

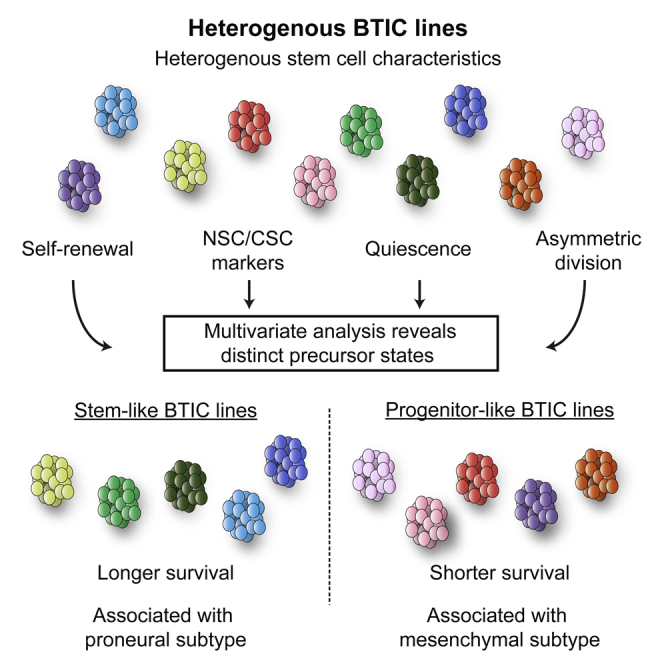
Summary
In glioblastoma multiforme (GBM), brain-tumor-initiating cells (BTICs) with cancer stem cell characteristics have been identified and proposed as primordial cells responsible for disease initiation, recurrence, and therapeutic resistance. However, the extent to which individual, patient-derived BTIC lines reflect the heterogeneity of GBM remains poorly understood. Here we applied a stem cell biology approach and compared self-renewal, marker expression, label retention, and asymmetric cell division in 20 BTIC lines. Through cluster analysis, we identified two subgroups of BTIC lines with distinct precursor states, stem- or progenitor-like, predictive of survival after xenograft. Moreover, stem and progenitor transcriptomic signatures were identified, which showed a strong association with the proneural and mesenchymal subtypes, respectively, in the TCGA cohort. This study proposes a different framework for the study and use of BTIC lines and provides precursor biology insights into GBM.
Graphical Abstract
Highlights
-
•
BTICs are extensively characterized using a stem cell approach
-
•
Two groups of BTIC lines are identified: stem-like and progenitor-like
-
•
Progenitor-like BTICs lead to strikingly shorter survival in xenografted mice
-
•
Stem- and progenitor-like profiles associate with proneural and mesenchymal subtypes
In this article, Weiss and colleagues show that multivariate analysis of otherwise heterogeneous stem cell characteristics allow segregation of BTIC lines into two groups: stem- and progenitor-like. Remarkably, these groups are predictive of survival, with progenitor-like lines leading to strikingly shorter survival in xenografts. Interestingly, stem- and progenitor-like groups also associated with proneural and mesenchymal GBM subtypes, respectively.
Introduction
Glioblastoma multiforme (GBM) is the most common adult primary brain tumor, with an extremely dismal prognosis and high rate of recurrence following standard therapy (Wen and Kesari, 2008). Populations of brain tumor initiating cells (BTICs), which express many of the properties of cancer stem cells (CSCs), have been identified in GBM (Singh et al., 2004). BTICs display CSC characteristics of long-term self-renewal, multilineage differentiation, and tumorigenicity, which may be collectively referred to as cancer “stemness” features (reviewed in Clevers, 2011). BTIC lines have become a valuable tool for modeling GBM (Westphal and Lamszus, 2011) and for the development of experimental therapeutics (Luchman et al., 2014).
In a recent review, Stopschinski and colleagues (Stopschinski et al., 2013) argued that a consensus standardization of the BTIC model will be essential to harness its full cell biologic and experimental therapeutic potential in a heterogeneous disease such as GBM. Initial studies aimed at examining single stemness characteristics, such as dye retention (Deleyrolle et al., 2011) and self-renewal (Campos et al., 2014), have provided insights as to the relationship between BTIC lines and tumor heterogeneity. Nevertheless, a comprehensive precursor cell analysis of multiple BTIC lines has yet to be performed.
It has been suggested (Binda et al., 2014; Vescovi et al., 2006) that understanding BTICs may best be achieved by examining them in relation to normal neural stem cells (NSCs). NSCs are organized in a hierarchical manner, with a quiescent cell (Codega et al., 2014) at the apex. Through asymmetric cell division (Shen et al., 2002; Sun et al., 2005), the NSC self-renews, giving rise to an NSC and transit-amplifying progenitor that proliferates rapidly and produces the bulk of the short-term neurogenic population (Doetsch et al., 1999a). Moreover, BTICs are most often cultured with techniques originally developed for NSCs (Reynolds and Weiss, 1992) and share many stemness characteristics with NSCs, including the expression of markers such as NESTIN, CD133, and SOX2 (Gangemi et al., 2009; Singh et al., 2004), label retention (Deleyrolle et al., 2011), asymmetric cell division (Lathia et al., 2011), and self-renewal (Campos et al., 2014). However, a single study employing multiple stemness characteristics to examine a large set of heterogeneous BTIC lines has yet to be reported.
Hypothesizing that heterogeneity among BTIC lines is related to differences in stemness features, we used a comprehensive and integrated stem cell biology approach to characterize 20 lines. We report the identification of two groups, with distinct precursor states, predictive of survival after xenografts and exhibiting strong gene expression profile associations with relevant subtypes of the Cancer Genome Atlas (TCGA).
Results
Heterogeneous Self-Renewal Ability and Marker Expression within BTIC Lines
A comprehensive phenotypic characterization of 20 GBM patient-derived BTIC lines, focusing on features of NSC biology, was performed. The BTIC lines express, although to variable degrees, NSC markers (NESTIN, SOX2, MUSASHI1, VIMENTIN, CD15, and CD133) (Figure S1); differentiate to express neuronal, astrocytic, and oligodendrocytic markers (Figure S1); and are tumorigenic, suggesting that they contain true CSCs. Furthermore, they exhibit mutations/deletions typically found in GBM (EGFR, TP53, PTEN, NF1, and CDKN2A) (Table S1).
BTIC self-renewal capacity was assessed by calculating the percentage of cells capable of giving rise to a new sphere after dissociation. A high degree of variability was observed across the lines, with sphere-forming cells ranging from 7.8% ± 0.2% (mean ± SEM) to 22.3% ± 0.3% (Figures 1A and 1B; inter-line differences were significant, p < 0.0001, one-way ANOVA). Expression of NSC and BTIC markers EGFR, CD133, and CD15 was analyzed by fluorescence-activated cell sorting (FACS) (Figures 1C–1H), and a remarkable inter-line diversity was again observed, with the percentage of positive cells ranging from 2.6% ± 0.2% to 93.3% ± 1.1% for EGFR, from 0.4% ± 0.2% to 81.5% ± 2.3% for CD133, and from 0.2% ± 0.1% to 93.7% ± 1.9% for CD15 (for all, p < 0.0001, one-way ANOVA).
Figure 1.

Complex Self-Renewal and Marker Expression Patterns in BTICs
(A) Representative phase-contrast micrograph shows BTIC spheres in self-renewal experiments.
(B) Quantification of sphere formation in lines is shown.
(C–H) Expression of cell surface markers common to BTICs and NSCs, EGFR (C and D), CD133 (E and F), and CD15 (G and H), was analyzed by FACS and quantified (D, F, and H). (C), (E), and (G) show examples of the FACS plots for BT147, BT134, and BT189. Scale bar, 100 μm.
Error bars represent SEM (B, D, F, and H). Each dot represents a single experiment. See also Table S2.
Quiescence and Asymmetry in BTICs
Quiescence, the reversible exit from the cell cycle, is a process common to NSCs and BTICs. Due to slower cellular turnover, quiescent cells can be detected by the retention of a dye or label (Cheung and Rando, 2013). The abundance of label-retaining cells was assayed with carboxyfluorescein diacetate succinimidylester (CFSE). While a large number of cells were CFSE negative, a substantial population remained clearly labeled after growth in standard conditions (Figures 2A and 2C). Cells that divided only once or twice, as determined by the intensity of fluorescence, were defined as CFSEHIGH. These cells were functionally quiescent, as confirmed by the ability of both CFSEHIGH- and CFSELOW-sorted cells to form new spheres (Figure S2). Analysis of the percentage of label-retaining cells in all BTIC lines showed different frequencies (Figure 2E), ranging from 0.4% ± 0.1% to 5.2% ± 1.1% (p < 0.0001, one-way ANOVA). The presence of label-retaining cells in each line showed a weak inverse correlation to sphere formation and a positive correlation with the percentage of CD133+ cells, but not with the other stemness parameters (Table S2).
Figure 2.

Heterogeneous Representation of Quiescent and Asymmetrically Dividing Cells in BTIC Lines
(A–E) CFSE-retaining cells (green in A and C) and by FACS (B and D; day 0, freshly labeled cells). Percentage of label-retaining cells is shown in (E); individual dots represent independent experiments; error bars represent SEM. DIC, differential interference contrast.
(F) Examples of asymmetric distribution of the markers EGFR, NUMB, NESTIN, and GFAP are shown.
(G) Quantification of asymmetric cell division across the 20 BTIC lines. Each bar is the average of at least four independent experiments. Errors bars represent SEM.
Scale bars, 50 μm (A and C) and 10 μm (F). See also Figure S2 and Table S2.
Asymmetric cell division is a key step in the homeostatic self-renewal of stem cells and has been previously described in BTICs (Lathia et al., 2011). Using key markers of NSC biology, we analyzed the frequency of asymmetry in BTIC lines by assaying patterned asymmetric distribution of NUMB, EGFR, NESTIN, and GFAP, after cell division. Single cells were plated at low density, synchronized, and analyzed 18–20 hr after the cell division block was removed. Clear examples of asymmetric cell division (Figure 2F) could be found for all four markers. Similar results were observed using additional NSC/BTIC markers (CD133, SOX2, MUSASHI1, and CD15; data not shown). Quantification showed that asymmetric cell division, while very common in some BTIC lines (up to 46.4% ± 5.4% of couples), was virtually non-existent in others (Figure 2G). The inter-line differences were significant for all markers (p < 0.0001, one-way ANOVA). Interestingly, the asymmetric division rate did not correlate with the expression of each individual marker, except for GFAP (Figure S3).
Cluster Analysis Reveals Two Distinct Precursor State Groups
To understand parameters relevant to the biology of BTIC lines, we first attempted to find direct correlations between all pairs of assays performed (Table S2). Although some associations were found, no unifying pattern was identified. Instead, using hierarchical clustering with Manhattan distance and Ward’s agglomeration method, we identified two clusters (Figure 3A). K-means clustering with Manhattan distance confirmed a two-cluster solution, and the same membership in these two clusters was found (data not shown). We next looked at which variables defined the clustering of BTIC lines in each of these groups. Cluster A was characterized (Z scores > 1.96) by stem cell features, such as higher levels of asymmetry, label-retaining cells, and CD133-expressing cells, suggesting that these BTICs may be more akin to classically defined stem cells (stem-like: SL). Cluster B was defined by higher sphere formation rate, resembling transit-amplifying progenitors found in normal neurogenesis (progenitor-like: PL). Given the resemblance of SL and PL cells to the NSC biology counterparts, we defined these features of the two BTIC groups as a difference in precursor state. Interestingly, neither of the two clusters associated with specific molecular alterations in any of the genes analyzed (Fisher’s exact test, p > 0.05 for each mutation).
Figure 3.

Cluster Analysis Defines Precursor States of BTICs, Associated with Survival In Vivo
(A) Heatmap representing unsupervised clustering of 20 BTIC lines based on the parameters studied above. The lines within the two clusters were found to be SL or PL.
(B and C) Representative images of tumor formation after xenograft. BTICs were visualized by human-specific nucleolin staining.
(D) Kaplan-Meier survival curves represent the individual BTICs (light lines) and the combined for each group (bold lines) (n = 5–10 mice per cell line; total SL, n = 42; total PL, n = 59; p < 0.0001 in log-rank test statistic; see also Table S3 for specific survival and number of animals per line).
(E) The growth rate of 12 BTIC lines in vitro was not correlated to median survival (Pearson’s correlation, p = 0.44, R = 0.23; for each line, n = 3 independent experiments).
(F) The frequency of sphere-forming cells showed an inverse correlation to the median survival in 15 BTIC lines (Pearson’s correlation, p = 0.03, R = −0.56; sphere formation data from experiments reported in Figure 1).
Scale bars, 1 mm (B and C) and 50 μm (insets).
Precursor States Associate with Survival in Xenografts
We next examined whether SL or PL features of BTICs play a defining role in tumor formation, by implanting 15 lines in immunocompromised mice. All BTIC lines were tumorigenic (Figures 3B and 3C), but, notably, animals xenografted with SL lines survived significantly longer than those implanted with PL lines (average median survival SL = 183.7 ± 24.5 versus PL = 67.4 ± 11.4 days, p < 0.0001, log-rank test; SL n = 42, PL n = 59) (Figure 3D; detailed survival times in Table S3).
To test whether the survival difference was due to variations in BTIC proliferation rates, we measured the growth kinetics of 16 lines in vitro (eight for each group). Although cells in SL lines divided slower (doubling time 4.58 ± 0.36 in SL versus 3.57 ± 0.19 days in PL, p < 0.0001), survival in vivo was not correlated with the mean doubling time observed in culture (Figure 3E; p = 0.41, R = 0.25), indicating that shorter survival was not solely due to a difference in proliferation rate. In contrast, median survival showed an inverse correlation with the abundance of sphere-forming cells (Figure 3F; p = 0.03, R = −0.56).
Transcriptome Analysis Identifies an Association between Precursor States and GBM Subtypes
RNA sequencing (RNA-seq) was performed on seven BTIC lines from each group. Unsupervised clustering based on the GBM subtype transcriptomic signatures (Verhaak et al., 2010) did not distinguish proneural, mesenchymal, classical, or neural BTICs. We then performed differential expression analysis and found that 1,110 genes were significantly upregulated in SL-BTICs and 269 genes were upregulated in PL-BTICs (Figure 4A). We then derived a signal-to-noise measure (DSN) to eliminate genes with high SD within either BTIC group, and ultimately we selected the top tenth percentile of differentially expressed genes based on DSN, generating a signature of 136 genes (Figure S4).
Figure 4.

Transcriptome Analysis Reveals an Association between Precursor States and Proneural and Mesenchymal Subtypes
(A) Initial selection of genes differentially expressed in our two groups of seven lines each is shown. RPKM, reads per kilobase per million.
(B) Combined average Z scores (Z′) of TCGA patient samples divided by GBM subtype are shown. ∗p < 0.05, ∗∗∗∗p < 0.0001.
(C) TCGA samples were ranked based on Z′ and divided into three groups (Z′ > 0.1, −0.1 < Z′ < 0.1, and Z′ < −0.1).
(D) Pie charts represent the distribution of GBM subtypes within the three groups.
(E) Representative gene set enrichment analysis comparing groups I and III for the enrichment of GBM subtype gene sets. All genes are ranked based on the expression in the dataset (bottom; subtype-specific genes are correlated to the ranking, resulting in the enrichment score. See also Table S4 for complete analysis of the three groups.
(F) Clustergram shows the expression of the 136-gene signature in proneural and mesenchymal samples.
TCGA data (B–F) were from 152 samples. See also Figure S4.
To further understand the relevance of this signature in the disease context, we used the publicly available GBM transcriptome dataset (Cancer Genome Atlas Research Network, 2008). To match the precursor state profiles with samples in the TCGA dataset, we calculated Z scores for genes overexpressed in SL- and PL-BTIC lines (referred to as SL- and PL-genes, respectively) for each GBM patient in the dataset. A combined average Z score, hereafter referred to as Z′, was calculated by subtracting the average PL-gene Z score from the average SL-gene Z score, for each TCGA patient. Z′ was used as a summary measure of similarity to either the SL or the PL group. Notably, in the 152 TCGA samples that were already classified into the four GBM subtypes described previously (Verhaak et al., 2010), there was a significant difference in Z′ between proneural and mesenchymal tumors (p < 0.0001, one-way ANOVA with Bonferroni post hoc analysis) (Figure 4B).
The patient samples were then separated into three groups, based on Z′ values (Z′ cutoff 0.1 and −0.1; groups I, II, and III, n = 44, 40, and 68, respectively; Figure 4C). Therefore, for the 136-gene profile, group I (Z′ > 0.1) had a pattern of expression similar to SL lines and group III (Z′ < −0.1) similar to PL lines. Group I was predominantly composed of proneural samples (45.5%) and group III had mostly mesenchymal (51.5%) (χ2 = 30.20, df = 6, p < 0.0001); no specific subtype was over-represented in group II (Figure 4D).
To further confirm the associations between groups defined by our 136-gene signature and GBM subtypes, we performed gene set enrichment analysis (GSEA) of genes that define the four GBM subtypes (Verhaak et al., 2010) within the TCGA dataset (Figure 4E). As expected, we observed a strong enrichment of proneural-signature genes in group I (p < 0.05, false discovery rate [FDR] < 0.10). Conversely, the mesenchymal gene set was enriched in group III (p < 0.05, FDR < 0.05). A complete list of associations is reported in Table S4. Patient survival was not significantly different across the three groups (p = 0.25, log-rank; data not shown).
Finally, we asked whether our 136-gene precursor state signature could effectively stratify the proneural (n = 38) and mesenchymal (n = 49) patients in the TCGA dataset. In unsupervised clustering (Figure 4F), our signature segregated proneural and mesenchymal GBM samples, further confirming the relationship between these subtypes and BTIC precursor states. SL genes significantly overlapped with the cluster of genes overexpressed in proneural patients (hypergeometric p = 0.001). Similarly, PL genes overlapped significantly with those overexpressed in mesenchymal patients (hypergeometric p = 0.001).
Discussion
By applying cluster analysis to the study of stemness properties of BTICs, we report the emergence of two distinct groups of patient-derived BTIC lines. Utilizing concepts taken from NSC biology, we defined these two groups as SL and PL. Mice xenografted with PL lines showed strikingly shorter median survival, which was not simply due to their higher proliferation rate. Given that SL- and PL-BTICs both contain CSCs, yet show remarkable differences in vitro and in vivo, we propose that they differ in precursor state. Our model provides a conceptual link between BTICs and NSCs, which may be critical in developing a better understanding of GBM. This is highlighted by the fact that the gene expression signatures derived from SL and PL lines were found to associate with GBM subtypes, further underscoring the differences between the two groups of BTIC lines.
The CSC hypothesis hinges on the presence of a hierarchical organization within a tumor (Kreso and Dick, 2014), reminiscent of normal stem cell biology, which results in a heterogeneous cell population. Furthermore, diverse genetic and mutational backgrounds, as well as differences in disease evolution, can result in heterogeneity among CSCs (Meacham and Morrison, 2013). BTICs are a widely used model for GBM but show great inter-line heterogeneity (Stopschinski et al., 2013), mirroring the inherent heterogeneity of GBM, recently observed even at the single-cell level within and between patient tumors (Patel et al., 2014).
Others previously have used in vitro growth characteristics in attempts to identify subgroups among BTIC lines. Günther and colleagues (Günther et al., 2008) classified BTICs based on free-floating or adherent growth, finding that they had different gene expression and in vivo behavior. Clonogenicity, measured in a colony formation assay, was the basis of the classification proposed by Campos et al. (2014). Interestingly, this group found that highly clonogenic lines had fewer label-retaining cells and were more aggressive, a pattern resembling our findings. Recently, Mao and colleagues (Mao et al., 2013) identified and characterized two mutually exclusive glioma stem cell subtypes. Transcriptomic analysis distinguished proneural and mesenchymal groups, and the latter displayed more aggressive phenotypes both in vitro and in vivo as well as increased resistance to radiation compared with the proneural group. These studies offer interesting insights in BTIC biology and help strengthen our conclusions. Importantly, by taking a multi-dimensional stem cell biology approach, our study also allowed the identification of two BTIC subgroups that appear highly relevant to the subtypes in the TCGA dataset.
Expression profiles from tumor tissue and derived cell lines are extremely different, thus the current transcriptomic signature for GBM subtyping may not apply in vitro. Using our precursor state signature, we were able to identify an association with the proneural and mesenchymal subtypes. Interestingly, it has been proposed that, upon recurrence, GBMs tend to shift toward the mesenchymal subtype (Phillips et al., 2006), suggesting that the latter might be downstream in hierarchy. At the same time, proneural GBMs show a worse response to standard therapeutic treatment (Verhaak et al., 2010), as would be expected for stem-like cells. Moreover, Ozawa et al. (2014) recently showed that most GBMs might derive from a common proneural-like precursor. These reports suggest that the mesenchymal subtype may be downstream of the proneural. Our study provides a possible interpretation of these data, in the context of stem cell biology. SL-BTICs would be less dysregulated, maintaining more normal stem cell features; this would be reflected in longer survival of xenografted mice and in the fact that they are associated with the proneural subtype. In addition, the SL lines may be more CSC-like and, hence, possibly have a larger fraction of tumor-initiating cells; while displaying less aggressive growth, they may be more tumorigenic in vivo. This premise needs be confirmed by xenografting smaller numbers of cells in tumorigenicity studies. Furthermore, our data suggest that the precursor state model will be essential to test hypotheses in cancer cell lineage and tumor evolution, currently assessed with mouse models (Ozawa et al., 2014).
The precursor state model offers the conceptual basis for developing more targeted GBM cell biology and experimental therapeutic studies. SL lines may be more useful to study tumor recurrence and hierarchy, while PL-BTICs might be best employed to investigate the pathways involved in rapid and aggressive growth. It is conceivable that the precursor state also could influence response to cytotoxic treatment in BTICs, as it does in NSCs. For example, neural progenitors in adult mouse brain are ablated easily by treatment with the chemotherapy agent cytarabine, while the quiescent stem cells are able to survive and repopulate the subventricular zone (Doetsch et al., 1999b).
Failure of several targeted therapies, both in experimental and clinical settings (Ohka et al., 2012), is a grim reminder of the complexity and heterogeneity of GBM. Here we provide a fresh classification of BTICs using a classic stem cell biology approach combined with cluster analysis. Improved characterization of BTICs, as a model of GBM, will allow for a better understanding of their response to treatment, both in vitro and in vivo, and ultimately open the way for findings of high relevance to GBM therapeutics.
Experimental Procedures
BTIC Cultures and Growth Assays
BTIC lines (n = 20) were isolated, established, and maintained as described previously (Kelly et al., 2009). Lines were used within 25–30 passages of establishment from primary cells. For sphere formation assays, 1,000 cells per well were plated (n = 6) and spheres were counted when they reached a diameter of 200–250 μm. For growth curves, cells were seeded at specific densities and alamarBlue (Life Technologies) conversion was measured every day over the course of 2 weeks.
Flow Cytometry
Single-cell suspensions were labeled with appropriately conjugated antibodies and analyzed with an ATTUNE flowcytometer (Life Technologies). For label retention studies, single cells were stained with 1 μM CFSE (Life Technologies) and grown in standard conditions.
Asymmetry Studies
Asymmetry was analyzed by plating cells at low density, synchronizing them with nocodazole or thymidine (Sigma); the block was released and the cells were fixed and stained after 18–20 hr. Asymmetric distribution of markers was counted in at least 80 couples of daughter cells.
In Vivo Studies
Cells (100,000) were implanted in the right striatum of 6- to 8-week-old female C17/SCID mice (n = 6–10 per line). Animals were euthanized upon demonstration of overt disease symptoms.
RNA-Seq and Analyses
RNA-seq was performed using HiSeq2000 (Illumina). The list of differentially expressed genes was derived using DEfine v.0.9.2 (FDR cutoff of 0.01). Ranking of the TCGA samples was performed by subtracting the average Z scores of SL genes by the average Z scores of PL genes (Z′), then dividing the dataset in three groups based on Z′ (TCGA data Ver.2014-08-28).
Statistical Analyses
All data reported for in vitro experiments are representative of at least three independent replicates and are illustrated in scatterplots or bar graphs, including mean ± SEM. Statistical analyses and graphing were performed with Prism 6.0 (GraphPad), SPSS (IBM), and R (version 3.0.1).
Author Contributions
C. Cusulin performed the experiments. C. Cusulin and C. Chesnelong designed the study, analyzed the data, and wrote the paper. P.B., M.B., S.J.J., and M.A.M. performed the RNA-seq and analyzed the data. K.K. performed the cluster analysis. J.A.C. and J.G.C. contributed to the acquisition and analysis of samples. H.A.L. and S.W. supervised the study and wrote the paper.
Acknowledgments
We thank Rozina Hassam, Dorothea Livingstone, and Orsolya Cseh for technical assistance; the Calgary Brain Tumour and Tissue Bank for providing patient samples; and the sequencing and bioinformatics teams at the Michael Smith Genome Science Center for data processing. Funding to S.W. and H.A.L. was from the Stem Cell Network of Canada and to S.W., H.A.L., J.A.C., J.G.C., S.J., and M.A.M. was from the Terry Fox Research Institute.
Published: June 18, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, four figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.05.010.
Accession Numbers
The accession number for the RNA-seq data reported in this paper is NCBI Sequence Read Archive (SRA): SRP057855.
Supplemental Information
References
- Binda E., Reynolds B.A., Vescovi A.L. Glioma stem cells: turpis omen in nomen? (The evil in the name?) J. Intern. Med. 2014;276:25–40. doi: 10.1111/joim.12254. [DOI] [PubMed] [Google Scholar]
- Campos B., Gal Z., Baader A., Schneider T., Sliwinski C., Gassel K., Bageritz J., Grabe N., von Deimling A., Beckhove P. Aberrant self-renewal and quiescence contribute to the aggressiveness of glioblastoma. J. Pathol. 2014;234:23–33. doi: 10.1002/path.4366. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung T.H., Rando T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013;14:329–340. doi: 10.1038/nrm3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. The cancer stem cell: premises, promises and challenges. Nat. Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- Codega P., Silva-Vargas V., Paul A., Maldonado-Soto A.R., Deleo A.M., Pastrana E., Doetsch F. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron. 2014;82:545–559. doi: 10.1016/j.neuron.2014.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleyrolle L.P., Harding A., Cato K., Siebzehnrubl F.A., Rahman M., Azari H., Olson S., Gabrielli B., Osborne G., Vescovi A., Reynolds B.A. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain. 2011;134:1331–1343. doi: 10.1093/brain/awr081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F., Caillé I., Lim D.A., García-Verdugo J.M., Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- Doetsch F., García-Verdugo J.M., Alvarez-Buylla A. Regeneration of a germinal layer in the adult mammalian brain. Proc. Natl. Acad. Sci. USA. 1999;96:11619–11624. doi: 10.1073/pnas.96.20.11619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangemi R.M., Griffero F., Marubbi D., Perera M., Capra M.C., Malatesta P., Ravetti G.L., Zona G.L., Daga A., Corte G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009;27:40–48. doi: 10.1634/stemcells.2008-0493. [DOI] [PubMed] [Google Scholar]
- Günther H.S., Schmidt N.O., Phillips H.S., Kemming D., Kharbanda S., Soriano R., Modrusan Z., Meissner H., Westphal M., Lamszus K. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene. 2008;27:2897–2909. doi: 10.1038/sj.onc.1210949. [DOI] [PubMed] [Google Scholar]
- Kelly J.J., Stechishin O., Chojnacki A., Lun X., Sun B., Senger D.L., Forsyth P., Auer R.N., Dunn J.F., Cairncross J.G. Proliferation of human glioblastoma stem cells occurs independently of exogenous mitogens. Stem Cells. 2009;27:1722–1733. doi: 10.1002/stem.98. [DOI] [PubMed] [Google Scholar]
- Kreso A., Dick J.E. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Lathia J.D., Hitomi M., Gallagher J., Gadani S.P., Adkins J., Vasanji A., Liu L., Eyler C.E., Heddleston J.M., Wu Q. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011;2:e200. doi: 10.1038/cddis.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchman H.A., Stechishin O.D., Nguyen S.A., Lun X.Q., Cairncross J.G., Weiss S. Dual mTORC1/2 blockade inhibits glioblastoma brain tumor initiating cells in vitro and in vivo and synergizes with temozolomide to increase orthotopic xenograft survival. Clin. Cancer Res. 2014;20:5756–5767. doi: 10.1158/1078-0432.CCR-13-3389. [DOI] [PubMed] [Google Scholar]
- Mao P., Joshi K., Li J., Kim S.H., Li P., Santana-Santos L., Luthra S., Chandran U.R., Benos P.V., Smith L. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA. 2013;110:8644–8649. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham C.E., Morrison S.J. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohka F., Natsume A., Wakabayashi T. Current trends in targeted therapies for glioblastoma multiforme. Neurol. Res. Int. 2012;2012:878425. doi: 10.1155/2012/878425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T., Riester M., Cheng Y.K., Huse J.T., Squatrito M., Helmy K., Charles N., Michor F., Holland E.C. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips H.S., Kharbanda S., Chen R., Forrest W.F., Soriano R.H., Wu T.D., Misra A., Nigro J.M., Colman H., Soroceanu L. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Reynolds B.A., Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- Shen Q., Zhong W., Jan Y.N., Temple S. Asymmetric Numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development. 2002;129:4843–4853. doi: 10.1242/dev.129.20.4843. [DOI] [PubMed] [Google Scholar]
- Singh S.K., Hawkins C., Clarke I.D., Squire J.A., Bayani J., Hide T., Henkelman R.M., Cusimano M.D., Dirks P.B. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Stopschinski B.E., Beier C.P., Beier D. Glioblastoma cancer stem cells—from concept to clinical application. Cancer Lett. 2013;338:32–40. doi: 10.1016/j.canlet.2012.05.033. [DOI] [PubMed] [Google Scholar]
- Sun Y., Goderie S.K., Temple S. Asymmetric distribution of EGFR receptor during mitosis generates diverse CNS progenitor cells. Neuron. 2005;45:873–886. doi: 10.1016/j.neuron.2005.01.045. [DOI] [PubMed] [Google Scholar]
- Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vescovi A.L., Galli R., Reynolds B.A. Brain tumour stem cells. Nat. Rev. Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- Wen P.Y., Kesari S. Malignant gliomas in adults. N. Engl. J. Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- Westphal M., Lamszus K. The neurobiology of gliomas: from cell biology to the development of therapeutic approaches. Nat. Rev. Neurosci. 2011;12:495–508. doi: 10.1038/nrn3060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.