

 A Rh-catalyzed desymmetrization of α,α-bis(allyl)aldehydes occurs by enantioselective isomerization followed by olefin-directed hydroacylation.
A Rh-catalyzed desymmetrization of α,α-bis(allyl)aldehydes occurs by enantioselective isomerization followed by olefin-directed hydroacylation.
Abstract
We describe a Rh-catalyzed desymmetrization of all-carbon quaternary centers from α,α-bis(allyl)aldehydes by a cascade featuring isomerization and hydroacylation. This desymmetrization competes with two other novel olefin functionalizations that are triggered by C–H bond activation, including carboacylation and bisacylation. A BIPHEP ligand promotes enantioselective formation of α-vinylcyclopentanones. Mechanistic studies support irreversible and enantioselective olefin-isomerization followed by olefin-hydroacylation.
Introduction
Desymmetrization has emerged as a way to access chiral quaternary-carbon motifs, which are among the most challenging stereocenters to generate with enantiocontrol.1–4 Strategies involving C–H bond activation are especially promising yet rare.5,6 Given this challenge, we propose that prochiral aldehyde 1 could isomerize to scaffolds bearing quaternary centers via two possible pathways triggered by aldehyde C–H bond activation (Fig. 1). Herein, we communicate Rh-catalyzed olefin functionalizations, including hydroacylation and carboacylation from a common aldehyde. This initial report focuses on hydroacylation of bis(allyl)aldehydes to generate α-vinylcyclopentanones 2 bearing quaternary stereocenters.7 Mechanistic studies reveal a cascade process featuring an enantioselective olefin-isomerization followed by olefin-hydroacylation.
Fig. 1. Two pathways to quaternary-carbon motifs from desymmetrization of 1.

The use of oxygen, nitrogen, and sulfur-based functional groups has allowed breakthroughs in enantioselective Rh-catalyzed hydroacylation.8 These heteroatoms act as directing groups by binding to rhodium and favoring C–H bond activation while accelerating hydroacylation over competitive pathways, such as decarbonylation or catalyst decomposition.9 Fu demonstrated intramolecular hydroacylation of alkynals bearing β-methoxy groups (Fig. 2a).10 Our laboratory reported intermolecular hydroacylation of cyclopropenes using chelating aldehydes, specifically salicylaldehyde derivatives (Fig. 2b).5 Given their ability to bind Rh, we reasoned that olefins could be used as directing groups for hydroacylation.11 We were encouraged that Tanaka and Suemune reported desymmetrization of β-bis(alkenyl) aldehydes (Fig. 2c).12 Although not proposed, we reason that the pendant olefin in their substrate could be acting as a directing group. These previous desymmetrizations by hydroacylation generate ketones bearing β-quaternary stereocenters. Given this limitation, we chose to develop a complementary desymmetrization of α-trisubstituted aldehydes, which represents a sterically hindered and thus, challenging substrate class.13b–c,14 If successful, our strategy would allow access to cyclopentanones bearing α-quaternary centers, whereby the pendant olefin serves as both a directing group and versatile handle for further elaboration.
Fig. 2. Previous desymmetrizations by hydroacylation result in ketones bearing β-quaternary stereocenters.

Results and discussion
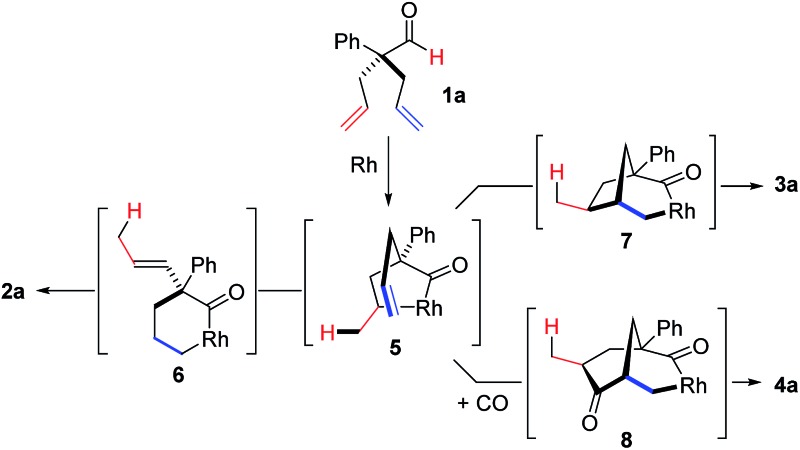
To test our proposal, we studied the desymmetrization of model 1a, which can be prepared in one-step from commercially available phenylacetaldehyde.15 Aldehyde 1a was subjected to cationic Rh(i)-catalysts and various bidentate phosphine ligands that are known to promote formyl C–H bond activation.16 We imagined that oxidative addition followed by alkene insertion would generate metallacycle 5, which could diverge into various scaffolds (Table 1). The choice of phosphine ligand had a dramatic impact on product outcome and enabled chemoselective formation of two major products, cyclopentanone 2a and bicyclo[2.2.1]heptanone 3a.
Table 1. Divergent pathways for desymmetrization of 1a based on ligand-choice a .
| |||
| Entry | Ligand | Major product, yield c | Minor product(s), yield d |
| 1 |
|
|
3a 19%, 33% ee |
| 2 |
|
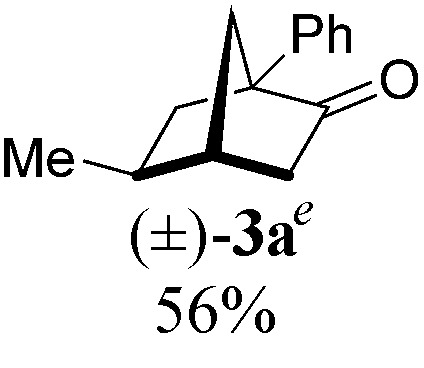
|
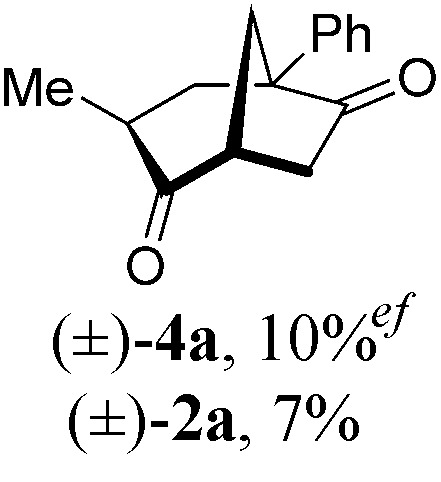
|
| 3 b |
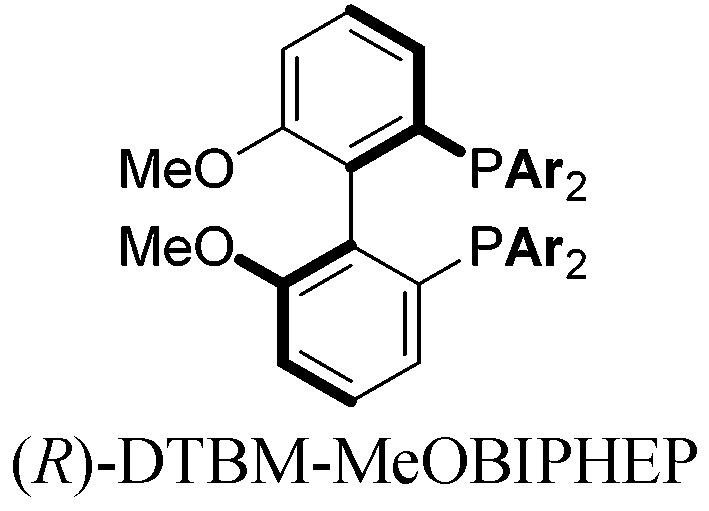
|
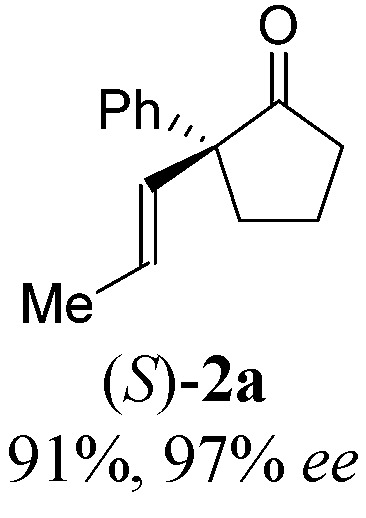
|
3a and 4a not observed |
aReaction conditions: [(coe)2RhCl]2 (5 mol%), ligand (10 mol%), AgBF4 (10 mol%), DCE (0.2 M), 40 °C, 36 h.
bReaction conditions: [(coe)2RhCl]2 (2.5 mol%), ligand (5 mol%), AgBF4 (5 mol%), 40 °C, 4 h.
cIsolated yield.
dDetermined by GC-FID.
e>20 : 1 dr, determined by 1H NMR.
fOne equivalent of 1a was used as a CO donor. DTBM: 3,5-di(tert-butyl)-4-methoxyphenyl.
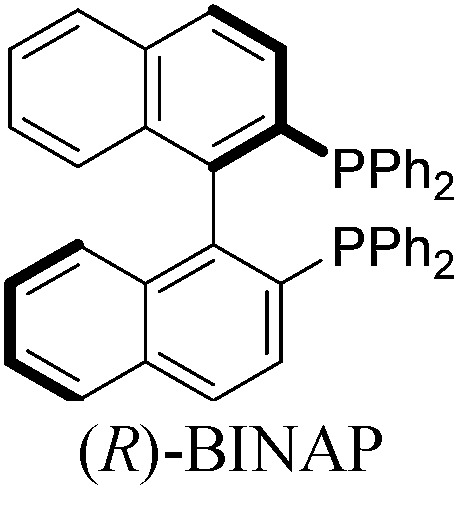
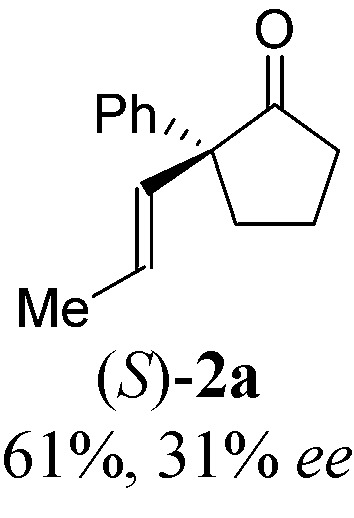
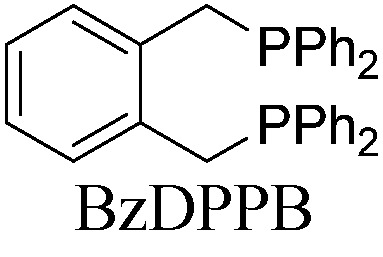
With a BINAP-ligated rhodium catalyst, we observed formation of both 2a and 3a in 61% and 19% yields, respectively (entry 1). We discovered that the hydroacylation product, cyclopentanone 2a, bears an internal olefin, which presumably results from isomerization of the terminal olefin. Carbometallation of the pendant olefin from 5 results in intermediate 7, which undergoes reductive elimination to form bicycloheptanone 3a as a minor product. Use of BzDPPB ligand, however, favors the carboacylation pathway to generate 3a as the major product in 56% yield with high diastereoselectivity (>20 : 1, entry 2). This unique olefin functionalization takes advantage of C–H activation rather than strained C–C activation to achieve carboacylation.17 While our study was in progress, Aïssa reported a related carbocyclization using pyridyl directing groups.18 We also observed bicyclo[3.2.1]octadione 4a as a minor product in 10% yield (entry 2). The molecular structure of this homologated ketone 4a was confirmed by X-ray crystallography (see ESI†). We believe that the second carbonyl arises from a disproportionation process where a second equivalent of aldehyde 1a undergoes decarbonylation to generate CO.
With these promising leads in hand, we plan to further study each pathway and develop enantioselective variants. Towards this goal, we realized that electron-donating aromatic groups on phosphines enhance selectivity for 2a. Among the ligands evaluated, (R)-DTBM-MeOBIPHEP provided the best reactivity and enantioselectivity for 2a (entry 3). The absolute configuration of cyclopentanone 2a was determined by elaboration with 2,4-dinitrophenylhydrazine to hydrazone 9, in which the molecular structure was established by X-ray crystallographic analysis (Fig. 3).
Fig. 3. Determination of the absolute configuration of 2a and X-ray crystal structure of (2S)-9.

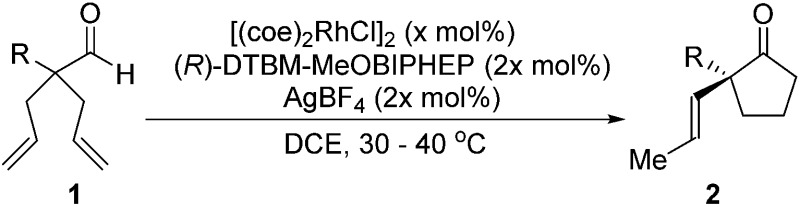
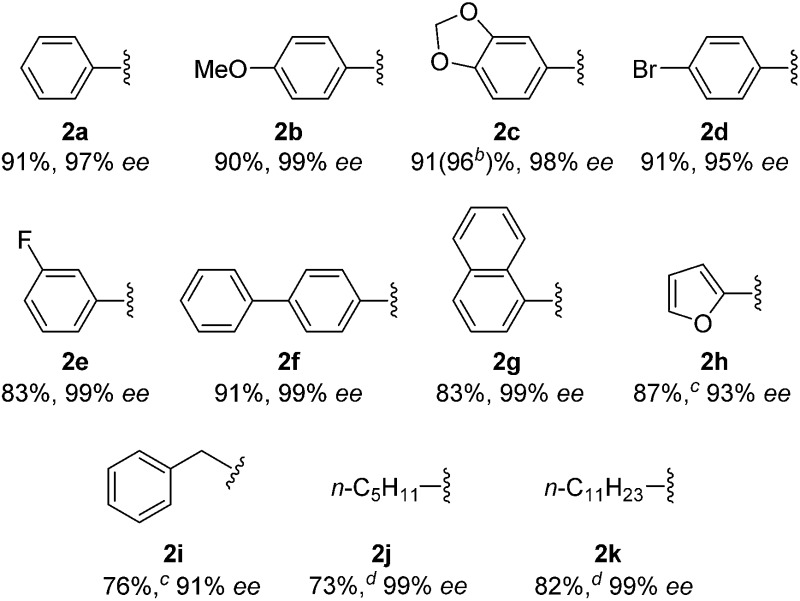
With this protocol, we prepared eleven cyclopentanones bearing various α-quaternary stereocenters (Table 2). Aldehydes with aromatic substituents (1a–1g) undergo desymmetrization in 83–91% yields and high enantioselectivities (95–99% ee). Ether, aryl halide, and acetal functional groups are well-tolerated. Heteroaromatic aldehyde (1h) as well as aldehydes bearing aliphatic substituents (1i–1k) rearrange to the corresponding cyclopentanones in excellent enantioselectivities albeit using increased catalyst loading at lower temperature.19,20
Table 2. Desymmetrization of α-quaternary aldehyde 1 by isomerization-hydroacylations a .
|
|
aReaction conditions: 0.1 mmol 1, x = 2.5, DCE (0.2 M), 40 °C, 4 h.
bReaction conditions: 1 mmol 1c used.
cReaction conditions: x = 5, DCE (0.33 M), 30 °C, 2 h.
dReaction conditions: x = 6, DCE (0.33 M), 30 °C, 2 h.
To understand the mechanism, we performed a deuterium-labelling study with d-1a. Desymmetrization of d-1a, under standard reaction conditions, led to exclusive formation of d-2a where the deuterium label was incorporated into the methyl group of the α-propenyl substituent (eqn (1)). This result indicates that isomerization of one allyl group takes place first through an endocyclic β-hydride elimination of a 5-membered rhodacycle d-5a.21 Our observations corroborate Aïssa's recent report on the isomerisation of 4-pentenals.22 Although β-hydride eliminations of this type are uncommon, it has been predicted that binding of a pendant alkene to the metal center significantly lowers the barrier to this process.23
When the reaction of d-1a was quenched at an early stage (40% conversion to d-2a), we recovered three deuterated aldehydes, d-1a, d-1a′ and d-1a′′ (eqn (2)). The observation of d-1a′ suggests that olefin-insertion is reversible. Yet, the deuterium is incorporated into only the methyl group of the α-propenyl unit in product d-2a. This lack of deuterium scrambling on the cyclopentanone ring suggests that Rh-D insertion occurs with high enantioselectivity (with the olefin shown in red). Thus, the insertion step is both reversible and highly enantioselective.24
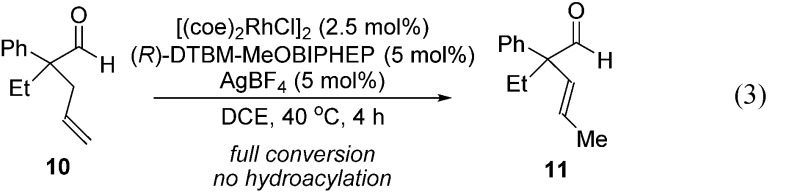
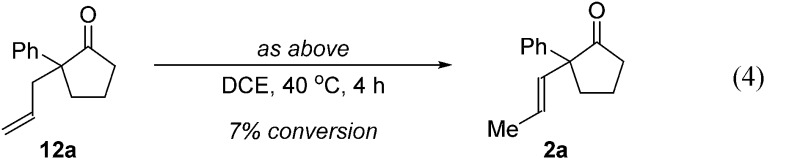
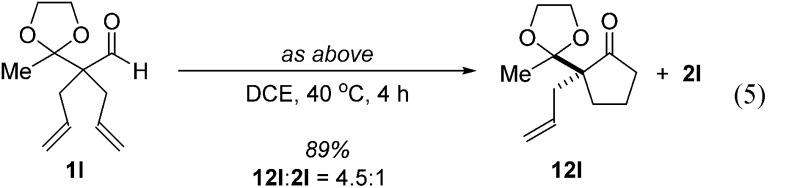
Further experiments support the notion that the α-vinyl group (formed from initial isomerization) directs hydroacylation. For example, α-trisubstituted aldehyde 10 (with only one allyl group) does not undergo hydroacylation. Instead, this aldehyde undergoes isomerization to generate α-vinyl aldehyde 11 (eqn (3)).22 In addition, subjecting α-allylcyclopentanone 12a to the optimized reaction conditions results in trace formation of α-vinylcyclopentanone 2a (eqn (4)). Thus, the cyclopentanones obtained in Table 2 must arise from an isomerization that occurs prior to hydroacylation. In contrast, we discovered that aldehyde 1l, containing an acetal group, yields α-allylcyclopentanone 12l as the major product (eqn (5)). In this case, we reason that the acetal acts as an oxygen-directing group which promotes hydroacylation over olefin isomerization.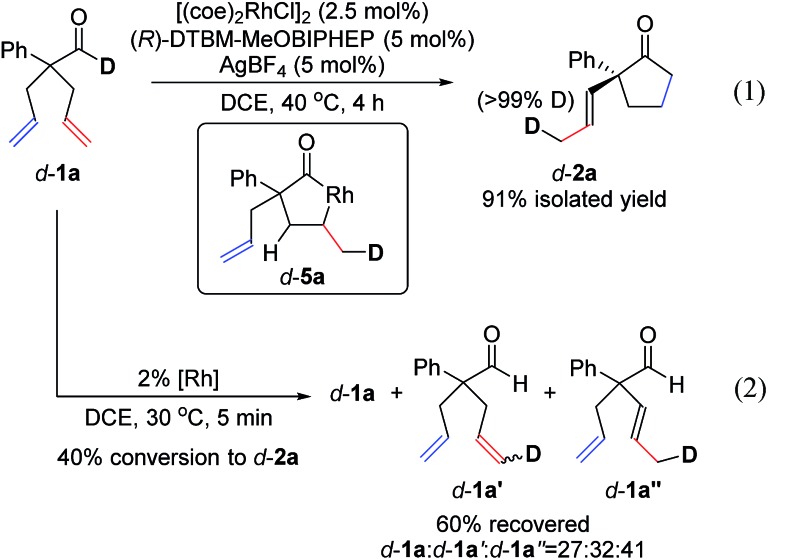
On the basis of literature reports and our own observations, we propose a mechanism starting with cationic Rh(i)-complex activating the aldehyde C–H bond of 1 to form acyl-Rh(iii)-hydride 13 (Scheme 1). Insertion of the olefin into Rh(iii)-hydride 13 leads to formation of the more thermodynamically stable 5-membered metallacycle 5.25 A rare endocyclic β-hydride elimination takes place to produce isomerized acyl-Rh(iii)-hydride 14. The allyl olefin inserts into the Rh(iii)-hydride to form a 6-membered rhodacycle 6. Finally, reductive elimination affords the cyclopentanone product 2 and regenerates the Rh(i)-catalyst.
Scheme 1. Proposed mechanism for Rh-catalyzed cascade.

Conclusions
We have demonstrated a Rh-catalyzed enantioselective synthesis of α-quaternary cyclopentanones. Studies on the scope and mechanism support an olefin-assisted isomerization23 and olefin-directed hydroacylation cascade. While endocyclic β-hydride elimination has been proposed in the literature on the basis of theoretical studies,21 our results provide experimental evidence for this elementary step. The use of a BIPHEP ligand enables high selectivity for one out of three possible rearrangements, all initiated by the activation of an aldehyde C–H bond. Insights from these studies will guide efforts to understand and expand the power of the related carboacylation and bisacylation as routes to scaffolds containing chiral all-carbon stereocenters.
Acknowledgments
Funding provided by UC Irvine and the National Institutes of Health (GM105938). We are grateful to Eli Lilly for a Grantee Award. K.G.M.K. thanks NSERC of Canada for a CGS-D fellowship. We thank Van Nguyen for initial ligand studies, Kaitlyn Kung for help with substrate synthesis, and Dr Joseph W. Ziller for X-ray crystallographic analysis.
Footnotes
References
- For reviews on enantioselective construction of quaternary stereocenters, including desymmetrization strategies, see: ; (a) Quasdorf K. W., Overman L. E. Nature. 2014;516:181. doi: 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Douglas C. J., Overman L. E. Proc. Natl. Acad. Sci. U. S. A. 2004;101:5363. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Y., Han S.-J., Liu W.-B., Stoltz B. M. Acc. Chem. Res. 2015;48:740. doi: 10.1021/ar5004658. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lebel H., Marcoux J.-F., Molinaro C., Charette A. B. Chem. Rev. 2003;103:977. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]
- For recent alkene functionalizations to establish enantioenriched quaternary stereocenters, see: Mei T.-S., Patel H. H., Sigman M. S., Nature, 2014, 508 , 340 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- For recent selected desymmetrizations to quaternary stereocenters, see: ; (a) Zhou F., Cheng G.-J., Yang W., Long Y., Zhang S., Wu Y.-D., Zhang X., Cai Q. Angew. Chem., Int. Ed. 2014;53:9555. doi: 10.1002/anie.201405575. [DOI] [PubMed] [Google Scholar]; (b) Roux C., Candy M., Pons J.-M., Chuzel O., Bressy C. Angew. Chem., Int. Ed. 2014;53:766. doi: 10.1002/anie.201308268. [DOI] [PubMed] [Google Scholar]; (c) Yao L., Liu K., Tao H.-Y., Qiu G.-F., Zhou X., Wang C.-J. Chem. Commun. 2013;49:6078. doi: 10.1039/c3cc42587h. [DOI] [PubMed] [Google Scholar]; (d) Lee J. Y., You Y. S., Kang S. H. J. Am. Chem. Soc. 2011;133:1772. doi: 10.1021/ja1103102. [DOI] [PubMed] [Google Scholar]
- For selected examples of using carbenoids to set quaternary stereocenters, see: ; (a) Briones J. F., Davies H. M. L. J. Am. Chem. Soc. 2013;135:13314. doi: 10.1021/ja407179c. [DOI] [PubMed] [Google Scholar]; (b) Spangler J. E., Davies H. M. L. J. Am. Chem. Soc. 2013;135:6802. doi: 10.1021/ja4025337. [DOI] [PubMed] [Google Scholar]; (c) Hyster T. K., Ruhl K. E., Rovis T. J. Am. Chem. Soc. 2013;135:5364. doi: 10.1021/ja402274g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan D. H. T., Kou K. G. M., Dong V. M. J. Am. Chem. Soc. 2010;132:16354. doi: 10.1021/ja107738a. [DOI] [PubMed] [Google Scholar]
- (a) Xiao K.-J., Lin D. W., Miura M., Zhu R.-Y., Gong W., Wasa M., Yu J.-Q. J. Am. Chem. Soc. 2014;136:8138. doi: 10.1021/ja504196j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shi B.-F., Zhang Y.-H., Lam J. K., Wang D.-H., Yu J.-Q. J. Am. Chem. Soc. 2010;132:460. doi: 10.1021/ja909571z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glorius recently reported cyclopentanones bearing α-quaternary centers by an organocatalyzed NHC hydroacylation: Janssen-Müller D., Schedler M., Fleige M., Daniliuc C. G., Glorius F., Angew. Chem., Int. Ed., 2015. 10.1002/anie.201412302 . [Google Scholar]
- ; For recent enantioselective intramolecular hydroacylations without chelation-assistance, see: ; (a) Stemmler R. T., Bolm C. Adv. Synth. Catal. 2007;349:1185. [Google Scholar]; (b) Coulter M. M., Kou K. G. M., Galligan B., Dong V. M. J. Am. Chem. Soc. 2010;132:16330. doi: 10.1021/ja107198e. [DOI] [PubMed] [Google Scholar]; (c) Coulter M. M., Dornan P. K., Dong V. M. J. Am. Chem. Soc. 2009;131:6932. doi: 10.1021/ja901915u. [DOI] [PubMed] [Google Scholar]; (d) Khan H. A., Kou K. G. M., Dong V. M. Chem. Sci. 2011;2:407. [Google Scholar]; (e) Osborne J. D., Randell-Sly H. E., Currie G. S., Cowley A. R., Willis M. C. J. Am. Chem. Soc. 2008;130:17232. doi: 10.1021/ja8069133. [DOI] [PubMed] [Google Scholar]; (f) Du X.-W., Ghosh A., Stanley L. M. Org. Lett. 2014;16:4036. doi: 10.1021/ol501869s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ghosh A., Stanley L. M. Chem. Commun. 2014;50:2765. doi: 10.1039/c4cc00210e. [DOI] [PubMed] [Google Scholar]
- Willis M. C. Chem. Rev. 2010;110:725. doi: 10.1021/cr900096x. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Fu G. C. J. Am. Chem. Soc. 2002;124:10296. doi: 10.1021/ja0266161. [DOI] [PubMed] [Google Scholar]
- ; Olefin-assisted transformations using other transition-metal catalysts, see: ; (a) Tobisu M., Hyodo I., Onoe M., Chatani N. Chem. Commun. 2008:6013. doi: 10.1039/b806285d. [DOI] [PubMed] [Google Scholar]; (b) Gülak S., von Wangelin A. J. Angew. Chem., Int. Ed. 2012;51:1357. doi: 10.1002/anie.201106110. [DOI] [PubMed] [Google Scholar]; (c) Gandeepan P., Cheng C.-H. J. Am. Chem. Soc. 2012;134:5738. doi: 10.1021/ja300168m. [DOI] [PubMed] [Google Scholar]
- (a) Tanaka M., Imai M., Fujio M., Sakamoto E., Takahashi M., Eto-Kato Y., Wu X. M., Funakoshi K., Sakai K., Suemune H. J. Org. Chem. 2000;65:5806. doi: 10.1021/jo000781m. [DOI] [PubMed] [Google Scholar]; (b) Tanaka M., Takahashi M., Sakamoto E., Imai M., Matsui A., Fujio M., Funakoshi K., Sakai K., Suemune H. Tetrahedron. 2001;57:1197. [Google Scholar]
- ; There is some evidence for alkynes promoting C–H activation: ; (a) Hoffman T. J., Carreira E. M. Angew. Chem., Int. Ed. 2011;50:10670. doi: 10.1002/anie.201104595. [DOI] [PubMed] [Google Scholar]; (b) Campbell Jr R. E., Lochow C. F., Vora K. P., Miller R. G. J. Am. Chem. Soc. 1980;102:5824. [Google Scholar]; (c) Larock R. C., Oertle K., Potter G. F. J. Am. Chem. Soc. 1980;102:190. [Google Scholar]; (d) Tanaka K., Fu G. C. Org. Lett. 2002;4:933. doi: 10.1021/ol017308v. [DOI] [PubMed] [Google Scholar]
- (a) Fairlie D. P., Bosnich B. Organometallics. 1988;7:936. [Google Scholar]; (b) Mukherjee S., List B. J. Am. Chem. Soc. 2007;129:11336. doi: 10.1021/ja074678r. [DOI] [PubMed] [Google Scholar]; (c) Rolandsgard M., Baldawi S., Sirbu D., Bjørnstad V., Rømming C., Undheim K. Tetrahedron. 2005;61:4129. [Google Scholar]; (d) Okamoto R., Tanaka K. Org. Lett. 2013;15:2112. doi: 10.1021/ol400574s. [DOI] [PubMed] [Google Scholar]
- Jiang G., List B. Adv. Synth. Catal. 2011;353:1667. [Google Scholar]
- (a) Shibata Y., Tanaka K. J. Am. Chem. Soc. 2009;131:12552. doi: 10.1021/ja905908z. [DOI] [PubMed] [Google Scholar]; (b) Tanaka K., Shibata Y., Suda T., Hagiwara Y., Hirano M. Org. Lett. 2007;9:1215. doi: 10.1021/ol070153s. [DOI] [PubMed] [Google Scholar]; (c) Kou K. G. M., Le D. N., Dong V. M. J. Am. Chem. Soc. 2014;136:9471. doi: 10.1021/ja504296x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Murphy S. K., Bruch A., Dong V. M. Angew. Chem., Int. Ed. 2014;53:2455. doi: 10.1002/anie.201309987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of carboacylation by C–C bond activation, see: ; (a) Ko H. M., Dong G. Nat. Chem. 2014;6:739. doi: 10.1038/nchem.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Souillart L., Cramer N. Angew. Chem., Int. Ed. 2014;53:9640. doi: 10.1002/anie.201405834. [DOI] [PubMed] [Google Scholar]; (c) Souillart L., Parker E., Cramer N. Angew. Chem., Int. Ed. 2014;53:3001. doi: 10.1002/anie.201311009. [DOI] [PubMed] [Google Scholar]; (d) Xu T., Ko H. M., Savage N. A., Dong G. J. Am. Chem. Soc. 2012;134:20005. doi: 10.1021/ja309978c. [DOI] [PubMed] [Google Scholar]; (e) Dreis A. M., Douglas C. J. J. Am. Chem. Soc. 2009;131:412. doi: 10.1021/ja8066308. [DOI] [PubMed] [Google Scholar]
- Aïssa C., Ho K. Y. T., Tetlow D. J., Pin-Nó M. Angew. Chem., Int. Ed. 2014;53:4209. doi: 10.1002/anie.201400080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Experiments with 5 mol% catalystat 40 °C gave a mixture of 2 and 12 (2 : 12 = 1 : 2)
- Preliminary experiments using phenylbis(methallyl)aldehyde resulted in 55% isolated yield for the non-isomerized hydroacylation product 12, and no hydroacylation product obtained using phenylbis(crotyl)aldehyde
- An endocyclic β-hydride elimination within a metallacyclopentanone structure has been computed by DFT in a study on the mechanism of rhodium-catalyzed decarbonylation: Hyatt I. F. D., Anderson H. K., Morehead Jr A. T., Sargent A. L., Organometallics, 2008, 27 , 135 . [Google Scholar]
- Yip S. Y. Y., Aïssa C., Angew. Chem., Int. Ed., 2015, 54 , 6870 , . In this work, isomerization by endocyclic β-hydride elimination was supported by a deuterium labelling study using phenyl(d2-allyl)aldehydes . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Zhu J., Lin Z. Organometallics. 2004;23:4154. [Google Scholar]
- Further studies are required to identify the turn-over limiting step. On the basis of the IUPAC Gold book (http://old.iupac.org/goldbook/P04862.pdf), the selectivity-determining step is identical to, or occurs later than, the rate-controlling step on the reaction coordinate. See also: Simmons E. M., Hartwig J. F., Angew. Chem., Int. Ed., 2012, 51 , 3066 . [Google Scholar]
- Omae I. J. Organomet. Chem. 2007;692:2608. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.