

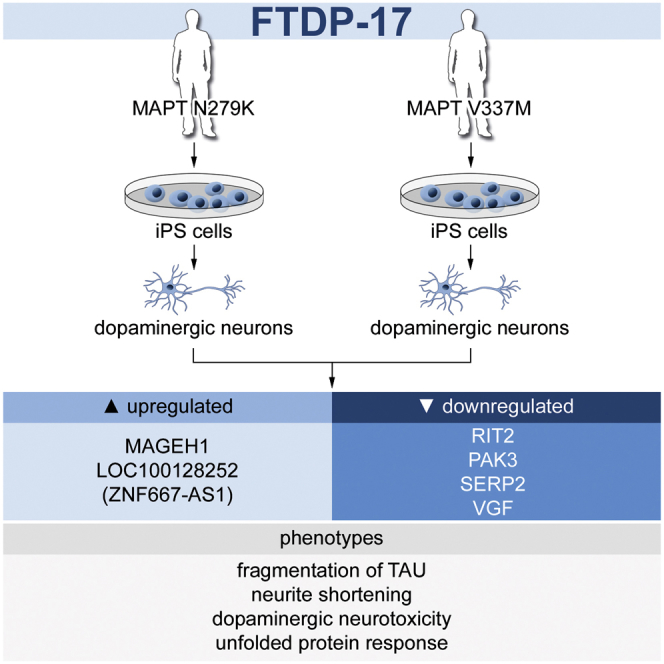
Summary
Frontotemporal dementia (FTD) is a frequent form of early-onset dementia and can be caused by mutations in MAPT encoding the microtubule-associated protein TAU. Because of limited availability of neural cells from patients’ brains, the underlying mechanisms of neurodegeneration in FTD are poorly understood. Here, we derived induced pluripotent stem cells (iPSCs) from individuals with FTD-associated MAPT mutations and differentiated them into mature neurons. Patient iPSC-derived neurons demonstrated pronounced TAU pathology with increased fragmentation and phospho-TAU immunoreactivity, decreased neurite extension, and increased but reversible oxidative stress response to inhibition of mitochondrial respiration. Furthermore, FTD neurons showed an activation of the unfolded protein response, and a transcriptome analysis demonstrated distinct, disease-associated gene expression profiles. These findings indicate distinct neurodegenerative changes in FTD caused by mutant TAU and highlight the unique opportunity to use neurons differentiated from patient-specific iPSCs to identify potential targets for drug screening purposes and therapeutic intervention.
Graphical Abstract
Highlights
-
•
FTD-iPSC-derived neurons show TAU pathology and disturbed neurite function
-
•
FTD neurons are vulnerable to oxidative stress and show an activation of the unfolded protein response
-
•
FTD neurons demonstrate distinct alterations in whole-genome gene expression
-
•
Validation of distinct disease phenotypes in post-mortem brain tissue of FTD patients
In this article, Hargus and colleagues generated iPSC-derived neurons from individuals carrying frontotemporal dementia-associated mutations in MAPT, the microtubule-associated protein TAU. These neurons showed pronounced TAU pathology, decreased neurite extension, an increased but reversible oxidative stress response, an activation of the unfolded protein response, and altered whole-genome expression profiles demonstrating distinct, mutant MAPT-associated neurodegenerative changes in these cells.
Introduction
Frontotemporal dementia (FTD) comprises a group of neurodegenerative diseases and is the second most common early-onset dementia after Alzheimer’s disease (AD), having a prevalence of ∼15–22/100,000 person-years (Boxer and Miller, 2005; Knopman and Roberts, 2011). FTD is characterized by cortical degeneration of the frontal and temporal lobes (frontotemporal lobar degeneration [FTLD]) leading to impairment of behavior, language, and cognition (Boxer and Miller, 2005; Goedert et al., 2012). About 20%–30% of patients have hereditary forms of FTD, and up to 15%–20% of these patients carry mutations in the MAPT gene located on chromosome 17q21, which encodes the microtubule-associated protein TAU (Goedert and Spillantini, 2011). FTD patients with MAPT mutations present with dementia and Parkinson-like motor impairment due to additional degeneration of subcortical brain areas, including the substantia nigra (SN). Thus, this form of FTD was termed FTD and Parkinsonism linked to chromosome 17 (FTDP-17; for review, see Ghetti et al., 2015).
TAU is a neuronal protein that stabilizes microtubules and axoplasmic transport, establishes neuronal polarity, mediates axonal outgrowth and dendritic positioning, and protects DNA from heat damage and oxidative stress (Noble et al., 2013). Six different TAU isoforms are expressed in the adult human brain as a result of alternative splicing of exons 2, 3, and 10 (Goedert et al., 1989). While exons 2 and 3 encode for amino-terminal TAU domains, exon 10 encodes for one of the four microtubule-binding domains in the carboxy-terminal half of TAU (Goedert et al., 1989). In the normal brain, the ratio of isoforms including exon 10 (4R isoforms) to those isoforms devoid of exon 10 (3R isoforms) is usually balanced, but in some FTDP-17 patients, this ratio is shifted toward 4R isoforms, leading to increased deposition of 4R TAU protein within neurons and glial cells (Goedert and Spillantini, 2011).
A pathological hallmark of FTDP-17 is the deposition of excessive amounts of hyperphosphorylated TAU (p-TAU) protein in neurons and glial cells in affected brain areas, including neural cells within the temporal cortex and dopaminergic (DA) neurons of the SN. Hyperphosphorylation of TAU is thought to suppress its ability to stabilize microtubules, resulting in axonal degeneration and eventual cell death (for review, see Noble et al., 2013). Deposition of p-TAU is not limited to FTD caused by mutant MAPT but is seen in about 40%–45% of all FTD patients, grouped together with FTDP-17 as FTLD-TAU, and includes patients with Pick’s disease, progressive supranuclear palsy (PSP), and corticobasal degeneration (Goedert et al., 2012; Irwin et al., 2015). Furthermore, excessive accumulation of p-TAU is found in patients with AD, which together with FTLD-TAU forms the large group of neurodegenerative tauopathies.
Despite thorough characterization of histopathological hallmarks in FTDP-17, mechanisms leading to neuronal degeneration remain largely elusive. This originates, at least in part, from the limited availability of viable neurons from patients’ brains. In this context, induced pluripotent stem cells (iPSCs) uniquely enable the identification of mechanisms of disease development in patient-derived neurons. As such, iPSCs have been widely used to study neurodegenerative changes in a variety of neurodegenerative disorders such as Parkinson’s disease (PD), AD, or FTD caused by mutant GRN or mutant C9ORF72 (Almeida et al., 2012, 2013; Cooper et al., 2012; Hargus et al., 2010; Reinhardt et al., 2013b; Soldner et al., 2009; for review, see Hargus et al., 2014b). Here, we generated iPSCs from individuals with MAPT mutations as an in vitro model of FTDP-17 to identify distinct neurodegenerative changes in FTDP-17.
Results
Derivation and Characterization of iPSCs
We derived iPSCs from individuals carrying the N279K MAPT mutation (referred to as FTDP-17-1) or the V337M MAPT mutation (referred to as FTDP-17-2), located in exons 10 and 12, respectively. iPSCs were generated by lentiviral transduction of fibroblasts with a polycistronic vector encoding OCT4, KLF4, SOX2, and C-MYC. The same procedure was applied to derive iPSCs from two healthy individuals (Ctrl-1 and Ctrl-2). An additional control iPSC line was also included in this study (Ctrl-3), which had been generated with the same four transcription factors (Reinhardt et al., 2013b). Altogether, four FTDP-17 iPSC clones (FTDP-17-1-I, FTDP-17-1-II, FTDP-17-2-I, and FTDP-17-2-II) and four Ctrl-iPSC clones (Ctrl-1-I, Ctrl-1-II, Ctrl-2-I, and Ctrl-3-I) were used in this study. Undifferentiated iPSCs expressed the pluripotency-associated markers OCT4, NANOG, SSEA-4, TRA 1-60, and TRA 1-81 (Figure 1A) and expressed OCT4, KLF4, SOX2, and C-MYC at levels comparable to those in H9 embryonic stem cells (ESCs) (Figure 1B). Sanger sequencing confirmed the N279K and the V337M MAPT mutation in FTDP-17-patient-derived stem cells (Figures 1C and 1D). Furthermore, we performed whole-genome microarray analysis on undifferentiated Ctrl- and FTDP-17 iPSCs (Figure 1E). This analysis revealed comparable expression profiles in all iPSC clones, which were similar to those of ESCs, but clearly differed from those of fibroblasts (Figure 1E). FTDP-17 and Ctrl cells carried normal karyotypes (Figure 1F), and teratoma formation assays confirmed pluripotency of iPSCs, which differentiated into mesodermal, endodermal, and ectodermal derivatives in vivo (Figure 1G).
Figure 1.

Derivation and Characterization of Ctrl and FTDP-17 iPSCs
(A) Immunostainings of FTDP-17-1 and FTDP-17-2 iPSCs demonstrating expression of pluripotency-associated markers. Scale bars represent 400 μm.
(B) qRT-PCR expression analysis of endogenous OCT4, SOX2, KLF4, and C-MYC in iPSC clones and H9 ESCs. Data are represented as mean of three replicates +SEM.
(C and D) DNA sequencing electropherograms from genomic DNA of differentiated Ctrl-1-I, Ctrl-2-I, FTDP-17-I-1, and FTDP-17-2-I iPSCs confirming the presence of the heterozygous N279K MAPT mutation in exon 10 in FTDP-17-1 cells and the heterozygous V337M MAPT mutation in exon 12 in FTDP-17-2 cells.
(E) Heatmap demonstrating whole-genome expression profiles of human fibroblasts (Fib), of undifferentiated Ctrl and FTDP-17 iPSCs, and of H1 and H9 ESCs.
(F) G-banded karyotype analysis of differentiated Ctrl and FTDP-17 iPSCs.
(G) H&E stainings of teratoma showing differentiation of FTDP-17-1 and FTDP-17-2 iPSCs into mesoderm, endoderm, and ectoderm in vivo. The scale bar represents 200 μm.
Neuronal Differentiation Is Not Impaired in FTDP-17 iPSCs
We have recently developed a protocol to efficiently generate neural progenitor cells and mature neurons from human pluripotent stem cells (Hargus et al., 2014a; Reinhardt et al., 2013a). Using this protocol, we observed efficient derivation of NESTIN+ and SOX1+ neural progenitor cells from both Ctrl- and FTDP-17 iPSCs (Figure 2A). Progenitor cells from both groups subsequently differentiated at similar efficiencies into mature βIII-TUBULIN+ neurons (Figure 2B). Approximately 45% of these neurons were γ-aminobutyric acid+ GABAergic neurons (Figures S1A and S1B), and about 50% were midbrain-like FOXA2+ and tyrosine hydroxylase+ (TH+) DA neurons, which are at significant risk in FTDP-17 (Figures 2B–2E; Slowinski et al., 2007); 2%–4% of all neurons were choline acetyltransferase+ cholinergic neurons (Figures S1C and S1D). To further characterize maturation of iPSC-derived neurons, we obtained electrophysiological recordings using the patch-clamp technique. Both Ctrl- and FTDP-17 neurons fired evoked action potentials with no obvious differences in size, shape, and amplitude (Figure 2F). Furthermore, whole cell voltage-clamp currents and tetrodotoxin (TTX) sensitivity of action potentials indicated functional voltage-gated Na+ channel inward currents in both Ctrl and FTDP-17 iPSC-derived neurons (Figure 2F). These data demonstrated proper maturation of iPSC-derived neurons in vitro and indicated that neuronal differentiation is not impaired in FTDP-17 iPSCs.
Figure 2.

Differentiation of Ctrl and FTDP-17 iPSCs into Mature Neurons
(A) Immunostainings for SOX1 (red) and NESTIN (green) demonstrating differentiation of Ctrl and FTDP-17 iPSCs into neural progenitor cells. Nuclei were counterstained with DAPI (blue). The scale bar represents 50 μm.
(B and C) Immunostainings for (B) TUJ1 (βIII-TUBULIN, red) and TH (green) as well as (C) for FOXA2 (red) and TH (green) showing differentiation of Ctrl and FTDP-17 iPSCs into mature midbrain-like DA neurons. Nuclei were counterstained with DAPI (blue). Scale bars represent 50 μm in (B) and 10 μm in (C).
(D and E) Quantification of immunostainings for (D) βIII-TUBULIN and (E) TH. Data are represented as mean of replicates from three independent differentiation experiments +SEM.
(F) Representative traces of evoked action potentials during current-clamp recordings of Ctrl and FTDP-17 neurons with inserts depicting single action potentials at −30 mV (n = 7; left). Whole-cell voltage-clamp currents (n = 7; middle) and TTX sensitivity of action potentials (n = 5; right) indicate functional voltage-gated Na+ channel inward currents. The indicated scales apply to the complete panel. All recordings were performed on cells from one differentiation experiment per line.
See also Figure S1.
Altered Expression of TAU in FTDP-17 iPSC-Derived Neurons
As mutant MAPT causes FTDP-17, we next set out to investigate whether FTDP-17 neurons exhibit altered expression of TAU on mRNA and protein levels. Thus, we first performed qPCR analysis for total TAU and for different TAU isoforms in differentiated Ctrl and FTDP-17 iPSCs (Figures 3A–3C and S2A). An analysis of total TAU expression revealed varying expression levels among the different iPSC clones but no significant differences between the Ctrl and FTDP-17 group (Figure 3A). Similarly, expression levels of TAU isoforms, including exon 2, did not differ between Ctrl and FTDP-17 neurons (Figure 3B). Isoforms containing exon 3 were not detected in any of the cultures. However, we observed mutation-specific differences in the expression of 4R TAU isoforms, which include exon 10 (Figures 3C, 3D, and S2A). FTDP-17-1-I/II neurons carrying the N279K mutation expressed significantly higher levels of 4R TAU, in contrast to FTDP-17-2-I/II neurons carrying the V337M mutation, which showed 4R TAU expression levels comparable to Ctrl neurons. This observation was important for the validation of our iPSC model of FTDP-17 since in brains of FTDP-17 patients, some MAPT mutations, including the N279K mutation, lead to missplicing of MAPT with an increased expression of 4R TAU, while others, including the V337M mutation, do not (Goedert and Spillantini, 2011).
Figure 3.

TAU Pathology in FTDP-17 iPSC-Derived Neurons
(A–C) qRT-PCR on Ctrl and FTDP-17 iPSC-derived neurons for (A) total TAU, for (B) TAU isoforms containing exon 2, and for (C) 4R TAU isoforms containing exon 10. Expression levels were normalized to Ctrl-1-I/II and GAPDH as housekeeping gene. Data are represented as mean of replicates from three independent experiments per indicated lines (n = 6) +SEM. One-way-ANOVA with post hoc Tukey test was performed for statistical analysis (∗∗∗p < 0.001).
(D) Electrophoretic separation of 3R and 4R TAU isoforms.
(E–I) Representative western blot analysis on Ctrl and FTDP-17 neurons for TAU protein using the HT7 (E–G) and TAU-5 (H and I) antibodies with the β-ACTIN antibody as loading control. Independent replicates are shown for each line. Quantification of full-length TAU (F) and TAU protein fragments (G) in Ctrl and FTDP-17 neurons using the HT7 antibody. Analysis of TAU-5 western blots showing quantification of ratios of TAU-fragments to full-length TAU (H) in Ctrl and FTDP-17 neurons.
(J and K) Representative western blot on CASPASE-cleaved TAU (J) and quantification of CASPASE-cleaved TAU normalized to full-length TAU (K) in Ctrl and FTDP-17 neurons.
(F–K) Blot quantifications are represented as mean of replicates from three independent differentiation experiments per indicated lines (n = 12 in F–H; n = 9 in K) +SEM. Student’s t test was performed for statistical analysis (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(L and M) Immunostainings of neurons for AT8 (red), TH (green), and DAPI (blue) (L) with quantification (M).
(N and O) Immunostainings of neurons for AT8 (red), MAP2 (green), and DAPI (blue) (N) with quantification (O).
(L–O) Data in (M) and (O) are represented as means of replicates from three independent differentiation experiments +SEM. One-way-ANOVA with post hoc Tukey test was performed for statistical analysis (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). Scale bars in (L) and (N) represent 50 μm.
See also Figure S2.
Next, we investigated the expression of TAU protein in Ctrl and FTDP-17 neurons by western blotting (Figures 3E–3K). By using the HT7 TAU antibody, we observed a band of around 52 kDA in both Ctrl- and FTDP-17 neurons, reflecting predominant expression of fetal TAU, as previously described for in vitro-differentiated human ESC-derived neurons (Iovino et al., 2010). Furthermore, we identified TAU fragments with a size of 25–27 kDa, which were only weakly expressed in Ctrl neurons but strongly expressed in FTDP-17 neurons (Figure 3E). Thus, it is possible that the N279K and the V337M MAPT mutations result in increased fragmentation of full-length TAU protein, as previously described for the A152T MAPT variant in differentiated neurons (Fong et al., 2013). To address this possibility, we quantified full-length TAU and fragments in Ctrl and FTDP-17 neurons and found increased amounts of TAU fragments in FTDP-17-1-I/II and FTDP-17-2-I/II neurons at the expense of full-length TAU (Figures 3F and 3G). These findings of increased TAU fragmentation were reproduced using three different TAU antibodies directed against the central core (TAU-5; Figures 3H and 3I), the C terminus (TAU-C17; Figures S2B and S2C), and the N terminus of TAU (TAU-A12; Figures S2D and S2E) as well as with an antibody that specifically recognizes CASPASE-cleaved TAU (TAU-C3; Figures 3J and 3K).
Next, we performed immunostainings on Ctrl and FTDP-17 neurons using the AT8 antibody, which detects p-TAU (Figures 3L–3O). In comparison to Ctrl cultures, we observed significantly increased numbers of AT8+/TH+ neurons in FTDP-17-1-I/II cultures (Figures 3L and 3M) and significantly increased numbers of AT8+/MAP-2+ neurons in FTDP-17-1-I/II and also FTDP-17-2-I/II cultures (Figures 3N and 3O). These findings are consistent with widespread expression of AT8+ p-TAU within the brains of FTDP-17 patients and mutation-specific deposition of AT8+ p-TAU in midbrain DA neurons of patients (Slowinski et al., 2007). In both Ctrl and FTDP-17 neurons, p-TAU showed a punctate expression pattern and was mainly localized to the axonal compartment with some additional perinuclear staining. An intracellular aggregation of p-TAU or a pathologic somatodendritic redistribution of p-TAU was not observed in our cells, as similarly seen in a previous study on iPSC-derived neurons carrying the A152T variant of MAPT (Fong et al., 2013).
Neurite Extension Is Impaired in FTDP-17 iPSC-Derived Neurons
Next, we wondered whether the pronounced TAU pathology in FTDP-17 neurons influenced microtubule function, as TAU is a microtubule-associated protein. Neurites from FTDP-17 neurons appeared much shorter and at smaller densities than neurites from Ctrl neurons. Thus, a neurite outgrowth assay, quantifying neurite length of βIII-TUBULIN+ neurons 24 and 48 hr after plating, was performed to identify any impairment of neurite extension (Figures 4A–4C). Already 24 hr after plating, the length of FTDP-17-1-I/II and FTDP-17-2-I/II neurons was mildly, but significantly, shorter than those of Ctrl neurons (Figure 4B). By 48 hr, Ctrl neurons had almost doubled their neurite extensions, while FTDP-17 neurons presented with similar neurite lengths as seen at 24 hr after plating (Figure 4B). Only FTDP-17-2-I/II neurons showed a mild but significant increase in neurite length between 24 and 48 hr after seeding (Figure 4B). We performed the same analysis on TH+ DA Ctrl and FTDP-17 neurons, which revealed similar results (Figure 4C). These observations indicated impaired neurite morphology and complexity in FTDP-17-iPSC-derived neurons.
Figure 4.

Decreased Neurite Outgrowth, Enhanced Vulnerability to Oxidative Stress, and Activation of the UPR in FTDP-17 Neurons
(A) Immunostainings of Ctrl and FTDP-17 neurons for TH (green) and TUJ1 (red) 48 hr after plating. Nuclei were stained with DAPI.
(B and C) Quantification of neurite outgrowth in (B) βIII-TUBULIN+ neurons and in (C) TH+ DA neurons 24 hr and 48 hr after plating. Neurite outgrowth was assessed by measurement of the longest neurite of each neuron. Data are represented as mean of at least 100 measurements per indicated lines from three independent experiments +SEM. Two-way ANOVA with post hoc Tukey test was used as statistical test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ###p < 0.001: denotes effect over time within groups).
(D) Effect of oxidative stress on FTDP-17 and Ctrl neurons analyzed by measurement of LDH release after 48 hr of rotenone treatment.
(E) Quantification of cleaved-CASPASE-3+ DA neurons in Ctrl and FTDP-17 cultures after 48 hr of rotenone treatment.
(F and G) Rescue of FTDP-17 neurons through treatment with (F) CoQ10 or (G) the GSK-3 inhibitor CHIR99021.
(D–G) Data are represented as mean of replicates from three independent experiments +SEM. One-way ANOVA with post hoc Tukey test was used as statistical test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(H) Western blot analyses of ER stress-associated markers PUMA, p-PERK, and BiP in Ctrl and FTDP-17 neurons. GAPDH or β-ACTIN was used as a loading control.
(I) Quantifications of protein expression in FTDP-17 neurons relative to expression in Ctrl neurons and to expression of GAPDH or β-ACTIN, respectively. Data are represented as mean of replicates from three independent differentiation experiments per indicated lines (n = 12) +SEM. Student’s t test was used as statistical test (∗p < 0.05; ∗∗∗p < 0.001).
Increased Oxidative Stress and Activation of the Unfolded Protein Response in FTDP-17 Neurons
FTDP-17 patients show pronounced and widespread neurodegeneration as a result of increased cellular vulnerability. Thus, we next investigated whether FTDP-17 neurons are more susceptible to oxidative stress. We measured responses of Ctrl and FTDP-17 neurons to rotenone, which induces oxidative stress by inhibiting complex I of the mitochondrial respiratory chain (Figures 4D–4G). This experiment revealed increased vulnerability of FTDP-17 neurons toward respiratory stress, as measured by enhanced lactate dehydrogenase (LDH) release 48 hr after application of either 100 or 200 nM rotenone (Figure 4D). Concomitantly, an increased number of cleaved-CASPASE-3+ neurons was detected in cultures of FTDP-17 neurons (Figure 4E). Notably, cell death in stressed FTDP-17 neurons significantly declined after application of the antioxidant coenzyme Q10 (0.5 and 1 μM) or the GSK-3 inhibitor CHIR99021 (0.5 and 1 μM; Figures 4F and 4G). These findings indicated that FTDP-17 neurons are more susceptible to oxidative stress and that reversal of oxidative stress is possible in FTDP-17 neurons.
In a variety of neurodegenerative diseases, misfolded proteins accumulate within the ER and activate an ER stress response, which eventually results in apoptosis (Schröder and Kaufman, 2005). Thus, we further investigated whether ER stress is involved in the pathogenesis of FTDP-17. ER stress response is known as the unfolded protein response (UPR) and involves an altered expression of the molecular chaperone binding immunoglobulin protein (BiP), which binds to the protein kinase RNA-like ER kinase (PERK) under unstressed conditions. Upon initiation of ER stress, BiP dissociates from PERK, leading to increased levels of activated phosphorylated PERK (p-PERK) with increased expression of pro-apoptotic proteins such as p53-upregulated modulator of apoptosis (PUMA; Tan et al., 2014). To investigate UPR activation in FTDP-17, we determined expression levels of BiP, p-PERK, and PUMA in FTDP-17 and Ctrl neurons by western blot analysis (Figures 4H and 4I). We found significantly increased levels of BiP, p-PERK, and PUMA in FTDP-17 neurons when compared with Ctrl neurons (Figures 4H and 4I). These findings indicated significant UPR activation in FTDP-17 neurons and pointed toward increased ER stress in FTDP-17.
Altered Gene Expression Profiles in FDTP-17 iPSC-Derived Neurons
We next performed whole-genome transcriptome analysis by microarrays on Ctrl- and FTDP-17 neurons to investigate whether gene expression profiles were altered in FTDP-17 neurons (Figure 5). As expected, expression profiles of both Ctrl- and FTDP-17 neurons differed significantly from undifferentiated iPSCs and formed a separate cluster in the hierarchical cluster analysis reflecting efficient neuronal maturation (Figures 5A and 5B). We investigated genes that were significantly upregulated or downregulated in FTDP-17-1 and FTDP-17-2 neurons in comparison to Ctrl cells (p values ≤ 0.01; Figures 5C–5F, S3A, and S3B; Table S1). This analysis revealed similar, overlapping gene expression profiles in FTDP-17-1 and FTDP-17-2 neurons with 59 upregulated genes in FTDP-17-1 and 42 upregulated genes in FTDP-17-2 neurons (Figure 5C). Similarly, compared with Ctrl neurons, 130 and 113 genes were downregulated in FTDP-17-1 and FTDP-17-2 neurons, respectively (Figure 5D). Functional annotation of these downregulated genes was associated with Gene Ontology (GO) terms such as altered monooxygenase activity (CYP46A1, TH), altered GTPase regulator activity (ARHGDIB, BCR, THY1, RAPGEF4), and disturbed cytoskeleton organization and biogenesis (ADRA2A, AMOT, ARHGDIB, THY1, RND1; Table S1). Downregulated genes also included PAK3, a kinase associated with cytoskeletal reorganization and mental retardation (Allen et al., 1998), and RIT2, a RAS family GTPase and a susceptibility factor for PD with decreased expression in the SN of patients (Pankratz et al., 2012). Furthermore, we observed significantly decreased expression of SERP2 (stress-associated ER protein family member 2) and VGF, which have been linked to ER and ER-associated stress (Figure 5E; Shimazawa et al., 2010). Functional annotation of upregulated genes was related to GO terms such as cell-cycle arrest (INHA) and cellular biosynthesis process (AK5, APOA1, INHA, RPSY4Y1, DMD; Table S1). Interestingly, particularly highly expressed genes in FTDP-17 neurons included LOC100128252 (ZNF667-AS1), a non-coding RNA transcript with currently unknown function, and MAGEH1, an intracellular protein, which interacts with the p75 receptor, a neurotrophin receptor with context-dependent functions promoting either cellular integrity or cellular death to neural cells (Figure 5F; Casaccia-Bonnefil et al., 1999; Tcherpakov et al., 2002).
Figure 5.

Gene Expression Profiles in FTDP-17 and Ctrl Neurons
Whole-genome transcriptome microarray analysis of Ctrl and FTDP-17 neurons derived from three independent differentiations.
(A and B) Whole genome expression profiles presented as (A) heatmap and (B) hierarchical clustering dendrogram.
(C and D) Venn diagram showing the overlap of genes significantly upregulated (C) or downregulated (D) in FTDP-17-1 and FTDP-17-2 neurons.
(E and F) Validation of differentially downregulated (E) and upregulated (F) genes by qRT-PCR. Data are represented as mean of replicates from three independent experiments per indicated lines +SEM. One-way ANOVA with post hoc Tukey test was used as statistical test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
(G) Relative gene expression of MAGEH1 in FTDP-17-1/2-I/IIMAGEH1-KD or FTDP-17-1/2-I/IIscr control neurons.
(H) Quantification of cleaved-CASPASE-3+ neurons in FTDP-17-1/2-I/IIMAGEH1-KD or FTDP-17-1/2-I/IIscr control neurons after 48 hr of rotenone treatment. At least 800 cells were counted per cell line and differentiation.
(I) Immunostainings of FTDP-17-1/2-I/IIMAGEH1-KD or FTDP-17-1/2-I/IIscr control neurons for βIII-TUBULIN 48 hr after plating as single cells. Nuclei were stained with DAPI.
(J) Quantification of neurite outgrowth in cultures of FTDP-17-1/2-I/IIMAGEH1-KD or FTDP-17-1/2-I/IIscr control neurons 48 hr after plating. At least 100 neurons were measured per cell line and differentiation.
(K) Relative expression of the indicated genes in FTDP-17-1/2-I/IIMAGEH1-KD or FTDP-17-1/2-I/IIscr control neurons.
(H, J, and K) Data are presented as mean of independent replicates from four different lines +SEM. Student’s t test was performed for statistical analysis (∗p < 0.05, ∗∗p < 0.01).
To investigate a potential role of MAGEH1 in the pathogenesis of FTDP-17, we examined the effects of a MAGEH1 gene knockdown in FTDP-17 neurons using a lentiviral shRNA expression system. We achieved MAGEH1 knockdown efficiencies of up to 80% compared with scrambled (scr)-shRNA-expressing FTDP-17 neurons (Figure 5G). We observed an increased susceptibility of FTDP-17 neurons with MAGEH1 knockdown (FTDP-17-1/2-I/IIMAGEH1-KD) to rotenone-induced oxidative stress, as evidenced by an increased number of cleaved-CASPASE-3 and βIII-TUBULIN double+ neurons compared with cultures of scr-shRNA expressing FTDP-17-1/2 neurons (FTDP-17-1/2-I/IIscr; Figure 5H). Concomitantly, FTDP-17-1/2-I/IIMAGEH1-KD neurons showed reduced neurite outgrowth compared with FTDP-17-1/2-I/IIscr neurons supporting a protective effect of MAGEH1 in FTDP-17 neurons (Figures 5I and 5J). We also found that knockdown of MAGEH1 in FTDP-17-1/2 neurons was followed by reduced expression of RIT2 and PAK3, which were already downregulated in FTDP-17-1/2 neurons compared with healthy Ctrl cells before the lentiviral treatment (Figures 5E and 5K).
To further elucidate the role of MAGEH1 in differentiated neurons, we overexpressed MAGEH1 in healthy control cells derived from Ctrl-1/2/3 iPSCs (Figures S3C–S3E). While we did not find any changes in neurite outgrowth after MAGEH1 overexpression, we observed a significant decrease in the number of cleaved-CASPASE-3+ neurons in rotenone-treated MAGEH1 overexpressing Ctrl cultures (Ctrl-1/2/3-I/IIMAGEH1) when compared with rotenone-treated healthy Ctrl cultures expressing red fluorescent protein (RFP; Ctrl-1/2/3-I/IIRFP). These data further supported a putative neuroprotective effect of MAGEH1 in mature neurons.
Some Pathological Changes Are Observed in the Brains of FTDP-17 Patients
Next, we investigated whether pathological changes in FTDP-17 iPSC-derived neurons were also observed in post-mortem brain tissue of FTDP-17 patients (Figures 6A–6H). We collected midbrain tissue from four patients carrying either the N279K mutation or the P301L mutation, another frequent, FTDP-17-causing mutation located in exon 10 of MAPT. Consistent with our findings in vitro, we found marked expression of AT8+ p-TAU in DA neurons of the SN and also in non-DA neurons in surrounding brain stem nuclei (Figures 6A and 6B). In contrast to FTDP-17 neurons in vitro, AT8+ p-TAU was deposited as cytoplasmic, perinuclear aggregates within neurons and as neurofibrillary tangles (NFTs) within axonal projections, which appeared morphologically disturbed (Figures 6A and 6B). AT8+ deposits were also identified in glial cells (Figure 6A). An analysis of AT8 distribution within the temporal cortex of FTDP-17 patients revealed similar results (Figure 6A). These changes were accompanied by significant neuronal cell loss.
Figure 6.

Identification of Pathological Changes in Post-mortem Tissue from FTDP-17 Patients
(A and B) Immunostaining for AT8 demonstrating pronounced p-TAU expression in neurons and glial cells in the midbrain (patients A–C) and temporal cortex (patient D) of FTDP-17 patients carrying the P301L MAPT mutation (patients A and B) or the N279K MAPT mutation (patients C and D). The scale bar represents 200 μm. Higher magnification image in (B) reveals AT8 immunoreactivity in FTDP-17 midbrain neurons with neuropil threads. The scale bar represents 20 μm.
(C) qRT-PCR for detection of 4R TAU isoforms in Ctrl individual and FTDP-17 patient midbrain samples.
(D) Immunostaining for the ER stress marker BiP in the midbrain (patients A–C) and temporal cortex (patient D) of FTDP-17 patients. The scale bar represents 200 μm.
(E) Western blot analysis of BiP expression in the midbrain of Ctrl individuals and FTDP-17 patients. GAPDH expression was used as loading control.
(F) Quantification of BiP expression in FTDP-17 patients relative to Ctrl A and normalized to GAPDH expression.
(G and H) qRT-PCR for detection of (G) LOC100128252 and (H) MAGEH1 in Ctrl individual and FTDP-17 patient midbrain samples.
(C, G, and H) Data were normalized to GAPDH expression and to Ctrl A and are represented as mean of triplicates from the same tissue preparation.
The expression of 4R TAU was increased in the patient’s brains (Figure 6C), and in line with our in vitro observations of increased UPR activation in FTDP-17 neurons, we found abundant expression of BiP protein within DA and non-DA neurons in histological specimen from all four FTDP-17 patients (Figure 6D). Since BiP is expressed to certain extents also under normal conditions, we quantified expression levels by western blot analysis and processed native, fresh-frozen midbrain tissue from two FTDP-17 patients and from two Ctrl individuals for molecular analyses. We observed increased levels of BiP protein in samples from FTDP-17 patients as similarly seen in in vitro-differentiated FTDP-17 iPSCs (Figures 6E and 6F).
Likewise, we found that some of the aforementioned dysregulated genes in FTDP-17 iPSC-derived neurons were also dysregulated in post-mortem tissue from FTDP-17 patients. Indeed, while PAK3, RIT2, SERP2, and VGF were not downregulated in post-mortem tissue of FTDP-17 patients, we observed that LOC100128252 and MAGEH1, which were particularly strongly expressed in FTDP-17 neurons, were also more strongly expressed in post-mortem tissue of FTDP-17 patients compared with Ctrl individuals (Figures 6G and 6H). These data further indicated that these two molecules with currently unknown function in the context of neurodegeneration could play a significant role during FTDP-17 pathogenesis.
Discussion
We used iPSC-derived neurons to identify distinct neurodegenerative changes in FTDP-17. We found that overall neuronal differentiation was not impaired in FTDP-17 iPSCs. However, FTDP-17 neurons presented with disturbed neurite morphology as well as with impaired neurite extension and exhibited distinct TAU pathology with increased fragmentation of full-length TAU protein and an increased immunoreactivity for AT8. FTDP-17 neurons displayed increased and reversible vulnerability toward oxidative stress and presented with activated UPR. Furthermore, we have identified genes, which were significantly dysregulated in FTDP-17 neurons, including an upregulation of LOC100128252 and MAGEH1. Notably, some of these pathologic changes could also be identified in post-mortem material from patients with FTDP-17.
FTDP-17 is caused by MAPT mutations, which lead to either increased expression of 4R TAU isoforms or to decreased interaction of TAU protein with microtubules (Goedert and Spillantini, 2011). As such, we reasoned to examine FTDP-17 iPSCs, which have been derived from individuals carrying MAPT mutations representing the former and the latter group, respectively. Only FTDP-17-1 neurons carrying the N279K mutation expressed increased amounts of 4R TAU transcripts, while FTDP-17-2 neurons carrying the V337M mutation showed normal 4R TAU expression levels, thus reflecting observations made in vivo. Despite these differences in 4R TAU expression, most of the observed FTDP-17 disease phenotypes were almost identical and appeared to similar extents in FTDP-17-1 and FTDP-17-2 neurons. The major mutation-specific difference comprised an increased expression of AT8+ p-TAU in FTDP-17-1 neurons, but not in FTDP-17-2 DA neurons. In contrast, non-DA neurons from both FTDP-17 patients presented with significantly increased AT8 positivity as compared with the Ctrl groups. These findings are in line with observations made in vivo, as deposition of AT8+ p-TAU is especially observed in SN DA neurons of patients carrying the N279K mutation. On the other hand, excessive and widespread AT8 positivity is found in non-DA neurons in brains of patients with the N279K mutation and in patients with the V337M mutation. Thus, findings in vivo were mimicked in vitro validating our iPSC-based model of FTDP-17.
Both the N279K and the V337M mutation render the TAU protein more susceptible to hyperphosphorylation (Alonso Adel et al., 2004). When tau becomes hyperphosphorylated, it detaches from microtubules and destabilizes microtubule assemblies, thereby affecting microtubule function (Noble et al., 2013). These findings could explain the impairment of neurite outgrowth in FTDP-17 neurons. Furthermore, we found an increased fragmentation of TAU protein in FTDP-17 neurons at the expense of full-length TAU, which most likely further contributed to disturbed neurite morphology. Increased TAU fragmentation and abnormal neurite morphologies were previously described in iPSC-derived neurons carrying the A152T variant of MAPT, which may increase the risk for the development of FTD, PSP, and AD (Fong et al., 2013). In this study, heterozygous A152T neurons showed a similar increase in TAU fragmentation as compared with gene-corrected control cells. This effect could further be potentiated by the introduction of a second mutation at the same site. Thus, these data support our observation that genetic changes in MAPT can have significant effects on the structural integrity of TAU. Fragmentation of TAU can occur through cleavage by several caspases including caspase-3 (Fasulo et al., 2000). We observed that increased TAU fragmentation in FTDP-17 neurons was at least in part mediated by CASPASE-driven proteolysis, as evidenced by the detection of a truncated TAU protein, which is recognized at its C terminus by a CASPASE-cleaved TAU-specific antibody. This CASPASE-associated fragmentation was not specific to the N279K and V337M mutation but also occurred to a similar extent in neurons carrying the A152T MAPT variant (Fong et al., 2013). Interestingly, however, our N279K and V337M mutant FTDP-17 neurons expressed an additional low-molecular-weight variant of CASPASE-cleaved TAU, indicating that a fraction of CASPASE-cleaved TAU was further truncated at its N terminus by additional proteases. These data encourage performing additional comparative studies on iPSC-derived neurons from patients with different MAPT mutations to better characterize mutation-specific expression profiles of proteases in FTDP-17.
TAU fragments carry an apoptosis-inducing activity, which is further potentiated when cells are exposed to TAU fragments carrying the N279K mutation (Fasulo et al., 2005). In addition, overexpression of full-length TAU carrying the N279K or the V337M mutation resulted in induction of apoptosis after serum deprivation (Furukawa et al., 2000). These findings are consistent with our data demonstrating an increased vulnerability of FTDP-17 neurons toward oxidative stress induced by the mitochondrial complex I inhibitor rotenone. This response was accompanied by an increased expression of cleaved-CASPASE-3. Rotenone-induced cytotoxicity has been observed in an iPSC-based model of PD (Reinhardt et al., 2013b), which is characterized by an increased vulnerability of midbrain DA neurons as similarly seen in FTDP-17. Increased cell death of FTDP-17 neurons could, at least in part and in addition to increased TAU fragmentation, be related to a direct impact of the N279K and V337M mutations on mitochondrial function. Such an effect was recently described for P301L mutant TAU, which when overexpressed in a neuroblastoma cell line led to reduced mitochondrial complex I activity and to an increased vulnerability toward H2O2-mediated oxidative stress (Schulz et al., 2012).
Stress responses were reversible, as the antioxidant coenzyme Q10 prevented cell death in challenged FTDP-17 neurons. Furthermore, GSK-3 inhibition in FTDP-17 neurons by CHIR99021 resulted in overall rescue of cell viability, consistent with a pro-apoptotic function of GSK-3 in the context of oxidative stress, as shown in SH-SY5Y cells (King and Jope, 2005). Furthermore, GSK-3-β showed increased activity in SH-SY5Y cells upon UPR activation (Song et al., 2002), which was readily observed in our FTDP-17 iPSC-derived neurons. These experiments provided proof of principle that an intervention is possible to prevent cell death in patients’ neurons at risk. At the same time, they highlight the unique potential of iPSCs to identify potentially beneficial compounds in FTDP-17, as human neurons can be produced efficiently and at high numbers for high-throughput drug screening purposes (Heilker et al., 2014).
While UPR is initially a protective response mechanism toward misfolded proteins, its prolonged activation can lead to the onset of apoptosis and eventually to cell death. We observed an upregulation of BiP protein in FTDP-17 neurons, which was accompanied by an increased expression of p-PERK and the apoptosis-inducing protein PUMA. These findings indicated increased ER stress as a contributing factor to FTDP-17. In line with this observation, we also found UPR activation in the brains of patients with FTDP-17. UPR activation has been described in brains of patients with AD and in cases of FTDL-TAU (Hoozemans et al., 2009; Nijholt et al., 2012). In neurons of AD patients, markers of UPR activation appeared at early stages of disease development and were co-expressed with AT8 prior to NFT formation (Hoozemans et al., 2009). Furthermore, a recent whole-genome association study for PSP identified a small nuclear polymorphism in EIF2AK3, the gene encoding PERK, as a risk factor for the development of PSP (Höglinger et al., 2011). These studies and our present data on FTDP-17 neurons thus demonstrate that TAU pathology is closely linked to ER stress in tauopathies. The mechanisms, however, by which TAU interferes with ER function and UPR are still unclear, as TAU is located in the cytoplasm and not in the ER (Abisambra et al., 2013).
Our whole-genome expression analysis identified dysregulated genes in FTDP-17 neurons. Downregulated genes included PAK3 and RIT2, which have been linked to mental retardation (Allen et al., 1998) and PD (Pankratz et al., 2012), respectively. Upregulated genes included LOC100128252 (ZNF667-AS1), a non-coding RNA transcript with currently unknown function, and the intracellular molecule MAGEH1, both of which were also highly expressed in the post-mortem brains of FTDP-17 patients. While further studies have to be performed to characterize the function of LOC100128252 in the context of FTDP-17, we could identify a beneficial role of MAGEH1 in patient-derived neurons. Our MAGEH1 knockdown experiments revealed increased susceptibility of FTDP-17 neurons to oxidative stress and demonstrated reduced neurite outgrowth. Accordingly, overexpression of MAGEH1 in healthy control neurons resulted in reduced vulnerability toward oxidative stress, suggesting that upregulation of MAGEH1 in FTDP-17 neurons represents a so far unknown endogenous neuroprotective mechanism. MAGEH1 can associate with the intracellular domain of the p75 receptor (Tcherpakov et al., 2002), which has been linked to cell survival and differentiation in neural progenitor cells (Casaccia-Bonnefil et al., 1999). It remains to be shown whether the beneficial effects of MAGEH1 in FTDP-17 neurons involve the p75 receptor and further studies are necessary to identify the mechanisms by which MAGEH1 promotes neuroprotection in FTDP-17 neurons.
In conclusion, our iPSC-based model of FTDP-17 is well suited for identifying pathways involved in neurodegenerative processes in FTDP-17 as it captures a large variety of disease-specific phenotypes and offers the possibility to analyze disease-affected cells in vitro. At the same time, it provides the unique opportunity to use these cells as a platform for drug screening purposes to establish therapeutic approaches in FTDP-17.
Experimental Procedures
Derivation of iPSCs
Fibroblasts from individuals with MAPT mutations, here referred to as FTDP-17 patients, were obtained from the Coriell Institute for Medical Research and were reprogrammed using a lentiviral SF-OSKM-tomato expression vector. See the Supplemental Experimental Procedures for further details, which also includes a list of antibodies used in this study (Table S2) and a list of the primers employed for qRT-PCR analyses (Table S3). The work with human ESCs has been approved by the Robert Koch Institute (Az. 1710-79-1-4-55-E1).
Neurite Outgrowth Assays
After 22 days of terminal differentiation, neurons were singularized by treatment with accutase (PAA) for approximately 10 min at 37°C. Cells were subsequently diluted in DMEM (PAA) and spun down at 200 × g for 5 min. The cell pellet was resuspended in neuronal maturation medium, and cells were plated on matrigel-coated 48-well plates at a density of 3 × 104 cells per well. At 24 and 48 hr after plating, cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 20 min at 4°C, and neurons were stained for βIII-TUBULIN and TH. Neurite outgrowth was analyzed in a double-blinded manner on random fluorescence images and on at least 100 neurons per iPSC clone using the Image J software.
Stress-Induced Cell Death and Rescue
Neurons were replated after 10 days of neuronal differentiation at a density of 8 × 103 cells per well into 96-well plates. After another 11 days in maturation medium, 100 and 200 nM rotenone (Sigma) in N2 medium consisting of DMEM-F12 with 1:100 N2 supplement and 1% penicillin/streptomycin/glutamine were added to the cells for 24 hr. Viability was assessed by measuring LDH leakage in the supernatant using the Cytotoxicity Detection Kit Plus (Roche). Absorbance was recorded at 490 nm and was expressed as the percentage of absorbance in control cells after subtraction of background absorbance. For rescue experiments, 0.5 and 1 μM CHIR99021 (Axon Medchem) was added to the medium 48 hr before and throughout rotenone treatment, and 0.5 and 1 μM Coenzyme Q10 (Sigma) was added to the medium 20 min before measurement.
Statistics
Data of at least three independent differentiation experiments are presented as mean + SEM. Statistical significance was determined by Student’s t test and with one-way ANOVA and two-way ANOVA with post hoc Tukey test, respectively.
Author Contributions
M.E., A.-L.H., H.R.S., J.S., T.K., and G.H. developed and designed the study. M.E., A.-L.H., P.R., S.K., A.R., O.E.P., A.L.O., J.R.M., B.G., H.Z., and G.H. performed experiments. M.E., A.-L.H., P.R., M.J.A.-B., S.K., A.R., M.G., P.E., S.G.M., B.G., A.R., H.Z., J.S., T.K., and G.H. analyzed data. M.E., A.-L.H., J.S., T.K., and G.H. wrote the manuscript. P.R. and M.J.-A.-B. contributed equally.
Acknowledgments
We thank Martina Radstaak, Ingrid Gelker, Elke Hoffmann, Claudia Kemming, Claudia Ortmeier, Martina Sinn, and Susanne Peetz-Dienhart for outstanding technical assistance and Axel Schambach and Christopher Baum, MHH, Hannover, Germany, for providing the lentiviral SFFV reprogramming vector. This study was supported by research funding from the IMF at University Hospital Münster to G.H. and P.E. (I-HA-111219 and AL121108), from the U.S. Public Health Service to B.G. (P30 AG10133), from the German Research Foundation (DFG) to J.S. and P.R. (STE 1835/1-1, GA 402/18-1, FZT 111 DFG, Center for Regenerative Therapies Dresden, Cluster of Excellence), from the DFG to T.K. (SFB-TRR128-B7), and from the German Federal Ministry of Education and Research to H.R.S. and H.Z. (BMBF 01GN0811 and 01GN1008B).
Published: July 2, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, three figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.06.001.
Accession Numbers
The accession number for the data discussed in this publication is GEO: GSE62935 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi$acc=GSE62935).
Supplemental Information
References
- Abisambra J.F., Jinwal U.K., Blair L.J., O’Leary J.C., 3rd, Li Q., Brady S., Wang L., Guidi C.E., Zhang B., Nordhues B.A. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013;33:9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K.M., Gleeson J.G., Bagrodia S., Partington M.W., MacMillan J.C., Cerione R.A., Mulley J.C., Walsh C.A. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 1998;20:25–30. doi: 10.1038/1675. [DOI] [PubMed] [Google Scholar]
- Almeida S., Zhang Z., Coppola G., Mao W., Futai K., Karydas A., Geschwind M.D., Tartaglia M.C., Gao F., Gianni D. Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2012;2:789–798. doi: 10.1016/j.celrep.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida S., Gascon E., Tran H., Chou H.J., Gendron T.F., Degroot S., Tapper A.R., Sellier C., Charlet-Berguerand N., Karydas A. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso Adel C., Mederlyova A., Novak M., Grundke-Iqbal I., Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem. 2004;279:34873–34881. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- Boxer A.L., Miller B.L. Clinical features of frontotemporal dementia. Alzheimer Dis. Assoc. Disord. 2005;19(Suppl 1):S3–S6. doi: 10.1097/01.wad.0000183086.99691.91. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P., Gu C., Khursigara G., Chao M.V. p75 neurotrophin receptor as a modulator of survival and death decisions. Microsc. Res. Tech. 1999;45:217–224. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<217::AID-JEMT5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Cooper O., Seo H., Andrabi S., Guardia-Laguarta C., Graziotto J., Sundberg M., McLean J.R., Carrillo-Reid L., Xie Z., Osborn T. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012;4:141ra190. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo L., Ugolini G., Visintin M., Bradbury A., Brancolini C., Verzillo V., Novak M., Cattaneo A. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J. Neurochem. 2000;75:624–633. doi: 10.1046/j.1471-4159.2000.0750624.x. [DOI] [PubMed] [Google Scholar]
- Fasulo L., Ugolini G., Cattaneo A. Apoptotic effect of caspase-3 cleaved tau in hippocampal neurons and its potentiation by tau FTDP-mutation N279K. J. Alzheimers Dis. 2005;7:3–13. doi: 10.3233/jad-2005-7102. [DOI] [PubMed] [Google Scholar]
- Fong H., Wang C., Knoferle J., Walker D., Balestra M.E., Tong L.M., Leung L., Ring K.L., Seeley W.W., Karydas A. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Reports. 2013;1:226–234. doi: 10.1016/j.stemcr.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K., D’Souza I., Crudder C.H., Onodera H., Itoyama Y., Poorkaj P., Bird T.D., Schellenberg G.D. Pro-apoptotic effects of tau mutations in chromosome 17 frontotemporal dementia and parkinsonism. Neuroreport. 2000;11:57–60. doi: 10.1097/00001756-200001170-00011. [DOI] [PubMed] [Google Scholar]
- Ghetti B., Oblak A.L., Boeve B.F., Johnson K.A., Dickerson B.C., Goedert M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015;41:24–46. doi: 10.1111/nan.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Spillantini M.G. Pathogenesis of the tauopathies. J. Mol. Neurosci. 2011;45:425–431. doi: 10.1007/s12031-011-9593-4. [DOI] [PubMed] [Google Scholar]
- Goedert M., Spillantini M.G., Jakes R., Rutherford D., Crowther R.A. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Goedert M., Ghetti B., Spillantini M.G. Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006254. doi: 10.1101/cshperspect.a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus G., Cooper O., Deleidi M., Levy A., Lee K., Marlow E., Yow A., Soldner F., Hockemeyer D., Hallett P.J. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc. Natl. Acad. Sci. USA. 2010;107:15921–15926. doi: 10.1073/pnas.1010209107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargus G., Ehrlich M., Araúzo-Bravo M.J., Hemmer K., Hallmann A.L., Reinhardt P., Kim K.P., Adachi K., Santourlidis S., Ghanjati F. Origin-dependent neural cell identities in differentiated human iPSCs in vitro and after transplantation into the mouse brain. Cell Rep. 2014;8:1697–1703. doi: 10.1016/j.celrep.2014.08.014. [DOI] [PubMed] [Google Scholar]
- Hargus G., Ehrlich M., Hallmann A.L., Kuhlmann T. Human stem cell models of neurodegeneration: a novel approach to study mechanisms of disease development. Acta Neuropathol. 2014;127:151–173. doi: 10.1007/s00401-013-1222-6. [DOI] [PubMed] [Google Scholar]
- Heilker R., Traub S., Reinhardt P., Schöler H.R., Sterneckert J. iPS cell derived neuronal cells for drug discovery. Trends Pharmacol. Sci. 2014;35:510–519. doi: 10.1016/j.tips.2014.07.003. [DOI] [PubMed] [Google Scholar]
- Höglinger G.U., Melhem N.M., Dickson D.W., Sleiman P.M., Wang L.S., Klei L., Rademakers R., de Silva R., Litvan I., Riley D.E., PSP Genetics Study Group Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans J.J., van Haastert E.S., Nijholt D.A., Rozemuller A.J., Eikelenboom P., Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iovino M., Patani R., Watts C., Chandran S., Spillantini M.G. Human stem cell-derived neurons: a system to study human tau function and dysfunction. PLoS ONE. 2010;5:e13947. doi: 10.1371/journal.pone.0013947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D.J., Cairns N.J., Grossman M., McMillan C.T., Lee E.B., Van Deerlin V.M., Lee V.M., Trojanowski J.Q. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2015;129:469–491. doi: 10.1007/s00401-014-1380-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T.D., Jope R.S. Inhibition of glycogen synthase kinase-3 protects cells from intrinsic but not extrinsic oxidative stress. Neuroreport. 2005;16:597–601. doi: 10.1097/00001756-200504250-00016. [DOI] [PubMed] [Google Scholar]
- Knopman D.S., Roberts R.O. Estimating the number of persons with frontotemporal lobar degeneration in the US population. J. Mol. Neurosci. 2011;45:330–335. doi: 10.1007/s12031-011-9538-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijholt D.A., van Haastert E.S., Rozemuller A.J., Scheper W., Hoozemans J.J. The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J. Pathol. 2012;226:693–702. doi: 10.1002/path.3969. [DOI] [PubMed] [Google Scholar]
- Noble W., Hanger D.P., Miller C.C., Lovestone S. The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol. 2013;4:83. doi: 10.3389/fneur.2013.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz N., Beecham G.W., DeStefano A.L., Dawson T.M., Doheny K.F., Factor S.A., Hamza T.H., Hung A.Y., Hyman B.T., Ivinson A.J., PD GWAS Consortium Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Ann. Neurol. 2012;71:370–384. doi: 10.1002/ana.22687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt P., Glatza M., Hemmer K., Tsytsyura Y., Thiel C.S., Höing S., Moritz S., Parga J.A., Wagner L., Bruder J.M. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS ONE. 2013;8:e59252. doi: 10.1371/journal.pone.0059252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt P., Schmid B., Burbulla L.F., Schöndorf D.C., Wagner L., Glatza M., Höing S., Hargus G., Heck S.A., Dhingra A. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 2013;12:354–367. doi: 10.1016/j.stem.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Schröder M., Kaufman R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Schulz K.L., Eckert A., Rhein V., Mai S., Haase W., Reichert A.S., Jendrach M., Müller W.E., Leuner K. A new link to mitochondrial impairment in tauopathies. Mol. Neurobiol. 2012;46:205–216. doi: 10.1007/s12035-012-8308-3. [DOI] [PubMed] [Google Scholar]
- Shimazawa M., Tanaka H., Ito Y., Morimoto N., Tsuruma K., Kadokura M., Tamura S., Inoue T., Yamada M., Takahashi H. An inducer of VGF protects cells against ER stress-induced cell death and prolongs survival in the mutant SOD1 animal models of familial ALS. PLoS ONE. 2010;5:e15307. doi: 10.1371/journal.pone.0015307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slowinski J., Dominik J., Uitti R.J., Ahmed Z., Dickson D.D., Wszolek Z.K. Frontotemporal dementia and Parkinsonism linked to chromosome 17 with the N279K tau mutation. Neuropathology. 2007;27:73–80. doi: 10.1111/j.1440-1789.2006.00742.x. [DOI] [PubMed] [Google Scholar]
- Soldner F., Hockemeyer D., Beard C., Gao Q., Bell G.W., Cook E.G., Hargus G., Blak A., Cooper O., Mitalipova M. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L., De Sarno P., Jope R.S. Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 2002;277:44701–44708. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- Tan S., Wei X., Song M., Tao J., Yang Y., Khatoon S., Liu H., Jiang J., Wu B. PUMA mediates ER stress-induced apoptosis in portal hypertensive gastropathy. Cell Death Dis. 2014;5:e1128. doi: 10.1038/cddis.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherpakov M., Bronfman F.C., Conticello S.G., Vaskovsky A., Levy Z., Niinobe M., Yoshikawa K., Arenas E., Fainzilber M. The p75 neurotrophin receptor interacts with multiple MAGE proteins. J. Biol. Chem. 2002;277:49101–49104. doi: 10.1074/jbc.C200533200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.