

Abstract
Chromatin factors have emerged as the most frequently affected family of proteins in cancer. We have previously identified the histone deacetylase SIRT6 as a key tumor suppressor, yet whether point mutations are selected for in cancer remains unclear. In this manuscript, we characterized naturally occurring patient-derived SIRT6 mutations. Strikingly, all the mutations significantly affected either stability or catalytic activity of SIRT6, indicating that these mutations were selected for in these tumors. Further, the mutant proteins failed to rescue SIRT6 KO cells, as measured by the levels of histone acetylation at glycolytic genes and their inability to rescue the tumorigenic potential of these cells. Notably, the main activity affected in the mutants was histone deacetylation rather than demyristoylation, pointing to the former as the main tumor suppressive function for SIRT6. Our results identified cancer-associated point mutations in SIRT6, cementing its function as a tumor suppressor in human cancer.
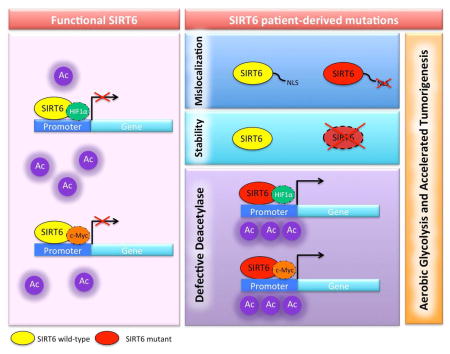
Graphical abstract
INTRODUCTION
The (NAD)+-dependent histone deacetylase, SIRT6 is a mammalian sirtuin with broad functions including glucose homeostasis, maintenance of genome stability, and suppression of cellular transformation (Mostoslavsky et al., 2006; Sebastian et al., 2012; Zhong et al., 2010). In this context, SIRT6 co-represses both HIF1α and MYC by deacetylating histone 3 (H3) lysine 9 (K9) and lysine 56 (K56) at the promoters of several glycolytic and ribosomal protein genes. Consequently, SIRT6-deficient cells display increased glycolysis even under normoxic conditions, a phenomenon termed aerobic glycolysis by Otto Warburg, who first described this phenotype in cancer cells (Warburg, 1956). Indeed, SIRT6 inhibits cancer growth, in a manner that depended on glycolytic metabolism (Sebastian et al., 2012). Importantly, we found SIRT6 commonly downregulated or deleted in human cancer, where lower SIRT6 expression is associated with poor prognosis. Thus, SIRT6 acts as a key tumor suppressor and critical node between cellular transformation and metabolism (Sebastian et al., 2012).
SIRT6-dependent phenotypes have been attributed to its intrisic histone deacetylase activity, which seems negligible in biochemical assays, but can be enhanced by binding to nucleosomes and/or long-chain fatty acids (Feldman et al., 2013; Gil et al., 2013; Kawahara et al., 2009; Michishita et al., 2008; Sebastian et al., 2012; Zhong et al., 2010). Recent studies have shown that SIRT6 can also function as a protein demyristoylase (Feldman et al., 2013; Jiang et al., 2013), introducing the possibility that SIRT6 may suppress tumorigenesis through the deacylation of long-chain fatty acyl groups rather than histone deacetylation. The lack of known SIRT6 point mutations selected for in human cancer has hindered progress in the molecular understanding of the tumor suppressive roles of SIRT6.
In this manuscript, we identify and characterize eight naturally occurring tumor-associated point mutations in SIRT6 that alter stability, localization and/or enzymatic activity and characterize their ability to repress HIF1a and MYC transcriptional activity, glycolytic metabolism and cellular transformation.
RESULTS
SIRT6 is mutated in a variety of human cancers
In order to determine whether SIRT6 could be inactivated in human tumors through point mutations, we analyzed somatic mutations obtained via exome sequencing of patient-derived tumor samples from 12 tumor types in the TCGA and found eight somatic mutations in SIRT6. These mutations were found in a variety of tumor types such as non-small cell lung cancer, renal clear cell carcinoma, cervical carcinoma and melanoma (Figure 1A). Although SIRT6 did not meet statistical significance due to the low frequency of mutations (Lawrence et al., 2014), tumorportal.org), all of the mutations were nonsynonymous; seven of them were missense mutations and one mutation was a nonsense mutation, suggesting that they may have functional relevance. The mutations occured throughout the protein and involved residues that are highly conserved from flies to humans (Figure 1B). Mutations occurring in the N-terminus include an aspartic acid at position 25 mutated to asparagine (D25N) and a glutamic acid at position 36 mutated to valine (E36V). Catalytic domain mutations include an aspartic acid at position 63 mutated to tyrosine (D63Y), an alanine at position 89 mutated to serine (A89S), an aspartic acid at position 116 mutated to asparagine (D116N), a threonine at position 263 mutated to a proline (T263P) and finally a glutamic acid at position 260 replaced with a stop codon (E260Term), leading to premature truncation of the protein and loss of the C-terminus and nuclear localization signal (NLS). Only one mutation involved the C-terminus, where a proline at position 274 was mutated to a lysine (P274L) (Figure 1A–B).
Figure 1. Identification of patient-derived SIRT6 loss-of-function mutations in cancer.

(A) Table of patient-derived SIRT6 point mutations, the disease, type of mutation and amino acid change. (B) Alignment of metazoan SIRT6 homologues. Red boxes highlight the position of the mutations in the N-terminus (red) catalytic core (yellow) or C-terminus (blue). (C) Western blot of chromatin fraction in SIRT6 KO MEFs with doxycycline inducible overexpression of wild-type (WT) SIRT6 and SIRT6 mutants. (D) Sub-cellular localization of WT and mutant SIRT6 proteins. Fluorescent images of fixed 293T cells transiently transfected with the indicated EGFP-tagged SIRT6 deletion and point mutants. Nuclei are co-stained with DAPI (40×). (E) Thermal denaturation assays were performed to determine the melting temperature (Tm) of WT (circle), D25N (square), D63Y (triangle), and D116N (upside-down triangle) SIRT6. A graph with representative experiments is shown. At least three trials were performed for each SIRT6 variant.
SIRT6 point mutations that alter localization or stability
Each of these SIRT6 mutations were cloned and expressed in SIRT6 knockout (KO) mouse embryonic fibroblasts (MEFs). We also included, as a control, a variant found in the human population, N46S. Three of the mutants (D25N, D116N and E260Term) demonstrated reduced chromatin-bound and whole cell lysate protein levels, despite equivalent mRNA expression, suggesting mislocalization or poor protein stability (Figure 1C & Supplemental Fig. 1A & B). We noted that the appearance of reduced expression with the D25N mutant may have been an artifact of reduced antibody affinity since the antibody epitope includes D25, rather than mislocalization or poor protein stability. Therefore, we used a GFP antibody to detect a GFP-tagged version of wild-type (WT) SIRT6 and each of the SIRT6 mutants to confirm that SIRT6 D25N expression and localization to chromatin was equivalent to WT (Supplemental Fig. 1C), while the D116N and E260Term mutants displayed reduced levels in chromatin, as observed with the non-GFP tagged constructs (Figure 1C). We also noted that each of the mutant alleles localized to the nucleus, except for the E260Term mutant, consistent with the lack of a nuclear localization signal (Fig. 1D). Interestingly, the levels of the D116N mutant were consistently reduced both in the chromatin fraction when expressed in mammalian cells, and when recombinantly overexpressed in E. coli, suggesting defective protein stability. To test this hypothesis, we determined the melting temperature of D116N, D25N, D63Y, and WT SIRT6. While, D25N (43.74±0.04 °C) and D63Y (45.8±0.1 °C) exhibited melting temperatures very similar to WT (46.08±0.05 °C), the melting temperature of D116N was ~13 °C lower (33.4±0.6 °C) (Figure 1E). At physiologic temperatures, this protein is expected to be largely unfolded and subject to degradation.
In silico analysis predicts functionally significant structural changes induced by cancer-associated SIRT6 mutations
To gain insight into the functional significance of these mutated residues, we analyzed the previously solved co-crystal structure of SIRT6 bound to ADP-ribose and an H3K9 myristoylated peptide (PDB: 3ZG6) (Jiang et al., 2013)(Figure 2A). Aspartic acid 25 is located in the N-Terminal Domain (NTD) and hydrogen bonds to the backbone amide nitrogen and carbonyl oxygen of H255 as well as to the ε-amino group of K33 (Figure 2B). The NTD is required for catalytic activity and chromatin association (Tennen et al., 2010), therefore a mutation to asparagine will likely disrupt the orientation and interactions between the loop and the Rossmann-fold domain. Aspartic acid 63 is located in the NAD+-binding pocket, ~3 Å from the adenosine moiety of NAD+, and forms hydrogen-bonding interactions with amino acids in the active site (Figure 2C). Therefore, a mutation to tyrosine is predicted to have a highly detrimental effect on the ability of SIRT6 to bind NAD+ and catalyze deacetylation. Alanine 89 and Aspartic acid 116 are located on loops in the back of the active site (Figure 2D–E). Alanine 89 forms a backbone hydrogen bond interaction with T85 and mutation to serine might affect the orientation of this loop. The invariant aspartic acid residue (D116) forms a hydrogen bond to the carboxamide amino group of nicotinamide and mutation to glutamine will disrupt this conserved interaction. Glutamic acid 36 and Threonine 263 are located in the Rossmann-fold domain and form hydrogen bonds with R39 and D259, respectively (Figure 2F–G). Mutation of E36 to valine will abolish this hydrogen bond, which could destabilize other interactions in the helix. A proline mutation at T263 will disrupt the structure of the helix in the Rossmann-fold. Proline 274 is located in the proline rich C-Terminal Domain (CTD) loop, and a mutation to threonine may affect the orientation of the loop (Figure 2H). Taken together, all of the identified mutations are likely to affect catalytic activity directly or through structural rearrangements.
Figure 2. Structural and enzymatic analysis of WT SIRT6 and SIRT6 point mutations.

(A) Locations of mutated residues mapped on the crystal structure of SIRT6 (PDB: 3GZ6). (B–H) Zoomed in view of the mutated residues highlighted in red. Also displayed are residues in close proximity to the mutated residues and hydrogen bonding interactions. (I) SIRT6 deacetylase activity measured in vitro. Recombinant WT and SIRT6 mutants (2 μM) were incubated with 50 μM H3K9Ac peptide and 0.5 mM NAD+. (J) Fold change in SIRT6-dependent deacetylation in the presence of 300 μM myristic acid. (K) SIRT6 demyristoylase activity measured in vitro. WT and SIRT6 mutants (0.5 μM) were incubated with 50 μM H3K9Myr peptide and 0.5 mM NAD+. (I–K n≥3, ± standard deviation) (** = p ≤ 0.01, *** = p ≤ 0.001, ns = p > 0.05) (L) Western blot of chromatin fraction in SIRT6 KO MEFs with doxycycline inducible overexpression of WT SIRT6 and SIRT6 mutants. SIRT6 and H3 total western blots from Fig. 1C are represented here for direct comparison. (M) Steady-state rates of demyristoylation were measured by varying NAD+ (25–1200 μM) in the presence of 0.5 μM D63Y SIRT6 and 50 μM H3K9Myr peptide. Calculated values determined from non-linear regression fits to Michaelis-Menton are shown below for WT, D63Y, A89S, T263P (n≥3, ± standard deviation). *WT SIRT6 values previously published (Feldman et al., 2015).
Deacetylase activity of SIRT6 mutants
We next performed a highly-sensitive, quantitative, in vitro deacetylase assay on purified recombinant SIRT6 mutants and compared the results obtained with WT enzyme. First, WT or SIRT6 mutants were reacted with 50 μM H3K9Ac peptide and 0.5 mM NAD+. Reaction substrates and products were separated by HPLC and quantified. Strikingly, all mutants displayed decreased deacetylase activity relative to WT SIRT6 (3.9±0.3×10−4 μmol min−1 mg SIRT6−1). D25N, E36V, A89S, T263P and P274L all exhibited approximately 50% of WT SIRT6 activity, while D116N and D63Y yielded nearly negligible deacetylase activity at ~2% of WT SIRT6 levels (Figure 2I). Importantly, the non-cancer associated N46S variant displayed deacetylase activity similar to WT. The decreased catalytic activity of D116N SIRT6 is due in part to decreased protein stability, as evidenced by its 13 °C lower melting temperature. Additionally, the invariant D116 residue is located in the nicotinamide-binding pocket of SIRT6 and plays a direct role in NAD+ binding (Fig. 2E). The corresponding aspartic acid residue in the crystal structure of bacterial Sir2Tm forms a hydrogen bond with the carboxamide amino group of the nicotinamide moiety. Mutation of the aspartic acid residue to an asparagine decreased the catalytic efficiency of Sir2Tm by two orders of magnitude (Avalos et al., 2005). Therefore, as predicted, a mutation of D116 to asparagine led to a similar reduction in SIRT6 activity.
We recently demonstrated that SIRT6 can be directly activated by long-chain fatty acids in vitro (Feldman et al., 2013). To assess the ability of fatty acids to activate these SIRT6 mutants, we analyzed the in vitro deacetylase activity in the presence of myristic acid. WT and SIRT6 mutants were reacted with 50 μM H3K9Ac peptide and 0.5 mM NAD+ in the presence of 300 μM myristic acid. The fold change in activity of each mutant in the presence of myristic acid was compared to WT. D25N (11±0.5) and E36V (11±2) were activated by myristic acid to the same level as WT SIRT6 (10±1), while P274L (7±1) and T263P (6±1) were activated to a slightly lesser extent than WT (Figure 2J). Interestingly, the activity of A89S SIRT6 decreased 2-fold in the presence of myristic acid, which resulted in a 20-fold decrease in activation relative to WT.
To determine whether cancer-associated mutations in SIRT6 affect the previously reported demyristoylase activity (Jiang et al., 2013), WT and SIRT6 mutants were reacted with 50 μM H3K9Myr peptide in the presence of 0.5 mM NAD+ and analyzed as described above. As predicted, the N46S variant, which was used as a control in the assay, displayed demyristoylase activity equal to WT. Surprisingly, the D25N, E36V, A89S, and P274L mutations displayed similar ability to remove myristoyl groups compared with WT SIRT6 (≥ 75%). The demyristoylase activity of T263P activity was greater than 70% of WT, whereas D63Y and D116N were the only two mutants that exhibited less than 5% demyristoylase activity (Figure 2K), which is consistent with loss of general catalytic function. Importantly, these data suggest that cancer-associated point mutations may specifically inactivate SIRT6 deacetylase activity, without affecting its ability to remove larger fatty acyl groups. Most dramatic is the A89S mutant, which displays similar demyristoylation activity to that of WT, but cannot be activated toward acetylated histone substrates (compare Figures 2J and 2K).
To examine the effect of SIRT6 mutations on histone acetylation in vivo, we analyzed total H3K9 and H3K56 acetylation levels in bulk chromatin of MEFs following short-term expression of either WT or mutant SIRT6 using a dox-inducible system. In this context, the D25N, D63Y, D116N, E260Term mutants failed to reduce levels of H3K56 and K9 acetylation when compared to WT SIRT6 (Figure 2L). This is consistent with the reduced deacetylase activity of the D25N and D63Y mutations and the reduced binding of the D116N and E260term mutants to chromatin. The D25N mutant behaves similarly to a previously described, catalytically inactive mutation where the highly conserved histidine 133, within the core sirtuin domain of SIRT6, is mutated to tyrosine (H133Y)(Mostoslavsky et al., 2006). The SIRT6 mutants E36V, A89S, T263P and P274L were able to reduce the levels of acetylated H3K56 and H3K9 in bulk chromatin when overexpressed, despite having decreased in vitro catalytic activity relative to WT (compare 2I and Figure 2L), suggesting that overexpression can overcome their reduced enymatic activity. However, these changes in bulk chromatin may not specifically reflect the effect of these SIRT6 mutants to deacetylate histones in the promoter regions of specific SIRT6 regulated genes.
The D63Y mutant displays limited deacetylase and demyristoylase activity and led to large increases in acetylation in vivo. As described earlier, D63 is located in the NAD+ binding pocket and is 3 Å from the adenosine moiety of NAD+ (Figure 2C). To test the hypothesis that D63Y decreases the affinity of SIRT6 for NAD+, we performed steady-state kinetic analysis with increasing NAD+ at saturating levels of H3K9Myr peptide. The H3K9Myr peptide was used in the assay due to the prohibitively high concentration of H3K9Ac peptide required for saturation (estimated at nearly 4.5 mM (Feldman et al., 2013)). The data were subjected to Michaelis-Menten analysis, and the kinetic constants were compared to WT SIRT6, as well as to A89S and T263P, which were expected to show no defects in NAD+ affinity (Figure 2M, Supplemental Fig. 1E). The kcat/Km value is the lowest for D63Y (7.2±0.9 s−1 M−1) and is 125 times slower than WT SIRT6 (9.0±0.6×102 s−1 M−1). Whereas the small decrease in the catalytic efficiency of A89S (6.3±0.5×102 s−1 M−1) and T263P (2.5±0.3×102 s−1 M−1) is caused by a decrease in the kcat, the decrease in D63Y is due to both a decrease in kcat and an increase in the Km for NAD+. The Km for NAD+ increased ~7 times (90±10 μM vs. 13±1 μM) and the kcat decreased ~19 times relative to WT SIRT6 (6.3±0.2×10−4 s−1M−1 vs. 1.20±0.02×10−2 s−1M−1). Together, the results indicate a mutation of D63 to tyrosine disrupts both the affinity of SIRT6 for NAD+ and catalysis, leading to dramatic loss of SIRT6-dependent deacetylation and the subsequent robust increase in acetylation observed in vivo.
SIRT6 mutants fail to repress glycolytic genes
We next determined the ability of the mutants to repress HIF1α and MYC transcriptional activity. As shown in Figure 3A–B, both HIF1α and MYC luciferase reporters were repressed by WT SIRT6, however the mutants were unable to repress either HIF1α or MYC luciferase activity. We next expressed each of these mutants as well as WT SIRT6 in SIRT6 KO MEFs. Consistent with their inablity to repress HIF1a and MYC transcriptional activity, the SIRT6 mutants were unable to decrease glucose uptake by SIRT6 KO MEFs (Figure 3C). Similarly, these mutants were unable to repress the glycolytic genes PFKm, PDK1 and LDHb (Figure 3D), and the ribosomal genes RPL3, RPL23, RPS15A (Supplemental Figure 2B). Interestingly, E36V, T263P and P274L were still able to partially inhibit glucose uptake. Thus, we tested additional genes that may explain why these SIRT6 mutants may be able to repress glucose uptake and found that, unlike other glycolysis-related genes, these mutants retain the ability to silence the Glucose transporter 1 (GLUT1), a SIRT6 target gene which is directly responsible for controlling glucose uptake (Supplemental Figure 2A). Taken together, these results suggest that these cancer-associated SIRT6 mutations are unable to repress MYC- and HIF1α-dependent transcription.
Figure 3. SIRT6 cancer-associated point mutations fail to repress HIF and MYC.

(A & B) Luciferase reporter gene assay for MYC (A) and HIF1α (B) in 293T cells transiently expressing the indicated SIRT6 constructs. Data are shown as mean ± s.e.m. between triplicates and are representative of four independent experiments. (C) NBDG-Glucose uptake in SIRT6 KO MEFs. Data are shown as mean ± s.e.m. between duplicates and are representative of two independent experiments. (D) Quantitative real-time PCR showing the expression of indicated genes. Data are shown as mean ± s.e.m. between duplicates in six independent experiments. (E&F) Chromatin immunoprecipitation for SIRT6 (E) and H3K56Ac (F) in SIRT6 KO MEFs expressing the indicated SIRT6 constructs followed by quantitative PCR amplification of the indicated glycolytic genes. Data are shown as mean ± s.e.m. between duplicates. *= p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001.
To further explore the mechanism by which SIRT6 regulates glycolytic gene expression, we performed chromatin immunoprecipitation (ChIP) for H3K9Ac, H3K56Ac and SIRT6 in cells expressing SIRT6 WT and mutant alleles H133Y, D63Y, A89S and D116N. We found that WT SIRT6 binds to PDK1, GLUT1, LDHa and LDHb and is able to deacetylate H3K56, as previously reported (Figure 3E–F; Zhong et al., 2010) thus leading to the reduced gene expression we observed in Figure 3D. The SIRT6 mutants D63Y and A89S were able to bind to the glycolytic genes while D116N was unable to bind, most likely due to its reduced protein stability (Figure 3E). Similar to our previous in vitro and cellular characterization data, H133Y, D63Y, A89S and D116N were unable to deacetylate H3K56 to the same degree as WT SIRT6 (Figure 3F). Interestingly, we also observed that expression of WT SIRT6 had a less dramatic impact on H3K9Ac levels both in bulk chromatin (Figure 2L) and over the promoters of specific SIRT6 target genes (Supplementary Figure 2C), suggesting that removal of H3K56Ac marks may be more critical for SIRT6-mediated gene repression in this system.
SIRT6 mutants fail to suppress anchorage independent growth and tumor formation
To assess the ability of these SIRT6 mutants to repress transformation, we first performed soft agar colony formation assays on our SIRT6 KO MEFs engineered to express either emtpy vector, WT SIRT6 or each of our cancer-associated mutations. Indeed, we found that all of the tumor-associated mutations were unable to suppress colony formation in soft agar to the same degree as WT SIRT6 (Figure 4A–B). The only exceptions were T263P and P274L, consistent with their ability to supress glucose uptake, indicating that overexpression of either mutant may be sufficient to overcome its reduced enzymatic activity. Thus, the histone deacetylase activity of SIRT6 seems critical to suppress cellular transformation. To test these mutants in a more stringent environment, we implanted D25N, A89S, D116N, E260Term, the variant N46S and WT SIRT6 subcutaneously into the flanks of SCID mice and monitored tumor growth over two months. Strikingly, the cells expressing the different mutants readily formed tumors, while cells re-expressing WT SIRT6 and the variant N46S formed negligible tumors (Figure 4C–D). These results indicate that these specific mutations eliminate the capacity of SIRT6 to protect against tumor formation in vivo.
Figure 4. SIRT6 cancer-associated point mutations fail to repress anchorage independent growth.

(A) Quantification of colonies grown in soft agar of SIRT6 KO MEFs expressing each mutant. Data are shown as mean ± s.e.m. between triplicates in four independent experiments. (B) Representative pictomicrographs of colonies. (C) Gross images of subcutaneous tumors grown in SCID mice of SIRT6 KO cells re-expressing the indicated SIRT6 constructs (n=5) (D) Quantification of tumor weight from (C). *= p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001.
DISCUSSION
Using data mining from The Cancer Genome Atlas (TCGA), we identified eight point mutations in SIRT6 which spontaneously arose in a variety of human cancers. Based on biochemical, biological and structural analysis, we find that these mutations render the protein unable to fully repress HIF1α and MYC transcriptional activity, resulting in a glycolytic switch and cellular transformation. Importantly, several of these mutations decrease SIRT6 deacetylase activity, while maintaining the ability to remove myristoyl groups, demonstrating that SIRT6 deacetylase activity, as opposed to the demyristoylase activity, is critical for its major tumor-suppressor functions (Supplemental Table 1). Thus, although rare, point mutations in SIRT6 are selected for in human cancers, further highlighting SIRT6 as a tumor suppressor and its molecular function as a histone deacetylase repressing both pro-growth and glycolytic phenotypes.
From a genetic perspective, tumor suppressors, in contrast to oncogenes, are frequently deleted, silenced and, less often, mutated (Vogelstein et al., 2013). Although in previous work we identified deletion and transcriptional silencing of SIRT6 as mechanisms through which tumors eliminate or reduce SIRT6 activity, in this study we aimed to identify whether specific point mutations in SIRT6 could provide tumors with a selective growth advantage. Also, such precise mutations might provide the opportunity to determine which catalytic activity of SIRT6 is the main driver of tumor suppression, as sirtuins are now recognized as general protein deacylases (Feldman et al., 2013). Using data mining from The Cancer Genome Atlas (TCGA), we found 8 naturally occuring mutations in SIRT6. Using in vitro, biochemical, cellular and in vivo experiments, we found that, strikingly, each one of these mutations clearly reduced SIRT6 function, either by affecting SIRT6 protein stability or catalytic activity.
The mutations were distributed broadly throughout the gene, suggesting that, likely, no particular hot spots evolved that selectively affect SIRT6 activity. Although the frequency of the mutations is low, each mutation affected the activity or protein stability of SIRT6, providing further evidence for SIRT6 as a tumor suppressor. When analyzed for protein stability, two specific mutations (D116N and E260Term) exhibited less chromatin binding and protein levels, despite normal RNA expression. E260Term was the only mutation that lacked nuclear staining, consistent with a deletion of the nuclear localization signal in this mutant. D116N, in turn, appears to directly affect stability of the protein, as confirmed by a strong decrease in melting temperature compare to WT SIRT6. At physiologic temperatures, this protein will mostly remain unfolded and be subjected to degradation. All the other mutations affected SIRT6 catalytic activity. D63Y significantly affects NAD+ binding, while D25N, E36V, A89S, T263P and P274L mutations are not directly involved in binding the acetylated substrate or NAD+ and likely decrease SIRT6 activity by imposing specific structural defects that alter the deacetylase function.
The SIRT6 A89S mutant is especially intriguing given that it exhibits deacetylase activity that is approximately 50% of WT in the absence of activation by myristic acid in our in vitro assays. While not dramatically inhibiting basal in vitro deacetylase activity of SIRT6, the A89S mutation prevents SIRT6 from reaching maximal activity level in the presence of free fatty acid. A89 is located on a loop in the back of the active site and forms a backbone hydrogen-bonding interaction with T85 in the loop (Figure 2D). Thus, mutation of A89 to a serine likely alters the orientation of the loop, disfavoring myristic acid binding. In this context, the activity of the A89S mutant is comparable with the D116N mutant. Therefore, we propose that both basal and enhanced (i.e., with free fatty acid) activity of SIRT6 may be important for its tumor suppressive effects. The A89S mutant reduces the levels of H3K9Ac and H3K56Ac in bulk chromatin similar to WT SIRT6. However, when looking at specific SIRT6 target genes, the A89S mutant is defective in removing H3K56Ac groups from specific SIRT6 target genes, such as PDK1, GLUT1, LDHa and LDHb. Additionally, the A89S mutation displays a more pronounced effect on tumor growth in vivo compared to its ability to form colonies in soft agar. This may reflect the inability of the A89S mutant to be activated by free fatty acids or a functionally related small molecule that is present in vivo, but not in vitro.
Interestingly, ChIP analysis revealed that SIRT6 deacetylated H3K56Ac more efficiently than H3K9Ac at the promoters of glycolytic genes. This is suggestive of a role for H3K56Ac in gene silencing, consistent with our previous publication (Sebastian et al., 2012). It is also important to note that we cannot completely rule out that some of the phenotypes observed could be due to the possibility that these mutations affect the binding of SIRT6 with other yet to be described partners, thus influencing their regulation and function. Finally, while all cancer-associated mutations had decreased deacetylase activity, the N46S variant found in the human population displayed WT level activity, providing additional evidence these mutations were specifically selected for in these cancers.
Recent studies identified a de-fatty acylase activity for SIRT6, by virtue of its capacity to modulate TNFα secretion by removing a myristoyl group (Jiang et al., 2013). This recently characterized activity has created mechanistic uncertainty as to whether SIRT6 functions predominantly as a long-chain deacylase or as a H3K9 and H3K56 deacetylase in order to regulate cellular activity. Notably, all the cancer mutants exhibit clear defective histone deacetylation activity, while their demyristoylase activity was similar to the WT enzyme, with the exception of D63Y and D116N, which affect NAD+ binding. Additionally, ADP-ribosylation activity has also been ascribed to SIRT6. However, consistent with published reports (Du et al., 2009; Feldman et al., 2012; Tanner et al., 2000), we were unable to detect significant evidence of any ADP-ribosylation activity by WT SIRT6 or any of the mutants we tested (data not shown), suggesting that such activity for SIRT6 may only be observed under specific circumstances (Mao et al., 2010). These results indicate that histone deacetylation is the main activity conferring tumor suppressive functions to SIRT6, likely through its ability to repress glycolytic and ribosomal protein gene expression, as we previously published (Sebastian et al., 2012).
Taken together, our results provide convincing mechanistic evidence for a tumor suppressive role for this enzyme and increase our understanding of the interplay between epigenetics, metabolism and cancer.
EXPERIMENTAL PROCEDURES
Cell Lines
SIRT6 knockout (KO) primary mouse embryonic fibroblast (MEFs) were generated from 13.5-day-old embryos as described (Mostoslavsky et al., 2006). These cells were immortalized by using the standard 3T3 protocol. Please see Supplemental Experimental procedures for culture conditions.
Computational analysis to identify SIRT6 mutations
Somatic mutations in SIRT6 were obtained from Lawrence et al. (Lawrence et al., 2014), which are available at tumorportal.org. Details on discovery of somatic mutations are found in Lawrence et al. (Lawrence et al., 2014).
Expression and purification of recombinant human SIRT6
His-tagged WT and mutant SIRT6 proteins were overexpressed in BL21(DE3) E. coli strain, as previously described (Feldman et al., 2015). Cells were harvested by centrifugation and stored at −80 °C. SIRT6 WT and mutant proteins were purified by nickel affinity resin chromatography as reported previously (Pan et al., 2011). Protein concentrations were determined by the Bradford reagent assay.
Gel Electrophoresis and Western Blotting
Chromatin fractions and western blot analysis was performed as previously described (Sebastian et al., 2012), details are listed in Supplemental Experimental procedures. Antibodies used were anti-SIRT6 (Abcam, ab62739), anti-H3K9Ac (Millipore 07-352), anti-H3K56Ac (ab76307), anti-GFP (ab290) or total-H3 (ab1791) as a loading control.
Immunofluorescence
293T cells were transfected using Trans-IT 293 (Mirus) with empty vector, pEGFP-SIRT6 or the SIRT6 mutant constructs. 24 hrs after transfection cells were trypsinized and seeded onto 8-well chamber slides and allowed to adhere overnight. Cells were then fixed using a 2% paraformaldehyde in a 1% PBS solution, permeabilized with 0.1% sodium citrate and 0.1% Triton X-100 and nuclei were stained using DAPI. Images were taken using a fluorescent microscope.
Statistics
For steady-state deacylation assays, real-time RT PCR analysis, glucose uptake, luciferase reporter assay, chromatin immunoprecipitation assay, anchorage independent growth and tumor size, significance was analyzed using 2-tailed Student’s t-test. A p-value of less than 0.05 was considered statistically significant.
Thermal denaturation assays
Thermal denaturation assays were performed to determine the melting temperature (Tm) of WT, D25N, A89S, and D116N SIRT6. Purified proteins were diluted to 10 μM (20 mM sodium phosphate, pH 7.5) containing 3.75x Sypro Orange (Invitrogen, delivered at 5000x). Samples (35 μL) were aliquoted into PCR strip tubes and placed in a Bio-Rad CFX96 Real-Time System C1000 thermal cycler. The temperature was increased at a rate of 0.5 °C/min over a range of 10–95 °C and fluorescence was monitored with the FRET channels. At least three trials were performed for each SIRT6 variant. The measured fluorescence was normalized so that the minimum fluorescence was set to 0 and the maximum fluorescence set to 1. The data were fitted to Equation 1 as previously utilized by Albaugh et al. (Albaugh et al., 2011) to obtain the Tm,
| (Eq. 1) |
where I is the normalized fluorescence value at temperature T and C is a slope factor.
HPLC deacylation assay
Peptides corresponding to residues 4–17 of histone H3 (Acetyl: AcQTARKacSTGGKAPR-WW-NH2 and Myristoyl: Ac-QTARKmyrSTGGKAPRWW-NH2) were synthesized as described previously (Feldman et al., 2013). Deacylation reactions were analyzed by reversed phase high-performance liquid chromatography on Kinetex C18 column (100 Å, 100 × 4.6 mm, 2.6 μm, Phenomenex) by monitoring the formation of the deacylated product at 214 nm (Feldman et al., 2013) and also described in Supplemental Experimental Procedures.
Steady-state Kinetic Analyses
Steady-state rates were measured by varying NAD+ (2–1200 μM) in the presence of 0.5 μM WT, D63Y, A89S, or T263P SIRT6 and 50 μM NAD+ in 20 mM sodium phosphate, pH 7.5, at 37 °C. Initial velocities were determined and data were fitted to the Michaelis-Menten equation.
Glucose Uptake Assay
Cells were grown under normal conditions for 24 hr and 100 μM 2-NBDG (Invitrogen) was added to the media for 2 hrs. Fluorescence was measured by flow cytometry using a FACSCalibur Analyzer (BD).
Luciferase Reporter Assay
MYC and HIF1α transcriptional activity was determined in 293T cells using pMYC-luc and mpGL3:HRE4 constructs respectively by luciferase reporter assay as previously described (Sebastian et al., 2012; Zhong et al., 2010), details are listed in Supplemental Experimental Procedures.
Real-Time RT-PCR Analysis
Total RNA was extracted with the TriPure Isolation Reagent (Roche) as described by the manufacturer. For cDNA synthesis, 1μg of total RNA was reverse-transcribed by using the QuantiTect Reverse Transcription Kit (Qiagen). Real-time PCR was performed as previously described (Sebastian et al., 2012), details are listed in Supplemental Experimental procedures. Data were expressed as relative mRNA levels normalized to the β-actin expression level in each sample. The primer sequences are listed in Supplemental Table 2.
Chromatin Immunoprecipitation Assays
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (Sebastian et al., 2008), with some modifications; details are listed in Supplemental Experimental Procedures. Antibodies used: 5 μl anti-SIRT6 (Abcam, ab62739), 5 μl anti-H3K9Ac (Millipore 07-352), 5 μl anti-H3K56Ac (ab76307). A control was performed with unspecific IgGs (AbCam). Real time RT-PCR was performed with primers listed in Supplemental Table 2.
Anchorage independent growth
5000 cells were resuspended in 0.4% agar and plated in triplicates in 6 well-plates containing a 0.8% base agar layer. Colonies were grown in the presence of doxycycline for 3 months and counted. Media was changed every three days.
Xenografts
4X106 cells in 100 μl of 50% matrigel were injected subcutaneously into the flanks of SCID mice (n=5) (Taconic Farms, Inc., Hudson, NY) that were maintained on doxycycline (200μg/ml) in their drinking water. Mice were checked for the appearance of tumors twice a week, and the tumors were harvested when they reached ~100 mm3 in size.
Supplementary Material
Highlights.
Eight loss-of-function SIRT6 mutations were identified in human cancers
SIRT6 mutants alter either SIRT6 stability, localization or enzymatic activity
SIRT6 mutants fail to repress glycolysis and cellular transformation
Deacetylase, not demyristoylase, activity is critical for SIRT6 tumor-suppressor function
Acknowledgments
We thank members of the Mostoslavsky and Denu labs for invaluable discussions and advise. This work was supported in part by NIH grants GM093072-01, DK088190-01A1 (to R.M.) and NIH GM065386 (to J.M.D). R.M. is the Kristine and Bob Higgins MGH Research Scholar, the Warshaw Institute Fellow, and a Howard Goodman Awardee. S.K. is the recipient of a Canadian Institutes of Health Research postdoctoral fellowship. C.S. is supported by the Visionary postdoctoral fellowship from the Department of Defense.
Footnotes
AUTHOR CONTRIBUTIONS: S.K., J.L.F., M.K., S.D., and A.R.C collected the data. S.K. J.L.F. and M.K. analyzed the data. R.M., D.M.S., C.S. and J.M.D. contributed to the experimental design. C.M. and G.G. performed computational analysis to identify SIRT6 mutations. S.K., J.L.F., J.M.D. and R.M. wrote the paper. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albaugh BN, Arnold KM, Lee S, Denu JM. Autoacetylation of the histone acetyltransferase Rtt109. The Journal of biological chemistry. 2011;286:24694–24701. doi: 10.1074/jbc.M111.251579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Molecular cell. 2005;17:855–868. doi: 10.1016/j.molcel.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Du J, Jiang H, Lin H. Investigating the ADP-ribosyltransferase activity of sirtuins with NAD analogues and 32P-NAD. Biochemistry. 2009;48:2878–2890. doi: 10.1021/bi802093g. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Baeza J, Denu JM. Activation of the Protein Deacetylase SIRT6 by Long-chain Fatty Acids and Widespread Deacylation by Mammalian Sirtuins. The Journal of biological chemistry. 2013 doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Dittenhafer-Reed KE, Denu JM. Sirtuin catalysis and regulation. The Journal of biological chemistry. 2012;287:42419–42427. doi: 10.1074/jbc.R112.378877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Dittenhafer-Reed KE, Kudo N, Thelen JN, Ito A, Yoshida M, Denu JM. Kinetic and Structural Basis for Acyl-Group Selectivity and NAD Dependence in Sirtuin-Catalyzed Deacylation. Biochemistry. 2015 doi: 10.1021/acs.biochem.5b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil R, Barth S, Kanfi Y, Cohen HY. SIRT6 exhibits nucleosome-dependent deacetylase activity. Nucleic acids research. 2013;41:8537–8545. doi: 10.1093/nar/gkt642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Khan S, Wang Y, Charron G, He B, Sebastian C, Du J, Kim R, Ge E, Mostoslavsky R, et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature. 2013;496:110–113. doi: 10.1038/nature12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, McCord RA, Ongaigui KC, Boxer LD, Chang HY, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell. 2009;136:62–74. doi: 10.1016/j.cell.2008.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature. 2008;452:492–496. doi: 10.1038/nature06736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell. 2006;124:315–329. doi: 10.1016/j.cell.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Pan PW, Feldman JL, Devries MK, Dong A, Edwards AM, Denu JM. Structure and biochemical functions of SIRT6. The Journal of biological chemistry. 2011;286:14575–14587. doi: 10.1074/jbc.M111.218990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian C, Serra M, Yeramian A, Serrat N, Lloberas J, Celada A. Deacetylase activity is required for STAT5-dependent GM-CSF functional activity in macrophages and differentiation to dendritic cells. Journal of immunology. 2008;180:5898–5906. doi: 10.4049/jimmunol.180.9.5898. [DOI] [PubMed] [Google Scholar]
- Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner KG, Landry J, Sternglanz R, Denu JM. Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennen RI, Berber E, Chua KF. Functional dissection of SIRT6: identification of domains that regulate histone deacetylase activity and chromatin localization. Mech Ageing Dev. 2010;131:185–192. doi: 10.1016/j.mad.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.