

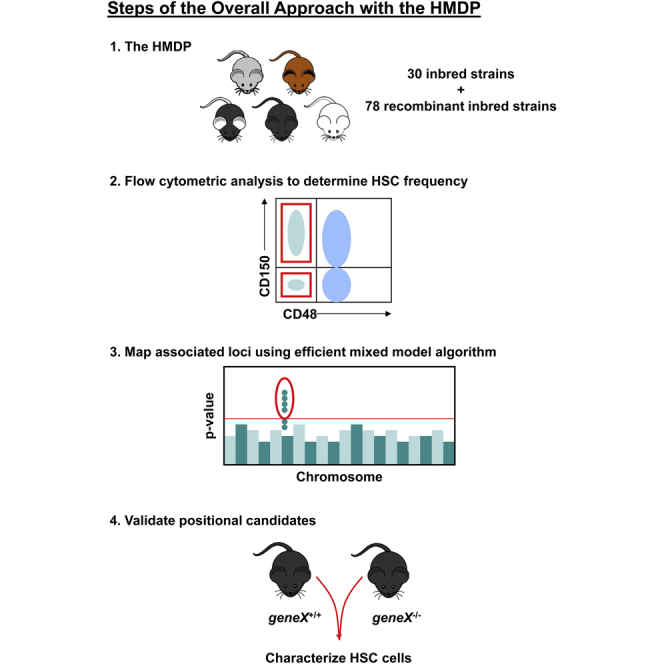
Summary
Prior efforts to identify regulators of hematopoietic stem cell physiology have relied mainly on candidate gene approaches with genetically modified mice. Here we used a genome-wide association study (GWAS) strategy with the hybrid mouse diversity panel to identify the genetic determinants of hematopoietic stem/progenitor cell (HSPC) frequency. Among 108 strains, we observed ∼120- to 300-fold variation in three HSPC populations. A GWAS analysis identified several loci that were significantly associated with HSPC frequency, including a locus on chromosome 5 harboring the homeodomain-only protein gene (Hopx). Hopx previously had been implicated in cardiac development but was not known to influence HSPC biology. Analysis of the HSPC pool in Hopx−/− mice demonstrated significantly reduced cell frequencies and impaired engraftment in competitive repopulation assays, thus providing functional validation of this positional candidate gene. These results demonstrate the power of GWAS in mice to identify genetic determinants of the hematopoietic system.
Graphical Abstract
Highlights
-
•
Genetic variation across mouse strains influences hematopoietic stem cell frequency
-
•
This variation can be exploited for genome-wide association studies
-
•
Hopx is a regulator of hematopoietic stem/progenitor cell function
-
•
This approach can be used to identify genetic determinants of other stem cell systems
In this article, Allayee, Adams, and colleagues use an unbiased strategy in mice to identify the genetic determinants of the hematopoietic system. A genome-wide association study with over 100 inbred mouse strains from the Hybrid Mouse Diversity Panel, followed by functional validation studies, revealed Hopx as a gene that controls the frequency of a sub-population of hematopoietic stem cells.
Introduction
In the hematopoietic stem cell (HSC) field, the identification of regulators of HSC physiology has relied mainly on candidate gene approaches with genetically modified mouse models (Rossi et al., 2012). However, these studies have not necessarily provided a complete picture of the complex network of signals that govern HSC proliferation, differentiation, and function. Therefore non-biased strategies, such as transcriptome analyses, have been used, but these approaches also sometimes are restricted by the screen itself (Hope et al., 2010; Karlsson et al., 2013). For example, many genes have been shown to be expressed at specific differentiation stages, including the most primitive HSCs (Chambers et al., 2007), but they do not provide information as to the key regulators of HSC physiology.
An alternative unbiased strategy to elucidate the determinants of HSC frequency/function is to leverage naturally occurring variation in a forward genetics approach. In this regard, prior studies have shown that hematopoietic stem/progenitor cell (HSPC) frequency in mice differs as a function of genetic background (de Haan et al., 1997), and attempts to identify the underlying genes have employed linkage analysis in crosses between inbred mouse strains. For example, using the long-term culture-initiating cell (LTC-IC; Müller-Sieburg and Riblet, 1996) or cobblestone area-forming cell (CAFC; de Haan and Van Zant, 1997) assays, quantitative trait loci (QTLs) for HSPC frequency have been identified on chromosomes 1 and 18 among a panel of recombinant inbred (RI) strains derived from C57Bl/6 × DBA/2 (BXD). This strategy also has been used to map loci for dynamic changes in CAFC frequency associated with aging (de Haan and Van Zant, 1999; Geiger et al., 2001). With the development of immunophenotypic markers that could identify functional HSPCs (Morrison and Weissman, 1994), flow cytometric analysis has been used to identify QTLs for HSPC frequency in the BXD RI panel, but this approach has only been successful in aged mice (Henckaerts et al., 2002, 2004). In addition, QTLs for HSPCs distinct from those mapped in the BXD RI panel have been identified using different inbred mouse strains (Morrison et al., 2002; van Os et al., 2006; Jawad et al., 2008), suggesting that the limited variation in crosses between two strains restricts the identification of other genetic factors that play a role in HSPC physiology. Furthermore, the classical QTL approach has inherently low mapping resolution, since the regions of interest typically span large chromosomal intervals and can contain hundreds to thousands of genes. To overcome these obstacles, a genetical genomics approach has been proposed, where transcriptomics analysis is combined with QTL mapping (de Haan et al., 2002; Bystrykh et al., 2005; Gerrits et al., 2008). However, to date, the incorporation of intermediate molecular traits into linkage analysis has led to the identification of only one gene, latexin, where differential levels of expression influence the pool of HSPCs in the bone marrow (BM) (Liang et al., 2007).
Genome-wide association studies (GWASs) represent another unbiased approach that has proven particularly successful in humans for gene discovery of numerous disease phenotypes (Hindorff et al., 2015). Encouraged by this success, GWASs also have been proposed in mice using a recently developed panel of classic inbred and RI mouse strains, termed the Hybrid Mouse Diversity Panel (HMDP). Our prior studies with the HMDP have identified key associations for a variety of complex traits that are highly relevant to human diseases, which collectively illustrate the power of this approach for gene discovery in mice (Bennett et al., 2010, 2012; Farber et al., 2011; Ghazalpour et al., 2011, 2012, 2014; Park et al., 2011; Orozco et al., 2012; Davis et al., 2013; Parks et al., 2013; Hartiala et al., 2014). In the present study, we used this mouse platform to carry out a comprehensive genetic screen for HSPC frequency in the adult BM. We identified multiple loci associated with the frequencies of three HSPC populations and functionally validated one positional candidate gene that previously had not been known to play a role in the hematopoietic system.
Results
Variation in HSPC Frequencies in the HMDP
While the competitive repopulation assay is the gold standard for measuring HSPC frequency in a given population of cells (Purton and Scadden, 2007), this method is influenced by a number of parameters, including radiation sensitivity of the host, homing and lodgment of the injected cells in the BM microenvironment, and differences in compensatory mechanisms involved in the differentiation of the mature hematopoietic cells. To limit variation due to any one or a combination of these parameters, we chose immunophenotypic analysis to characterize the frequency of primitive HSPC subsets in the adult BM. We obtained BM mononuclear cells (MNCs) from 12-week-old male mice (Table S1) and used flow cytometry to determine the frequency of several HSPC populations. These included Lineage[Lin]−Sca-1+c-Kit+ (LSK) HSPCs, the more immature LSKCD150−CD48− multipotent progenitors (MPPs), which have been shown to be able to differentiate into all lineages of the hematopoietic system but have limited potential to self-renew, and LSKCD150+CD48− cells, which are the most primitive long-term HSCs (Kiel et al., 2008; Oguro et al., 2013). In addition, we chose CD150 and CD48 for our analyses, since these Slam markers have been validated to identify the different sub-populations of HSPCs across multiple inbred mouse strains (Kiel et al., 2005). Representative plots that illustrate the gating strategy used for these analyses are shown in Figure S1.
Among the 108 HMDP strains screened, the frequency of LSK, LSKCD150−CD48−, and LSKCD150+CD48− cells varied by approximately 120- to 300-fold (Figure 1). Since all mice were age- and sex-matched and kept under identical environmental conditions, we attributed these differences, at least in part, to naturally occurring genetic variation. This notion was confirmed by calculating the heritability for each of the three HSPC sub-populations, which yielded values of 0.90, 0.92, and 0.70 for LSK, LSKCD150−CD48−, and LSKCD150+CD48− cells, respectively. We note, however, that these heritability estimates are somewhat higher than what would be typically expected for complex traits in humans, since phenotype measurements in the HMDP are obtained from multiple animals of the same genotype (strain).
Figure 1.

Variation in Three HSPC Populations in the HMDP
The frequency of LSK, LSKCD150−CD48−, and LSKCD150+CD48− cells exhibits 120- to 300-fold variation among 108 HMDP strains. Each dot represents an individual mouse from the respective strain and the mean values are indicated by the horizontal black bars. BM MNCs were isolated from the femurs and tibias of 12-week-old male mice (n = 3–8 per strain; N = 467), and the frequency of different HSPC sub-populations was determined by flow cytometry. Data are expressed as a percentage of BM MNCs. See also Tables S1–S3 and Figures S1 and S2.
Relationship between HSPC Frequencies and Other Hematological Parameters
We next explored the relationship between LSK, LSKCD150−CD48−, and LSKCD150+CD48− cells and other hematological parameters. The three types of primitive HSPCs were all significantly correlated with each other (Figure S2), with a particularly strong association between LSK and LSKCD150−CD48− cells (r = 0.70; p < 0.0001). LSK cells exhibited modestly positive, but significant, correlations with total white blood cell (WBC) count and with the numbers of lymphocytes and monocytes (Table S2). By comparison, LSKCD150−CD48− cells were negatively correlated with lymphocyte and monocyte counts and positively associated with granulocytes. With the exception of a weakly positive association with WBC count and a negative relationship with mean corpuscular hemoglobin, no correlations were observed with the most primitive LSKCD150+CD48− cells. Furthermore, no significant correlations were observed between any of the three HSPC populations and other red blood cell (RBC) traits, such as hemoglobin and hematocrit levels (Table S2). These data suggest that variation in LSK and LSKCD150−CD48− cells and mature WBCs could be controlled, in part, by similar genetic mechanisms, whereas variation in LSKCD150+CD48− cells as well as RBC parameters may be driven by distinct factors.
GWAS for HSPC Frequencies
To identify the genetic determinants of HSPC frequency, we used the phenotype data to carry out a GWAS for the three cell populations (Figures 2A–2C). One significantly associated locus for LSKCD150+CD48− cells was identified at the distal end of chromosome 18 (Figure 2A; Table 1), where the lead SNP (rs36866074; p = 3.2 × 10−6) mapped to intron 1 of the mitogen-activated protein kinase 4 (Mapk4) gene (Figure 3A). On chromosome 11 (Figure 2A; Table 1), we also identified suggestive association with an intergenic region located ∼464 kb downstream of the neuromedin-U receptor 2 (Nmur2) and ∼507 kb upstream of the glutamate receptor 1 isoform 1 precursor (Gria1) genes (rs29434264; p = 2.3 × 10−5).
Figure 2.

Manhattan Plots of GWAS Results for HSPC Frequency in the HMDP
(A) The frequency of LSKCD150+CD48− cells was significantly associated with a locus on chromosome 18 harboring Mapk4 and suggestively associated with a region on chromosome 11 near Nmur2 and Gria1.
(B) A GWAS for LSK cells revealed several suggestively associated loci on chromosomes 2, 4, 6, and 18 as well as a highly significant locus on chromosome 15 containing Ly6e-Ly6a-Ly6f.
(C) Three significantly associated loci on chromosomes 1, 5, and 15 were identified in the GWAS analysis for LSKCD150−CD48− cells, as well as several suggestively associated regions on chromosomes 3, 11, 17, and 18. For each significantly associated locus, the gene(s) nearest to the peak SNP is indicated. The GWAS analyses for each of the three HSPCs included 3–8 mice per strain (N = 467) and 880,924 SNPs, whose genomic positions are shown along the x axis with their corresponding −log10 p values indicated by the y axis. The genome-wide thresholds for significant (p = 4.1 × 10−6) and suggestive (p = 4.1 × 10−4) evidence of association are indicated by the horizontal red and blue lines, respectively.
See also Figure S3.
Table 1.
Loci Identified in GWAS for HSPCs in the HMDP
| HSPC Population | Chromosome | Position (bp)a | Lead SNP | Nearest Gene(s) | Minor Allele Frequency | p Value | p Valueb |
|---|---|---|---|---|---|---|---|
| LSKCD150+CD48− | 11 | 56318423 | rs29434264 | Nmur2-Gria1 | 0.06 | 2.3 × 10−5 | NA |
| 18 | 74202262 | rs36866074 | Mapk4 | 0.13 | 3.2 × 10−6 | NA | |
| LSK | 15 | 75203270 | rs31675052 | Ly6e-Ly6a-Ly6f | 0.47 | 3.7 × 10−14 | NA |
| 18 | 14494409 | rs30267408 | Zfp521-Ss18 | 0.41 | 4.3 × 10−4 | 9.4 × 10−6 | |
| LSKCD150−CD48− | 1 | 173168046 | rs8242728 | Ndufs2-Cd48-Slamf1 | 0.50 | 9.3 × 10−10 | 4.4 × 10−12 |
| 5 | 77528483 | rs29633853 | Hopx | 0.15 | 5.1 × 10−7 | 1.1 × 10−6 | |
| 15 | 74669094 | rs32350275 | Ly6e-Ly6a-Ly6f | 0.40 | 3.0 × 10−7 | NA | |
| 18 | 14474199 | rs31073841 | Zfp521-Ss18 | 0.11 | 3.8 × 10−4 | 5.7 × 10−5 |
NA, not applicable.
Base pair position of lead SNP given according to NCBI build 37 of the reference mouse genome sequence.
p value obtained from GWAS analysis that excluded strains carrying the uninformative haplotype of Sca-1 at the chromosome 15 locus. See also Figures S3 and S4.
Figure 3.

Regional Plots of Loci Significantly Associated with HSPCs in the HMDP
(A) The lead SNP at the chromosome 18 locus for LSKCD150+CD48− cells (rs36866074) maps to intron 1 of Mapk4 (boxed in red).
(B) The lead SNP on chromosome 15 for LSKs (rs31675052) maps to a region harboring Ly6e, Ly6a, and Ly6f (boxed in red), which are part of a family of genes at this locus that encode one of the surface markers used to immunophenotypically quantitate HSPC frequency (Sca-1).
(C) The chromosome 1 locus identified for LSKCD150−CD48− cells encompasses a large ∼2-Mb LD block containing dozens of genes and numerous SNPs that yielded equivalently significant p values. Although the lead SNP (rs8242728) is specifically located in Ndufs2 (boxed in red), this interval also includes Cd48 and Slamf1 (boxed in red), which encode two of other flow cytometry markers used for quantitating HSPCs (CD48 and CD150).
(D) The peak SNP at the chromosome 5 locus (rs29633853) associated with LSKCD150−CD48− cells is located within a small LD block and localizes to Hopx (boxed in red). For each plot, a 1–2-Mb region is shown and the lead SNP is indicated by a red diamond.
(A–D) (Top) SNP positions are shown on the x axis with their corresponding −log10 p values indicated by the y axis. (Bottom) The locations of genes in the selected intervals are given. See also Figure S4.
A GWAS for LSK cells revealed several suggestively associated loci, including those on chromosomes 2, 4, 6, and 18, as well as a highly significant (p = 3.7 × 10−14) locus on chromosome 15 (Figure 2B; Table 1), where the lead SNP (rs31675052) is located approximately 100 kb downstream of Ly6f (Figure 3B). Ly6f is part of a family of genes located at this locus that encode Sca-1, which is one of the surface markers used to immunophenotypically quantitate HSPC frequency. While Sca-1 is known to play a role in the function of HSPCs (Ito et al., 2003), some studies have suggested that it is not an informative cell surface marker for flow cytometry analysis in certain mouse strains (Spangrude and Brooks, 1993). To address this potential issue and remove the effect of the chromosome 15 locus, we re-performed the GWAS analysis after excluding strains carrying the low Sca-1-expressing haplotype (Table S1). Importantly, exclusion of these strains did not appreciably decrease the heritability for variation in LSK cells (0.90 versus 0.82). Furthermore, the Sca-1 locus did not yield an association signal in this analysis, as expected, but the suggestive peak on chromosome 18 increased in significance from p = 4.3 × 10−4 to just below the threshold for genome-wide significance with p = 9.4 × 10−6 (Table 1; Figure S3A). The peak SNP (rs30267408) on chromosome 18 is not located within a known gene, but it maps ∼363 kb distal to the zinc-finger protein 521 (Zfp521) and ∼290 kb proximal to the synovial sarcoma-associated Ss18-alpha (Ss18) genes (Figure S3B).
We next performed a GWAS for LSKCD150−CD48− cells and identified three significantly associated loci on chromosomes 1 (rs8242728; p = 9.3 × 10−10), 5 (rs29633853; p = 5.1 × 10−7), and 15 (rs32350275; p = 3.0 × 10−7), as well as several suggestively associated loci on chromosomes 3, 11, 17, and 18 (Figure 2C; Table 1). Since the chromosome 15 locus for LSKCD150−CD48− cells was the same as that identified for LSKs, we re-performed the GWAS after excluding the same strains carrying the low Sca-1-expressing haplotype. Similar to LSK cells, excluding these strains only had a marginal effect on the heritability of LSKCD150−CD48− cells (0.92 versus 0.88). This analysis strengthened the association signal on chromosome 1 (p = 4.4 × 10−12), but had no effect on chromosome 5 (p = 1.1 × 10−6) nor revealed any additional regions associated with LSKCD150−CD48− cells (Table 1; Figure S4A). The lead SNP on chromosome 1 (rs8242728) fell within a large ∼2-Mb linkage disequilibrium (LD) block containing dozens of genes and numerous SNPs that yielded equivalently significant p values (Figure 3C). Given this LD pattern, it would be difficult to narrow down the underlying causative gene without extensive follow-up. For example, rs8242728 is specifically located within the NADH dehydrogenase [ubiquinone] iron-sulfur protein 2 (Ndufs2) gene. However, this interval also encompasses the Slam locus and contains the Cd48 and Slamf1 genes, which encode two of the other flow cytometry markers used for quantitating HSPCs (CD48 and CD150). By comparison, the most significantly associated SNPs on chromosome 5 are located within a small LD block, with the peak SNP (rs29633853) localizing to the homeodomain-only protein (Hopx) gene (Figure 3D). Interestingly, the suggestive association with the peak SNP on chromosome 18 (rs31073841), which maps ∼20 kb proximal to the peak SNP of the same Zfp521-Ss18 locus identified for LSKs, also increased in significance by nearly one order of magnitude from p = 3.8 × 10−4 to p = 5.7 × 10−5 after exclusion of the strains carrying the low Sca-1-expressing haplotype (Table 1; Figure S4A). This observation suggests that this chromosome 18 locus may exert pleiotropic effects on multiple HSPC subsets.
Prioritization of Positional Candidate Genes at Chromosome 5 Locus for LSKCD150−CD48− Frequency
Of the loci identified, we focused on the chromosome 5 locus because of the increased mapping resolution and the possibility that this region contains a previously unknown gene(s) for LSKCD150−CD48− cells. In particular, rs29633853 is located within intron 1 of Hopx, which has been shown to play important roles in cardiac development (Chen et al., 2002; Shin et al., 2002) and lymphoid regulation (Albrecht et al., 2010) and to serve as a marker for quiescent intestinal epithelial stem cells (Takeda et al., 2011) and multipotent hair follicle stem cells (Takeda et al., 2013). However, Hopx has otherwise not been implicated in the biological regulation of HSPCs. To evaluate Hopx as a positional candidate, we first searched the publicly available genomic sequences of ∼60 inbred mouse strains, but we did not identify any amino acid substitutions and/or other protein structure-altering variations. We next used gene expression data across multiple tissues in the HMDP (http://geneeqtl.genetics.ucla.edu/) to determine whether the chromosome 5 locus exhibited any cis expression QTL (eQTL), defined as those mapping within a 2-Mb interval centered on the lead GWAS SNP (rs29633853). Of the ∼20 genes in this region (Figure 3D), Srd5a3, Tmem165, Exoc1, Cep135, and Hopx exhibited cis eQTLs in either bone, macrophages, liver, or heart (Table 2). However, only variants in Hopx yielded both significant associations with LSKCD150−CD48− cells and cis eQTLs across all four tissues examined. In particular, the lead eQTL SNP in liver (rs33117479; p = 3.4 × 10−18) was located ∼8 kb proximal to the lead SNP for LSKCD150−CD48− cells (rs29633853), whereas the lead eQTL SNP in heart (rs33175967; p = 6.2 × 10−19) was ∼17 kb distal. The overlap between the association signals for LSKCD150−CD48− frequency and hepatic Hopx mRNA expression at the chromosome 5 locus is illustrated in Figure S4B.
Table 2.
Genes Exhibiting cis eQTLs across Multiple Tissues at the Chromosome 5 Locus for LSKCD150−CD48− Cells
| Gene | Gene Position (bp)a | SNP | SNP Position (bp)a | GWAS p Valueb | eQTL p Value |
|||
|---|---|---|---|---|---|---|---|---|
| Bone | Macrophage | Liver | Heart | |||||
| Srd5a3 | 76569296-76584529 | rs13478330 | 75857166 | 0.26 | 5.8 × 10−7 | ns | ns | ns |
| rs3153753 | 76574923 | 0.99 | 4.0 × 10−15 | ns | ns | ns | ||
| rs4225290 | 76583956 | 0.76 | ns | ns | ns | 5.0 × 10−8 | ||
| rs33559924 | 76254389 | 0.47 | ns | ns | 9.0 × 10−12 | ns | ||
| rs3697530 | 76575062 | 0.82 | ns | ns | 3.9 × 10−16 | ns | ||
| rs13478335 | 76955455 | 0.41 | ns | 2.0 × 10−17 | ns | ns | ||
| Tmem165 | 76612905-76638270 | rs4225290 | 76583956 | 0.76 | ns | 1.5 × 10−11 | ns | ns |
| Exoc1 | 76958336-76999319 | rs4225290 | 76583956 | 0.76 | 9.4 × 10−10 | ns | ns | ns |
| rs13478334 | 76817411 | 0.64 | ns | 9.4 × 10−12 | ns | ns | ||
| rs33139946 | 76913636 | 0.64 | ns | 9.4 × 10−12 | ns | ns | ||
| rs31555016 | 76915601 | 0.64 | ns | 9.4 × 10−12 | ns | ns | ||
| rs31555860 | 76921199 | 0.64 | ns | 9.4 × 10−12 | ns | ns | ||
| rs29561143 | 77013954 | 0.86 | ns | ns | 5.2 × 10−7 | ns | ||
| Cep135 | 77075352-77075401 | rs29824030 | 75977692 | 0.36 | 6.2 × 10−8 | ns | ns | ns |
| rs13478331 | 76154830 | 0.38 | 1.7 × 10−9 | ns | ns | ns | ||
| Hopx | 77516011-77544181 | rs33077631 | 77509306 | 6.1 × 10−6 | 4.0 × 10−8 | ns | ns | ns |
| rs33175967 | 77511463 | 2.6 × 10−5 | ns | ns | ns | 6.2 × 10−19 | ||
| rs33750358 | 77534348 | 6.1 × 10−6 | 4.0 × 10−8 | ns | ns | ns | ||
| rs33117479 | 77536186 | 4.8 × 10−5 | ns | 1.6 × 10−6 | 3.4 × 10−18 | ns | ||
A genomic region ±1 Mb around the peak SNP for LSKCD150−CD48− cells on chromosome 5 (rs29633853; bp position 77528483) was interrogated for the presence of cis eQTLs in multiple tissues using the UCLA Systems Genetics Resource (http://systems.genetics.ucla.edu/). Only genes exhibiting cis eQTLs in bone, macrophage, liver, or heart are listed. Ns, not significant.
Base pair (bp) positions of genes and SNPs are given according to NCBI build 37 of the reference mouse genome sequence.
p values are given from GWAS results that included all HMDP strains. See also Figure S4.
To further evaluate whether Hopx plays a role in the regulation of HSPCs, we performed gene expression analysis by RT-PCR in BM MNCs of 25 different mouse strains with varying LSKCD150−CD48− frequencies. These analyses demonstrated that Hopx was expressed in BM and that its mRNA levels were both variable and correlated (r = 0.44) with LSKCD150−CD48− frequency in these strains (Figure 4A). This latter observation is not entirely surprising since the coincident mapping of cis eQTLs for Hopx across multiple tissues and LSKCD150−CD48− frequency to the same chromosome 5 could be expected to result in a correlation between Hopx gene expression and cell number in BM. We next purified various HSPC subsets and other hematopoietic-derived lineages from C57BL/6 mice and determined the cell-specific expression pattern of Hopx. These analyses revealed that Hopx was expressed in all hematopoietic cells and at particularly high levels in mature hematopoietic lineages, such as CD3+ T lymphocytes (Figure 4B). To confirm these findings, we used flow cytometry to analyze BM of genetically modified mice that express GFP under the control of the endogenous Hopx promoter (Takeda et al., 2013). As shown in Figure 4C, Hopx was expressed highest in the LSKCD150−CD48− population of HSPCs and in CD3+ cells, which is consistent with the RT-PCR results. Collectively, these data point to Hopx as a strong positional candidate gene since (1) genetic variation at this locus is functional with respect to Hopx gene expression in multiple tissues, (2) mRNA levels of Hopx in BM MNCs are positively associated with LSKCD150−CD48− number, and (3) Hopx is expressed at relatively high levels in the HSPC subset whose frequency maps to the chromosome 5 locus.
Figure 4.

Relationship between Hopx Gene Expression and HSPCs
(A) Among 25 HMDP strains, Hopx expression levels in BM MNCs are positively correlated (r = 0.44) with LSKCD150−CD48− frequency. Each data point represents the mean expression of each mouse strain (n = 2–3 mice/strain) across two independent experiments. LSKCD150−CD48−frequency is expressed as a percentage of BM MNCs and expression levels are shown relative to the expression of Hprt, as an endogenous control, in relative units (ru).
(B) Hopx is expressed in various hematopoietic cell subsets of C57Bl/6 mice (n = 3), including all three HSPC populations, but at very low levels in mesenchymal stem cells (MSCs). Expression is also abundant in mature leukocytes, particularly in T lymphocytes (CD3+) compared with B lymphocytes (B220+) or monocytes (CD11b+). Total RNA was isolated from BM and RT-PCR was carried out as described in the Experimental Procedures. Expression levels are shown relative to the expression of Hprt, as an endogenous control, in relative units (ru) with error bars representing SEM.
(C) As determined by relative GFP fluorescence intensity, Hopx expression was highest in the LSKCD150−CD48− population of HSPCs and especially abundant in CD3+ cells, consistent with the RT-PCR results. Fluorescence was quantitated by flow cytometry in different HSPC sub-populations and mature hematopoietic lineages in BM MNCs that were isolated from the femurs and tibias of 12-week-old genetically modified mice (n = 3) expressing GFP under the control of the endogenous Hopx promoter (Takeda et al., 2013). Data are expressed in a.u. Note the different x axes in the plots.
Biological Validation of Hopx as a Gene that Regulates LSKCD150−CD48− Cells
To biologically validate Hopx as a genetic determinant of HSPCs, we characterized previously generated Hopx knockout (Hopx−/−) mice (Chen et al., 2002) for differences in HSPC frequency and function. Flow cytometric analysis revealed that Hopx−/− mice had significantly reduced numbers of LSKCD150−CD48− cells compared to wild-type Hopx+/+ littermates, but no differences in the frequency of LSK (Figure 5A) or LSKCD150+CD48− cells (data not shown). Since the reduced frequency of LSKCD150−CD48−cells in Hopx−/− mice could be due to either intrinsic defects of the cells or extrinsic factors, we performed competitive repopulation assays with HSCs from Hopx−/− mice and wild-type littermates. These experiments demonstrated that transplantation of HSCs from Hopx−/− mice did not affect levels of circulating CD45.2 cells at early time points, but led to significantly impaired engraftment at 16 weeks after transplantation and extending to 24 weeks (Figure 5B). However, analysis of multi-lineage engraftment in these mice did not reveal significant differences in the relative abundance of circulating B lymphocytes, T lymphocytes, or myeloid cells (Figure S5).
Figure 5.

Functional Validation of the Effects of Hopx on HSPC Physiology
(A) The frequency of LSKCD150−CD48− cells, as a percentage of BM MNCs (left y axis), are significantly reduced in Hopx−/− mice compared to wild-type littermate controls (Hopx+/+), whereas LSKs are not affected (right y axis). BM MNCs were isolated from the femurs and tibias of 12-week-old male mice and the frequency of different HSPC sub-populations was determined by flow cytometry.
(B) Competitive repopulation assay shows significantly decreased levels of engraftment starting at 16 weeks after transplantation of HSCs isolated from BM of Hopx−/− mice compared to wild-type Hopx+/+ littermates. Mice were bled from the tail vein every 4 weeks after transplantation and the level of engraftment, indicated by the percentage of circulating CD45.2+ cells, was quantified by flow cytometry.
(C) Frequency of LSKCD150−CD48− cells in BM, as a percentage of MNCs (left y axis), is significantly reduced in mice transplanted with HSCs from Hopx−/− mice compared to Hopx+/+ littermate controls 6 months following transplantation. There is no difference in the frequency of LSKs (right y axis).
(D) LSKCD150−CD48− cells of Hopx−/− mice exhibit altered cell-cycle status, with decreased and increased percentage of cells in the G0 and G1 phases, respectively, compared to wild-type littermates. No effects were observed in LSKs.
For each experiment in (A)–(D), error bars represent SEM and n = 3–5 mice per group. ∗p < 0.05; ∗∗p < 0.01. See also Figure S5.
We next analyzed the composition of BM MNCs at the 24-week time point. Compared to transplantation with Hopx+/+ HSCs, mice transplanted with HSCs from Hopx−/− mice had a significant and specific reduction in the frequency of LSKCD150−CD48− cells, but not LSK (Figure 5C) or LSKCD150+CD48− cells (data not shown). To investigate a potential biological mechanism for the functional effects of Hopx deficiency on HSPC frequency, we analyzed the cell-cycle status of LSK and LSKCD150−CD48− cells. Hopx−/− mice had significantly reduced numbers of quiescent LSKCD150−CD48− cells and increased numbers of cells in the G1 phase (Figure 5D). However, Hopx deficiency had no cell-cycle effects on LSKs (Figure 5D) or LSKCD150+CD48− cells (data not shown). Taken together, these data validate the GWAS findings and provide functional evidence that Hopx is a positive regulator of HSPC frequency in mice.
Discussion
In the present study, we carried out a phenotypic screen with over 100 inbred strains from the HMDP to investigate how the frequency of three HSPC populations varies as a function of naturally occurring genetic variation. The results revealed substantial variability across this panel of mouse strains, at least based on the specific cell surface markers we used to quantify cell number. However, this may not necessarily reflect the true variation in the pool size of all stem cells, since, for example, the number of HSPCs in strains carrying the Sca-1 null allele could be underestimated. Despite this possibility, we still observed relatively strong correlations between certain HSPC subsets, but, interestingly, not between HSPCs and the number of circulating mature blood cells or other hematological parameters.
Using the strain survey data, we also carried out a GWAS analysis and identified multiple distinct and overlapping loci that were significantly or suggestively associated with HSPC sub-populations. At one particular locus on chromosome 5, we provide strong evidence that LSKCD150−CD48− cell frequency and function is regulated by Hopx, a gene that was previously not known to influence HSPC biology. The collective results of the functional validation studies with Hopx−/− mice and competitive transplantation assays demonstrated that Hopx deficiency led to a specific and intrinsic functional defect in the MPP subset of HSCs (LSKCD150−CD48−), possibly related to cell-cycle status, but not in the most primitive long-term subset of cells or more committed progenitor populations. These observations were also entirely consistent with the gene expression analyses, showing that Hopx mRNA levels in the BM were positively correlated with LSKCD150−CD48− cells, and the GWAS results, where the Hopx locus was only associated with this HSPC subset and not with LSKs or LSKCD150+CD48− cells. Of direct relevance to our findings, a recent study demonstrated how different HSC populations, and LSKCD150−CD48− cells in particular, play distinct temporal roles in the maintenance of the hematopoietic system (Busch et al., 2015). More specifically, it is the LSKCD150−CD48− sub-population of cells that sustains adult hematopoiesis under homeostatic conditions through multiple rounds of self-renewal. Furthermore, proliferation of this cell population accounts for the differentiation of committed progenitors, while only receiving limited input from the more primitive LSKCD150+CD48− sub-population of cells.
Hopx is a homeodomain-only protein that lacks DNA-binding activity yet is predominantly localized to the nucleus (Chen et al., 2002; Shin et al., 2002). There are many different functional roles proposed for this protein depending on the cell type examined. Through direct protein-protein interactions, Hopx was shown to repress the expression of serum response factor (SRF)-dependent genes (Kook and Epstein, 2003), and conditional deletion of Srf in mouse BM leads to an approximate 3-fold increase in the frequency of all HSPC sub-populations (Ragu et al., 2010). Therefore, one plausible mechanism for why Hopx deficiency leads to decreased numbers of LSKCD150−CD48− cells could be as a result of increased Srf-mediated transcription of downstream targets. Alternatively, Hopx may affect HSPC frequency through its effects on cell-cycle status. For example, we demonstrated that Hopx−/− mice have a decreased proportion of LSKCD150−CD48− cells in the quiescent G0 state and increased numbers in the G1 phase. Other groups have shown that Hopx promoter methylation frequently is observed in many malignancies, including esophageal, gastric, pancreatic, and colorectal cancers, where it has been suggested that Hopx functions as a tumor suppressor gene (Yamashita et al., 2013). Since it is known that deficiency of a number of genes in the hematopoietic system leads to increased entry into cell cycle, with a consequent loss of HSPC frequency and function (Orford and Scadden, 2008), it is possible that perturbation of Hopx expression/activity affects the intrinsic homeostasis of HSPCs by altering cellular quiescence and, thus, cell frequency.
Two other interesting observations from our GWAS results were the overlap of loci that previously had been mapped for HSPC phenotypes using linkage analysis and identification of genomic regions that harbored genes encoding cell surface markers used for flow cytometry. In this regard, we identified a major locus for LSK cells over the genes encoding Sca-1 (Ly6e-Ly6a-Ly6f) on chromosome 15, which confirms previous reports that certain strains carry a Sca-1 haplotype associated with decreased expression (Spangrude and Brooks, 1993). Exclusion of these strains from the analysis did not significantly decrease the heritability of LSK variation or reveal additional loci, but yielded highly suggestive evidence for association with a chromosome 18 locus containing Zfp521. QTL analysis with BXD RI strains also identified a locus for HSC frequency on chromosome 18 near Zfp521, although the peak of that linkage signal mapped ∼20 Mb distal from our lead GWAS SNP (de Haan and Van Zant, 1997). Zfp521 encodes a transcription factor protein with 30 zinc fingers that was originally identified as being specifically expressed in primitive human CD34+ cells compared to more mature hematopoietic cells (Bond et al., 2004). More recently, Zfp521 also has been implicated in bone formation (Hesse et al., 2010; Kiviranta et al., 2013), but validation of Zfp521 as a gene directly influencing HSPCs will require additional functional studies. Notably, the loci on chromosomes 15 (Ly6e-Ly6a-Ly6f) and 18 (Zfp521-Ss18) were identified in the GWAS analyses for LSKCD150−CD48− cells as well. However, aside from these two regions, there was no overlap between the loci identified for the three HSPC populations or with those previously reported for blood cell traits in the HMDP (Davis et al., 2013). In addition to Ly6e-Ly6e-Ly6f, our GWAS analysis revealed highly significant association of LSKCD150−CD48− cells with the Slam locus on chromosome 1, the strength of which increased by two orders of magnitude after excluding the low Sca-1-expressing strains. Consistent with our results, Müller-Sieburg and Riblet (1996) also mapped a QTL for HSCs directly over the Slam locus using the CAFC assay and linkage analysis in the BXD RI panel. Although we cannot determine with certainty whether Cd48 and Slamf1 account for the signal on chromosome 1 due to extensive LD across this region, Cd48-deficient mice do exhibit increased numbers of short-term HSCs (Boles et al., 2011).
While GWAS in humans has been successful in identifying genes for many different disease phenotypes, these types of analysis have had limited success for HSPCs. This is due, in part, to the logistical challenges of obtaining the relevant tissue in humans (i.e., BM) in large numbers of subjects in order to have sufficient mapping power. For example, a GWAS in the Framingham Heart Study for levels of circulating CD34+ cells only identified suggestive loci, none of which could be functionally validated (Cohen et al., 2013). With respect to the present results, the relevance of the identified genes and pathways to humans is still to be determined. However, it is encouraging that recent studies in the HMDP were able to directly correlate mouse association signals with those identified in human GWAS (Davis et al., 2013; Parks et al., 2013). For instance, four of the five loci identified for RBC parameters in the HMDP (Davis et al., 2013) correspond to loci recently reported for analogous phenotypes in a large human GWAS (van der Harst et al., 2012). These observations reinforce the concept that the underlying biological pathways for complex traits are likely to be conserved between mice and humans.
In summary, we demonstrate the power of the HMDP as a platform to identify the genetic determinants of highly relevant biomedical traits that would otherwise not be feasible or logistically challenging to investigate in humans. While our studies only investigated the genetic determinants of HSPC frequency in the adult BM, they provide a proof of principle that similar unbiased approaches can be applied to comparable or more complex phenotypes in other stem cell systems. Such gene discovery efforts may lead to targets that could be studied toward further understanding the biological regulation of stem cells and/or manipulated for therapeutic development.
Experimental Procedures
Animals
The 12-week-old male mice of 108 inbred strains (n = 3–8) from the HMDP were obtained (The Jackson Laboratory) and used in accordance with the USC Institutional Animal Care and Use Committee guidelines. Mice were maintained on a 12-hr light/dark cycle in sterilized microisolator cages and received autoclaved food and water ad libitum. Biological validation of Hopx was carried out using previously generated genetically targeted mice (Chen et al., 2002; Takeda et al., 2013).
Flow Cytometry
BM MNCs were incubated in PBS with fluorescent labeled anti-mouse antibodies. The labeled cells were then analyzed by flow cytometry using an LSR II flow cytometer (Becton Dickinson), and HSPC frequencies were calculated according to established cell immunophenotypes using FlowJo flow cytometry analysis software. Figure S1 illustrates the gating strategy used for the flow cytometry analyses in selected mouse strains that were chosen to represent the range of variation in HSPC frequency. Details regarding the flow cytometric analyses are described in the Supplemental Experimental Procedures and a list of all antibodies used is given in Table S3.
Complete Blood Count Analysis
Mice (n = 3/strain) were fasted for 4–5 hr and bled from the retro-orbital plexus under isoflurane anesthesia 2–3 hr after the beginning of the light cycle. Blood was collected in 20-μl EDTA-coated glass capillaries, processed using standard procedures, and profiled with respect to hematological parameters using the Heska HemaTrue Veterinary Hematology Analyzer, as described previously (Davis et al., 2013).
Statistical Genetics Analysis
GWAS analyses utilized ∼880,000 genotyped and imputed SNPs and implemented an efficient mixed model algorithm (EMMA) that takes into account the underlying population structure and genetic relatedness across the strains, as described previously (Kang et al., 2008; Bennett et al., 2010). Details regarding the statistical genetics methodology are described in the Supplemental Experimental Procedures.
Gene Expression Analysis
Total RNA was extracted using RNA Miniprep Kits (Stratagene) and was reverse-transcribed into cDNA using SuperScript VILO cDNA synthesis kits (Invitrogen). Taqman gene expression assay primers and probe sets (Applied Biosystems and Roche Diagnostics) were used to quantify expression of Hopx (assay ID Mm00558630_m1) and Hprt (assay ID Mm01545399_m1) as an endogenous control. Gene expression levels were measured in triplicate using a 7900HT real-time PCR system (Applied Biosystems). Standard curves in these experiments were created with the use of qPCR mouse reference total RNA (Stratagene).
Competitive Repopulation Assays
BM MNCs were obtained from Hopx+/+ and Hopx−/− mice and the primitive LSKCD150+CD48− HSCs were isolated using cell sorting. To quantify the ability to reconstitute hematopoiesis, 100 LSKCD150+CD48− cells from Hopx+/+ or Hopx−/− mice, along with 250,000 competitor BM MNCs from a B6.SJL congenic mouse, were injected into the tail veins of B6.SJL mice that were lethally irradiated with 9 Gy approximately 24 hr prior to transplantation. Peripheral blood samples were obtained from the tail vein of hosts starting at 4 weeks after transplantation and every 4 weeks thereafter, for a total of 24 weeks. PE anti-mouse CD45.1, fluorescein isothiocyanate (FITC) anti-mouse CD45.2, APC anti-mouse CD3e, APC-Cy7 anti-mouse CD11b, and biotinylated anti-mouse B220 antibodies (all from Becton Dickinson) were used to assess the degree of multi-lineage reconstitution.
Cell-Cycle Experiments
For cell-cycle analysis, cells were incubated with 10 μg/ml Hoechst 33342 (Sigma-Aldrich) at 37°C for 45 min, then stained with lineage and stem cell antibodies as described above. The stained cells were resuspended and fixed in 10% buffered formalin and incubated at 4°C overnight. To stain for RNA content, pyronin Y (Polysciences) was added to the cells at a final concentration of 0.75 μg/ml and incubated at 4°C for 30 min. Cell-cycle status was examined using an LSR II flow cytometer (Becton Dickinson) and FlowJo software.
Author Contributions
G.B.A. and H.A. conceived the study. X.Z., A.L.C., J.H., T.J.S., A.G., L.W.B., B.B.B., B.W.P., and R.J. performed experiments, collected data, and analyzed data. E.E., J.A.E., A.J.L., G.B.A., and H.A. contributed to experimental design and data analysis and interpretation. X.Z., A.L.C., G.B.A., and H.A. wrote the paper.
Acknowledgments
This work was supported in part by California Institute for Regenerative Medicine grant TG2-01161; NIH grants T32ES013678, K99HL102223, K99HL123021, R01ES022282, R01HL071546, P01HL30568, P01HL28481, R01ES021801, 3R01ES021801-03S1, UL1TR000130, and P30ES007048; and the Margaret E. Early Medical Research Trust.
Published: June 4, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.05.008.
Contributor Information
Gregor B. Adams, Email: gregorad@med.usc.edu.
Hooman Allayee, Email: hallayee@usc.edu.
Supplemental Information
References
- Albrecht I., Niesner U., Janke M., Menning A., Loddenkemper C., Kühl A.A., Lepenies I., Lexberg M.H., Westendorf K., Hradilkova K. Persistence of effector memory Th1 cells is regulated by Hopx. Eur. J. Immunol. 2010;40:2993–3006. doi: 10.1002/eji.201040936. [DOI] [PubMed] [Google Scholar]
- Bennett B.J., Farber C.R., Orozco L., Kang H.M., Ghazalpour A., Siemers N., Neubauer M., Neuhaus I., Yordanova R., Guan B. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010;20:281–290. doi: 10.1101/gr.099234.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B.J., Orozco L., Kostem E., Erbilgin A., Dallinga M., Neuhaus I., Guan B., Wang X., Eskin E., Lusis A.J. High-resolution association mapping of atherosclerosis loci in mice. Arterioscler. Thromb. Vasc. Biol. 2012;32:1790–1798. doi: 10.1161/ATVBAHA.112.253864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles N.C., Lin K.K., Lukov G.L., Bowman T.V., Baldridge M.T., Goodell M.A. CD48 on hematopoietic progenitors regulates stem cells and suppresses tumor formation. Blood. 2011;118:80–87. doi: 10.1182/blood-2010-12-322339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond H.M., Mesuraca M., Carbone E., Bonelli P., Agosti V., Amodio N., De Rosa G., Di Nicola M., Gianni A.M., Moore M.A. Early hematopoietic zinc finger protein (EHZF), the human homolog to mouse Evi3, is highly expressed in primitive human hematopoietic cells. Blood. 2004;103:2062–2070. doi: 10.1182/blood-2003-07-2388. [DOI] [PubMed] [Google Scholar]
- Busch K., Klapproth K., Barile M., Flossdorf M., Holland-Letz T., Schlenner S.M., Reth M., Höfer T., Rodewald H.R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518:542–546. doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- Bystrykh L., Weersing E., Dontje B., Sutton S., Pletcher M.T., Wiltshire T., Su A.I., Vellenga E., Wang J., Manly K.F. Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics’. Nat. Genet. 2005;37:225–232. doi: 10.1038/ng1497. [DOI] [PubMed] [Google Scholar]
- Chambers S.M., Boles N.C., Lin K.-Y.K., Tierney M.P., Bowman T.V., Bradfute S.B., Chen A.J., Merchant A.A., Sirin O., Weksberg D.C. Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell. 2007;1:578–591. doi: 10.1016/j.stem.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Kook H., Milewski R., Gitler A.D., Lu M.M., Li J., Nazarian R., Schnepp R., Jen K., Biben C. Hop is an unusual homeobox gene that modulates cardiac development. Cell. 2002;110:713–723. doi: 10.1016/s0092-8674(02)00932-7. [DOI] [PubMed] [Google Scholar]
- Cohen K.S., Cheng S., Larson M.G., Cupples L.A., McCabe E.L., Wang Y.A., Ngwa J.S., Martin R.P., Klein R.J., Hashmi B. Circulating CD34(+) progenitor cell frequency is associated with clinical and genetic factors. Blood. 2013;121:e50–e56. doi: 10.1182/blood-2012-05-424846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R.C., van Nas A., Bennett B., Orozco L., Pan C., Rau C.D., Eskin E., Lusis A.J. Genome-wide association mapping of blood cell traits in mice. Mamm. Genome. 2013;24:105–118. doi: 10.1007/s00335-013-9448-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan G., Van Zant G. Intrinsic and extrinsic control of hemopoietic stem cell numbers: mapping of a stem cell gene. J. Exp. Med. 1997;186:529–536. doi: 10.1084/jem.186.4.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haan G., Van Zant G. Dynamic changes in mouse hematopoietic stem cell numbers during aging. Blood. 1999;93:3294–3301. [PubMed] [Google Scholar]
- de Haan G., Nijhof W., Van Zant G. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 1997;89:1543–1550. [PubMed] [Google Scholar]
- de Haan G., Bystrykh L.V., Weersing E., Dontje B., Geiger H., Ivanova N., Lemischka I.R., Vellenga E., Van Zant G. A genetic and genomic analysis identifies a cluster of genes associated with hematopoietic cell turnover. Blood. 2002;100:2056–2062. doi: 10.1182/blood-2002-03-0808. [DOI] [PubMed] [Google Scholar]
- Farber C.R., Bennett B.J., Orozco L., Zou W., Lira A., Kostem E., Kang H.M., Furlotte N., Berberyan A., Ghazalpour A. Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS Genet. 2011;7:e1002038. doi: 10.1371/journal.pgen.1002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger H., True J.M., de Haan G., Van Zant G. Age- and stage-specific regulation patterns in the hematopoietic stem cell hierarchy. Blood. 2001;98:2966–2972. doi: 10.1182/blood.v98.10.2966. [DOI] [PubMed] [Google Scholar]
- Gerrits A., Dykstra B., Otten M., Bystrykh L., de Haan G. Combining transcriptional profiling and genetic linkage analysis to uncover gene networks operating in hematopoietic stem cells and their progeny. Immunogenetics. 2008;60:411–422. doi: 10.1007/s00251-008-0305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazalpour A., Bennett B., Petyuk V.A., Orozco L., Hagopian R., Mungrue I.N., Farber C.R., Sinsheimer J., Kang H.M., Furlotte N. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 2011;7:e1001393. doi: 10.1371/journal.pgen.1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazalpour A., Rau C.D., Farber C.R., Bennett B.J., Orozco L.D., van Nas A., Pan C., Allayee H., Beaven S.W., Civelek M. Hybrid mouse diversity panel: a panel of inbred mouse strains suitable for analysis of complex genetic traits. Mamm. Genome. 2012;23:680–692. doi: 10.1007/s00335-012-9411-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazalpour A., Bennett B.J., Shih D., Che N., Orozco L., Pan C., Hagopian R., He A., Kayne P., Yang W.P. Genetic regulation of mouse liver metabolite levels. Mol. Syst. Biol. 2014;10:730. doi: 10.15252/msb.20135004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartiala J., Bennett B.J., Tang W.H., Wang Z., Stewart A.F., Roberts R., McPherson R., Lusis A.J., Hazen S.L., Allayee H., CARDIoGRAM Consortium Comparative genome-wide association studies in mice and humans for trimethylamine N-oxide, a proatherogenic metabolite of choline and L-carnitine. Arterioscler. Thromb. Vasc. Biol. 2014;34:1307–1313. doi: 10.1161/ATVBAHA.114.303252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henckaerts E., Geiger H., Langer J.C., Rebollo P., Van Zant G., Snoeck H.W. Genetically determined variation in the number of phenotypically defined hematopoietic progenitor and stem cells and in their response to early-acting cytokines. Blood. 2002;99:3947–3954. doi: 10.1182/blood.v99.11.3947. [DOI] [PubMed] [Google Scholar]
- Henckaerts E., Langer J.C., Snoeck H.W. Quantitative genetic variation in the hematopoietic stem cell and progenitor cell compartment and in lifespan are closely linked at multiple loci in BXD recombinant inbred mice. Blood. 2004;104:374–379. doi: 10.1182/blood-2003-12-4304. [DOI] [PubMed] [Google Scholar]
- Hesse E., Kiviranta R., Wu M., Saito H., Yamana K., Correa D., Atfi A., Baron R. Zinc finger protein 521, a new player in bone formation. Ann. N Y Acad. Sci. 2010;1192:32–37. doi: 10.1111/j.1749-6632.2009.05347.x. [DOI] [PubMed] [Google Scholar]
- Hindorff, L.A., Junkins, H.A., Hall, P.N., Mehta, J.P., and Manolio, T.A. (2015). A catalog of published genome-wide association studies. http://www.genome.gov/gwastudies/.
- Hope K.J., Cellot S., Ting S.B., MacRae T., Mayotte N., Iscove N.N., Sauvageau G. An RNAi screen identifies Msi2 and Prox1 as having opposite roles in the regulation of hematopoietic stem cell activity. Cell Stem Cell. 2010;7:101–113. doi: 10.1016/j.stem.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Ito C.Y., Li C.Y., Bernstein A., Dick J.E., Stanford W.L. Hematopoietic stem cell and progenitor defects in Sca-1/Ly-6A-null mice. Blood. 2003;101:517–523. doi: 10.1182/blood-2002-06-1918. [DOI] [PubMed] [Google Scholar]
- Jawad M., Cole C., Zanker A., Giotopoulos G., Fitch S., Talbot C.J., Plumb M. QTL analyses of lineage-negative mouse bone marrow cells labeled with Sca-1 and c-Kit. Mamm. Genome. 2008;19:190–198. doi: 10.1007/s00335-008-9097-x. [DOI] [PubMed] [Google Scholar]
- Kang H.M., Zaitlen N.A., Wade C.M., Kirby A., Heckerman D., Daly M.J., Eskin E. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson C., Baudet A., Miharada N., Soneji S., Gupta R., Magnusson M., Enver T., Karlsson G., Larsson J. Identification of the chemokine CCL28 as a growth and survival factor for human hematopoietic stem and progenitor cells. Blood. 2013;121:3838–3842. doi: 10.1182/blood-2013-02-481192. [DOI] [PubMed] [Google Scholar]
- Kiel M.J., Yilmaz O.H., Iwashita T., Yilmaz O.H., Terhorst C., Morrison S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Kiel M.J., Yilmaz O.H., Morrison S.J. CD150- cells are transiently reconstituting multipotent progenitors with little or no stem cell activity. Blood. 2008;111:4413–4414. doi: 10.1182/blood-2007-12-129601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiviranta R., Yamana K., Saito H., Ho D.K., Laine J., Tarkkonen K., Nieminen-Pihala V., Hesse E., Correa D., Määttä J. Coordinated transcriptional regulation of bone homeostasis by Ebf1 and Zfp521 in both mesenchymal and hematopoietic lineages. J. Exp. Med. 2013;210:969–985. doi: 10.1084/jem.20121187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kook H., Epstein J.A. Hopping to the beat. Hop regulation of cardiac gene expression. Trends Cardiovasc. Med. 2003;13:261–264. doi: 10.1016/s1050-1738(03)00107-5. [DOI] [PubMed] [Google Scholar]
- Liang Y., Jansen M., Aronow B., Geiger H., Van Zant G. The quantitative trait gene latexin influences the size of the hematopoietic stem cell population in mice. Nat. Genet. 2007;39:178–188. doi: 10.1038/ng1938. [DOI] [PubMed] [Google Scholar]
- Morrison S.J., Weissman I.L. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1:661–673. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- Morrison S.J., Qian D., Jerabek L., Thiel B.A., Park I.K., Ford P.S., Kiel M.J., Schork N.J., Weissman I.L., Clarke M.F. A genetic determinant that specifically regulates the frequency of hematopoietic stem cells. J. Immunol. 2002;168:635–642. doi: 10.4049/jimmunol.168.2.635. [DOI] [PubMed] [Google Scholar]
- Müller-Sieburg C.E., Riblet R. Genetic control of the frequency of hematopoietic stem cells in mice: mapping of a candidate locus to chromosome 1. J. Exp. Med. 1996;183:1141–1150. doi: 10.1084/jem.183.3.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H., Ding L., Morrison S.J. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. 2013;13:102–116. doi: 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orford K.W., Scadden D.T. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat. Rev. Genet. 2008;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- Orozco L.D., Bennett B.J., Farber C.R., Ghazalpour A., Pan C., Che N., Wen P., Qi H.X., Mutukulu A., Siemers N. Unraveling inflammatory responses using systems genetics and gene-environment interactions in macrophages. Cell. 2012;151:658–670. doi: 10.1016/j.cell.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C.C., Gale G.D., de Jong S., Ghazalpour A., Bennett B.J., Farber C.R., Langfelder P., Lin A., Khan A.H., Eskin E. Gene networks associated with conditional fear in mice identified using a systems genetics approach. BMC Syst. Biol. 2011;5:43. doi: 10.1186/1752-0509-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks B.W., Nam E., Org E., Kostem E., Norheim F., Hui S.T., Pan C., Civelek M., Rau C.D., Bennett B.J. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 2013;17:141–152. doi: 10.1016/j.cmet.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purton L.E., Scadden D.T. Limiting factors in murine hematopoietic stem cell assays. Cell Stem Cell. 2007;1:263–270. doi: 10.1016/j.stem.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Ragu C., Elain G., Mylonas E., Ottolenghi C., Cagnard N., Daegelen D., Passegué E., Vainchenker W., Bernard O.A., Penard-Lacronique V. The transcription factor Srf regulates hematopoietic stem cell adhesion. Blood. 2010;116:4464–4473. doi: 10.1182/blood-2009-11-251587. [DOI] [PubMed] [Google Scholar]
- Rossi L., Lin K.K., Boles N.C., Yang L., King K.Y., Jeong M., Mayle A., Goodell M.A. Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell. 2012;11:302–317. doi: 10.1016/j.stem.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C.H., Liu Z.P., Passier R., Zhang C.L., Wang D.Z., Harris T.M., Yamagishi H., Richardson J.A., Childs G., Olson E.N. Modulation of cardiac growth and development by HOP, an unusual homeodomain protein. Cell. 2002;110:725–735. doi: 10.1016/s0092-8674(02)00933-9. [DOI] [PubMed] [Google Scholar]
- Spangrude G.J., Brooks D.M. Mouse strain variability in the expression of the hematopoietic stem cell antigen Ly-6A/E by bone marrow cells. Blood. 1993;82:3327–3332. [PubMed] [Google Scholar]
- Takeda N., Jain R., LeBoeuf M.R., Wang Q., Lu M.M., Epstein J.A. Interconversion between intestinal stem cell populations in distinct niches. Science. 2011;334:1420–1424. doi: 10.1126/science.1213214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N., Jain R., Leboeuf M.R., Padmanabhan A., Wang Q., Li L., Lu M.M., Millar S.E., Epstein J.A. Hopx expression defines a subset of multipotent hair follicle stem cells and a progenitor population primed to give rise to K6+ niche cells. Development. 2013;140:1655–1664. doi: 10.1242/dev.093005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Harst P., Zhang W., Mateo Leach I., Rendon A., Verweij N., Sehmi J., Paul D.S., Elling U., Allayee H., Li X. Seventy-five genetic loci influencing the human red blood cell. Nature. 2012;492:369–375. doi: 10.1038/nature11677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Os R., Ausema A., Noach E.J., van Pelt K., Dontje B.J., Vellenga E., de Haan G. Identification of quantitative trait loci regulating haematopoietic parameters in B6AKRF2 mice. Br. J. Haematol. 2006;132:80–90. doi: 10.1111/j.1365-2141.2005.05835.x. [DOI] [PubMed] [Google Scholar]
- Yamashita K., Katoh H., Watanabe M. The homeobox only protein homeobox (HOPX) and colorectal cancer. Int. J. Mol. Sci. 2013;14:23231–23243. doi: 10.3390/ijms141223231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.