell strainer to remove any cell clumps. The single-cell suspensions were then cultured at 37 °C for 1 h, and non-adherent cells were collected and re-plated in complete DMEM with 25% L929 conditioned media. To fully differentiate BMDMs, the cells were cultured for an additional 8 days with media change every 2 days. All cells, when analysed by FACS, were CD11b+F4/80+.
For neutrophils, single-cell suspensions prepared from mouse bone marrow were subjected to Percoll (GE Healthcare Life Sciences, Piscataway, NJ, USA) gradient separation (81%-62.5%) by centrifugation at 1,600 rpm for 30 min at RT. Neutrophils were collected during interphase, washed twice, re-suspended in HBSS and used for further analysis. Cell purity was more than 90% as stained for CD11b+Gr1+ and analysed by FACS.
Cytokine measurement
Cell supernatants or sera were collected at indicated time points after stimulation with LPS or after CLP induction. The concentrations of mouse IL-6, TNF or IL-1β were measured with ELISA kits (Biolegend) or CBA kits (BD Bioscience) according to the manufacturers' instructions.
Cecal ligation and puncture
Mice were anesthetized, and an abdominal incision was made to identify the cecum. The distal one-third of the cecum was ligated with silk suture and punctured once using a 21-gauge needle. A small amount of cecal contents was extruded through the perforation. The peritoneum and skin was closed with a continuous suture after returning the cecum into the abdomen. 1 ml saline was injected i.p. for resuscitation. For sham-treated mice, all of the same steps were performed, except for ligation and puncture of the cecum.
Determination of bacterial loads in tissues
Bacterial counts were performed on aseptically obtained peritoneal fluid, liver, lung, kidney and spleen. 24 hours after CLP and sham surgery, for peritoneal fluid mice were euthanized and the skin of abdomen was cut open in the midline without injury to the muscle. Peritoneal fluid sample was serially diluted in PBS and cultured on tryptose soy agar (TSA) blood agar plates. Colony-forming units (CFUs) were counted after 18–24h incubating 37°C and the results were expressed as log10 of the number of CFU/mL peritoneal fluid. Similar procedures were used on homogenates of liver, lung, kidney and spleen obtained 24 h after sham surgery or after CLP. Values for CFU were expressed as colony counts per gram of tissue.
Construction of GFP-expressing E. coli
Constitutive prokaryotic promoter BBa_J23100 was inserted upstream of the GFP generator BBa_E0840 in the vector pSB1A2. The product was transformed into DH5α-competent cells to acquire a GFP stably expressing E. coli strain (GFP-E.coli). GFP generator BBa_E0840 was a gift from the iGEM team from the Ocean University of China (OUC-iGEM). The sequence of the promoter BBa_J23100 was obtained from partsregistry.org.
Phagocytosis and bacterial killing assay
For FACS-based measurement of phagocytosis, a total of 1 × 106 WT and cDKO BMDMs or neutrophils in PBS were cooled down to 4°C for 30 min. Then, the cells were left uninfected or infected with heat-killed, FITC-labelled and fresh mouse serum-opsonized L. monocytogenes (MOI, 10) or E. coli (MOI, 20) for 20 min at 37°C or 4°C, after which they were washed extensively with cold PBS twice and fixed with 4% paraformaldehyde. The fluorescence of extracellular particles was quenched by replacing the medium with 0.2% Trypan blue in PBS, pH 5.5, shortly before the actual measurement with FACS.
For immunofluorescence measurements of phagocytosis and killing, a total of 2 × 105 BMDMs were grown on glass cover slips in 35 mm dishes. Cells were infected with GFP-E. coli (MOI 20) for 1 h followed by several washes with PBS. Infected cells were further cultured in media containing 10 μg/ml gentamicin. At the indicated time points, the cells were washed in PBS and fixed in 4% (vol/vol) paraformaldehyde for 10 min at RT. Slides were mounted with the mounting medium Vectashield (H1200, Vector laboratories, CA, USA) containing 4′,6-diamidino-2-phenylindole (DAPI) and were imaged with a confocal microscope (Zeiss LSM 780; Zeiss, Germany). The mean pixel intensities of GFP were measured using the Zeiss ZEN lite 2012 software. The ratios of the total areas of the GFP dots to the overall cell number are plotted.
For the in vitro bacterial killing assay, fresh overnight cultures of E. coli and L. monocytogenes were suspended in PBS and opsonized with fresh mouse serum. The BMDMs or neutrophils were incubated with E. coli (MOI, 20) or L. monocytogenes (MOI, 10) for the time points as described at 37°C with intermittent shaking. After each time period, cells were lysed by adding distilled H2O and diluted aliquots were spread on LB agar (E. coli) or Brain Heart Infusion agar (L. monocytogenes) plates. The CFUs were counted after incubating the plates overnight at 37°C.
Mitochondrial membrane potential assay
The mitochondrial membrane potential was measured using a dual-emission potential-sensitive probe; i.e., JC-1 dye according to the manufacturers' instructions. Briefly, WT and cDKO BMDMs in a 6-well plate were incubated with 1 ml of mitochondrial staining solution containing the 50% medium used for cell growth and 2.5 μg/ml of JC-1 dye in a 5% CO2 incubator at 37 °C for 20 min. The cells were then washed twice with warm PBS. Lastly, images were viewed and scanned by a confocal microscopy (Zeiss LSM 780) at 490 nm excitation and 530 nm emission for green and at 540 nm excitation and 590 nm emission for red.
SDS–PAGE, Phos-tag SDS-PAGE and immunoblotting
SDS-PAGE or Phos-tag SDS-PAGE gels were prepared according to the manufacturer's instructions. The proteins were separated by SDS-PAGE or Phos-tag SDS-PAGE, transferred onto a PVDF membrane and then identified by immunoblotting using the appropriate primary antibodies with 1:1000 dilutions or as specifically indicated. Antibodies against HA (3724), P-p38 (9211), P-Jnk (9251), P-Erk (9101), GAPDH (2118), IkBa (4814), P-MARCKS (2741), Hsp60 (4870), COX IV (4850), CYCS (4280), PHB1 (2426), pyruvate dehydrogenase (3205), SDHA (5839), SOD1 (4266), VDAC (4661), NRF1 (12381), K63-polyubiquitin (5621), ubiquitin (3936), PKCα (2056), PKCδ (9616), PKCζ (9372) were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against LyGDI (16122-1-AP), TRAF6 (12809-1-AP), HSP60 (66041-1-Ig), MTERF (16957-1-AP), TFAM (19998-1-AP) were obtained from Proteintech Group Inc. (Wuhan, China). Antibodies against ACO2 (AB61224a) were obtained from Sangon Biotech. (Shanghai, China). Antibodies specific for TRAF6 (ab181622), Rac1 (ab33186), and PGC-1a (ab106814) were obtained from Abcam (Cambridge, MA, USA). Antibodies against Rac1 (sc-217), HA (sc-7392), Myd88 (sc-11356) and ECSIT (sc-79473) were obtained from Santa Cruz (Santa Cruz, CA, USA). Antibodies against β-actin (a2066, 1:5000) and Flag (F1804, 1:5000) were purchased from Sigma. Antibodies against Mst1, Mst2, and phospho-Mob1 (Thr35) were obtained as described previously 49. Horseradish peroxidase-conjugated antibodies against rabbit or mouse IgG were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA, 1:3000). The protein bands were visualized using a SuperSignal West Pico Kit (Thermo Fisher Scientific Pierce, IL, USA) according to the manufacturer's instructions.
In vitro kinase assays
Mst2 kinase was expressed as a Flag-tagged protein in HEK293T cells. The Mst2 kinase assay was performed by incubating Flag-tagged Mst2 immunoprecipitate with the recombinant (purified in bacteria) GST-PKCα NT (1-338aa), GST-PKCα NT (1-338aa) S226A in a kinase assay buffer containing 2 mM dithiothreitol (DTT), 10 mM MgCl2, 50 μM ATP, 40 mM HEPES (pH 7.4), 1 mM EDTA. The reactions were incubated for 30 min at 30°C prior to SDS-PAGE and immunoblotting.
H&E staining
The tissue specimens were fixed in 10% neutral-buffered formalin for 24 h and then dehydrated in increasing concentrations of isopropyl alcohol followed by clearing of alcohol by xylene. The specimens were subsequently embedded in paraffin wax in cassettes to facilitate tissue sectioning. Standard H&E staining was performed on 5 μm sections from each specimen block.
Assay of endogenous and recombinant Rac1 GTP charging
293T cells seeded in a 60 mm dish with 85% confluence were transfected with the indicated constructs. Forty-eight hours after transfection, the cells were harvested and lysed in binding buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 10 mg/ml leupeptin, 10 mM NaF, 2 mM Na3VO4 and 1 mM PMSF). For BMDMs, cells seeded in a 60 mm dish with 80% confluence were harvest using binding buffer. Twenty micrograms of E. coli-purified GST-PAK1-CRIB preloaded on GSH Sepharose was incubated with the precleared cell lysates at 4°C for 2 h. After three washes with binding buffer, the amount of Rac-GTP bound to the beads was determined by Flag or Rac1 immunoblot after SDS-PAGE.
In vivo and in vitro ubiquitination assay
For the in vivo ubiquitination assay, 293T cells were transfected with the indicated plasmids for 36 h and lysed in ice-cold lysis buffer TNTE 0.5% (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, and 0.5% Triton X-100, containing 10 mM NaF, 2 mM Na3VO4, 10 mg/ml leupeptin and 1 mM PMSF). The cell lysates were then subjected to anti-Flag IP, eluted by boiling 10 min in 1% SDS, diluted 10 times in lysis buffer TNTE 0.5% and then re-immunoprecipitated with anti-Flag antibody (2 × IP). The ubiquitin-conjugated proteins were detected by immunoblotting with appropriate antibodies. For in vitro ubiquitination reactions, bacterially expressed GST-Rac1-Flag and GST-TRAF6 proteins were purified using glutathione sepharose beads in TNTE 0.5% buffer. Ubiquitination reactions were carried out with 0.3 mg Rac1, 0.35 mg rabbit E1 ubiquitin activating enzyme (Boston Biochem, Cambridge, MA, USA), 0.4 mg UBE2N/UEV1A recombinant E2 complex (Boston Biochem), and 10 mg of ubiquitin (Boston Biochem) in 20 μl of ubiquitin conjugation reaction buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 0.05 mM DTT, supplemented with 5 mM ATP). Rac1 was loaded with GTPγS or GDP or left in its Mg2+-free GDP/GTP-empty form, as described previously 50.
shRNA and lentiviral infection
Lentivirus was produced by co-transfecting 293T cells with the shRNA in the pll3.7 vector, VSV-G and Δ8.9 plasmids using Lipofectamine 2000 (Invitrogen). TRAF6-shRNA-1 (5′-GGTTGCCGAAATGGAAGCA-3′), TRAF6-shRNA-2 (5′-GAGAACAGATGCCTAATCA-3′), TRAF6-shRNA-3 (5′-CAGAACTGCTTGCCTTTCA-3′) and control shRNA (5′-GCAAGCTGACCCTGAAGTTC-3′). The most efficient shRNA was TRAF6-shRNA-1. Viral supernatant was harvested at 48-72 h post-transfection, passed through a 0.45 μm filter, diluted 2:3 with fresh medium containing 8 μg/ml polybrene and used to infect the target cells at 80% confluence. The protein expression was visualized by immunoblotting.
Latex bead phagocytosis assay
Yellow-green 3 micron Fluoresbrite carboxy latex microparticles (Polysciences, Warrington, PA, USA) were left untreated or coated with 30 μg/ml LPS or Pam3CSK4 overnight at 4 °C in PBS. The beads were washed ten times in large volumes of PBS containing 1% FBS to remove unbound TLR agonists. BMDMs were cultured on coverslips in 6-well dishes and incubated on ice for 10 min. Microparticles were added at a concentration of approximately 4-8 beads per cell and allowed to settle on the cells for an additional 10 min on ice. Warm medium was added to the cells, and the plates were warmed to 37 °C to allow bead phagocytosis for the indicated times. Cells were then fixed with 3.7% paraformaldehyde at room temperature (RT) for 20 min, permeabilized with 0.1% Triton X-100 and stained with the indicated primary and appropriately labelled secondary antibodies for confocal microscopy (Zeiss LSM 780).
CFSE labelling
BMDMs were labelled with 2.5 μM CFSE dye at 37°C for 8 mins according to manufacturer's instructions (Invitrogen), washed three times in complete DMEM, trypsinized and mixed with unlabelled cells as indicated.
Confocal fluorescence microscopy
For fluorescence analysis of F-actin, cells on coverslips were fixed with 3.7% paraformaldehyde for 10 min and washed with 0.1% Triton X-100 in PBS for 5 min 3 times. F-actin in cells was stained by using Actin-Tracker Green (FITC-labelled phalloidin, Beyotime, China) in a solution containing 0.1% Triton X-100 and 5% BSA in PBS. After incubation for 50 min at RT, the cells were extensively washed, mounted and imaged using a fluorescence microscope (Zeiss LSM 780).
For fluorescence analysis of other molecules, BMDMs seeded on glass coverslips in 6-well dishes were incubated with GFP-E. coli or FITC-labelled heat-inactivated L. monocytogenes for 30 min. HeLa cells seeded on coverslips in 6-well dish with 30% confluence were transfected with indicated constructs shown in the figures and cultured for another 24 h. The cells were washed three times with PBS and fixed with 4% (vol/vol) paraformaldehyde for 15 min at RT, after which additional immunofluorescence staining was applied. For staining of Hsp60 (1:100; Proteintech 66041-1), Rac1 (1:100; Abcam ab33186), TRAF6 (1:100; Proteintech 12809), ECSIT (1:100; Santa Cruz sc-79473), OctA (Flag)-tag (1:250; Santa Cruz sc-807), Myc-tag (1:250; Santa Cruz sc-40), and HA-tag (1:250; Santa Cruz sc-7392), fixed cells were rinsed with PBS and then incubated with 0.2% Triton® X-100, 0.2% BSA in PBS on ice for 10 min. Following permeabilization, the cells were blocked with 0.02% Triton® X-100, 5% BSA in PBS at RT for 30 min and were incubated with specific primary antibodies for 1 h. After three washes with PBS, the cells were incubated with secondary antibodies (Alexa Fluor 488-conjugated anti-mouse IgG (A21202), 488-conjugated anti-rabbit IgG (A21206), 555-conjugated anti-mouse IgG (A31570), 555-conjugated anti-rabbit IgG (A31572) or 488-conjugated donkey anti-goat IgG (A11055) purchased from Invitrogen as needed for another 1 h. Subsequently, the cells were washed three times with PBS and mounted with mounting medium Vectashield containing DAPI. All images were collected using a confocal microscope (Zeiss LSM 780).
Statistical analysis
All statistical analyses were performed using Prism5 (GraphPad Software, Inc. CA, US). The data are presented as the mean ± standard deviation (s.d.), and Student's t-test was used for comparisons between two groups. Survival data were analysed using the Kaplan–Meier statistical method. A p value of less than 0.05 was considered statistically significant. In the graphed data, *, **, and *** denote p values of less than 0.05, 0.01, and 0.001, respectively.
Supplementary Material
Acknowledgments
This work was supported by grants from National Basic Research Program (973) of China (2015CB910502, L.C.), China's 1000 Young Talents Program (D.Z., L.C.), the 111 Projects (numbers B12001 and B06016), the Fundamental Research Funds for the Central Universities of China-Xiamen University (numbers CXB2014004 (J.Z.), 20720140551 (L.C.), 2013121034 (D.Z.) and 20720140537 (D.Z.)), the National Natural Science Foundation of China (grant numbers 31270918 (D.Z.), 81222030(D.Z.), 81372617(L.C.), J1310027(D.Z.), 81422018 (L.C.), 81472229 (L.H.), 81302529 (X.L.) and U1405225 (L.C.)), the Natural Science Foundation of Fujian (grant numberd 2013J06011 (DZ) and 2014D007 (X.L.). JA was in part supported by the RO1 awards CA136567 and institutional funds. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author Contributions: D.Z., L.C., J.G., and X.S. designed the research and helped with data analysis; J.G., X.S., P.W., S.Z., X.W., H.W., C.X., L.H., X.L., H.Z., Q.L., M.J., K.J., Q.C., J.Z., and Y.L. performed the experiments and helped with data analysis; S.S., H.R.W., R.Z., R.J., S.C.L., J. H., and J.A. contributed to discussions and provided critical reagents; D.Z., L.C., and J.A. wrote the paper.
Comompeting Financial Interests: The authors declare no competing financial interests.
References
- 1.West AP, Koblansky AA, Ghosh S. Recognition and signaling by toll-like receptors. Annual review of cell and developmental biology. 2006;22:409–437. doi: 10.1146/annurev.cellbio.21.122303.115827. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B, et al. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annual review of immunology. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 3.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nature reviews Immunology. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 4.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological reviews. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Bokoch GM, Diebold BA. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood. 2002;100:2692–2696. doi: 10.1182/blood-2002-04-1149. [DOI] [PubMed] [Google Scholar]
- 6.Mehta D, Rahman A, Malik AB. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. The Journal of biological chemistry. 2001;276:22614–22620. doi: 10.1074/jbc.M101927200. [DOI] [PubMed] [Google Scholar]
- 7.DerMardirossian C, Schnelzer A, Bokoch GM. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Molecular cell. 2004;15:117–127. doi: 10.1016/j.molcel.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 8.Ambruso DR, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4654–4659. doi: 10.1073/pnas.080074897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams DA, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. 2000;96:1646–1654. [PubMed] [Google Scholar]
- 10.Gu Y, et al. Biochemical and biological characterization of a human Rac2 GTPase mutant associated with phagocytic immunodeficiency. The Journal of biological chemistry. 2001;276:15929–15938. doi: 10.1074/jbc.M010445200. [DOI] [PubMed] [Google Scholar]
- 11.West AP, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Molecular cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emre Y, et al. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. The Biochemical journal. 2007;402:271–278. doi: 10.1042/BJ20061430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu W, Hsu YM, Bi L, Songyang Z, Lin X. CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex. Nature immunology. 2009;10:1208–1214. doi: 10.1038/ni.1788. [DOI] [PubMed] [Google Scholar]
- 15.Arsenijevic D, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nature genetics. 2000;26:435–439. doi: 10.1038/82565. [DOI] [PubMed] [Google Scholar]
- 16.Bulua AC, et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) The Journal of experimental medicine. 2011;208:519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Creasy CL, Ambrose DM, Chernoff J. The Ste20-like protein kinase, Mst1, dimerizes and contains an inhibitory domain. The Journal of biological chemistry. 1996;271:21049–21053. doi: 10.1074/jbc.271.35.21049. [DOI] [PubMed] [Google Scholar]
- 18.Creasy CL, Chernoff J. Cloning and characterization of a human protein kinase with homology to Ste20. The Journal of biological chemistry. 1995;270:21695–21700. doi: 10.1074/jbc.270.37.21695. [DOI] [PubMed] [Google Scholar]
- 19.Rawat SJ, Chernoff J. Regulation of mammalian Ste20 (Mst) kinases. Trends in biochemical sciences. 2015;40:149–156. doi: 10.1016/j.tibs.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avruch J, et al. Protein kinases of the Hippo pathway: regulation and substrates. Seminars in cell & developmental biology. 2012;23:770–784. doi: 10.1016/j.semcdb.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou D, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 2004;306:2267–2270. doi: 10.1126/science.1103233. [DOI] [PubMed] [Google Scholar]
- 23.Del Re DP, et al. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Molecular cell. 2014;54:639–650. doi: 10.1016/j.molcel.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maejima Y, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nature medicine. 2013;19:1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan D. The hippo signaling pathway in development and cancer. Developmental cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nature reviews Cancer. 2013;13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- 27.Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes & development. 2013;27:355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nature reviews Drug discovery. 2014;13:63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer cell. 2008;13:188–192. doi: 10.1016/j.ccr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 30.Zhou D, et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E1312–1320. doi: 10.1073/pnas.1110428108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdollahpour H, et al. The phenotype of human STK4 deficiency. Blood. 2012;119:3450–3457. doi: 10.1182/blood-2011-09-378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nehme NT, et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood. 2012;119:3458–3468. doi: 10.1182/blood-2011-09-378364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du X, et al. Mst1/Mst2 regulate development and function of regulatory T cells through modulation of Foxo1/Foxo3 stability in autoimmune disease. J Immunol. 2014;192:1525–1535. doi: 10.4049/jimmunol.1301060. [DOI] [PubMed] [Google Scholar]
- 34.Dong Y, et al. A cell-intrinsic role for Mst1 in regulating thymocyte egress. J Immunol. 2009;183:3865–3872. doi: 10.4049/jimmunol.0900678. [DOI] [PubMed] [Google Scholar]
- 35.Ueda Y, et al. Mst1 regulates integrin-dependent thymocyte trafficking and antigen recognition in the thymus. Nature communications. 2012;3:1098. doi: 10.1038/ncomms2105. [DOI] [PubMed] [Google Scholar]
- 36.Katagiri K, Imamura M, Kinashi T. Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nature immunology. 2006;7:919–928. doi: 10.1038/ni1374. [DOI] [PubMed] [Google Scholar]
- 37.Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nature immunology. 2003;4:741–748. doi: 10.1038/ni950. [DOI] [PubMed] [Google Scholar]
- 38.Katagiri K, et al. Crucial functions of the Rap1 effector molecule RAPL in lymphocyte and dendritic cell trafficking. Nature immunology. 2004;5:1045–1051. doi: 10.1038/ni1111. [DOI] [PubMed] [Google Scholar]
- 39.Zhou D, et al. The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naive T cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20321–20326. doi: 10.1073/pnas.0810773105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mou F, et al. The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. The Journal of experimental medicine. 2012;209:741–759. doi: 10.1084/jem.20111692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raab M, et al. T cell receptor “inside-out” pathway via signaling module SKAP1-RapL regulates T cell motility and interactions in lymph nodes. Immunity. 2010;32:541–556. doi: 10.1016/j.immuni.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasai M, Yamamoto M. Pathogen recognition receptors: ligands and signaling pathways by Toll-like receptors. International reviews of immunology. 2013;32:116–133. doi: 10.3109/08830185.2013.774391. [DOI] [PubMed] [Google Scholar]
- 43.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 44.Jiang X, Chen ZJ. The role of ubiquitylation in immune defence and pathogen evasion. Nature reviews Immunology. 2012;12:35–48. doi: 10.1038/nri3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiao S, et al. The kinase MST4 limits inflammatory responses through direct phosphorylation of the adaptor TRAF6. Nature immunology. 2015;16:246–257. doi: 10.1038/ni.3097. [DOI] [PubMed] [Google Scholar]
- 46.Lehtinen MK, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 47.Yuan Z, et al. Regulation of neuronal cell death by MST1-FOXO1 signaling. The Journal of biological chemistry. 2009;284:11285–11292. doi: 10.1074/jbc.M900461200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao L, et al. The c-Abl-MST1 signaling pathway mediates oxidative stress-induced neuronal cell death. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:9611–9619. doi: 10.1523/JNEUROSCI.0035-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Praskova M, Xia F, Avruch J. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Current biology : CB. 2008;18:311–321. doi: 10.1016/j.cub.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torrino S, et al. The E3 ubiquitin-ligase HACE1 catalyzes the ubiquitylation of active Rac1. Developmental cell. 2011;21:959–965. doi: 10.1016/j.devcel.2011.08.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.