

Abstract
Background
The cinereous vulture, Aegypius monachus, is the largest bird of prey and plays a key role in the ecosystem by removing carcasses, thus preventing the spread of diseases. Its feeding habits force it to cope with constant exposure to pathogens, making this species an interesting target for discovering functionally selected genetic variants. Furthermore, the presence of two independently evolved vulture groups, Old World and New World vultures, provides a natural experiment in which to investigate convergent evolution due to obligate scavenging.
Results
We sequenced the genome of a cinereous vulture, and mapped it to the bald eagle reference genome, a close relative with a divergence time of 18 million years. By comparing the cinereous vulture to other avian genomes, we find positively selected genetic variations in this species associated with respiration, likely linked to their ability of immune defense responses and gastric acid secretion, consistent with their ability to digest carcasses. Comparisons between the Old World and New World vulture groups suggest convergent gene evolution. We assemble the cinereous vulture blood transcriptome from a second individual, and annotate genes. Finally, we infer the demographic history of the cinereous vulture which shows marked fluctuations in effective population size during the late Pleistocene.
Conclusions
We present the first genome and transcriptome analyses of the cinereous vulture compared to other avian genomes and transcriptomes, revealing genetic signatures of dietary and environmental adaptations accompanied by possible convergent evolution between the Old World and New World vultures.
Electronic supplementary material
The online version of this article (doi:10.1186/s13059-015-0780-4) contains supplementary material, which is available to authorized users.
Keywords: Cinereous vulture, Old world vulture, New world vulture, Transcriptome, Genome, Next-generation sequencing
Background
Vultures play an important role in the ecosystem by consuming animal carcasses, which helps prevent the spread of disease [1]. The term vulture refers to two independently evolved groups of birds of prey, namely the Old World (16 species) and New World (7 species) vultures. While the two vulture groups are members of different avian families (Accipitridae and Cathartidae), they are phenotypically similar and are both obligate scavengers. The cinereous vulture, also known as the monk vulture, or the Eurasian black vulture, is a member of the family Accipitridae [2], and is a carrion bird [3]. The cinereous vulture (Aegypius monachus), the largest bird of prey [4], is distributed throughout Eurasia and is an iconic bird in the Far East. Its population is estimated to number 7,200–10,000 pairs globally, with 5,500–8,000 pairs residing in Asia. Over the past two centuries, its numbers have declined across most of its range leading to this species being classified as ‘near threatened’ on the International Union for Conservation of Nature (IUCN) Red List [5]. Recently, several protection and conservation efforts for this species have been developed at a national [6] and international level [7, 8].
Vultures are suspected to have strong immune defense systems compared to other vertebrates due to their scavenging lifestyle [9], suggesting that they have evolved mechanisms to prevent infection by the microbes found in their diet. Despite the potential interest in the immune system of scavenger birds, little is known about the genetic variations and molecular mechanisms involved in the regulation of immune processes in vultures.
The Avian Phylogenomics Project recently sequenced and analyzed 48 avian species to provide insights into avian evolution [10]. Among the species included were two members of the Accipitridae family, the bald eagle (Haliaeetus leucocephalus) and white-tailed eagle (H. albicilla). Additionally, the project included the turkey vulture (Cathartes aura), which is a member of the Cathartidae (New World vulture). However, no whole genome or transcriptome analyses have been reported for any of the Old World vultures. In order to identify the genetic adaptations and possible convergent evolution linked to obligate scavenging, we provide a whole genome and transcriptome analysis of the cinereous vulture which we compared to other avian genomes; including other birds of prey belonging to the family Accipitridae, as well as New World vultures from the family Cathartidae.
Results and discussion
Whole genome sequence
The genomic DNA from a cinereous vulture was sequenced using the Illumina HiSeq2000. We obtained 595 million paired reads (total length, 119 Gb) with a read length of 100 bp and an insert size of 346 bp (Additional file 1: Table S1). We performed a K-mer analysis (K = 17) using the vulture whole genome sequences (Additional file 2: Fig. S1), and we estimate the genome to be approximately 1.13 Gb (Additional file 1: Table S2), which is consistent with the estimates for other birds (1.0–1.2 Gb) [11–15]. As there is no cinereous vulture reference genome, we aligned the DNA reads to the bald eagle reference, which (as a member of the Accipitridae) is one of the closest species to the cinereous vulture and of higher coverage than the white-tailed eagle (another Accipitridae species) [10]. The mapping rate was relatively high (89.61 %), and the mapped reads covered 91.45 % and 89.84 % of the reference genome and protein coding region at 15× or greater depth, respectively (Additional file 1: Table S1). Compared to the bald eagle reference, there were approximately 34 million single nucleotide variations (SNVs) and 2.0 million small insertions or deletions (indels) in the cinereous vulture. The SNVs were composed of 32,093,779 (95.6 %) homozygous and 1,482,652 (4.4 %) heterozygous SNVs (Additional file 1: Table S3).
Comparative evolutionary analyses
A set of close but distinct species with clear trait differences can let us predict variants that may contain geno-phenotype association information if their whole genomes are compared [16, 17]. For our comparative evolutionary analyses, we generated a DNA consensus sequence of the cinereous vulture by substituting the vulture SNVs detected from the whole genome sequencing data to the bald eagle reference genome. We then identified orthologous genes between the cinereous vulture consensus sequence and the 3,710 orthologous gene clusters from 17 avian genomes (adelie penguin, american crow, bald eagle, chicken, chimney swift, common cuckoo, crested ibis, downy woodpecker, hoatzin, killdeer, little egret, mallard, ostrich, peregrine falcon, rock pigeon, turkey vulture, and white-tailed eagle) previously reported [18], by matching and adding the cinereous vulture genes to orthologous clusters of the bald eagle genes. A total of 48,081 four-fold degenerate sites of the orthologous gene families were used to calculate the divergence time between the cinereous vulture and the other avian species. The divergence time between the cinereous vulture and bald eagle was estimated to be 18 million years ago (MYA) (Additional file 2: Fig. S2). This split is older than the divergence time between the bald eagle and white-tailed eagle (6–7 million years), and much younger than that of the Old World vultures and New World vultures (about 60 million years) [19]; confirming that the cinereous vulture is more closely related to the eagles.
The identification of positively selected genes (PSGs) is a key genomics tool to understand the evolution of species, especially under distinctive environmental pressures. We identified genes evolving under positive selection by performing tests for deviations in the dN/dS ratio (non-synonymous substitutions per non-synonymous site to synonymous substitutions per synonymous site, ω) and likelihood ratio tests (LRTs) (P ≤0.05) [20, 21]. We successfully discovered a suite of positively selected genes in the Accipitrimorphae clade (Accipitriformes and Cathartiformes orders: bald eagle, cinereous vulture, white-tailed eagle, and turkey vulture). A total of seven and 136 genes were identified as PSGs using, respectively, a branch-site model with a 10 % of false discovery rate (FDR) and a branch model (1 < dN/dS ≤5 was interpreted as positive selection) (Additional file 1: Tables S4 and S5). To investigate the functional enrichments of these PSGs, we used the DAVID functional annotation tool (Additional file 1: Table S6) [22].
In order to investigate the biologically meaningful PSGs of the Accipitrimorphae, we selected the PSGs associated with the digestive system in the KEGG database (Table 1) [23], since it is known that the Accipitrimorphae have extremely acidic stomachs [24, 25]. Among the PSGs of the Accipitriformes, Somatostatin (SST) (ωaverage: 0.04840, ωAccipitriformes: 1.4515) was involved in the digestive system pathway. SST is a strong inhibitor of gastrin release and plays a role in regulating gastric acid secretion [26]. We suggest that SST has been functionally selected in the Accipitrimorphae clade.
Table 1.
Positively selected genes involved in immune defense and digestive system pathways
| KEGG category | KEGG pathways | Accipitrimorphae | Cinereous vulture | Turkey vulture | |||
|---|---|---|---|---|---|---|---|
| Branch model | Branch-site model | Branch model | Branch-site model | Branch model | Branch-site model | ||
| Immune system | Hematopoietic cell lineage | DNTT | GP9 | ||||
| Complement and coagulation cascades | C1QB | ||||||
| Platelet activation | RAP1B | GP9 | |||||
| Toll-like receptor signaling pathway | TAB2 | LY96 | |||||
| NOD-like receptor signaling pathway | TAB2 | ||||||
| Cytosolic DNA-sensing pathway | POLR1D | ||||||
| Natural killer cell mediated cytotoxicity | FAS | ||||||
| Fc gamma R-mediated phagocytosis | ARPC2 | ||||||
| Leukocyte transendothelial migration | RAP1B | ||||||
| Chemokine signaling pathway | RAP1B | ||||||
| Digestive system | Salivary secretion | ADRA1B | |||||
| Gastric acid secretion | SST | ||||||
| Pancreatic secretion | RAP1B | ||||||
| Vitamin digestion and absorption | BTD | RBP2 | |||||
| Mineral absorption | CYBRD1 | SLC31A1, CYBRD1 | SLC31A1 | ||||
The cinereous vulture belongs to the family of Accipitridae together with the bald eagle and white-tailed eagle, while the turkey vulture (Cathartes aura) belongs to the family of Cathartidae, which is an obligate scavenger like the cinereous vulture [27]. To identify the convergent and unique adaptations to a scavenging lifestyle [28], we compared the PSGs of the cinereous and turkey vultures using other birds of prey as a reference (bald eagle, peregrine falcon, and white-tailed eagle). Six and 167 genes were identified as PSGs in the cinereous vulture and 196 and 85 PSGs in the turkey vulture using a branch-site model and a branch model, respectively (Additional file 1: Tables S7-S10). For the PSGs of the cinereous and turkey vultures, we also performed a functional clustering analysis using the DAVID tool (Additional file 1: Tables S11 and S12).
We detected nine PSGs that were shared in both the cinereous vulture (Accipitridae) and turkey vulture (Cathartidae) (Table 2). Among the nine PSGs, several genes were associated with immune functions. The AHSG gene is involved in endocytosis, which is a key process that allows cells to internalize particulate matters such as micro-organisms and apoptotic cells. The Alpha2-HS glycoprotein (AHSG) promotes endocytosis and possesses opsonic properties, through which a pathogen is marked for ingestion and eliminated by a phagocyte. In addition, the programmed cell death protein six (PDCD6), which encodes a calcium-binding protein required for glucocorticoid-induced cell death, was positively selected in the both vultures. It is known that interaction between PDCD6 and DAPK1 (Death-associated protein kinase 1) can accelerate apoptotic cell death. Viral induction of apoptosis occurs when one or several cells of a living organism are infected with a virus, and many viruses encode proteins that can inhibit apoptosis [29]. We speculate that these positively selected genes may play a role in helping the two vulture species combat pathogens encountered in their diet, complementing the role of gastric secretion discussed previously. Taken together, our genome scale variation analyses have provided interesting new targets for further investigation at the transcriptome, proteome, and even epigenome levels; and will enable future experimental studies to conclusively examine the roles of these genes in the adaptive evolution of these species.
Table 2.
Positively selected genes by branch and branch-site model that were shared in both the cinereous vulture (Accipitridae) and turkey vulture (Cathartidae)
| Gene name | Cinereous vulture | Turkey vulture | ||||||
|---|---|---|---|---|---|---|---|---|
| Branch models | Branch-site models | Branch models | Branch-site models | |||||
| M0: one-ratio | M1: free-ratio | 2ΔL | P value | M0: one-ratio | M1: free-ratio | 2ΔL | P value | |
| ZDBF2 | 0.64881 | 2.2872 | 0.72895 | 1.3267 | ||||
| MIIP | 0.61631 | 2.1433 | 0.62069 | 1.7923 | ||||
| TSPO2 | 0.42486 | 1.704 | 0.45792 | 1.2814 | ||||
| AHSG | 0.51914 | 1.5601 | 0.67974 | 1.8824 | 8.166016 | 4.27E-03 | ||
| CRYZL1 | 0.14953 | 1.4475 | 7.21743 | 7.22E-03 | ||||
| C14orf159 | 0.31294 | 1.2295 | 7.897072 | 4.95E-03 | ||||
| PDCD6 | 0.04673 | 1.1766 | 17.16492 | 3.43E-05 | ||||
| C8orf33 | 0.59386 | 1.1212 | 0.70879 | 1.9528 | ||||
| CYBRD1 | 0.37151 | 1.0177 | 0.61098 | 1.7848 | ||||
M0 denotes dn/ds for all the species in the study; M1 denotes dn/ds for the lineage leading to the target species; Likelihood ratio tests (LRT) were used to detect positive selection
If adaptive evolution is episodic and affects only a few crucial amino acids, the application of the PSG criterion is likely to result in the omission of important genes [30]. To overcome this limitation, we investigated the cinereous vulture and turkey vulture-specific amino acid changes in the immune system pathways (Fig. 1) (Additional file 1: Tables S13 and S14). A total of 17 and 24 genes had amino acid changes specific to the cinereous vulture and the turkey vulture, respectively. Among these genes, three were shared only by the cinereous vulture and turkey vulture (Table 3). The Protein Variation Effect Analyzer (PROVEAN) software predicted that the vulture-specific amino acid substitutions observed in all three genes (PIK3AP1, TBK1, and TNFAIP3) are likely to cause alterations to protein function [31]. Of particular interest is TANK-binding kinase-1 (TBK1), which is essential for regulating the response to viral and microbial agents and promotes the development of a cellular antiviral state by activating interferon regulatory factors [32] (Fig. 2). Two of the three vulture-specific amino acid polymorphisms present in TBK1 were predicted as function altering amino acid changes and are located in the ubiquitin-like domain on the exposed surface of the protein. Namely, the positively charged side chain (His) at residue 327 in the cinereous vulture has been replaced by an uncharged side chain (Gln), while the polar side chain (Thr) at residue 364 in the turkey vulture has been substituted with a hydrophobic side chain (Ala). It is known that TBK1 forms several different complexes with a variety of scaffolding molecules, and that the deletion or mutation of the ubiquitin-like domain in TBK1 impairs kinase activation and substrate phosphorylation in cells [33–35]. Therefore, we speculate that the amino acid changes in this gene may contribute to these vultures’ enhanced immune systems. Additionally, the PIK3AP1 and TNFAIP3 genes, which also contain functional altering amino acid changes, are involved in B-cell development, antigen presentation, auto-inflammation, and NF-kappa B activation, respectively [36, 37].
Fig. 1.
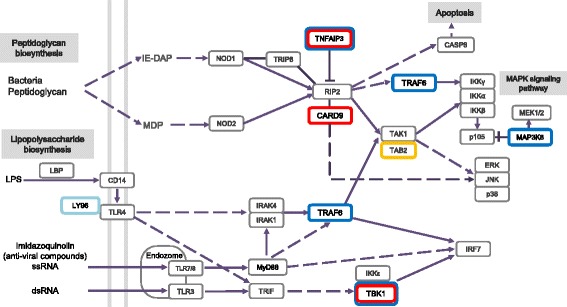
Vulture-specific genetic change in immune system. Genes with vulture-specific amino acid changes (TNFAIP3, CARD9, TRAF6, MAP3K8, and TBK1) are shown in red (cinereous vulture) and blue (turkey vulture) rectangles. Upregulated gene in the cinereous vulture (TAB2) is shown in yellow rectangle. A positively selected gene (LY96) in the turkey vulture is shown in a light blue rectangle. The pathway was drawn according to KEGG pathway (NOD-like receptor and Toll-like receptor signaling pathways). The solid lines indicate direct relationships between enzymes and metabolites. The dashed lines indicate that more than one step is involved in a process
Table 3.
Unique amino acid site changes on sites between two vultures in immune system-related proteins
| Protein | Species | Position | Residue | Other species residue | PROVEAN score (Prediction) |
|---|---|---|---|---|---|
| PIK3AP1 | Cinereous vulture | 1 | A | T | 0.757 |
| Turkey vulture | 68 | L | H | −4.913 (Deleterious) | |
| 208 | F | Y | −1.206 | ||
| 232 | P | S | −1.225 | ||
| 398 | N | K | −1.674 | ||
| TBK1 | Cinereous vulture | 327 | Q | H | −5.692 (Deleterious) |
| 528 | S | P | −1.76 | ||
| Turkey vulture | 374 | A | T | −3.225 (Deleterious) | |
| TNFAIP3 | Cinereous vulture | 446 | A | G | −0.857 |
| 742 | Q | A,P,L,V | −0.044 | ||
| Turkey vulture | 243 | G | E | −5.456 (Deleterious) | |
| 270 | G | E,V | −4.024 (Deleterious) | ||
| 590 | S | P,L | 0.871 | ||
| 636 | T | A | 0.385 | ||
| 643 | S | R | 0.132 | ||
| 677 | M | T | −2.323 |
Fig. 2.
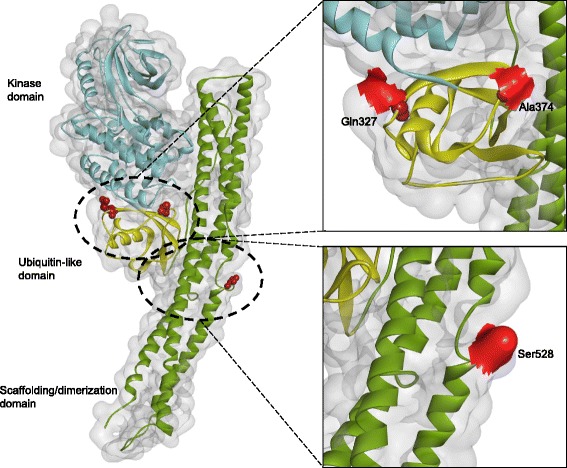
Positions of two vulture-specific amino acid changes in the three-dimensional structure of TBK1. The kinase, ubiquitin-like, and scaffolding/dimerization domains are colored cyan, yellow, and green, respectively. Structural positions of the three amino acids changes in the two vultures are shown in red
We also investigated the Accipitridae (cinereous vulture, bald eagle, and white-tailed eagle) and Cathartidae (turkey vulture)-specific amino acid changes in the digestive system pathways (Additional file 1: Table S15). A total of eight and 10 genes were identified as unique amino acid changes in the Accipitridae and the Cathartidae, respectively. Among these genes, seven were shared by both the Accipitridae and Cathartidae (Additional file 1: Table S16). Of these, two genes (KCNJ16 and SLC26A7) are involved in the gastric acid secretion pathways (Additional file 2: Fig. S3). KCNJ16 is an inward-rectifier potassium channel which regulates fluid and pH balance, and SLC26A7 is known to play an important role in gastric acidification [38]. Among the five amino acid differences in these genes, two were predicted as function-altering using Polyphen2 [39] and PROVEAN (Additional file 1: Table S17). Additionally, two genes (KCNC1 and KCTD18), which are involved in potassium channel activity, were positively selected in the cinereous vulture, although it was not the case in the turkey vulture. It is known that potassium ions are required for gastric acid secretion [40]. Recently, a vulture metagenome study showed that the facial microbial community was significantly more diverse than that of the hindgut, reflecting the breakdown of dietary DNA in the vulture gastrointestinal tract and pointing to extraordinarily harsh chemical conditions [41]. Taken together, we propose that these vulture-specific amino acid changes in the immune system and gastric acid-associated genes are evidence of the vultures’ enhanced defenses against microbial/viral infections from carcasses and are good candidates for future functional studies.
Transcriptome analysis
To extend our genomic inferences, we constructed a cDNA library from total RNA isolated from the blood sample of a second cinereous vulture from the same population. Using an Illumina HiSeq2500, a total of 30,118,314 paired reads (approximately 6.0 Gb in length) were produced, with a read length of 100 bp and an insert size of 280 bp. To reduce the effect of sequencing errors, 51,537,752 (85.6 %, approximately 5.15 Gb in length) reads were selected after filtering out ambiguous and low-quality reads. All filtered short-read transcriptome data were assembled using the Trinity de novo assembler program [42] with an optimized K-mer length of 25. The filtered reads were assembled into 423,702 contigs with a mean length of 3,670 bp (N50 of 6,110 bp), and the contigs were clustered into 352,872 non-redundant unigenes with a mean length of 3,469 bp and an N50 of 6,015 bp (Additional file 1: Table S18).
To validate and annotate the 352,872 assembled unigenes, we searched the GenBank non-redundant (NR) database for homologs of all the assembled unigenes, using the BLASTx program [43] with the criterion E-value ≤ 1.0E-5. A total of 241,347 (68.4 %) out of 352,872 unigenes were homologous to sequences in the NR database. As expected, the BLASTx top-hit species distribution of the vulture unigene was highly enriched in vertebrates, especially among avian species (Additional file 2: Figs. S4 and S5). For a further evaluation of the assembled transcripts, we also examined the expression levels of the assembled vulture unigenes. The number of mapped reads for each annotated unigene was normalized to fragments per kilobase of transcript per million mapped fragments (FPKM). A total of 170,990 unigenes were determined to be expressed (FPKM >0); of those, 73,984 unigenes could be functionally annotated by matching with sequences in the databases. Ultimately, 33,302 functionally annotated and highly expressed unigenes (FPKM ≥1.0) were selected and used in the transcriptome analysis.
A total of 29,774 out of the 33,302 unigenes were functionally clustered into 52 subcategories under three non-mutually exclusive ontologies (cellular components, molecular functions, and biological process) using WEGO [44] (Additional file 2: Fig. S6): 27,632 were assigned to cellular components, 25,572 to molecular functions, and 25,145 to biological processes (Additional file 1: Table S19). Of the cellular components, most unigenes were classified as components of cells (or cell parts), with only a small portion classified as components of virions (or virion parts). Importantly, a considerable number of unigenes were assigned to immune system processes (2,305 unigenes, 7.7 %) and responses to stimulus (5,425 unigenes, 18.2 %) subcategories that could be linked to the vultures’ immune defenses. The numbers are higher than those found in blood lymphocytes transcriptome from the Chinese goose (Anser cygnoides) [45], which are 0.3 % and 2.8 % for the immune system processes and the responses to stimulus, respectively. We also analyzed the annotated unigenes using the KEGG automatic annotations server (KAAS) [46]. We obtained KEGG orthology-based annotations for the vulture unigenes from the human, chicken, turkey, and zebra finch. A total of 327 KEGG pathways were identified in the vulture unigenes. The vulture unigenes were most over-represented in metabolic pathways (649 unigenes), biosynthesis of secondary metabolites (162 unigenes), and pathways in cancer (153 unigenes) (Additional file 1: Table S20), which showed a similar pattern found in a crow lung transcriptome [47] (top three over-represented pathways: metabolic pathways, pathways in cancer, and PI3K-Akt signaling pathway).
The Toll-like receptors (TLRs), together with interleukin-1 receptor type 1 (IL-1R) gene family, recognize pathogen components and trigger a signaling cascade through the recruitment of different combinations of toll/interleukin receptor (TIR)-domain containing adaptor proteins [48]. TLRs are essential in pathogen recognition [49], and a previous study reported that TLR1 in the Griffon vulture (Gyps fulvus), an Old World vulture, has a unique gene structure (with respect to the length of the ectodomain, number and position of leucine-rich repeat [LRRs] motifs, and N-glycosylation sites) relative to orthologous genes from other organisms [9]. These features have possible functional implications and we confirmed that the TLR1 unigene of the cinereous vulture has identical LRR motifs and N-glycosylation sites (Additional file 2: Fig. S7).
Additionally, we compared the gene expression levels of the cinereous vulture to those of blood lymphocytes from a Chinese goose and blood from four Eurasian siskins (Spinus spinus) [50] (Additional file 1: Table S21). We examined the expression levels of immune system associated genes and identified differentially expressed genes that satisfy the log2 fold-change of at least 2.0 and FDR less than or equal to 0.1. A total of four immune system associated genes (TAB2, STAT3, CYLD, and NFYA) were significantly highly expressed in the cinereous vulture, compared to the others (Additional file 1: Table S22). The TAB2 protein is a mediator of TAK1 activation in the interleukin-1 signaling transduction pathway. Expression of TAB2 induces JNK (c-Jun N-terminal kinase) and NF-kappaB activation, which upregulate the expression of many proinflammatory genes [51]. The CYLD protein plays an important role in the regulation of pathways leading to NF-kappaB activation [52]. Also, it is known that STAT3 mediates cellular responses to interleukins and growth factors, and STAT3 mediates the expression of a variety of genes in response to cell stimuli, and thus plays a key role in apoptosis.
Genetic diversity and population history
The demographic history of a species can be reconstructed from deeply sequenced genomes [53]. We first calculated the level of the genomic diversity of cinereous vulture by measuring the rate of heterozygous SNVs found in the genome. The genomic diversity was found to be 0.00132, which is comparable to the diversity observed in the turkey vulture (0.00148) and much higher than that of humans (0.00069) [54] and bald eagle (0.00053). We also inferred the demographic history of the cinereous vulture using the pairwise sequentially Markovian coalescent (PSMC) model [55] (Fig. 3). Between 1,000,000 and 200,000 years ago, the cinereous vulture and the bald eagle showed similar effective population sizes. However, during the late Pleistocene (approximately 110,000 to 12,000 years ago), the effective population size of the cinereous fluctuated markedly, whereas the bald eagle and the turkey vulture were relatively stable. In particular, the cinereous vulture population experienced a strong bottleneck between 110,000 and 70,000 years ago, whereas the bald eagle and the turkey vulture showed relatively stable and increasing population sizes, respectively.
Fig. 3.
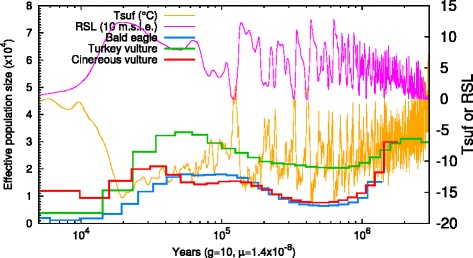
Demographic history of the cinereous vulture. g, generation time (years); μ, mutation rate per site per generation time; Tsuf, atmospheric surface air temperature; RSL, relative sea level; 10 m.s.l.e., 10 m sea level equivalent
Conclusions
Our study provides the first whole genome and de novo transcriptome analyses of the cinereous vulture, along with comparative evolutionary analyses with other avian genomes. The comparative analyses revealed likely signatures of selection in cinereous vulture genes associated with the immune response and gastric acid secretion. Together with previous analysis of vulture metagenomes [41], our results suggest that the acidic gastrointestinal tract of vultures is a strong filter of the microbiota ingested from decaying carcasses, which likely plays a role in enabling the vulture’s scavenging lifestyle. A comparison to the turkey vulture revealed a number of possible instances of convergent evolution between Old and New World vultures, in line with the recent observation of convergent evolution (adaptive convergence) based on comparative genomics data [10, 19]. At the same time, it is important to note that convergence might be non-adaptive [56], and simply driven co-evolutionary interactions between sites, with the process at one site changing other coupled sites [57]. Therefore, with additional genomic data, the candidate genes identified in this study can be further investigated to test the possible roles of adaptive versus non-adaptive convergence in this system. While functional studies of candidate genes will be needed to confirm the role of individual genes, this first analysis of the cinereous vulture genome and transcriptome provides insights into the genetic adaptations that have allowed this lineage to successfully occupy such a challenging niche.
Methods
Sample preparation and sequencing
Blood samples from two cinereous vultures were acquired from the Korean Association for Bird Protection under the Cultural Heritage Administration (Korea) Permit. DNA was extracted using a QIAamp DNA Mini Kit (Qiagen, CA, USA). The amount of DNA was quantified by fluorometry using a Qubit dsDNA Assay Kit (Invitrogen, CA, USA) and an Infinite F200 Pro NanoQuant (TECAN, Männedorf, Germany). High-molecular weight genomic DNA was sheared to approximately 500 bp using a Covaris S2 Ultrasonicator, and then used in the preparation of whole-genome shotgun libraries according to Illumina’s library preparation protocols (Illumina, CA, USA). The efficacy of each step of library construction was ascertained using a 2100 Bioanalyzer (Agilent Technologies, CA, USA). A final dilution of the library was then sequenced on a HiSeq 2000 (Illumina), using the TruSeq Paired-End Cluster Kit v3 (Illumina) and a TruSeq SBS HS Kit v3 (Illumina) for 200 cycles.
Total RNA was extracted from the blood of an additional vulture using the Trizol LS Reagent (Ambion, TX, USA). RNA quality was assessed by analysis of rRNA band integrity on an Agilent RNA 6000 LabChip kit (Agilent Technologies). Prior to cDNA library construction, 2 μg of total RNA was treated with DNase I, and magnetic beads with Oligo (dT) were used to enrich poly (A) mRNA. Next, the purified mRNAs were disrupted into short fragments, and double-stranded cDNAs were immediately synthesized. The cDNAs were subjected to end-repair and poly (A) addition, and then connected with sequencing adapters using the TruSeq RNA Sample Prep Kit (Illumina). Suitable fragments, automatically purified using a BluePippin 2 % agarose gel cassette (Sage Science, MA, USA), were selected as templates for PCR amplification. The final library sizes and qualities were evaluated electrophoretically using an Agilent High Sensitivity DNA kit (Agilent Technologies); the fragment size range was 350–450 bp. Subsequently, the library was sequenced using a HiSeq 2500 sequencer (Illumina) in rapid run mode. Cluster generation was performed, followed by 2 × 100 cycle sequencing reads separated by a paired-end turnaround. Image analysis was performed using the HiSeq control software version 1.8.4.
Vulture DNA read alignment to bald eagle reference genome
A DNA read was filtered out when average quality was less than Q20 and/or ambiguous nucleotide ratio was higher than 10 %. The clean reads were aligned to the bald eagle genome sequence using BWA ver. 0.6.2 [58] with default options. The rmdup command of SAMtools 0.1.18 [59] was used to remove PCR duplicates from the reads. The raw SNVs and indels were called using the mpileup command of SAMtools ver. 0.1.19 with the -Q 30 option followed by the view command with the -cg option. From the raw SNVs, the consensus genome sequences of the cinereous vulture were generated using vcf2fq with -d 8 and -l 2 options. The final SNVs were defined by comparing the consensus sequence to the reference sequence. The raw indels were filtered using varFilter of vcfutils.pl with -d 8 to produce the final indel dataset. To remove gene sequences possibly derived from paralog genes in the vulture or low sequencing depth regions, we filtered out coding sequences (CDSs) using the following criteria; (1) CDSs that contain ambiguous nucleotides ‘N’; (2) CDSs that contain premature stop codons. Among the 16,526 protein coding genes, 8,546 genes were obtained and used in the further analyses. For the K-mer analysis, JELLYFISH [60] was used with K = 17. Genome size was estimated from the total number of K-mers divided by the peak depth of K-mer graph.
Transcriptome analysis and functional gene annotation
The raw sequencing reads contained low-quality reads and adaptors, which were removed as follows: First, reads with >10 % ambiguous bases (represented by the letter ‘N’) or with Q20 < 20 % were removed. If the average quality of the reads was under Q20, reads were also removed. The bases under Q20 were removed at the both end of a read.
De novo transcriptome assembly was performed on the remaining high-quality reads using the short-read assembly software Trinity [42]. These resulting contigs were further processed with the sequence clustering software TGICL [61] to produce unigenes. Coding sequence (CDS) were predicted using TransDecoder [62], and functions of unigenes were predicted by DNA and protein sequence homology searches using BLAST and InterProScan v5 [63]. The NCBI Blast 2.2.28+ and NR database were used to search for protein sequence homology. InterProScan v5, consisting of five protein databases (Pfam, Panther, SMART, SuperFamily, and Gene3D) was used to predict the function of unigenes. The FPKM method [64] was used to calculate unigene expression and gene expression levels were analyzed using the RSEM software [65]. The WEGO software was then used to perform GO functional classification of all unigenes and view the distribution of gene functions. Pathway assignments were performed according to the KEGG pathway database, using KAAS.
To compare the gene expression level of the cinereous vulture, Chinese goose, and Eurasian siskins, the RNA reads from the cinereous vulture, Chinese goose, and Eurasian siskins were aligned to the bald eagle, duck, and American crow reference genome using TopHat2 [66], respectively. The number of reads that were mapped to orthologous gene regions were counted using HTSeq-0.6.1 [67], and then normalized by gene length and transcripts per million (TPM) [68]. The genes having less than 1.0 TPM were filtered out. The TPM values were normalized by Trimmed Mean of M-values (TMM) [69] using edgeR [70]. The significance of differential expression between the vulture and siskins was calculated by the exact negative binomial test, and corrected for multiple testing by the Benjamini-Hochberg false discovery rate.
Molecular evolutionary analyses
To estimate divergence times, we used four-fold degenerate sites of avian one-to-one orthologous families using RelTime-CC version 2.0 [71] with the phylogenetic tree topology of published previously [19]. The evolutionary rates of the each branch were calculated by the resulting tree. The program used a maximum likelihood statistical method and time reversible model. The date of the node between duck and chicken was constrained to 65.8 MYA [19].
PRANK [72] was used to construct multiple sequence alignment among ortholog genes, and the CODEML program in PAML 4.5 was used to estimate the rates of synonymous (dS) and non-synonymous substitutions (dN) and the dN/dS ratio (ω) [20]. The one-ratio model, which allows only a single dN/dS ratio for all branches, was used to estimate the general selective pressure acting among all species. A free-ratios model was used to analyze the dN/dS ratio along each branch. To further examine potential positive selection, the branch-site test of positive selection was conducted [21]. Statistical significance was assessed using likelihood ratio tests (LRTs) with a conservative 10 % false discovery rate (FDR) criterion [73]. The Accipitrimorphae were used as the foreground branch and the other species as background branch. To identify the convergent evolution of the two vultures, the each vulture as foreground was used and the rest of the bird of prey as the background were used.
Function-altering amino acid changes were predicted using PolyPhen2 [39] and PROVEAN v1.1 [31] using the default cutoff values. A 3D modeling structure of the TBK1 was predicted by SWISS-MODEL [74–76].
Demographic history analysis
To infer the population history of the cinereous vulture, we used the PSMC program using scaffold length ≥50 kb sequence. We performed 100 rounds of bootstrapping and used 1.4 × 10−8 substitutions per site per generation [10]. We used 10 years as a generation time as previously reported [77].
Availability of supporting data
Whole-genome sequence data and RNA sequence data were deposited in the SRA database at NCBI with BioSample accession numbers SAMN02728242 and SAMN02739871, respectively. The SRA of whole genome sequencing can be accessed via reference numbers SRX518112, and the RNA sequence can be accessed as SRX529363. The data can also be accessed through BioProject accession number PRJNA244786 for the whole-genome sequence and PRJNA245779 for the RNA sequence data. The Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GDQP00000000. The version described in this paper is the first version, GDQP01000000.
Acknowledgments
This work was supported by National Research Foundation of Korea (2008–2004707 and 2013M3A9A5047052), and the 2014 Research fund (1.140077.01) of UNIST (Ulsan National Institute of Science & Technology). SJO was supported by Russian Ministry of Science Mega-grant no. 11.G34.31.0068. The authors thank the Ministry of Science, ICT, and Future Planning. The authors express special thanks to Chungcheongbuk-do, Osong Medical Innovation Foundation, and Chungbuk Industry University Cooperation Institute for providing laboratory space. The authors thank Maryana Bhak for editing.
Additional files
Sequencing and analysis statistics of the cinereous vulture’s WGS relative to the bald eagle genome. Table S2 17-mer statistics. Table S3 Summary of SNVs and small indel in the cinereous vulture. Table S4 PSGs list of the Accipitrimorphae using a branch-site model. Table S5 PSGs list of the Accipitrimorphae using a branch model. Table S6 Functional annotation chart of PSGs of the Accipitrimorphae. Table S7 PSGs list of the cinereous vulture using a branch-site model. Table S8 PSGs list of the cinereous vulture using a branch model. Table S9 PSGs list of the turkey vulture using a branch-site model. Table S10 PSGs list of the turkey vulture using a branch model. Table S11 Functional annotation chart of PSGs of the cinereous vulture. Table S12 Functional annotation chart of PSGs of the turkey vulture. Table S13 Unique amino acid changes of the turkey vulture. Table S14 Unique amino acid changes of the cinereous vulture Table S15 Unique amino acid changes of the Accipitridae. Table S16 Unique amino acid changes on sites between Accipitridae and Cathartidae of the digestive system-related proteins. Table S17 Unique amino acid changes on sites between Accipitridae and Cathartidae of the gastric acid secretion-related proteins. Table S18 Statistics regarding whole-transcriptome sequences and unigene construction. Table S19 GO analysis for the blood transcriptome of the cinereous vulture. Table S20 KEGG pathway analysis for the blood transcriptome of the cinereous vulture. Table S21 Gene expression in the cinereous vulture compared to the other avian species. Table S22 Immune related genes expression in the cinereous vulture compared to the other avian species. (XLSX 635 kb)
Estimation of genome size using 17-mers. Fig. S2 The divergence time of avian species. Fig. S3 Specific amino acid changes in the gastric acid secretion associated genes. Fig. S4 Species distribution of BLASTx top hits of cinereous vulture transcripts. Fig. S5 NR protein database properties for the assembled unigenes. Fig. S6 Gene Ontology classifications of cinereous vulture unigenes. Fig. S7 Amino-acid sequence comparison of toll-like receptor 1 of the Griffon and cinereous vultures. (DOCX 2228 kb)
Footnotes
Oksung Chung, Seondeok Jin and Yun Sung Cho contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SJ, JB, and WKP conceived and designed the experiments. OC, HK, SJho, HMK, JHJ, and JK analyzed the data. SJ, JL, JB, and WKP performed the study design, subject recruitment, and sample preparation. OC, YSC, SJho, JHJ, and HMK carried out data interpretation. OC, YSC, HMK, AC, JHJ, HJL, JE, JAW, KH, JL, SJO, AM, and JB write, edited, and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Oksung Chung, Email: okokookk219@gmail.com.
Seondeok Jin, Email: withbirds@daum.net.
Yun Sung Cho, Email: joys0406@gmail.com.
Jeongheui Lim, Email: jeongheuilim@gmail.com.
Hyunho Kim, Email: pgi.kimhh@gmail.com.
Sungwoong Jho, Email: sungwoong.jho@gmail.com.
Hak-Min Kim, Email: howmany2@gmail.com.
JeHoon Jun, Email: junjh0701@gmail.com.
HyeJin Lee, Email: leehj08111@gmail.com.
Alvin Chon, Email: chon.alvin@gmail.com.
Junsu Ko, Email: junsu.ko@therabio.kr.
Jeremy Edwards, Email: jeremy.scott.edwards@gmail.com.
Jessica A. Weber, Email: weberj@unm.edu
Kyudong Han, Email: kyudong.han@gmail.com.
Stephen J. O’Brien, Email: lgdchief@gmail.com
Andrea Manica, Email: am315@cam.ac.uk.
Jong Bhak, Email: jongbhak@genomics.org.
Woon Kee Paek, Email: paekwk@naver.com.
References
- 1.Parra J, Tellería JL. The increase in the Spanish population of Griffon Vulture Gyps fulvus during 1989–1999: effects of food and nest site availability. Bird Conserv Int. 2004;14:33–41. doi: 10.1017/S0959270904000048. [DOI] [Google Scholar]
- 2.del Hoyo J, Elliott A, Sargatal J. Handbook of the birds of the world. Barcelona: Lynx Edicions; 1994. [Google Scholar]
- 3.Ruxton GD, Houston DC. Obligate vertebrate scavengers must be large soaring fliers. J Theor Biol. 2004;228:431–6. doi: 10.1016/j.jtbi.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Ferguson-Lees J, Christie DA. Raptors of the world. Boston, MA: Houghton Mifflin Harcourt; 2001. [Google Scholar]
- 5.BirdLife International. Aegypius monachus. IUCN Red List of Threatened Species. Version 2014.3. Available at: www.iucnredlist.org (accessed 25 May 2015).
- 6.NIBR (National Institute of Biological Resources) Korean red list of threatened species (Mammals, Birds, Reptiles, Amphibians, Fishes and Vascular Plants. Seoul: Ministry of Environment of Korea; 2012. [Google Scholar]
- 7.de la Puente J, Moreno-Opo R, del Moral JC. El buitre negro en España. Censo Nacional 2006. Madrid: SEO/BirdLife; 2007. [Google Scholar]
- 8.Eliotout B, Lecuyer P, Duriez O. Premiers résultats sur la biologie de reproduction du Vautour Moine Aegypius monachus en France. Alauda. 2007;75:253–64. [Google Scholar]
- 9.de la Lastra JM, de la Fuente J. Molecular cloning and characterisation of the griffon vulture (Gyps fulvus) toll-like receptor 1. Dev Comp Immunol. 2007;31:511–9. doi: 10.1016/j.dci.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science. 2014;346:1311–20. doi: 10.1126/science.1251385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.International Chicken Genome Sequencing Consortium Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 12.Dalloul RA, Long JA, Zimin AV, Aslam L, Beal K, Le Blomberg A, et al. Multi-Platform Next-Generation Sequencing of the Domestic Turkey (Meleagris gallopavo): Genome Assembly and Analysis. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Li Y, Burt DW, Chen H, Zhang Y, Qian W, et al. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat Genet. 2013;45:776–83. doi: 10.1038/ng.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Künstner A, et al. The genome of a songbird. Nature. 2010;464:757–62. doi: 10.1038/nature08819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhan X, Pan S, Wang J, Dixon A, He J, Muller MG, et al. Peregrine and saker falcon genome sequences provide insights into evolution of a predatory lifestyle. Nat Genet. 2013;45:563–6. doi: 10.1038/ng.2588. [DOI] [PubMed] [Google Scholar]
- 16.Cho YS, Hu L, Hou H, Lee H, Xu J, Kwon S, et al. The tiger genome and comparative analysis with lion and snow leopard genomes. Nat Commun. 2013;4:2433. doi: 10.1038/ncomms3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yim HS, Cho YS, Guang X, Kang SG, Jeong JY, Cha SS, et al. Minke whale genome and aquatic adaptation in cetaceans. Nat Genet. 2014;46:88–92. doi: 10.1038/ng.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang G, Li B, Gilbert MTP, Jarvis E. The avian phylogenomic project data. GigaScience Database. Available at: http://gigadb.org/dataset/101000 (accessed 24 November 2014).
- 19.Jarvis ED, Mirarab S, Aberer AJ, Li B, Houde P, Li C, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346:1320–31. doi: 10.1126/science.1253451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–9. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- 22.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 23.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wink M. Phylogeny of Old and New World vultures (Aves: Accipitridae and Cathartidae) inferred from nucleotide sequences of the mitochondrial cytochrome b gene. Z Naturforsch C. 1995;50:868–82. doi: 10.1515/znc-1995-11-1220. [DOI] [PubMed] [Google Scholar]
- 25.Duke GE, Jegers AA, Loff G, Evanson OA. Gastric digestion in some raptors. Comp Biochem Physiol A Comp Physiol. 1975;50:649–56. doi: 10.1016/0300-9629(75)90121-8. [DOI] [PubMed] [Google Scholar]
- 26.Krejs GJ. Physiological role of somatostatin in the digestive tract: gastric acid secretion, intestinal absorption, and motility. Scand J Gastroenterol Suppl. 1986;119:47–53. doi: 10.3109/00365528609087431. [DOI] [PubMed] [Google Scholar]
- 27.Ogada DL, Torchin ME, Kinnaird MF, Ezenwa VO. Effects of vulture declines on facultative scavengers and potential implications for mammalian disease transmission. Conserv Biol. 2012;26:453–60. doi: 10.1111/j.1523-1739.2012.01827.x. [DOI] [PubMed] [Google Scholar]
- 28.Sibley CG, Ahlquist JE. Phylogeny and classification of birds: a study in molecular evolution. New Haven, CT: Yale University Press; 1990. [Google Scholar]
- 29.Teodoro JG, Branton PE. Regulation of apoptosis by viral gene products. J Virol. 1997;71:1739–46. doi: 10.1128/jvi.71.3.1739-1746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7 doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, et al. Activation of TBK1 and IKKε kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol. 2004;78:10636–49. doi: 10.1128/JVI.78.19.10636-10649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikeda F, Hecker CM, Rozenknop A, Nordmeier RD, Rogov V, Hofmann K, et al. Involvement of the ubiquitin-like domain of TBK1/IKK-i kinases in regulation of IFN-inducible genes. EMBO J. 2007;26:3451–62. doi: 10.1038/sj.emboj.7601773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goncalves A, Bürckstümmer T, Dixit E, Scheicher R, Górna MW, Karayel E, et al. Functional dissection of the TBK1 molecular network. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, et al. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U S A. 2012;109:9378–83. doi: 10.1073/pnas.1121552109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamazaki T, Takeda K, Gotoh K, Takeshima H, Akira S, Kurosaki T. Essential immunoregulatory role for BCAP in B cell development and function. J Exp Med. 2002;195:535–45. doi: 10.1084/jem.20011751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc Natl Acad Sci U S A. 1996;93:6721–5. doi: 10.1073/pnas.93.13.6721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, Song P, Nakamura S, Miller M, Barone S, Alper SL, et al. Deletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem. 2009;284:29470–9. doi: 10.1074/jbc.M109.044396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He W, Liu W, Chew CS, Baker SS, Baker RD, Forte JG, et al. Acid secretion-associated translocation of KCNJ15 in gastric parietal cells. Am J Physiol Gastrointest Liver Physiol. 2011;301:G591–600. doi: 10.1152/ajpgi.00460.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roggenbuck M, Schnell IB, Blom N, Bælum J, Bertelsen MF, Pontén TS, et al. The microbiome of New World vultures. Nat Commun. 2014;25:5. doi: 10.1038/ncomms6498. [DOI] [PubMed] [Google Scholar]
- 42.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:293–7. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tariq M, Chen R, Yuan H, Liu Y, Wu Y, Wang J, et al. De novo transcriptomic analysis of peripheral blood lymphocytes from the Chinese goose: gene discovery and immune system pathway description. PLoS One. 2015;10 doi: 10.1371/journal.pone.0121015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–5. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vijayakumar P, Raut AA, Kumar P, Sharma D, Mishra A. De novo assembly and analysis of crow lungs transcriptome. Genome. 2014;57:499–506. doi: 10.1139/gen-2014-0122. [DOI] [PubMed] [Google Scholar]
- 48.Loiarro M, Ruggiero V, Sette C. Targeting TLR/IL-1R signalling in human diseases. Mediators Inflamm. 2010;2010:674363. doi: 10.1155/2010/674363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–8. doi: 10.1016/S0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 50.Videvall E, Cornwallis CK, Palinauskas V, Valkiūnas G, Hellgren O. The avian transcriptome response to malaria infection. Mol Biol Evol. 2015;32:1255–67. doi: 10.1093/molbev/msv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takaesu G, Kishida S, Hiyama A, Yamaguchi K, Shibuya H, Irie K, et al. TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol Cell. 2000;5:649–58. doi: 10.1016/S1097-2765(00)80244-0. [DOI] [PubMed] [Google Scholar]
- 52.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature. 2003;424:793–6. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 53.Miller W, Schuster SC, Welch AJ, Ratan A, Bedoya-Reina OC, Zhao F, et al. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc Natl Acad Sci U S A. 2012;109:E2382–90. doi: 10.1073/pnas.1210506109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–5. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–6. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goldstein RA, Pollard ST, Shah SD, Pollock DD. Nonadaptive amino acid convergence rates decrease over time. Mol Biol Evol. 2015;32:1373–81. doi: 10.1093/molbev/msv041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pollock DD, Thiltgen G, Goldstein RA. Amino acid coevolution induces an evolutionary Stokes shift. Proc Natl Acad Sci U S A. 2012;109:E1352–9. doi: 10.1073/pnas.1120084109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. 1000 Genome Project Data Processing Subgroup: The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, et al. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19:651–2. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 62.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–20. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 65.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2010;26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tamura K, Battistuzzi FU, Billing-Ross P, Murillo O, Filipski A, Kumar S. Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci U S A. 2012;109:19333–8. doi: 10.1073/pnas.1213199109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Löytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci. 2005;30:10557–62. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, Hubisz MJ, et al. A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 75.Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–5. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Swiss-Model. Available at: http://swissmodel.expasy.org. Accessed 2015.
- 77.Gautschi B. Conservation genetics of the bearded vulture (Gypaetus barbatus). Diss. Univ. Zürich. Erbgut-Analyse bei Museumsbartgeiern–eine genetische Zeitreise. Infodienst Wildbiologie. 2001;4:15.