

Summary
Frequent tumor relapse in hepatocellular carcinoma (HCC) has been commonly attributed to the presence of residual cancer stem cells (CSCs) after conventional treatments. We have previously identified and characterized CD133 to mark a specific CSC subset in HCC. In the present study, we found endogenous and secretory annexin A3 (ANXA3) to play pivotal roles in promoting cancer and stem cell-like features in CD133+ liver CSCs through a dysregulated JNK pathway. Blockade of ANXA3 with an anti-ANXA3 monoclonal antibody in vitro as well as in human HCC xenograft models resulted in a significant reduction in tumor growth and self-renewal. Clinically, ANXA3 expression in HCC patient sera closely associated with aggressive clinical features. Our results suggest that ANXA3 can serve as a novel diagnostic biomarker and that the inhibition of ANXA3 may be a viable therapeutic option for the treatment of CD133+ liver-CSC-driven HCC.
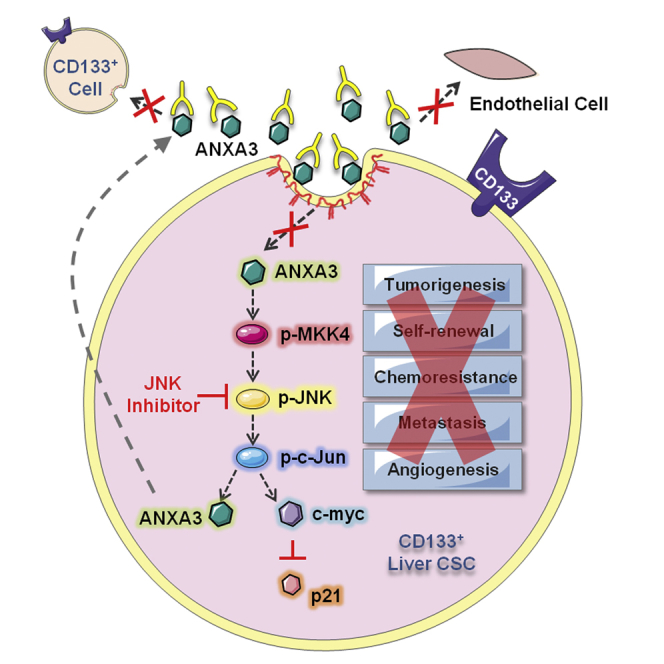
Graphical Abstract
Highlights
-
•
ANXA3 potentiates cancer and stem cell-like properties in CD133+ liver CSCs
-
•
ANXA3 is internalized via caveolae-mediated endocytosis and activates the JNK pathway
-
•
A novel neutralizing anti-ANXA3 antibody is developed
-
•
ANXA3 is a novel biomarker for HCC diagnosis
Ma and colleagues report the role of enhanced endogenous and secretory ANXA3 in the maintenance of cancer stem cell-like properties in CD133+ liver CSCs. ANXA3 is internalized via a caveolae-dependent endocytosis and activates the JNK pathway. Treatment of HCC with ANXA3 monoclonal antibody resulted in decreased tumor growth, self-renewal, and liver CSC proportions.
Introduction
Hepatocellular carcinoma (HCC) is the most common form of liver cancer. Resection and liver transplantation is remedial for early-stage HCC. Yet, since most patients are diagnosed at an advanced stage, therapy is rarely curative and the prognosis for the disease is poor. Despite advances in diagnosis and treatment, the disease remains a major health concern due to the infiltrative nature of these tumors, their resistance to chemotherapy, their high rate of recurrence, and our limited understanding of the mechanisms underlying initiation and progression of the disease. This dismal situation motivates the search for new therapies and better diagnostic biomarkers for detection of the disease at an earlier stage.
The cancer stem cell (CSC) model has helped explain why tumor eradication has not been achieved despite advances in treatment. The model suggests that a cellular hierarchy exists in some cancers, with self-renewing CSCs generating progeny constituting the tumor bulk. CSCs possess both tumor and stem cell-like properties (Pardal et al., 2003). Studies have shown that CSCs bear the exclusive ability to regenerate tumors. Treatment of bulk cancer cell populations within tumors with chemotherapy has been shown to select for the outgrowth of therapy-resistant cancer cells that are more tumorigenic, invasive, and stem-like. Hence, cancer therapies may be rendered ineffective because the bulk of cancer cells within a tumor may be eliminated while leaving behind CSC-enriched cells that proceed to regenerate tumors. This underscores the need for a detailed understanding of the molecular differences between CSCs and non-CSCs to discover cell-state-specific features that may render CSCs susceptible to selective therapeutic intervention.
The perpetuation of many cancer types has been suggested to stem from CSCs. We have found HCC to be driven by a liver CSC subset marked by the CD133 phenotype. CD133+ HCC cells display sustained self-renewal, differentiate toward multiple lineages, and phenocopy the original tumor upon xenotransplantation (Ma et al., 2007, 2010). These cells also possess an enhanced ability to resist chemotherapy through activated AKT/BCL-2 (Ma et al., 2008). CD133 is not simply a marker of liver CSCs; it also plays a functional role in regulating HCC tumorigenesis (Tang et al., 2012). Increased CD133 expression in HCC is associated with worse overall survival and higher recurrence rates (Ma et al., 2010). Our results are consistent with studies by other groups where CD133 was also found to be an important risk factor for overall survival of the disease, demonstrating the prominence of CD133 in HCC. Despite our growing understanding of the importance of a CD133+ liver CSC population, the functional paths by which these cells promote hepatocarcinogenesis remains limited.
Since the intrinsic molecular mechanisms by which CSCs sustain tumor growth is believed to be inter-related with its tumor microenvironment, our present study aims at investigating the mechanism by which CD133+ liver CSCs mediate tumor formation, self-renewal, and interaction with its niche. Toward this goal, RNA sequencing (RNA-seq) profiling was carried out to compare the differential gene expressions between CD133+ liver CSCs and CD133− differentiated counterparts. Many of the differentially expressed genes common to the two samples encoded for secretory proteins, which we know represent major means of communication between cancer cells and the microenvironment. From our profiling, the most significantly deregulated gene that encodes for a secretory protein is annexin A3 (ANXA3), a gene we now show to be critical in promoting CSC-like properties in CD133+ liver-CSC-driven HCC through both an autocrine and paracrine manner. ANXA3 belongs to the annexin family of Ca2+-dependent phospholipid-binding proteins (Raynal and Pollard, 1994). It has been shown to possess the ability to promote angiogenesis (Park et al., 2005) and rat liver regeneration (Harashima et al., 2008). Upregulation of ANXA3 expression is detected in various tumor types including prostate, ovarian, and lung cancers (Köllermann et al., 2008; Schostak et al., 2009; Liu et al., 2009; Yan et al., 2010). In ovarian cancer, serum ANXA3 levels were significantly upregulated in diseased patients compared with healthy individuals (Yin et al., 2012). Further, overexpression of ANXA3 was found to contribute to platinum resistance in ovarian cancer (Yan et al., 2010). In HCC, ANXA3 was also found to be overexpressed in 5-fluorouracil (5-FU)-resistant cell lines (Yin et al., 2012) and to play a role in promoting tumorigenesis and resistance to chemotherapy (Pan et al., 2013). Nevertheless, the role of endogenous and secretory ANXA3 in the context of CD133+ liver CSCs or HCC and the mechanism by which ANXA3 regulates CSC-like features has not been explored. Here, we investigated the clinical significance, functional role, and therapeutic implications of ANXA3 in CD133+ liver-CSC-driven HCC. We identified caveolin-1-dependent endocytosis to mediate internalization of secretory ANXA3 into HCC cells, thereby activating a dysregulated JNK pathway to promote CSC-like properties. We also developed a monoclonal antibody specific against ANXA3 (anti-ANXA3 mAb) and showed in vivo that the use of this antibody alone or in combination with cisplatin could efficiently lead to a reduced ability of HCC cells to initiate tumor growth and self-renewal, concomitant with a decrease in liver CSC proportions.
Results
Transcriptome Sequencing Profiling Identifies ANXA3 to Be Preferentially Expressed in the CD133+ Liver CSC Subset
To detect differential gene expression profiles between CD133+ liver CSCs and their CD133− differentiated counterparts, we applied RNA-seq to investigate the sorted subsets isolated from two HCC cells, Huh7 and PLC8024. 95.61% of the reads mapped to the reference human genome (GRCh37/hg19) (Table S1). Using a stringent fold-change cutoff of >2 and <0.5 and a p value ≤ 0.05, 38 genes were found to be commonly de-regulated (Table S2). Pathway enrichment analysis identified critically over-represented pathways related to cancer, focal adhesion, extracellular matrix (ECM)-receptor interaction, drug metabolism, and ATP-binding casette (ABC) transporters in the deregulated gene set (Figure S1A). The same gene set was also surveyed using GSEA where MAPK signaling was found to be exclusively enriched in the CD133+ liver CSC subset while CD133− cells was enriched for genes associated with hepatocyte differentiation (Figure S1B). Of the commonly differentially expressed genes, a good proportion of them (13/38; 34.2%) encode for secretory proteins (Table S2, red). And of these 13, annexin A3 (ANXA3) was the most significantly upregulated in the CD133+ subset (Figure S1C) and was thus chosen for studies. Subsequent validation by qPCR confirmed preferential overexpression of ANXA3 in CD133+ liver CSCs isolated from a larger cohort of HCC cell lines and clinical samples (n = 8, Figure 1A). Endogenous and secretory proteomic ANXA3 levels were also likewise found elevated in the CD133+ liver CSC subset (Figures 1B and 1C). Dual-color immunofluorescence (IF) confirmed a high degree of ANXA3 and CD133 co-localization in Huh7 (Figure 1D) and PLC8024 (Figure S1D). Concordant with this finding, expressions of ANXA3 and CD133 were also positively correlated across a panel of HCC cell lines (Figure 1E; Figure S1E).
Figure 1.

Endogenous and Secretory Overexpression of ANXA3 Is Strongly Associated with HCC Pathogenesis
(A) Relative ANXA3 expression in sorted CD133 subsets isolated from HCC cells and clinical samples by qPCR. Results represent mean ± SD of duplicate wells in three independent experiments.
(B) Western blot showing expression of ANXA3 in sorted CD133 subsets isolated from HCC cells.
(C) Secretory ANXA3 expression levels in sorted CD133 subsets isolated from HCC cells as detected by ELISA.
(D) Dual-color IF images of CD133 (red) and ANXA3 (green) in Huh7. Nuclei stained with DAPI (blue).
(E) Western blots showing expression of CD133 and ANXA3 in a panel of liver cancer cell lines.
(F) Western blot showing expression of ANXA3 in HCC (T) and matched non-tumor liver (N) specimens from six individual patients. Association between tumor stage and ANXA3 overexpression in HCC. ∗p < 0.05. Results represent mean ± SD from three independent experiments.
(G) Boxplots showing expression of ANXA3 in the sera collected from healthy individuals (normal), HBV carriers (HBV+), patients with liver cirrhosis, and patients with either early HCC (I & II) or advanced HCC (III & IV). Results represent mean ± SD of duplicate wells in three independent experiments.
(H) ROC curve analysis of the sensitivity and specificity of using AFP, ANXA3, or a combination of both for HCC diagnosis. Summary of ROC curve analysis with area under the curve (AUC) and 95% CI values.
See also Figure S1.
Endogenous and Secretory ANXA3 Overexpression Is Tightly Associated with HCC Pathogenesis
We investigated endogenous ANXA3 expression in 83 matched primary HCC and non-tumor liver tissues. Approximately 50.6% (42/83) of the HCC specimens had ANXA3 upregulated (>1.5-fold) compared to non-tumor specimens. ANXA3 overexpression in HCC was significantly associated with advanced tumor stages (p = 0.027; Figure 1F; Table S3). Interestingly, secretory ANXA3 was also found to be progressively elevated from non-HCC patients (healthy subjects, hepatitis B virus [HBV] carriers, and patients with liver cirrhosis) to patients with early- and advanced-stage HCC, with ANXA3 overexpression in HCC sera tightly correlated with advanced tumor stages (p < 0.001), tumor sizes (p = 0.005), number of nodules (p = 0.036), and gender (p = 0.011) (n = 132). ANXA3 was found only at very low levels and was at times completely undetectable in sera collected from healthy individuals, HBV carriers, or patients with cirrhosis but without HCC (Figure 1G; Table S4). Alpha-fetoprotein (AFP) is the most widely used biomarker for HCC diagnosis. By receiver-operating characteristic (ROC) curve analysis, ANXA3 was found to be superior to AFP in distinguishing non-HCC from HCC (Figure 1H). The ROC curve analysis revealed that at the optimal cutoff value of 2.30 for ANXA3 alone, the sensitivity was 84.7% and the specificity was 78.8%, with an area under the curve (AUC) of 0.88 (95% confidence interval [CI], 0.82-0.93). The sensitivity was 42.3% and the specificity was 95.0% at the optimal cutoff value of 56.3 for AFP alone, with an AUC of 0.53 (95% CI, 0.41-0.64). The sensitivity was 93.2% and the specificity was 70% for the combination of ANXA3 and AFP, with an AUC of 0.90 (95% CI, 0.85-0.96).
Endogenous ANXA3 Regulates Both Cancer and Stem Cell-like Properties in HCC
To ascertain whether there is a causative relationship between ANXA3 overexpression and altered CSC-like phenotype in HCC, we generated stably overexpressed ANXA3 MIHA and MHCC97L cells and stably repressed ANXA3 Huh7 and PLC8024 cells (Figures S2A–S2C; sh-ANXA3 clones 244 and 246). ANXA3 knockdown in Huh7 and PLC8024 resulted in a significant decrease in the efficiency of the cells to form colonies, migrate, and invade. Conversely, stable ANXA3 overexpression resulted in opposing effects (Figures S2D–S2F). In addition, cells with ANXA3 suppressed displayed diminished abilities to induce hepatosphere formation in primary, secondary, and tertiary passages (Figure 2A; Figure S3A) and to potentiate resistance toward staurosporine (STS)-induced apoptosis, 5-FU, and cisplatin, chemotherapeutic agents commonly used in HCC therapy (Figures 2B and 2D; Figures S3B and S3D). Treatment of stably repressed ANXA3 cells with STS resulted in a marked reduction in cleaved caspase-3 and cleaved PARP (Figure 2C; Figure S3C). Treatment of Huh7 and PLC8024 cells with 5-FU and cisplatin resulted in an enriched ANXA3 subpopulation (Figure 2E; Figure S3E). Further, HCC cells with ANXA3 suppressed displayed an attenuated ability to mediate angiogenesis, as evidenced by capillary tube formation in endothelial cells (Figure 2F; Figure S3F). As a complementary model, similar assays were also performed in MIHA and MHCC97L cells with ANXA3 stably overexpressed, where enhanced abilities of the cells to confer cancer, stem cell-like, and metastatic traits were observed in the overexpressing clones (Figure 2; Figures S2 and S3).
Figure 2.

Endogenous ANXA3 Confers Enhanced Cancer and Stem Cell-like Properties in HCC
(A) Quantification of hepatospheres in HCC cells with ANXA3 stably expressed (MIHA EV and ANXA3 O/E) or repressed (Huh7 NTC and ANXA3 knockdown clones 244 and 246). ∗p < 0.05. EV, empty vector; O/E, overexpression; NTC, non-target control. Scale bar, 100 μm. Results represent mean ± SD of 12 replicates in three independent experiments.
(B) Percentage of Annexin-V-positive cells in MIHA and Huh7 with ANXA3 stably expressed and repressed, respectively, following STS treatment. Results represent mean ± SD of three independent experiments.
(C) Western blot showing expression of total and cleaved caspase-3 and PARP after STS treatment.
(D) Percentage of Annexin-V-positive cells in MIHA and Huh7 with ANXA3 stably expressed and repressed, respectively, following 5-FU and cisplatin treatment. Results represent mean ± SD of three independent experiments.
(E) Western blot showing expression of ANXA3 in Huh7 following treatment with increasing concentrations of 5-FU and cisplatin.
(F) Quantification of capillary tubes formed by human umbilical vein endothelial cells (HUVECs) following treatment with supernatant collected from MIHA and Huh7 with ANXA3 stably expressed and repressed, respectively. ∗p < 0.05. Scale bar, 100 μm. Results represent mean ± SD of duplicate wells in three independent experiments.
See also Figures S2 and S3.
ANXA3 Contributes to Augmented Tumorigenic and Metastatic Phenotype and Is Critical for Growth and Maintenance of the CD133+ Liver CSC Subset
We then extended our studies in vivo, where HCC cells with ANXA3 manipulated were injected subcutaneously into nude mice to determine the role of ANXA3 in HCC tumor formation. Overexpression of ANXA3 in MHCC97L resulted in a profound increase in the ability of cells to initiate tumor growth, while knockdown of ANXA3 in Huh7 led to the complete inhibition of tumor growth (Figure 3A). Histological analysis revealed that the tumor xenografts displayed an HCC phenotype (Figure 3A), whereas immunohistochemistry (IHC) staining for ANXA3 confirmed preferential ANXA3 expression in the overexpressing clones (Figure 3B). MHCC97L-ANXA3 xenografts also displayed an enhanced proliferation rate, as confirmed by proliferating cell nuclear antigen (PCNA) staining (Figure 3B). Next, we examined the roles of ANXA3 in HCC metastasis in vivo by orthotopic implantation of the luciferase-labeled MHCC97L-EV- and MHCC97L-ANXA3-overexpressing cells into the livers of immunodeficient mice. Extrahepatic lung metastasis was monitored by ex vivo bioluminescence imaging. Lung metastasis was detected in six out of six mice in the ANXA3-overexpressing group, in contrast to only two out of six mice in the EV group (Figure 3C). Metastasis of tumor cells from the liver to lung was further confirmed by H&E staining with the presence of tumor nodules in the lungs (Figure 3C). Two cardinal properties of CSCs are tumor initiation and self-renewal, which can only be tested in vivo by limiting dilution and serial transplantation. To assess the degree by which enhanced ANXA3 supports growth and maintenance of CD133+ liver CSCs, Huh7 cells with or without ANXA3 repressed were sorted into CD133+ and CD133− subpopulations and injected subcutaneously into non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice at limited dilutions. CD133+ANXA3low cells exhibited attenuated tumor initiation at limiting dilutions when compared with CD133+ANXA3high cells, as evidenced by reduced tumor incidence, delayed tumor latency, and a lower estimated tumor-initiating cell frequency (Figure 3D). The reduced tumor-initiating ability of CD133+ANXA3low cells was comparable to that exhibited by CD133−ANXA3high cells, indicating the critical role of ANXA3 in mediating tumor initiation in vivo. Self-renewal ability of cells was then examined by serial transplantation of primary xenografts into secondary mouse recipients. Single cells isolated from the four groups were resorted into CD133+ and CD133− subsets and injected subcutaneously into NOD/SCID mice at limited dilutions for secondary transplantation. CD133+ANXA3low cells had a diminished ability to reconstitute tumor formation in secondary transplantations compared with CD133+ANXA3high cells. Note that the estimated tumor-initiating cell number was only found enriched in secondary tumors generated from CD133+ANXA3high cells, but not in the other groups. After serial propagation, only one out of four mice in the CD133−ANXA3high group and none of the cells in the CD133−ANXA3low group were able to form secondary tumors, indicating that CD133+ liver CSCs mediate self-renewal through ANXA3 (Figure 3D). This demonstrates that ANXA3 plays a critical role in driving tumor initiation, self-renewal, and metastasis in HCC.
Figure 3.

Endogenous ANXA3 Contributes to Augmented Tumor-Initiating, Self-Renewal, and Metastatic Potential In Vivo
(A) Top: representative xenograft tumors derived from MHCC97L-EV and -ANXA3 O/E or Huh7-NTC and -ANXA3 knockdown clones 244 and 246 cells 4 weeks after subcutaneous injection (n = 5–6). Scale bar, 1 cm. Bottom: H&E- images of tumors derived from MHCC97L-EV, -ANXA3 O/E, and Huh7-NTC cells. Scale bar, 200 μm.
(B) IHC staining for expression of ANXA3 and PCNA in the resected xenograft tumors derived from MHCC97L-EV and -ANXA3 O/E cells. Scale bar, 200 μm.
(C) Ex vivo imaging of lungs harvested from mice that received orthotopic injections of MHCC97L-EV or -ANXA3 O/E cells in the liver. Luciferase signals shown as dot plot. H&E images of lung tissue harvested. Scale bars represent 1 cm (top) and 100 μm (bottom). Results represent mean ± SD of six mice from one independent experiment.
(D) Top: images of tumors (black arrows) formed in NOD/SCID mice injected subcutaneously with CD133+ANXA3+, CD133+ANXA3−, CD133−ANXA3+, and CD133−ANXA3− cells isolated from Huh7 (picture representative of a 5,000-cell injection) (n = 8 for primary and n = 4 for secondary implantations). Scale bar, 1 cm. Bottom: engraftment rates of CD133+ and CD133− subsets with or without ANXA3 repressed in Huh7.
See also Figures S2 and S3.
Secretory ANXA3 Confers Tumorigenic, Metastatic, and Self-Renewal Abilities in HCC
Given that we found secretory ANXA3 to be detected in HCC clinical sera samples, we further sought to examine the functions of secretory ANXA3 in the maintenance of CSC-like properties in HCC. Conditioned medium from ANXA3-overexpressing MIHA or MHCC97L cells were harvested for co-culture with both CD133 and ANXA3 absent parental HCC cells (Figure 4A). In addition, to provide direct evidence that it is indeed secretory ANXA3, and not other factors in the conditioned medium, that is responsible for promoting these properties, ANXA3-absent HCC cells were also treated with recombinant ANXA3 proteins (Figure 4A). HCC cells co-cultured with conditioned medium collected from ANXA3-overexpressing cells exhibited a greater ability to migrate, invade (Figure 4B), form hepatospheres (Figure 4C), and resist apoptosis as well as chemotherapy (Figure 4D), as compared to HCC cells co-cultured with conditioned medium collected from controls. Similar results were obtained when MIHA cells, absent of both ANXA3 and CD133, showed enhanced ability to migrate, form hepatospheres, and induce capillary tube formation following treatment with recombinant ANXA3 proteins (Figure 4E). Our in vitro observations were also substantiated in vivo. MHCC97L cells resuspended in either conditioned medium collected from ANXA3-overexpressing or EV controls were injected subcutaneously into immunodeficient mice. For 2 weeks following inoculation of cells, concentrated conditioned media were injected subcutaneously at the site of the tumor inoculation. Tumor-initiating ability was significantly potentiated in MHCC97L cells co-cultured with ANXA3-containing conditioned medium, where tumors were detected in all eight animals. In contrast, only four out of eight mice inoculated with MHCC97L cells mixed with EV-control conditioned medium developed tumor nodules. ANXA3-containing conditioned medium not only enhanced the tumor-initiating capacity of these cells but also promoted tumor growth, as evidenced by the larger tumors that were formed (Figure 4F). Collectively, this suggests that secretory ANXA3 is indeed pivotal for tumor initiation, growth, metastasis, and chemoresistance.
Figure 4.

Secretory ANXA3 Confers Enhanced Cancer and Stem Cell-like Properties in HCC In Vitro and In Vivo
(A) Secretory ANXA3 overexpression and recombinant ANXA3 model systems.
(B) Quantification of number of MIHA and MHCC97L cells that migrated or invaded following co-culture with empty vector control medium (EV CM) or ANXA3-containing conditioned medium (ANXA3 O/E CM). ∗p < 0.05. Scale bar, 200 μm. Results represent mean ± SD from three independent experiments.
(C) Quantification of hepatospheres formed following co-culture with EV CM or ANXA3 O/E CM. ∗∗∗p < 0.001. Scale bar, 100 μm. Results represent mean ± SD of 12 replicates in three independent experiments.
(D) Percentage of Annexin-V-positive cells in MIHA and MHCC97L co-cultured with EV CM or ANXA3 O/E CM, following treatment with STS, 5-FU, or cisplatin. Results represent mean ± SD of three independent experiments.
(E) Top and bottom: quantification of number of MIHA that migrated and formed hepatospheres following treatment with recombinant ANXA3 protein (rANXA3). ∗p < 0.05 and ∗∗p < 0.01. Results represent mean ± SD from three independent experiments. Middle: quantification of capillary tubes formed by HUVECs following treatment with rANXA3. ∗∗∗p < 0.001. Scale bars represent 200 μm (top) and 100 μm (middle and bottom). Results represent mean ± SD of duplicate wells in three independent experiments.
(F) Representative xenograft tumors derived from MHCC97L cells co-cultured with EV CM or ANXA3 O/E CM 4 weeks after subcutaneous injection (n = 8). Dot plot shows the tumor weight of each xenograft. Results represent mean ± SD of eight mice from one independent experiment.
Exogenous ANXA3 Is Internalized through Caveolin-1-Dependent Endocytosis
Next, we delineated the mechanism by which exogenous ANXA3 binds to and enters cells to exert its oncogenic effects to promote HCC. We first confirmed that exogenous ANXA3 was indeed internalized. Recombinant SNAP-tagged ANXA3 protein was first labeled with fluorophore (red). PLC8024 HCC cells were also labeled with Vybrant carboxyfluorescein diacetate (CFDA) succinimidyl ester cell tracer dye (green). Following co-culture of fluorophore-tagged recombinant ANXA3 protein with fluorescein-isothiocyanate-labeled PLC8024 cells, SNAP-tagged ANXA3 proteins were clearly detected on both the membrane and in the cytoplasm of PLC8024 cells by confocal microscopy (Figure 5A). This observation was further substantiated by western blot where subcellular fractionation of HCC cells co-cultured with recombinant ANXA3 protein was analyzed. FLAG-tagged ANXA3 proteins present in the two fractions were pulled down by immunoprecipitation with FLAG-conjugated antibodies and subjected to SDS-PAGE. A 35-kDa band, concordant with the size of ANXA3 protein, was identified solely in the cytoplasm in the Coomassie-blue-stained gel, but not in the untreated control. Intensity of this band increased in a dose-dependent manner when increasingly more ANXA3 protein was added (Figure 5B). The identity of the band was confirmed to be ANXA3 by western blot and mass spectrometry analysis (data not shown). Recombinant ANXA3 was detected in both cytoplasmic and membrane subfractions, suggesting accumulating levels of the protein inside the cell over time (Figure 5C).
Figure 5.

Exogenous ANXA3 Is Internalized through Caveolin-1-Dependent Endocytosis
(A) Representative 2D and 3D confocal microscopy images of CFDA-stained PLC8024 following co-culture with SNAP-tagged ANXA3 proteins (SNAP-ANXA3).
(B) Coomassie-blue-stained SDS-PAGE gel showing cytoplasmic and membrane sub-fractions of PLC8024 treated with 45 or 90 μg of recombinant FLAG-ANXA3 protein (rANXA3). Arrow depicts 35-kDa band, which corresponds to rANXA3.
(C) Western blot showing expression of ANXA3 in cytoplasmic and membrane sub-factions of PLC8024 treated with 90 μg of rANXA3 and immunoprecipitated by FLAG antibody.
(D) Western blot showing expression of FLAG, ANXA3, and CAV1 in PLC8024 and MHCC97L with or without CAV1 stably repressed and with or without rANXA3 treatment.
(E) Western blot showing expression of FLAG, ANXA3, and CAV1 in PLC8024 with or without Myc-CAV1 stably overexpressed and with or without rANXA3 treatment.
(F) Western blot showing expression of FLAG and ANXA3 in PLC8024, MIHA, and MHCC97L co-treated with rANXA3, heparinase I, heparinase II, or their combination.
Representative images from three independent experiments are shown for all experiments.
Endocytosis represents a major mechanism for internalization of extracellular cargoes. Caveolin-1 proteins are main components of caveolae that can facilitate internalization of glycosylphosphatidylinositol (GPI)-anchor proteins and receptors into target cells through caveolar endocytosis. In view of the ability of ANXA3 protein to bind phospholipid membrane, we hypothesized that exogenous ANXA3 proteins are recruited to the caveolae and internalized through this mechanism. To this goal, caveolin-1 (CAV1) was stably repressed by small hairpin RNA knockdown. PLC8024 and MHCC97L cells with CAV1 depleted and co-cultured with recombinant FLAG-tagged ANXA3 protein had a reduced ability to bind and internalize exogenous ANXA3 as compared to HCC cells transfected with non-target controls, as indicated by decreased FLAG and ANXA3 expression levels (Figure 5D; sh-CAV1 clones 8001 and 8002). In contrast, overexpression of CAV1 in PLC8024 cells resulted in an increased internalization of FLAG-tagged ANA3 proteins (Figure 5E). Since previous reports have revealed other members of the annexin family (ANXA1, A2, A4, A5, and A6) to commonly bind to glycosaminoglycans (GAGs) such as heparin sulfate chains, we also explored whether ANXA3 could also be internalized via heparin sulfate proteoglycan (HSPG)-mediated endocytosis. PLC8024, MIHA, and MHCC97L cells treated with heparinase I, heparinase III, or their combination and co-cultured with recombinant FLAG-tagged ANXA3 protein did not alter the amount of ANXA3 proteins internalized (Figure 5F), suggesting that entry of exogenous of ANXA3 in HCC is dependent on caveolin-1-mediated, but not HSPG-mediated, endocytosis.
ANXA3 Induces a Feed-Forward Loop that Is Mediated by the MKK4/JNK Signaling Cascade
In an effort to characterize the molecular mechanism by which ANXA3 drives CSC-like properties in HCC, mRNA expression profiling was performed to compare the gene expression profiles of PLC8024 cells with or without ANXA3 repressed (Figure S4A; NTC versus sh-ANXA3 clone 246). Using a fold-change cutoff of >3, 2,372 differentially expressed genes were identified. Subsequent pathway analysis found many of the deregulated genes to be closely associated with JNK/AP-1 and MAPK, pathways, which have previously been shown to play a critical role in HCC pathogenesis (Figure 6A; Figure S4B) (Hagiwara et al., 2012; Jin et al., 2013). Deregulation of key players of the JNK pathway, including p-MKK4, JNK kinase activity, c-MYC, and p21, was subsequently validated by western blot in HCC cells with ANXA3 repressed (Huh7 and PLC8024) or overexpressed (MIHA) (Figure 6B). Similar to results obtained from ANXA3-overexpressing cells, the addition of exogenous ANXA3 proteins in CD133- and ANXA3-absent MIHA cells or culturing of the cells with conditioned medium collected from ANXA3-overexpressing cells resulted in JNK pathway activation, as evidenced by an increase in JNK activity, increased c-MYC expression, and reduced p21 expression (Figure 6C). A luciferase-reporter construct consisting of a consensus sequence of AP-1 binding site was transfected into Huh7 cells with or without ANXA3 repressed. Activity of AP-1, a known downstream target of JNK, was significantly reduced in the ANXA3-knockdown clones, further suggestive of the critical role of the ANXA3-JNK axis in CD133+ liver-CSC-driven HCC (Figure 6D). Consistent with this, we also found p-JNK to be preferentially expressed in CD133+ HCC cells (Figure S4C), while dual-color IF confirmed co-localization of ANXA3 and p-JNK, as well as CD133 and p-JNK in both Huh7 and PLC8024 cells (Figure 6E; Figures S4D and S4E). To substantiate the importance of the JNK/AP-1 pathway in ANXA3-driven HCC, we performed rescue experiments using the JNK-specific inhibitor (JNKi) SP600125. JNKi suppressed the oncogenic properties conferred by ANXA3 overexpression, as evidenced by the diminished abilities of HCC cells to form colonies, migrate, invade, induce angiogenesis, form hepatospheres, and resist apoptosis and chemotherapy (Figures 6F–6J). Interestingly, treatment of parental HCC cells or HCC cells overexpressing ANXA3 with JNKi resulted in not only a reduction in JNK activity and modulation of downstream target genes (c-MYC and p21) but also a marked decrease in ANXA3 expression, suggesting that ANXA3 induces a feed-forward loop that is mediated by MKK4/JNK signaling (Figures 6K–6L).
Figure 6.

Increased ANXA3 Expression Enhances JNK Signaling in CD133+ Liver CSCs
(A) Gene expression profiling coupled with GeneGo Metacore analysis in PLC8024 with or without ANXA3 repressed identified a deregulated JNK/AP-1 signaling network.
(B) Western blot showing expression of ANXA3, a key player in the JNK pathway (p-MKK4, JNK kinase activity, total JNK), and its downstream targets (c-MYC and p21) in Huh7, PLC8024, and MIHA with or without ANXA3 stably repressed or overexpressed.
(C) JNK kinase assay and western blot analysis of activity and expression of JNK-related proteins in MIHA co-cultured with ANXA3 O/E CM, rANXA3, or their controls.
(D) Luciferase reporter assay for activity of AP-1 transcription factor in Huh7 with or without ANXA3 stably repressed. ∗p < 0.05. Results represent mean ± SD from triplicate wells in three independent experiments.
(E) Dual-color IF images of ANXA3 (green) and p-JNK (red) in Huh7. Nuclei stained with DAPI (blue).
(F) Quantification of number of colonies formed in the indicated stable cell lines with or without JNK inhibitor. ∗∗∗p < 0.001. Scale bar, 5 mm. Results represent mean ± SD from triplicate wells in three independent experiments.
(G) Quantification of number of cells that migrated or invaded following co-culture with or without JNK inhibitor. ∗p < 0.05 and ∗p < 0.01. Scale bar, 100 μm. Results represent mean ± SD from three independent experiments.
(H) Quantification of capillary tubes formed by HUVECs following treatment with supernatant collected from the indicated cell lines with or without JNK inhibitor. ∗p < 0.05. Scale bar, 100 μm. Results represent mean ± SD of duplicate wells in three independent experiments.
(I) Quantification of hepatospheres with or without JNK inhibitor. Scale bar, 100 μm. Results represent mean ± SD of 12 replicates in three independent experiments.
(J) Percentage of Annexin-V-positive cells in the indicated stable cell lines with or without JNK inhibitor, following treatment with STS, 5-FU, or cisplatin. Results represent mean ± SD of three independent experiments.
(K and L) Western blot showing expression of ANXA3, c-MYC, and p21 in ANXA3-overexpressing MIHA (K) and parental Huh7 and PLC8024 (L) treated with 25 or 50 μM JNK inhibitor (SP600125).
See also Figure S4.
ANXA3 Neutralization Suppresses Growth and Self-Renewal of HCC In Vivo, Sensitizes HCC to Chemotherapy, and Eradicates the CD133 Liver CSC Subset
In light of the implications of ANXA3 in CD133+ liver CSCs and in HCC, we subsequently developed a monoclonal antibody specific against ANXA3 (anti-ANXA3 mAb) and tested for its application as a therapeutic treatment against HCC. Specificity of the antibody was confirmed by western blot on a panel of HCC cell lines where a single 33-kDa band was detected, corresponding to ANXA3 (Figure 7A). The expression levels of ANXA3 obtained were concordant with findings using a commercially validated ANXA3 antibody. Cell proliferation rates of CD133- and ANXA3-positive Huh7 cells were significantly inhibited with the addition of anti-ANXA3 mAb, while the CD133- and ANXA3-absent immortalized normal liver cell line MIHA remained unresponsive (Figure 7B). Treatment of Huh7 with anti-ANXA3 mAb resulted in a marked increase in apoptotic cells, suggesting that cell death was a factor causing the inhibition of cell proliferation (Figure 7C). In addition, anti-ANXA3 mAb also reduced the abilities of HCC cells to migrate, invade, induce angiogenesis, and form hepatospheres (Figures 7D–7F). HCC cells treated with a combination of anti-ANXA3 mAb and cisplatin synergistically inhibited cell proliferation in vitro and sensitized HCC cells to cisplatin (Figure 7G). Consistently, a similar trend was also observed when anti-ANXA3 mAb was used for treatment of secretory ANXA3. MIHA and MHCC97L cells, both negative for CD133 and ANXA3, were stably transfected with ANXA3 or EV control. Conditioned medium from these cells were then collected and used for various functional experiments. Addition of ANXA3 mAb in HCC cells treated with conditioned medium collected from ANXA3-overexpressing cells resulted in a reduced ability of the cells to migrate, invade, induce angiogenesis, form hepatospheres, and resist apoptosis and chemotherapy (Figures S5A and S5B). This observation was confirmed in vivo when treatment of Huh7 xenografts in immunodeficient mice with anti-ANXA3 mAb alone or in combination with cisplatin resulted in a marked reduction in tumor volume (Figure 7H; Figure S5C). Tumor growth was markedly suppressed in mice treated with increasing concentrations of anti-ANXA3 mAb. When anti-ANXA3 mAb was administered in combination with cisplatin, the growth of the tumor engraftments was inhibited by as much as 90% compared with the controls (PBS or immunoglobulin G [IgG]). Although cisplatin treatment alone led to a bigger reduction in tumor volume as compared to anti-ANXA3 mAb (Figure 7H), the residual xenografts, when serially transplanted into secondary mouse recipients, formed the largest tumors (Figure 7I). Residual xenografts from anti-ANXA3 mAb or mAb and cisplatin treatment failed to give rise to tumors in serial transplantations (Figure 7I), suggesting that a subset of CSCs is enriched by chemotherapy treatment. Subsequent analysis of the residual xenografts by flow cytometry for CD133 confirmed this hypothesis, where proportion of CD133+ cells was found enriched after chemotherapy, while the proportion of CD133+ cells decreased following treatment with anti-ANXA3 mAb alone or in combination with cisplatin (Figure 7J; Figure S5E). In addition to CD133, other liver CSC markers known to have overlapping expression with CD133, including CD24 (Lee et al., 2011) and EpCAM (Yamashita et al., 2009), were also found to be diminished upon antibody treatment (Figure S5F). H&E staining and IHC were performed on tissue sections from the resected tumor residuals (Figure 7K; Figure S5D). Necrosis was only observed in the xenografts treated with anti-ANXA3 mAb alone or in combination with cisplatin. ANXA3 expression was increased in cisplatin-treated xenografts but was markedly reduced in anti-ANXA3 mAb-treated mice. A concomitant decrease in PCNA was also observed in xenografts treated with anti-ANXA3 mAb with or without cisplatin, indicative of attenuated proliferative potential. None of the mice showed signs of disability, behavior abnormalities, or significant changes in body weight. Compared to untreated control or IgG-treated mice, no additional tissue damage was observed in other vital organs (Figure S5G). Mechanistically, treatment of HCC cells with anti-ANXA3 mAb in vitro and in vivo similarly led to a suppressed JNK pathway (Figure 7L).
Figure 7.

ANXA3 Ablation Reduces Cancer and Stem Cell-like Properties in HCC In Vitro, Attenuates Tumor Formation In Vivo, and Decreases Liver CSC Subpopulations
(A) Western blot showing specificity of the ANXA3 monoclonal antibody in a panel of liver cell lines.
(B) XTT assay showing growth rates of Huh7 and MIHA in the absence or presence of ANXA3 mAb. ∗p < 0.05. Results represent mean ± SD of triplicate wells in three independent experiments.
(C) Percentage of Annexin-V-positive cells in Huh7 following treatment with 25 μg/ml IgG control or ANXA3 mAb. Results represent mean ± SD from three independent experiments.
(D) Quantification of cells that migrated or invaded following treatment with IgG control or ANXA3 mAb. ∗p < 0.05 and ∗∗∗p < 0.001. Scale bar, 100 μm. Results represent mean ± SD from three independent experiments.
(E) Quantification of capillary tubes formed by HUVECs following treatment with supernatant collected from HCC cells treated with IgG control or ANXA3 mAb. ∗p < 0.05. Scale bar, 100 μm. Results represent mean ± SD of duplicate wells in three independent experiments.
(F) Quantification of hepatospheres in Huh7 and clinical samples following treatment with IgG control or ANXA3 mAb. ∗p < 0.05 and ∗∗∗p < 0.01. Scale bar, 100 μm. Results represent mean ± SD of 12 replicates in three independent experiments.
(G) XTT assay showing percentage growth inhibition in Huh7 following treatment with ANXA3 mAb, cisplatin, their combination, or their controls. ∗p < 0.05. Results represent mean ± SD of triplicate wells in three independent experiments.
(H) Representative xenograft tumors resected from mice treated with PBS, IgG control, ANXA3 mAb, cisplatin, or a combination of ANXA3 mAb and cisplatin (n = 5). Graph of average tumor volumes of mice along treatment course. Red arrows indicate the days when treatment was administered. Scale bar, 1 cm. Results represent mean ± SD of five mice from one independent experiment.
(I) Representative images of secondary tumors (black arrows) formed in NOD/SCID mice injected subcutaneously with cells harvested from residual primary tumors shown in (A).
(J) Flow cytometry for CD133 in residual xenografts of the indicated treatment groups.
(K) H&E and IHC staining for expression of ANXA3 and PCNA in the resected xenograft tumors. Scale bar, 200 μm.
(L) JNK kinase assay and western blot analysis of activity and expression of JNK-related proteins in Huh7 or HCC xenografts resected from mice treated with IgG control or ANXA3 mAb. Images shown of data gathered from n = 5 mice for in vivo studies shown in (H)–(K).
See also Figure S5.
Discussion
Frequent tumor relapse in multiple tumor types has now been attributed to the presence of residual CSCs after conventional treatments. We and others have previously identified CD133 to mark a liver CSC subpopulation in HCC (Ma et al., 2007, 2010). Yet, the functional paths by which these cells promote hepatocarcinogenesis remain limited, significantly impeding our efforts in developing CSC-specific therapies. We characterized the mRNA transcriptome of CD133+ and CD133− subpopulations in HCC by RNA-seq. Pathway enrichment analysis found the CD133+ subset to be tightly associated with an activated MAPK pathway, which is in concordant with our previous study, where we found CD133 to promote angiogenesis through IL-8-activated ERK (Tang et al., 2012). In addition, our CD133− non-CSC subset was also found to be enriched for genes critical in hepatocyte differentiation. This result is also consistent with our finding where CD133− cells were found to be unable to differentiate into skeletal and cardiac lineages, while in contrast, CD133+ liver CSCs could efficiently differentiate into non-hepatocyte-like, angiomyogenic-like cells following cell-directed differentiation in vitro (Ma et al., 2007), suggesting that CD133− cells exist in a terminally differentiated state. Of interest, one-third of the differentially expressed genes identified were found to encode for secretory proteins. There is accumulating data to show that maintenance of cancer and stemness properties is dependent on the microenvironment in which deregulated secreting factors can communicate through paracrine or autocrine signaling. Given the importance of secretory factors in modulating CSC features and that they can be found accessible in the conditioned media of cells or serum in patients, thus making them prime druggable targets for novel therapy development, we focused our studies in this area.
The focus of ANXA3 research in the past has centered on its expression in various diseases. A number of studies have found ANXA3 to be frequently overexpressed in ovarian, breast, colon, lung, gastric, gallbladder, testicular, and urothelial cancers (Köllermann et al., 2008; Schostak et al., 2009; Liu et al., 2009; Yan et al., 2010; Wu et al., 2013). In contrast, ANXA3 was also found to be downregulated in prostate and papillary thyroid cancers (Wu et al., 2013). In recent years, a small number of studies have identified ANXA3 to be secreted, where low levels of ANXA3 present in the urine were found to have diagnostic significance for early prostate cancer (Schostak et al., 2009; Yin et al., 2012) and secretion of ANXA3 from ovarian cancer cells is associated with platinum resistance. Here, we found ANXA3 to represent the most significantly upregulated gene that encoded for a secretory protein in the CD133+ liver CSC subset. Haraguchi et al. also likewise found ANXA3 to be preferentially expressed in the side population (SP) isolated from Huh7 cells (Haraguchi et al., 2006). SP cells from HCC were subsequently characterized to also express CD133. Specifically in HCC, Tong et al. found ANXA3 to be upregulated in 5-FU-resistant HCC cells and that silencing of ANXA3 by RNAi resulted in enhanced sensitivity of HCC cells to chemotherapy (Tong et al., 2012). By mass-spectrometry-based profiling, Tsai et al. identified several altered proteins, including ANXA3, to be highly expressed in Huh7 CD133+ liver CSCs (Tsai et al., 2012). Pan et al. also reported the preferential upregulation of endogenous ANXA3 in CD133+ cells isolated from Huh7 and that ANXA3 is required for sphere formation, tumor initiation, migration, invasion, and chemoresistance in HCC cells via a deregulated HIF1α/NOTCH pathway (Pan et al., 2013, 2015). These studies do suggest the importance of ANXA3 in CD133+ liver CSCs and in HCC. However, since the authors did not perform in vivo transplantation and serial propagations at limited dilutions, which remains the gold standard to test tumorigenicity and self-renewal, the reports are at best suggestive. Further, their work only looked at cytoplasmic ANXA3. Our present study definitively reports the functional role of ANXA3 in mediating CSC-like properties in HCC. Our work also demonstrates the clinical relevance and functional significance of secretory ANXA3 in HCC. Whether ANXA3 can be developed as a standalone biomarker or used in combination with AFP for clinical use warrants further investigation in a larger patient sample cohort. Our functional studies identified the role of endogenous and exogenous ANXA3 in conferring CSC-like properties.
Next, we unraveled a mechanism by which exogenous ANXA3 proteins are internalized in HCC cells and characterized the mechanism by which ANXA3 promotes CSC-like properties following its internalization. There has been no study regarding the route of entry of exogenous ANXA3 in any cell type thus far. We found exogenous ANXA3 to be internalized into HCC cells through caveolin-1-mediated, but not HSPG-mediated, endocytosis. Consistently, data obtained from our previous mRNA expression profiles (Tang et al., 2012) also found caveolin-1 to be upregulated in the CD133+ subsets, suggesting an autocrine regulation through the secretion and internalization of ANXA3. Through gene expression profiling coupled with functional rescue experiments, we found ANXA3 to activate the JNK pathway in CD133+ liver CSCs. Multiple studies have found JNK to drive HCC (Hagiwara et al., 2012; Jin et al., 2013). One recent study also found JNK activation to regulate self-renewal and tumor initiation in CD133+ glioblastoma stem cells (Yoon et al., 2012). Through rescue experiments, we substantiated the importance of JNK pathway in mediating ANXA3-driven CSC-like features. Blockade of the JNK pathway in turn caused a reduction of ANXA3 expression, suggesting the existence of a positive feedback loop regulating ANXA3 expression in HCC. Our group has also initiated some studies to clarify the mechanism by which ANXA3 is secreted, and we have pilot data (not shown) to suggest that ANXA3 is secreted from CD133-expressing HCC cells as exosomes. However, more work is needed to validate this observation.
The last part of our study centered on the therapeutic potential of targeting secretory ANXA3 through the use of a neutralizing antibody. Here, we provide data to show that ANXA3 sequestration by our newly developed mAb resulted in attenuation of CSC-like properties via the suppression of JNK. Not only did the anti-ANA3 mAb suppress CSC properties, but also CSC content was depleted with a marked reduction in the expression of CD133, CD24, and EpCAM. We also found that when administered in combination with cisplatin, the mAb would exert a synergistic inhibitory effect against HCC. Emerging studies have suggested the possibility of CSC replenishment through dedifferentiation of cancer cells. Therefore, combination treatment therapy that targets both the CSC subsets and the differentiated cancer cells represents an ideal therapeutic regimen to attain complete eradication of cancer. Collectively, findings presented in this study provide evidence to show the clinical relevance, functional significance, and therapeutic implication of both endogenous and secretory ANXA3 in CD133+ liver CSCs and HCC. We believe that ANXA3 can be used as a novel biomarker for the better detection of HCC and that targeting ANXA3 can be potentially developed as a novel treatment regime for this disease.
Experimental Procedures
Cell lines, patient samples, reagents, plasmids, in vitro and in vivo assays, antibody production, microscopy, and statistical analyses are described in Supplemental Experimental Procedures.
Author Contributions
M.T. and S.M. conceived the project. M.T., K.-Y.N., S.T.L., T.K.L., and S.M. performed the experiments and analyzed the data. M.T., T.-M.F., and C.-H.L. performed statistical analysis. J.W.Y. provided reagents for caveolin-1 studies. K.W.C. provided advice on histology. F.N. and B.-J.Z. provided help with sorting. C.M.L, K.M., X.-Y.G., Y.-F.Y., and D.X. obtained consent from patients and provided the clinical samples and patient information. M.T. and S.M. wrote the manuscript.
Acknowledgments
We thank the Faculty Core Facility at the Faculty of Medicine, HKU for providing and maintaining the equipment needed for flow cytometry, sorting, animal imaging, and confocal microscopy. We also thank Yuen-Piu Chan and Pak-Shing Kwan for their assistance and help with statistical analysis. This study is supported by the RGC GRF (HKU_774513M, HKU_773412M) and CRF (C7027-14G), HMRF (12110792), the NSFC Science Fund for Young Scholars (81302171), and a Croucher Innovation Award (to S.M.). A patent application has been filed for the anti-ANXA3 mAb (US14/485,206).
Published: June 18, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, five figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.05.013.
Accession Numbers
The GEO accession number for the RNA-seq data reported in this paper is GSE62905.
Supplemental Information
References
- Hagiwara S., Kudo M., Nagai T., Inoue T., Ueshima K., Nishida N., Watanabe T., Sakurai T. Activation of JNK and high expression level of CD133 predict a poor response to sorafenib in hepatocellular carcinoma. Br. J. Cancer. 2012;106:1997–2003. doi: 10.1038/bjc.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi N., Utsunomiya T., Inoue H., Tanaka F., Mimori K., Barnard G.F., Mori M. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells. 2006;24:506–513. doi: 10.1634/stemcells.2005-0282. [DOI] [PubMed] [Google Scholar]
- Harashima M., Harada K., Ito Y., Hyuga M., Seki T., Ariga T., Yamaguchi T., Niimi S. Annexin A3 expression increases in hepatocytes and is regulated by hepatocyte growth factor in rat liver regeneration. J. Biochem. 2008;143:537–545. doi: 10.1093/jb/mvm248. [DOI] [PubMed] [Google Scholar]
- Jin Y., Mao J., Wang H., Hou Z., Ma W., Zhang J., Wang B., Huang Y., Zang S., Tang J., Li L. Enhanced tumorigenesis and lymphatic metastasis of CD133+ hepatocarcinoma ascites syngeneic cell lines mediated by JNK signaling pathway in vitro and in vivo. Biomed. Pharmacother. 2013;67:337–345. doi: 10.1016/j.biopha.2013.02.006. [DOI] [PubMed] [Google Scholar]
- Köllermann J., Schlomm T., Bang H., Schwall G.P., von Eichel-Streiber C., Simon R., Schostak M., Huland H., Berg W., Sauter G. Expression and prognostic relevance of annexin A3 in prostate cancer. Eur. Urol. 2008;54:1314–1323. doi: 10.1016/j.eururo.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Lee T.K., Castilho A., Cheung V.C., Tang K.H., Ma S., Ng I.O. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9:50–63. doi: 10.1016/j.stem.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Liu Y.F., Xiao Z.Q., Li M.X., Li M.Y., Zhang P.F., Li C., Li F., Chen Y.H., Yi H., Yao H.X., Chen Z.C. Quantitative proteome analysis reveals annexin A3 as a novel biomarker in lung adenocarcinoma. J. Pathol. 2009;217:54–64. doi: 10.1002/path.2429. [DOI] [PubMed] [Google Scholar]
- Ma S., Chan K.W., Hu L., Lee T.K., Wo J.Y., Ng I.O., Zheng B.J., Guan X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–2556. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Ma S., Lee T.K., Zheng B.J., Chan K.W., Guan X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–1758. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- Ma S., Tang K.H., Chan Y.P., Lee T.K., Kwan P.S., Castilho A., Ng I., Man K., Wong N., To K.F. miR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell. 2010;7:694–707. doi: 10.1016/j.stem.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Pan Q.Z., Pan K., Weng D.S., Zhao J.J., Zhang X.F., Wang D.D., Lv L., Jiang S.S., Zheng H.X., Xia J.C. Annexin A3 promotes tumorigenesis and resistance to chemotherapy in hepatocellular carcinoma. Mol. Carcinog. 2013 doi: 10.1002/mc.22126. Published online December 23, 2013. [DOI] [PubMed] [Google Scholar]
- Pan Q.Z., Pan K., Wang Q.J., Weng D.S., Zhao J.J., Zheng H.X., Zhang X.F., Jiang S.S., Lv L., Tang Y. Annexin A3 as a potential target for immunotherapy of liver cancer stem-like cells. Stem Cells. 2015;33:354–366. doi: 10.1002/stem.1850. [DOI] [PubMed] [Google Scholar]
- Pardal R., Clarke M.F., Morrison S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- Park J.E., Lee D.H., Lee J.A., Park S.G., Kim N.S., Park B.C., Cho S. Annexin A3 is a potential angiogenic mediator. Biochem. Biophys. Res. Commun. 2005;337:1283–1287. doi: 10.1016/j.bbrc.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Raynal P., Pollard H.B. Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim. Biophys. Acta. 1994;1197:63–93. doi: 10.1016/0304-4157(94)90019-1. [DOI] [PubMed] [Google Scholar]
- Schostak M., Schwall G.P., Poznanović S., Groebe K., Müller M., Messinger D., Miller K., Krause H., Pelzer A., Horninger W. Annexin A3 in urine: a highly specific noninvasive marker for prostate cancer early detection. J. Urol. 2009;181:343–353. doi: 10.1016/j.juro.2008.08.119. [DOI] [PubMed] [Google Scholar]
- Tang K.H., Ma S., Lee T.K., Chan Y.P., Kwan P.S., Tong C.M., Ng I.O., Man K., To K.F., Lai P.B. CD133(+) liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology. 2012;55:807–820. doi: 10.1002/hep.24739. [DOI] [PubMed] [Google Scholar]
- Tong S.W., Yang Y.X., Hu H.D., An X., Ye F., Hu P., Ren H., Li S.L., Zhang D.Z. Proteomic investigation of 5-fluorouracil resistance in a human hepatocellular carcinoma cell line. J. Cell. Biochem. 2012;113:1671–1680. doi: 10.1002/jcb.24036. [DOI] [PubMed] [Google Scholar]
- Tsai S.T., Tsou C.C., Mao W.Y., Chang W.C., Han H.Y., Hsu W.L., Li C.L., Shen C.N., Chen C.H. Label-free quantitative proteomics of CD133-positive liver cancer stem cells. Proteome Sci. 2012;10:69. doi: 10.1186/1477-5956-10-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N., Liu S., Guo C., Hou Z., Sun M.Z. The role of annexin A3 playing in cancers. Clin. Transl. Oncol. 2013;15:106–110. doi: 10.1007/s12094-012-0928-6. [DOI] [PubMed] [Google Scholar]
- Yamashita T., Ji J., Budhu A., Forgues M., Yang W., Wang H.Y., Jia H., Ye Q., Qin L.X., Wauthier E. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X., Yin J., Yao H., Mao N., Yang Y., Pan L. Increased expression of annexin A3 is a mechanism of platinum resistance in ovarian cancer. Cancer Res. 2010;70:1616–1624. doi: 10.1158/0008-5472.CAN-09-3215. [DOI] [PubMed] [Google Scholar]
- Yin J., Yan X., Yao X., Zhang Y., Shan Y., Mao N., Yang Y., Pan L. Secretion of annexin A3 from ovarian cancer cells and its association with platinum resistance in ovarian cancer patients. J. Cell. Mol. Med. 2012;16:337–348. doi: 10.1111/j.1582-4934.2011.01316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon C.H., Kim M.J., Kim R.K., Lim E.J., Choi K.S., An S., Hwang S.G., Kang S.G., Suh Y., Park M.J., Lee S.J. c-Jun N-terminal kinase has a pivotal role in the maintenance of self-renewal and tumorigenicity in glioma stem-like cells. Oncogene. 2012;31:4655–4666. doi: 10.1038/onc.2011.634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.