

Abstract
Human mutations that truncate the massive sarcomere protein titin (TTNtv) are the most common genetic cause for dilated cardiomyopathy (DCM), a major cause of heart failure and premature death. Here we show that cardiac microtissues engineered from human induced pluripotent stem (iPS) cells are a powerful system for evaluating the pathogenicity of titin gene variants. We found that certain missense mutations, like TTNtv, diminish contractile performance and are pathogenic. By combining functional analyses with RNAseq, we explain why truncations in the A-band domain of TTN cause DCM while truncations in the I-band are better tolerated. Finally, we demonstrate that mutant titin protein in iPS-cardiomyocytes results in sarcomere insufficiency, impaired responses to mechanical and β-adrenergic stress, and attenuated growth factor and cell signaling activation. Our findings indicate that titin mutations cause DCM by disrupting critical linkages between sarcomerogenesis and adaptive remodelling.
Dilated cardiomyopathy (DCM) is characterized by progressive left ventricular (LV) dilation, systolic dysfunction, and ultimately heart failure. Occurring in 1 of 250 adults (1), DCM arises from underlying cardiovascular conditions or as a primary genetic disorder. We recently identified dominant mutations that truncate the sarcomere protein titin (TTNtv) as the most common genetic cause of DCM, occurring in ~20% of familial or sporadic cases (2).
TTN is a massive protein that spans half of the sarcomere (1 μm) and is comprised of >34,000 amino acids within four functionally distinct segments (Fig. 1A): an amino-terminus that is anchored at the Z-disk; a distensible I-band (~1 MDa) composed of repeating immunoglobulin-like domains and disordered regions, an inextensible, thick filament-binding A-band (~2 MDa), and a carboxyl M-band with a kinase domain. TTNtv have been identified in each protein segment, but TTNtv in DCM patients are markedly enriched in the A-band (2, 3). In addition, numerous rare missense variants in TTN have been identified, most with unknown medical significance (2, 3). Both TTN’s size and incomplete knowledge of the protein’s function in cardiomyocyte biology have hindered traditional approaches for elucidating why some TTN mutations produce clinical phenotypes. To address this, we harnessed recent advances in stem cell reprogramming (4), gene editing (5), and tissue engineering (6) to produce human cardiac microtissue (CMT) models of TTNtv.
Fig. 1. Engineered iPS-CM microtissues with TTN mutations have impaired intrinsic contractility and responses to stress.
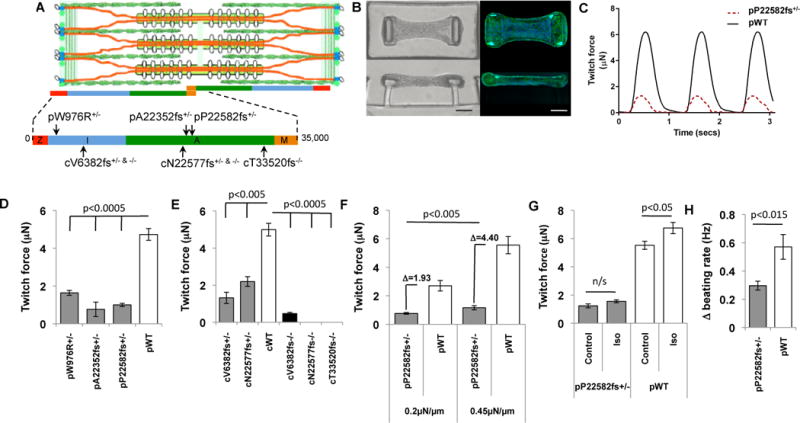
(A) Schematic of the cardiac sarcomere with TTN (orange), thick filaments (gold rods with white globular heads), and thin filaments (green, coiled ovals). TTN protein segments (z-disc, red; I-band, blue; A-band, green; M-band, gold) with location of human (“p” patient-derived; “c” CRISRP/CAS9-derived) mutations. (B) Images (bright field (left) and fluorescent (right); green, Phalloidin, Alexa Fluor® 488; blue, DAPI) of iPS-CMT suspended between two polydimethylsiloxane (PDMS) pillars from top-down (upper) and side (lower) views. Scale bar, 50 μm. (C) Representative force tracing of pWT (black) and pP22582fs+/− (red) CMT over three twitch cycles. (D) Mean force of pW976R+/−, pA22352fs+/−, and pP22582fs+/− iPS-CMTs compared to pWT iPS-CMTs (N=5). (E) Mean force produced by isogenic iPS-CMTs with heterozygous or homozygous I-band (cV6382fs) or A-band (cN22577fs) TTNtv (N > 4 CMTs). (F) Mean force produced by pWT and pP22582fs+/− CMTs in response to increased pillar stiffness (0.2 to 0.45 μN/μm; N>11 CMTs). Difference (Δ) in force generation between pP22528fs+/− and pWT measured at low and high stiffness. (G) Isoproterenol-induced force (N>5) and (H) spontaneous beating rate (N >5, Hz) in pP22582fs+/− versus pWT CMTs (N>5). Significance assessed by Student’s t test (D–H); data are means +/− SEM (D-H).
We generated iPS cell-derived cardiomyocytes (iPS-CMs) from patients (“p” preceding genotype). Cryopreserved blood samples from one unaffected and three DCM patients with dominant TTN mutations (Fig. 1A and table S1A) were reprogrammed, and high-quality iPS clones (figs. S1A–C) were expanded, differentiated (7), and enriched by metabolic selection (8) to achieve cultures with >90% iPS-CMs (figs. S2A–C). We produced iPS-CMs with two A-band TTNtvs (pA22352fs+/− or pP22582fs+/−) and a missense mutation (pW976R+/−) within the Z/I junction that co-segregated with DCM in a large family (9). Single cell assays of contractile function on microarray post detectors (mPADS) (10) showed no significant difference between wild-type (WT) and TTNtv iPS-CMs (fig. S2D). As three-dimensional CMTs (Fig. 1B) better recapitulate native cardiomyocyte architecture and mechanics, improving sarcomere alignment, expression of contractile proteins, and iPS-CMs maturity (6, 11, 12), we assessed the contractile function of iPS-CMTs containing WT or mutant iPS-CMs. We observed minor variation in contractile function between biological replicates of CMTs or between CMTs made from independent clones from the same patient (figs. S2E, F). However, CMTs expressing either A-band TTNtv or W976R+/− variants exhibited less than half the contractile force (Figs. 1C, D and Suppl. Videos) or the stress (force normalized to tissue area; fig. S2I) generated by pWTs, and function did not improve over time (fig. S2G). As static force in pWT and pP22582fs+/− CMTs were similar (fig. S2H), we conclude that the contractile deficits observed in mutant CMTs are not due to non-myocyte factors. In addition, the comparable force deficits observed in CMTs with both A-band TTNtv and the Z/I junction missense mutation demonstrates that W976R+/− is a pathogenic TTN missense mutation.
To ensure that the observed functional abnormalities did not reflect background genetic differences in patient-derived iPS-CMs, we also introduced TTNtv into an independent, isogenic iPS cell using scarless, CRISPR/CAS9 technology (5) to target the I- or A-band exons (Fig. 1A and table S1B). Mutant isogenic iPS lines (“c“ preceding genotype) were differentiated into iPS-CMs and incorporated into CMTs. cN22577fs+/− creates an A-band TTNtv in exon 322 (similar to patient-derived pP22582fs+/−; Fig 1A). cN22577fs+/− CMTs had significantly reduced contractile force (2.19 μN) compared to isogenic cWT-CMTs (Fig. 1E, p<0.003), but not to the extent observed in pA22352fs+/− or pP22582fs+/− CMTs (0.767μN and 1.001μN, respectively, p<0.02). Tissue stress was similarly reduced in TTNtv CMTs compared to WT (fig. S2J). These data confirm that A-band TTNtv markedly reduced contractile function in both patient-derived and isogenic CMTs, and raise the possibility that genetic background can modify the functional severity of TTNtv.
Premature protein truncation at any location within a molecule is generally assumed to result in loss of function and comparable deleterious consequences. However I-band TTNtv have been identified in healthy individuals and in the general population without DCM (2, 3). Two models have been proposed to explain this dichotomy: a) alternative splicing excludes I-band exons from most mature TTN transcripts and thereby reduces the functional consequences of I-band TTNtv, while the inclusion of A-band exons with TTNtv is deleterious; and b) A-band TTNtv transcripts produce longer, stable mutant protein with dominant negative effects on sarcomere biology. To distinguish these models we compared the functional consequences of isogenic heterozygous (cV6382fs+/−) and homozygous (cV6382fs−/−) I-band TTNtv in exon 66 to A-band TTNtv (cN22577fs+/−, cN22577fs−/−, cT33520fs−/−). TTN RNA splicing was assessed in iPS-CMs and compared to normal adult left ventricle (LV) using RNAseq analyses. TTN transcripts from iPS-CMs incorporated more I-band exons than adult LV (figs. S3A, B). Importantly, >80% of iPS-CM TTN transcripts included exon 66, 5-fold more than adult LV (18% of TTN transcripts contained exon 66).
cV6382fs+/− CMTs had markedly impaired force generation, similar to the deficits observed in isogenic CMTs with A-band cN22577fs+/− or patient-derived CMTs with A-band pP22582fs+/− (Figs. 1D, E). Functional comparisons of CMTs with homozygous TTNtv in the I- or A-band were also similar. Homozygous A-band CMTs (cN22577fs−/− and cT33520fs−/−) produced no force while homozygous I-band CMTs (cV6382fs−/−) generated small but demonstrable force (0.472 μN), presumably due to 18% of TTN transcripts that excluded exon 66 and the TTNtv (fig. S3A). Based on these functional and RNAseq data we suggest that alternative exon splicing is the predominant mechanism for reduced penetrance of I-band TTNtv.
As TTN is responsible for sensing and responding to myocardial stresses (13), we posited that TTNtv mutants would exhibit aberrant stress responses. To model low and high mechanical load, we assessed contractile performance of pWT and pP22582fs+/− CMTs grown on flexible (0.2 μN/μm) and rigid (0.45 μN/μm) cantilevers (Fig. 1F). pP22582fs+/− CMTs produced less force than pWT CMTs at low load. At higher load, pWT CMTs produced a two-fold increase in force, greater than four-fold more than that produced by pP22582fs+/− CMTs. In response to isoproterenol treatment to mimic β-adrenergic stimulation, force and beating rate were increased in pWT CMTs, but pP22582fs+/− CMTs had markedly blunted responses (Figs. 1G, H). Together these data demonstrate that TTNtv had both basal and stress-induced inotropic and chronotropic deficits that are expected to impair cardiac adaptation to increased mechanical load and β-adrenergic signalling.
We considered whether A-band TTNtv impacted the organization of the sarcomere, where TTN is localized, which could cause or contribute to functional defects. Using antibodies to TTN (9D10 recognizes amino-terminus to TTNtv, Fig. 2A) and alpha-actinin (Figs. 2B,C) we identified well-formed parallel arrays of repeating sarcomeres in myofibrils from WT iPS-CMs. pP22582fs+/− iPS-CMs had fewer myofibrils and abnormal, irregular sarcomeres, a phenotype that was even more pronounced in homozygous cT33520fs−/− iPS-CMs (Figs. 2A, D and S4A). Sarcomere disorganization was observed regardless of whether the iPS-CMs were aligned on micro-patterned lines (Fig. 2B) or three-dimensional CMTs (Fig. 2C). In addition, sarcomere length was shorter in pP22582fs+/− CMs compared to pWT CMs (Fig. 2E), indicating both a quantitative and qualitative defect in sarcomerogenesis. Analogous to these results, LV tissue from the patient with the P22582fs+/− mutation showed disorganized myofibrils compared to control tissue (Figs. 2G, H and S4B).
Fig. 2. Sarcomere abnormalities in TTNtv iPS-CMs.
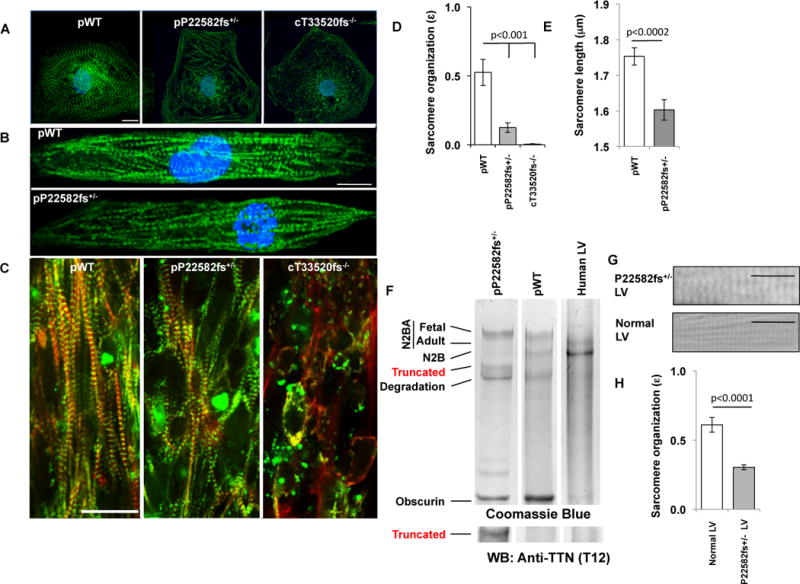
(A) pWT, pP22582fs+/− and cT33520fs−/− iPS-CMs stained with TTN specific antibody (9D10; green) and nuclei (DAPI; blue). (40X; Scale bar=20 μm). (B) pWT and pP22582fs+/− iPS-CMs were patterned on 25 μm × 25 μm grids, stained for z-discs (α-actinin A; green) and nuclei (DAPI; blue). (40X; Scale bar=20 μm). (C) pWT, pP22582fs+/−, and cT33520fs−/− CMTs stained for α-actinin A (green) and F-actin (red) (40X; Scale bar, 20 μm). (D) Sarcomere organization (ɛ) quantified by 2-dimensional Fourier Transform (FFT) analysis of pWT, pP22582fs+/− and cT33520fs−/− iPS-CMs (N>5 per genotype). (E) Sarcomere length (μm) measured by intensity profiles of α-actinin in pWT and pP22582fs+/− iPS-CMs (N>22). (F) Protein electropherograms of lysates from pP22582fs+/− and pWT iPS-CMs and human LV stained with Coomassie Blue (For additional blots, see fig S6). Sizes of TTN isoforms and fragments are: N2BA fetal, ~3700 kDa; N2BA adult, ~3300 kDa; N2B, ~3000kDa; TTNtv, ~2500 kDa; degraded TTN, ~1800–2200 kDa. Obscurin size is ~700 kDa. Western blots (WB) probed with amino-terminus TTN antibody (T12, fig. S6). TTNtv protein (~2500 kDa) was detected in pP22582fs+/− iPS-CMs. (G) Representative micrographs of H&E stained tissue from the left ventricles (LV) of control and P22582fs+/− patients (arrow highlights region of disorganized sarcomere; Scale bar=20 μm). (H) Sarcomere organization (ɛ) quantified by FFT analysis of LV tissue from control and P22582fs+/− patients (n>15 per genotype). Significance assessed by Student’s t test (D, E and H)); data are means +/− SEM (D, E and H).
To determine if sarcomere deficits reflected insufficient TTN levels, we performed RNAseq (14) and protein analyses. cWT and cN22577fs+/− iPS-CMs showed comparable levels of TTN transcripts (fragments per Kb of transcript per million mapped reads; FPKM) and similar patterns of TTN splicing (figs. S3B, C). RNAseq (table S1D) and Sanger sequencing of RT-PCR products (fig. S5) demonstrated equal amounts of mutant and WT transcripts. Consistent with RNAseq data, protein gels of iPS-CM extracts showed expression of the larger fetal TTN isoforms, which include more I-band exons than the adult TTN isoforms (Fig. 2F). We next sought to identify truncated TTN protein (Figs. 2F, S6A–I) in protein lysates from iPS-CMs with an A-band (pP22582fs+/−, pA22353fs+/−) or I-band (pS6394fs+/− (benign variant)) TTNtv. Both iPS-CMs and adult LV contained N2BA (~3300-3700kDa) and N2B TTN (~3000kDa) isoforms; however iPS-CM lysates also contained the larger fetal N2BA isoforms (~3700kDa). In addition, a smaller fragment (~2500kDa) was present in only pP22582fs+/−iPS-CM extracts, but not in pWT (Fig. 2F), pA22353fs+/−, or pS6394fs+/− iPS-CMs (figs. S6B–C). This smaller fragment reacted with TTN T12 antibody, and based on size and immunoreactivity, is likely a stable truncated TTN protein. Given both the detection of mutant TTNtv protein and the paucity of sarcomeres in heterozygous pP22582fs+/− and homozygous cT33520fs−/− iPS-CMs (Figs. 2A, C), we deduced that TTNtv protein, even if stable within CMs, is unable to promote sarcomerogenesis.
RNAseq analyses of cWT, cN22577fs+/−, and cN22577fs−/− iPS-CMs also indicated that TTNtv impacted CM signalling and RNA expression. The significantly altered transcripts in iPS-CMs with TTNtv (Fig. 3A and table S1C) inferred increased activity of three critical upstream microRNA regulators: miR-124 (15), miR-16 (16) and miR-1 (17). Consistent with this prediction, cN22577fs+/− and cN22577fs−/− iPS-CMs had diminished expression of miR-124 targets (15) including lower MYH7: MYH6 transcript and protein ratios (Figs 3B, C and S6J, K) and reduced atrial (NPPA) and brain (NPPB) natriuretic peptide transcript levels compared to WTiPS-CMs (Figs. 3B, C).
Fig. 3. TTN regulates iPS-CM signalling and RNA expression.
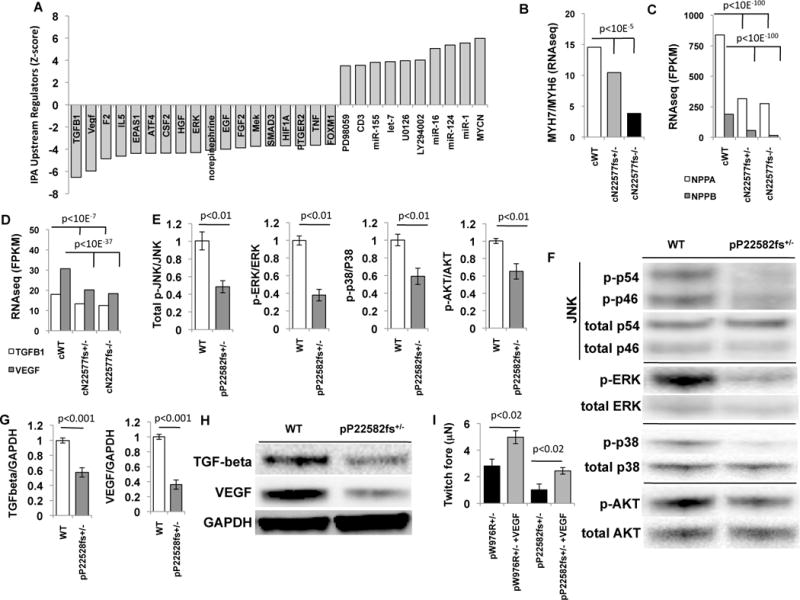
(A)Upstream transcriptional regulators were identified by Ingenuity pathway analysis of differentially regulated genes (normalized ratio >1.2 and <0.8 and p<0.01; table S1C) using RNAseq from cWT, cN22577fs+/−, and cN22577fs−/− iPS-CMs. Data is plotted as Z-score of enrichment (Z-scores cut-off ≥3.5 and≤3.5). (B–D) Comparison of cWT, cN22577fs+/−, and cN22577fs−/−iPS-CMs normalized expression (FPKM) of (B) β(MYH7) and α(MYH6) myosin heavy chain ratios; (C) atrial (NPPA) and brain natriuretic (NPPB) peptides; (D) TGF-β1 and VEGF-A in cWT, cN22577fs+/−, and cN22577fs−/− iPS-CMs. (E) Densitometry of western blots (N>4) of pWT and pP22582fs+/− lysates, normalized for protein loading, and probed with antibodies to phosphorylated JNK (p46, p54), ERK, p38, and AKT. (F) Representative western blots of pWT and pP22582fs+/− iPS-CMs lysates probed for (p)-JNK(T183/Y185), p-ERK(T202/Y204), p-p38(T180/Y182) and p-AKT(T308) and total JNK, ERK, p38 and AKT. (G) Densitometry of western blots (N>4) normalized to protein loading of TGF-β1-3 and VEGF. (H) Representative lanes from western blots from pWT and pP22582fs+/− iPS-CMs probed for TGF-β1-3, VEGF, and GAPDH. (I) Mean twitch force (μN) generated by pP22582fs+/− iPS-CMTs pre-treated with 50ng/ml VEGF (N>4 CMTs). Significance assessed by Bayesian p-values (B-D) or Student’s t test (E, G and I); data are means +/− SEM (E, G and I).
Pathway analysis of RNAseq data on cN22577fs+/− and cN22577fs−/− compared to WT iPS-CMs (fig. S3E) also implied diminished activation of factors regulating growth (TGFβ1, VEGF, HGF, EGF and FGF2), responses to hypoxia (HIF1A and EPAS1 or HIF2A), and MAP kinases (MEK and ERK). Quantification of transcripts, proteins, and phosphorylation levels confirmed that TTNtv iPS-CMs exhibited significant attenuation (all p<0.01) in the levels or phosphorylation of TGFβ, VEGF, MAPKs and AKT (Fig. 3D–H), but not HGF, EGF or FGF2 (fig. S3E). To determine if these signalling deficits contributed to force deficits, we pre-treated pP22582fs+/− and pW976R+/− iPS-CMs with VEGF (50ng/ml) or TGF-beta (0.5ng/ml) for four days prior to studying CMTs. In contrast to the failed augmentation in response to mechanical load (Fig. 1F) or β-adrenergic stimulation (Fig. 1G), VEGF supplementation but not TGF-beta (fig. S3F) improved force production in pP22582fs+/− and pW976R+/− CMTs (Fig. 3I).
We conclude that the use of patient-derived or gene-editing technologies to produce iPS-CMs, coupled with engineered biomimetic culture systems provides robust functional genomic insights. Our studies of iPS-CM with different TTN mutations revealed that some missense variants like TTNtv are pathogenic, while comparisons of contractile function in patient-derived and isogenic CMTs implied a role for genetic modifiers in clinical manifestations of TTNtv. We found that both I- and A-band TTNtv can cause substantial contractile deficits, but that alternative exon splicing (figs. S3A, B) mitigates the pathogenicity of I-band TTNtv. Additional factors, including that iPS-CMs do not fully recapitulate the cell biology of adult cardiomyocytes and CMT tissues lack in vivo compensatory responses, may also account for different degrees of contractile deficits in iPS-CMTs and human hearts with TTNtv. Surprisingly some TTNtv produced stable truncated protein but this mutant peptide failed to assemble with other contractile proteins into well-organized functional sarcomeres. The resultant sarcomere insufficiency (fig. S7) caused both profound baseline contractile deficits and attenuated signalling that limited cardiomyocyte reserve in response to mechanical and adrenergic stress, parameters that are critical to DCM pathogenesis. Remarkably, the consequences of TTNtv are strikingly different from truncating mutations in another sarcomere protein, myosin-binding protein-C, which causes LV hypertrophy and enhanced contractile force (18). Our findings also suggest potential therapeutic targets for TTNtv, including strategies to enhance TTN gene expression, diminish miRNAs that inhibit sarcomerogenesis (15, 19) or that stimulate cardiomyocyte signals that improve function (20).
Supplementary Material
Acknowledgments
For technical assistance, we thank Marion von Frieling-Salewsky for titin gels and Innolign Biomedical for CMTs. C.S.C. is the scientific founder for Innolign Biological, which is developing CMTs for commercial applications. This work was supported in part by grants from the LaDue Fellowship (J.T.H., J.H.), the American Heart Association (A.C.), the Sarnoff Foundation (N.N., C.C.S.), the Leducq Foundation (S.A.C., N.H., S.S., J.G.S., C.E.S.), the German Research Foundation (W.A.L. (SFB 1002 TPB3)), the National Institutes of Health (L.Y. & G.C. (HG005550), C.S.C (EB017103 and HL115553), C.C.B. (HL007374), J.T.H. (HL125807), C.E.S., and J.G.S.), the NIHR CV BRU of Royal Brompton and Harefield NHS Foundation Trust (S.A.C.), the RESBIO Technology Resource for Polymeric Biomaterials (C.S.C.) and Howard Hughes Medical Institute (C.E.S., A.H.).
Footnotes
Supplemental content:
Materials and Methods
Captions for Figures S1 to S7
Captions for Table S1
Captions for Movies S1 to S6
Supplemental References (#21–34)
References and Notes
- 1.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013 Sep;10:531. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 2.Herman DS, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012 Feb 16;366:619. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts AM, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015 Jan 14;7:270ra6. doi: 10.1126/scitranslmed.3010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loh YH, et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010 Jul 2;7:15. doi: 10.1016/j.stem.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mali P, et al. RNA-Guided Human Genome Engineering via Cas9. Science. 2013 Jan 3; doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boudou T, et al. A microfabricated platform to measure and manipulate the mechanics of engineered cardiac microtissues. Tissue Eng Part A. 2012 May;18:910. doi: 10.1089/ten.tea.2011.0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lian X, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat Protoc. 2012 Dec 20;8:162. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tohyama S, et al. Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2013 Jan 3;12:127. doi: 10.1016/j.stem.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 9.Gerull B, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002 Feb;30:201. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 10.Tan JL, et al. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci U S A. 2003 Feb 18;100:1484. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thavandiran N, et al. Design and formulation of functional pluripotent stem cell-derived cardiac microtissues. Proc Natl Acad Sci U S A. 2013 Dec 3;110:E4698. doi: 10.1073/pnas.1311120110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang D, et al. Tissue-engineered cardiac patch for advanced functional maturation of human ESC-derived cardiomyocytes. Biomaterials. 2013 Jul;34:5813. doi: 10.1016/j.biomaterials.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2008 Mar 1;77:637. doi: 10.1016/j.cardiores.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 14.Christodoulou DC, Gorham JM, Herman DS, Seidman JG. Construction of normalized RNA-seq libraries for next-generation sequencing using the crab duplex-specific nuclease. Curr Protoc Mol Biol. 2011 Apr;:12. doi: 10.1002/0471142727.mb0412s94. Chapter 4, Unit4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai B, et al. microRNA-124 regulates cardiomyocyte differentiation of bone marrow-derived mesenchymal stem cells via targeting STAT3 signaling. Stem Cells. 2012 Aug;30:1746. doi: 10.1002/stem.1154. [DOI] [PubMed] [Google Scholar]
- 16.Liu JL, et al. MicroRNA 16 enhances differentiation of human bone marrow mesenchymal stem cells in a cardiac niche toward myogenic phenotypes in vitro. Life Sci. 2012 Jun 27;90:1020. doi: 10.1016/j.lfs.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 17.Ikeda S, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009 Apr;29:2193. doi: 10.1128/MCB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watkins H, et al. Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat Genet. 1995 Dec;11:434. doi: 10.1038/ng1295-434. [DOI] [PubMed] [Google Scholar]
- 19.van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012 Nov;11:860. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zentilin L, et al. Cardiomyocyte VEGFR-1 activation by VEGF-B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB J. 2010 May;24:1467. doi: 10.1096/fj.09-143180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.