

Summary
Adult stem cells and tumor-initiating cells (TICs) often employ different mechanisms of DNA damage response (DDR) as compared to other tissue cell types. However, little is known about how mammary stem cells (MaSCs) and mammary TICs respond to DNA damage. Using the mouse mammary gland and syngeneic p53-null tumors as models, we investigated the molecular and physiological consequences of DNA damage in wild-type MaSCs, p53-null MaSCs, and p53-null TICs. We showed that wild-type MaSCs and basal cells are more resistant to apoptosis and exhibit increased non-homologous end joining (NHEJ) activity. Loss of p53 in mammary epithelium affected both cell-cycle regulation and DNA repair efficiency. In p53-null tumors, we showed that TICs are more resistant to ionizing radiation (IR) due to decreased apoptosis, elevated NHEJ activity, and more-rapid DNA repair. These results have important implications for understanding DDR mechanisms involved in both tumorigenesis and therapy resistance.
Graphical Abstract
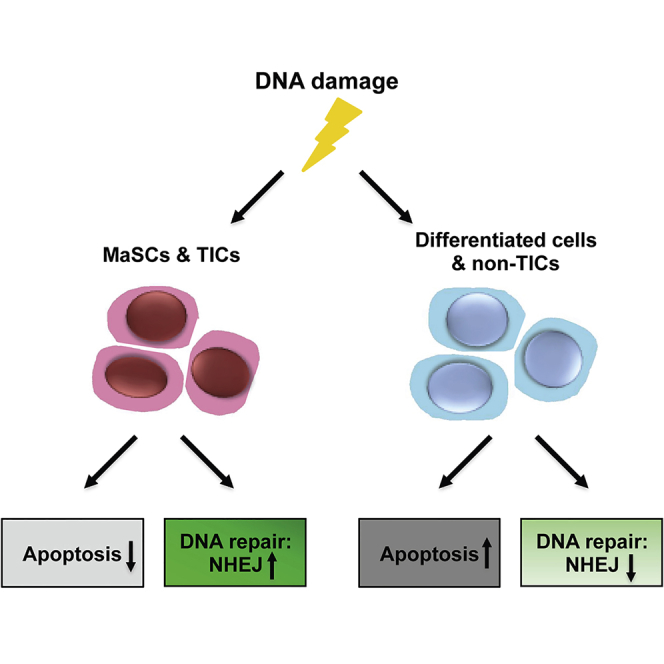
Highlights
-
•
MaSCs are more resistant to apoptosis and exhibit increased NHEJ activity
-
•
Loss of p53 in MECs impairs cell-cycle regulation and DNA repair efficiency
-
•
TICs exhibit decreased apoptosis, increased DNA repair efficiency, and NHEJ activity
-
•
TICs are highly proliferative and exhibit improper cell-cycle regulation after IR
In this article, Rosen and colleagues show that stem cells from both the mammary gland and mammary tumors are more resistant to apoptosis and exhibit increased NHEJ activity. Loss of p53 in the mammary epithelium disrupts both cell-cycle regulation and DNA repair efficiency. These results have important implications for understanding DDR mechanisms involved in both tumorigenesis and therapy resistance.
Introduction
DNA damage may occur at a rate of 100,000 lesions per cell per day due to internal and external insults (Hoeijmakers, 2009). Thanks to evolution, mammalian cells employ a sophisticated and highly conserved DNA damage response (DDR), which regulates cell cycle, damage repair, gene expression, and, alternatively, apoptosis or senescence (Harper and Elledge, 2007) to protect genome integrity and prevent mutations. Among all kinds of DNA damage, double-strand breaks (DSBs) are probably the most deleterious type of lesion, which is repaired through either the homologous recombination (HR) or non-homologous end joining (NHEJ) pathways (Khanna and Jackson, 2001). DDR mechanisms are especially important for long-lived tissue stem cells because they may accumulate more mutations throughout their lifetime. Indeed, a recent study showed that the total number of lifetime stem cell divisions is highly correlated with cancer risk in a particular tissue (Tomasetti and Vogelstein, 2015), further suggesting the importance of maintaining genome integrity in stem cells. Previous studies have shown that mouse hair follicle bulge stem cells and hematopoietic stem cells exhibit increased NHEJ activity and decreased apoptosis, resulting in their resistance to ionizing radiation (IR) (Mohrin et al., 2010; Sotiropoulou et al., 2010). However, little is known about how mammary stem cells (MaSCs) respond to IR treatment.
The mammary epithelium is composed of basal and luminal cell compartments. Although the existence and precise localization of bipotent MaSCs, which can give rise to both basal and luminal cells, are still controversial, most evidence suggests that MaSCs reside in the basal compartment (Rios et al., 2014; Shackleton et al., 2006; Stingl et al., 2006), and exhibit properties of myoepithelial cells (Prater et al., 2014), a cell type predominant in basal compartment. MaSCs can be further enriched using fluorescence-activated cell sorting (FACS) with the cell surface markers CD24 and either CD49f or CD29 (Shackleton et al., 2006; Stingl et al., 2006). MaSCs play a critical role in ensuring mammary gland homeostasis during puberty, pregnancy, lactation, and involution (Visvader and Stingl, 2014). Hence, it is important to understand how MaSCs maintain their genome integrity and how they react to DNA damage. In addition, mutation or loss of function of p53, a tumor suppressor gene that plays a major role in DDR (Meek, 2009), is correlated not only with mammary tumorigenesis but also with poor prognosis and treatment response in breast cancer (Bergh et al., 1995; Berns et al., 2000; Gasco et al., 2002; Sørlie et al., 2001). Therefore, dissecting the effects of p53 loss on DDR in mammary epithelium, especially in MaSCs, is particularly important for understanding breast cancer tumorigenesis.
In previous tumor studies, we have used a p53-null syngeneic mouse model to mimic p53 loss of function in human breast cancer. This model was developed by transplanting p53-null mammary epithelium into the cleared mammary fat pads of wild-type, syngeneic Balb/c-recipient mice, resulting in spontaneous tumor development (Jerry et al., 2000). Previously, we demonstrated that this tumor model mimics several of the different subtypes known to occur in human breast cancer (Herschkowitz et al., 2012; Zhang et al., 2008). Using this tumor model, we have identified tumor-initiating cells (TICs), also often referred to as tumor-propagating or cancer stem cells, based upon their expression of the cell surface markers CD24 and CD29, and we further demonstrated that these TICs are more resistant to IR (Zhang et al., 2008, 2010). However, similar to several other studies demonstrating that TICs from mammary tumors are more resistant to conventional therapies (Creighton et al., 2009; Diehn et al., 2009; Li et al., 2008), the DDR mechanisms underlying this therapeutic resistance are still largely unknown.
In this study, we comprehensively analyzed DDR mechanisms in stem cells and non-stem cells from wild-type and p53-null mammary epithelium and from p53-null tumors. We demonstrated that wild-type MaSCs and basal cells exhibited increased NHEJ activity and resistance to apoptosis as compared to luminal cells. MaSCs also exhibited increased G2 arrest after IR treatment. Loss of p53 in the mammary gland disrupted not only G1 cell-cycle arrest, damage-induced quiescence, but also DNA repair efficiency. Importantly, p53-null TICs exhibited mixed characteristics of wild-type and p53-null MaSCs, including decreased apoptosis, elevated NHEJ activity, and more-rapid DNA repair, but lacked the cell-cycle arrest and quiescence following DNA damage. These results suggest that inhibition of survival or the NHEJ pathway could offer novel treatment options to selectively sensitize TICs to IR treatment.
Results
Wild-Type MaSCs Are More Resistant to DNA Damage-Induced Apoptosis
To study the effects of DNA damage on apoptosis in different mammary epithelial cell compartments, 5- to 6-week-old virgin mice were treated with or without 6 Gy whole-body IR. Freshly dissociated mammary epithelial cells (MECs) were then stained with the cell-surface markers CD24 and CD49f, the apoptosis marker annexin V, and the dead cell marker Sytox Red, followed by flow cytometry analysis (Figure S1A). MaSCs, basal cells, and luminal cells were enriched in the Lin−CD49fhighCD24+, Lin−CD49fhighCD24low, and Lin−CD49flowCD24+ subpopulations, respectively. Apoptotic cells were represented by annexin-V-positive and Sytox-Red-negative cells (Figures 1A and S1A). In the absence of IR treatment, MaSCs and basal cells exhibited significantly decreased annexin V positivity as compared to untreated luminal cells, indicating they are intrinsically more resistant to apoptosis (Figure 1B). To understand the kinetics of apoptosis after IR, we analyzed annexin V positivity at 1 hr, 12 hr, and 24 hr post-IR. In luminal compartment, we observed a trend of increasing apoptosis after IR and a significant increase of apoptosis at the 24 hr time point as compared to non-IR, whereas MaSCs and basal cells maintained a level of apoptosis similar to that observed in untreated mice at all the different time points (Figure 1C). These data suggest that MaSCs and basal cells are also more resistant to DNA damage-induced apoptosis. To rule out the possibility that the enzymatic dissociation of mammary tissue specifically damaged luminal cells, we performed tissue immunofluorescence (IF) staining with cleaved caspase-3 (CC3) antibody in combination with the myoepithelial cell marker smooth muscle actin (SMA) or the luminal cell marker keratin 8 (K8). A similar difference in apoptosis was observed by direct tissue staining as compared to flow cytometry analysis. As shown in Figures S2A–S2Q, luminal cells contained more CC3-positive cells before IR as compared to myoepithelial cells, and after IR, only luminal cells exhibited increased apoptosis.
Figure 1.

MaSCs and Basal Cells Are More Resistant to DNA-Damage-Induced Apoptosis
(A) Representative FACS plots of annexin V and Sytox Red staining in different subpopulations 24 hr after IR.
(B) MaSCs and basal cells exhibit significantly decreased apoptosis as compared to luminal cells before IR (data are shown as mean ± SEM; n = 7; ∗∗∗p < 0.001; ns, non-significant).
(C) Percentage of annexin-V-positive cells in different subpopulations at different time points (data are shown as mean ± SEM; nonIR and 24 hr, n = 7; 1 hr and 12 hr, n = 4; ∗p < 0.05).
(D and E) RPPA analysis shows that pro-apoptotic proteins are downregulated in MaSCs and basal cells as compared to luminal cells before IR (n = 3).
(F) After IR, BAD and caspase-3 were further downregulated in MaSCs (n = 3).
(G) Western blot analysis of BAX and caspase-3 levels in different subpopulations before and 24 hr after IR.
To determine the difference of protein levels in different subpopulations, MECs were sorted followed by reverse phase protein array (RPPA) analysis (Figures S1A and S1B). Consistent with the results from annexin V staining, RPPA analysis showed that, before IR, pro-apoptotic proteins, including BAX, caspase-3, and caspase-7, were significantly decreased in MaSCs and basal cells as compared to luminal cells, whereas no differences were observed in these protein levels between MaSCs and basal cells (Figures 1D and 1E). After IR, BAD and caspase-3 were further downregulated in MaSCs, indicating stem cells are more protected from apoptosis following DNA damage (Figure 1F). Conversely, RPPA showed an upregulation of BAX in the luminal compartment after IR, which corresponded with their increased level of apoptosis (data not shown). These data were confirmed by western blot analysis as shown in Figure 1G. Furthermore, in the absence of IR, stress-responsive proteins, such as phospho-p38 (p-p38), phospho-p44 (p-p44), and phospho-AKT (p-AKT), were increased in MaSCs and basal cells as compared to luminal cells (Figures S2R–S2T), corresponding with their enhanced survival.
Wild-Type MaSCs Exhibited Increased G2 Arrest after DNA Damage
Regulation of cell-cycle progression may play a central role in DDR (Zhou and Elledge, 2000). To elucidate the effects of DNA damage on cell cycle in different subpopulations, we fixed and permeabilized freshly dissociated MECs after surface marker staining, stained the cells with propidium iodide (PI), and performed flow cytometry analysis (Figure S1A). Before IR, the majority of cells from all three subpopulations were in the G0/G1 phase of cell cycle, whereas a small, but not significantly higher, proportion of MaSCs resided in the G2/M phase as compared to basal and luminal cells (Figure 2A). Twelve hours after IR, the percentage of cells in the G2/M phase was increased more than 2-fold in MaSCs (Figures 2B and 2E) but began to decrease by 24 hr post-IR treatment (data not shown). In contrast, the basal and luminal cells exhibited a similar cell-cycle status (Figures 2C–2E), indicating that the increased G2 arrest after DNA damage is unique to the MaSC population.
Figure 2.

Wild-Type MaSCs Exhibit Increased G2 Arrest and Evade Damage-Induced Quiescence after IR
(A) Cell-cycle distributions of different subpopulations from MECs before IR were examined using PI staining (data are represented as mean ± SEM; n = 5).
(B) A significant increase of cells in G2/M was observed in MaSCs 12 hr after IR (data are shown as mean ± SEM; n = 3; ∗∗p < 0.01; ∗p < 0.05).
(C and D) The cell-cycle profiles of basal and luminal compartments before and 12 hr after IR (data are shown as mean ± SEM; n = 3).
(E) The fold change of cells in G2/M phase 12 hr after IR as compared to non-IR samples (data are shown as mean ± SEM; n = 3; ∗∗p < 0.01).
(F) Percentage of Ki67-positive MECs before and after IR (data are shown as mean ± SEM; n = 3).
(G) Representative FACS plots of Ki67 in different subpopulations before and after IR.
(H) Quantification of Ki67 positivity shows that basal and luminal cells became significantly more quiescent after IR (data are shown as mean ± SEM; n = 3; ∗∗∗p < 0.001; ∗p < 0.05).
We next investigated the quiescence and proliferation status of these three cell populations by staining of Ki67, which is absent in the G0 and present in the interphase (G1-S-G2/M) of cell cycle (Scholzen and Gerdes, 2000). Similar to the PI staining protocol, we used permeabilized cells with surface marker staining, followed by intracellular staining. This technique permitted the analysis of proliferation in the infrequent MaSCs (Figure S1A). In the total MECs after IR, as shown in Figure 2F, we observed a trend of a decrease in the percentage of Ki67-positive cells, indicating the presence of DNA-damage-induced quiescence in the wild-type mammary epithelium. We then analyzed the proliferation rate in each subpopulation. Before IR, we observed a trend toward decreased proliferation in MaSCs as compared to basal and luminal cells (Figure 2G), whereas after IR, the number of Ki67-positive cells was slightly increased in MaSCs and decreased significantly in the other two compartments (Figures 2G and 2H). These data suggest that basal and luminal cells undergo damage-induced quiescence after IR, whereas more stem cells entered interphase from G0, possibly to initiate a damage-induced injury response to maintain tissue homeostasis.
Wild-Type MaSCs and Basal Cells Exhibited Increased NHEJ Activity
Another key process regulated by DDR, other than cell cycle and apoptosis, is the repair of DNA damage. In mammary glands, the repair efficiency among different epithelial cell populations is still largely unknown. For this study, phospho-Histone H2AX (Ser139) (γH2AX) was employed as a marker of DSBs, and γH2AX intracellular staining was performed on freshly prepared MECs in combination with CD49f and CD24 staining, followed by flow cytometry analysis (Figure S1A). As shown in Figures 3A and 3B, 30 min after IR, more than 90% of the cells from each subpopulation were γH2AX positive, indicating IR induces DSBs in the majority of cells. By 12 hr post-IR treatment, most of the damage was repaired in all three populations. Slightly less γH2AX staining was observed in MaSCs after 12 hr as compared to luminal cells; however, this difference was not statistically significant (Figure 3B). Overall, these results suggest that all three subpopulations from wild-type glands exhibit a similar efficiency of DSB repair.
Figure 3.

Wild-Type Basal/MaSC Compartment Exhibits Increased NHEJ Activity
(A) Representative FACS plots of γH2AX intracellular staining 0.5 and 12 hr after IR in different subpopulations.
(B) All three subpopulations exhibit similar DSB repair efficiency (data are shown as mean ± SEM; n = 3).
(C) A schematic depiction of the high-throughput NHEJ assay.
(D) NHEJ activity was significantly higher in the basal/MaSC compartment as compared to luminal cells (data are shown as mean ± SEM; n = 4; ∗p < 0.05).
(E) Western blot analysis shows a 1.5-fold increase of 53BP1 level in basal cells as compared to luminal cells.
Because most of the MECs were residing in the G0/G1 phase, indicating they mainly use NHEJ to repair DSBs (Rothkamm et al., 2003), we then analyzed the NHEJ activity in each subpopulation of MECs. Using an ex vivo high-throughput NHEJ assay, we quantified the NHEJ activity by measuring the repair of an enzymatically cleaved GFP reporter. The cells were also electroporated with a linearized tdtomato plasmid as an internal transfection efficiency control (Figure 3C). By measuring the number of GFP-positive cells within the tdtomato-positive cell population, we showed that the basal/MaSC population (Lin−CD24+CD29high) exhibits a significantly increased NHEJ activity as compared to luminal cells (Lin−CD24+CD29low; Figure 3D). We also observed an increased level (1.5-fold) of 53BP1 in basal cells as compared to luminal cells using western blot analysis (Figure 3E). Whereas this difference alone may not affect NHEJ activity directly, it may facilitate the potential of cells to utilize NHEJ, because 53BP1 is an important positive regulator of NHEJ pathway (Escribano-Díaz et al., 2013; Gupta et al., 2014).
Loss of p53 Disrupted G1 Cell Cycle Arrest, Damage-Induced Quiescence, and DSB Repair Efficiency in MECs
To study the effects of loss of p53 on DDR in mammary epithelium, we analyzed the levels of apoptosis, cell-cycle regulation, and DNA repair mechanisms in MECs isolated from p53-null mice with and without IR. Surprisingly, the apoptosis rate in p53-null MECs was similar to that in wild-type MECs, indicating p53-independent apoptotic pathways play an important role in mammary epithelium. Similar to wild-type (see Figure 1B), p53-null MaSCs and basal cells had lower rates of apoptosis before and after IR as compared to luminal cells, and only the luminal cells showed increased apoptosis in response to IR (Figures S3A and S3B). In addition, p53-null MECs exhibited cell-cycle profiles similar to those of wild-type MECs. However, the difference in the G2/M phase between MaSCs and the other cell types was more significant (Figure 4A). Strikingly, after IR, we observed a dramatic increase in the percentage of cells in the G2/M phase in all three subpopulations, accompanied by a decreased percentage of cells in the G1 phase (Figures 4B–4D). This result suggests that loss of p53 impairs G1 arrest in the mammary epithelium, but no effect was observed on G2 arrest, as has been shown in other cell types (Kuerbitz et al., 1992; Wahl et al., 1996). On the other hand, total MECs from p53-null mammary glands after IR treatment exhibited the same ratio of Ki67-positive cells as compared to their non-IR counterparts (Figure S3C), indicating that loss of p53 impairs the damage-induced quiescence observed in wild-type mammary epithelium. This phenomenon was seen in all three subpopulations, and p53-null MaSCs exhibited an increased Ki67 positivity after IR, similar to what was observed in wild-type MaSCs (Figure S3D).
Figure 4.

Loss of p53 Disrupts G1 Cell-cycle Arrest and DSB Repair Efficiency in MECs
(A) Cell-cycle distributions of different subpopulations from p53-null MECs before IR (data are represented as mean ± SEM; n = 4; ∗∗p < 0.01).
(B–D) All three subpopulations exhibit higher percentage of G2/M and lower percentage of G1 12 hr after IR (data are represented as mean ± SEM; n = 3; ∗∗∗p < 0.001).
(E) Representative FACS plots of γH2AX staining 0.5 and 12 hr after IR in wild-type and p53-null MECs.
(F) Percentage of γH2AX-positive cells in wild-type and p53-null MECs before and after IR (data are shown as mean ± SEM; n = 3; ∗∗p < 0.01).
(G) The decreased repair efficiency was observed in all three subpopulations from p53-null MECs (data are shown as mean ± SEM; 0.5 hr, n = 1; non-IR and 12 hr, n = 3).
Previous studies have shown that p53 deficiency results in delayed DNA repair in cell lines (Zheng et al., 2014). However, it is still unclear whether loss of p53 affects the DNA repair efficiency in the mammary gland. Using the same γH2AX intracellular staining strategy as above (Figure S1A), we observed that the proportion of γH2AX-positive cells was significantly higher in p53-null MECs as compared to wild-type MECs 12 hr after IR, suggesting that the repair of DSBs is delayed in the absence of p53 (Figures 4E and 4F). A slight increase of γH2AX-positive staining was also observed in non-IR p53-null MECs, indicating there were more unrepaired DSBs accumulated in cells even without IR (Figure 4F). Furthermore, the delay in DSB repair was universal in all three subpopulations (Figure 4G). Using the high-throughput NHEJ assay to study the DSB repair mechanisms in p53-null MECs, we demonstrated that loss of p53 didn’t affect NHEJ activity in the mammary epithelium (Figure S3E). This result suggests that the delay in DSB repair in p53-null epithelium is not due to alterations in NHEJ activity. Overall, the data from p53-null mammary epithelium indicate that loss of p53 affects cell-cycle regulation and DSB repair efficiency, which may lead to increased accumulation of mutations and tumorigenesis.
TICs Exhibited Increased Resistance to Apoptosis as Compared to Non-TICs
To further understand how transformed p53-null MECs react to DNA damage, we used T7 tumors from the p53-null syngeneic mouse model. Previously, we demonstrated that TICs from T7 tumors are more resistant to IR (Zhang et al., 2008). Here, we studied the physiological and molecular mechanisms underlying this resistance. After dissociation, followed by CD24, CD29, and lineage marker staining, TICs and non-TICs can be enriched in Lin−CD29+CD24high and Lin−CD29−CD24low subpopulations, respectively (Figures S4A and S4B). To investigate the difference in apoptosis between TICs and non-TICs, we performed CC3 IF staining instead of annexin V staining, because we observed false-positive annexin V staining in the tumor cells, which had been reported previously to occur in solid tumors (Pozarowski et al., 2003). After sorting and CC3 staining, we observed significantly decreased apoptosis in TICs both before and after IR treatment as compared to non-TICs (Figures 5A and 5B). Interestingly, IR didn’t induce apoptosis in either subpopulation. We confirmed this result with the TdT-mediated dUTP nick end labeling (TUNEL) assay, in which we observed a similar pattern of apoptosis between TICs and non-TICs (Figure S5). Using western blot analysis, we observed that there were no differences in the levels of BAX and caspase-3 between the tumor subpopulations (data not shown). However, increased levels of p-AKT and p-p38 were observed in TICs (Figure 5C), which likely contribute to their enhanced survival.
Figure 5.

TICs Exhibit Decreased Apoptosis as Compared to Non-TICs
(A) Representative images of CC3 staining in TICs and non-TICs before and 24 hr after IR (bars, 10 μm).
(B) TICs exhibited significantly decreased apoptosis before and after IR (data are shown as mean ± SEM; n = 3; ∗p < 0.05).
(C) Western blot analysis shows that, before IR, p-AKT and p-p38 were upregulated in TICs as compared to non-TICs.
p53-Null Tumor Cells Are Highly Proliferative and Exhibit Improper Cell Cycle Regulation after IR
To investigate IR-induced alterations in cell-cycle regulation in p53-null tumors, PI staining was performed on the fixed cells with surface marker staining (Figure S4A). Before IR, there was no significant difference in the cell-cycle profiles between TICs and non-TICs, although we observed a slightly higher percentage of TICs residing in the G2/M phase (Figure 6A), similar to our observation in MaSCs. At three different time points after IR (0.5 hr, 12 hr, and 24 hr), both TICs and non-TICs exhibited very similar cell-cycle distributions as compared to their non-IR counterparts (Figures 6B and 6C; data not shown), indicating that cell-cycle arrest after DNA damage was disrupted in both subpopulations. Similar to the lack of cell-cycle arrest, p53-null tumor cells also failed to undergo damage-induced quiescence. As shown in Figures 6D and 6E, total tumor cells were highly proliferative, and the proliferation rates were unaffected by DNA damage at any time point after IR. Within the tumors, TICs exhibited a significantly higher proliferation rate as compared to non-TICs, and IR didn’t induce quiescence in either subpopulation (Figures 6D and 6F). Twenty-four hours after IR, we observed a slight increase of proliferation in TICs (Figure 6F), which was similar to our observation in MaSCs. These data indicate that p53-null tumor cells exhibit not only impaired G1 and G2 arrest but also disrupted damage-induced quiescence.
Figure 6.

p53-Null Tumor Cells Are Highly Proliferative and Fail to Exhibit Proper Cell-cycle Regulation after IR
(A) Cell-cycle distribution of TICs and non-TICs before IR (data are shown as mean ± SEM; n = 4).
(B and C) Twelve hours after IR, both subpopulations exhibit similar cell-cycle profiles as compared to their non-IR counterparts (data are shown as mean ± SEM; n = 4).
(D) Representative FACS plots of Ki67 staining in total tumor cells, TICs, and non-TICs before and after IR.
(E) Percentage of Ki67-positive cells in total tumor cells before and after IR (data are shown as mean ± SEM; n = 3).
(F) Percentage of Ki67-positive cells in TICs and non-TICs before and after IR (data are shown as mean ± SEM; n = 3; ∗p < 0.05; ∗∗p < 0.01).
TICs Exhibited More-Rapid DSB Repair and Increased NHEJ Activity as Compared to Non-TICs
To study the DSB repair efficiency in p53-null tumors, we sorted for TICs and non-TICs and performed γH2AX IF staining. Similar to the results from MECs, more than 90% of the cells exhibited γH2AX foci 0.5 hr after IR, demonstrating that DSBs occurred in the majority of cells. Twenty-four hours after IR, less than 25% of TICs showed γH2AX foci, whereas nearly 40% of non-TICs showed γH2AX foci, suggesting that TICs have the capacity for significantly faster DSB repair as compared to non-TICs (Figures 7A and 7B). To confirm this result, we performed a neutral comet assay to quantify DSBs by measuring the percentage of DNA in the comet tails. Similar to our observations in γH2AX IF staining, immediately after IR, there was a significant increase in DSBs in both subpopulations; however, by 24 hr after IR, TICs showed DSB levels similar to their non-IR counterparts, whereas non-TICs still had significantly higher DSB levels, suggesting TICs are able to repair the DNA damage faster (Figures 7C and 7D). To investigate the mechanisms of DSB repair in tumor cells, we performed the high-throughput NHEJ assay on freshly digested tumor cells. We observed a significant increase in NHEJ activity in TICs as compared to non-TICs (Figure 7E). Using western blot analysis, we observed that the level of 53BP1 was increased nearly 20% and Ku80, a crucial regulator in NHEJ pathway (Grundy et al., 2014), was increased nearly 40% in TICs as compared to non-TICs (Figure 7F). These endogenous protein levels may reflect the NHEJ potential in TICs.
Figure 7.

TICs Exhibit More-Rapid DSB Repair and Increased NHEJ Activity as Compared to Non-TICs
(A) γH2AX IF staining shows that TICs exhibited less damage foci as compared to non-TICs 24 hr after IR (bars, 10 μm).
(B) Percentage of γH2AX-positive cells in TICs and non-TICs before and after IR (data are shown as mean ± SEM; n = 4; ∗∗p < 0.01).
(C) Representative images from the neutral comet assay. Comet tails represent damaged DNA (bars, 25 μm).
(D) Percentage of DNA in the comet tails in TICs and non-TICs at different time points of IR treatment (data are shown as mean ± SEM; 70–100 cells were measured in each group; ∗∗p < 0.01; ∗p < 0.05).
(E) NHEJ activity is significantly higher in TICs as compared to non-TICs (data are shown as mean ± SEM; n = 5; ∗∗p < 0.01).
(F) Western blot analysis shows that 53BP1 and Ku80 were upregulated in TICs as compared to non-TICs.
Discussion
For more than three decades, p53 has been well studied in its roles of DDR and tumorigenesis. However, in the mammary gland, it is still largely unknown how loss of p53 affects DDR in vivo and how it leads to mammary tumorigenesis. In addition, the DDR in MaSCs has been difficult to study in vivo mainly because of their low frequency. In culture systems, it has been shown that MaSCs and mammary progenitor cells are more resistant to DNA damage due to upregulation of p21 and WNT signaling, respectively (Insinga et al., 2013; Woodward et al., 2007). However, it is still largely unknown how MaSCs react to DNA damage in vivo within the intact microenvironment and their appropriate niche. In this study, we investigated the mechanisms of DDR, including apoptosis, cell- cycle regulation, DSB repair, and gene expression, in different epithelial subpopulations from wild-type and p53-null mammary glands and p53-null mammary tumors. First, we demonstrated that, in wild-type MECs, MaSCs and basal cells exhibited significantly decreased apoptosis and increased NHEJ activity as compared to luminal cells, possibly due to downregulation of pro-apoptotic proteins, such as BAX and caspase-3, and upregulation of the NHEJ-regulating protein 53BP1. Wild-type MaSCs also exhibited increased G2 arrest after IR, accompanied by an increased number of cells entering interphase. Second, we showed that loss of p53 in MECs impaired G1 cell-cycle arrest, damage-induced quiescence, and DNA repair efficiency; however, p53-null MaSCs still exhibited decreased apoptosis. Lastly, we demonstrated that, in p53-null tumors, TICs were more resistant to apoptosis and they repaired DSBs more rapidly with increased NHEJ activity; however, they failed to exhibit the usual cell-cycle arrest and quiescence after DNA damage.
In the epidermis and gastrointestinal epithelium, tissue stem cells have been reported to exhibit differential sensitivity to IR-induced cell death (Barker et al., 2007; Inomata et al., 2009; Potten et al., 1997), and this sensitivity was not necessarily related to cell proliferation (Sotiropoulou et al., 2010). Here, we provide evidence that, in the mammary epithelium, MaSCs are more resistant to IR-induced apoptosis, even though they exhibit decreased quiescence after IR. Interestingly, we observed levels of apoptosis in p53-null MEC subpopulations similar to those in their wild-type MEC counterparts, indicating that p53-independent apoptosis plays a more-important role in the mammary gland. These results correspond with an earlier observation that apoptosis is not dependent on p53 during mammary gland ductal morphogenesis and instead might be regulated by members of the Bcl-2 family (Humphreys et al., 1996). Similar to MaSCs, TICs exhibited decreased apoptosis as compared to non-TICs, even though they are more proliferative. Notably, IR didn’t increase apoptosis in either subpopulation within the p53-null tumors, indicating that p53 is important for IR-induced apoptosis in mammary tumors.
The cycling and quiescence of MaSCs has been especially difficult to study (Visvader and Stingl, 2014). It has been shown that long-lived, label-retaining cells are enriched in the MaSC/basal compartment, indicating they are slowly dividing (Shackleton et al., 2006). However, it is still unclear how DNA damage affects the cycling and quiescence properties of MaSCs. In our study, we observed a slightly lower proliferation rate in wild-type MaSCs before IR as compared to other subpopulations. Interestingly, after IR, there was an increase in Ki67-positive cells in MaSCs and TICs, indicating more cells were entering the cell cycle from G0 into interphase. These data suggest that stem cells enter the cell cycle to initiate wound repair in the mammary gland and tumors to maintain tissue homeostasis, similar to what has been observed previously in stem cells in epidermis and bladder cancers (Kurtova et al., 2015; Mascré et al., 2012). In addition, we observed that non-stem cells in p53-null MECs and tumors failed to induce quiescence after IR, indicating that p53 regulates, at least partially, quiescence in the mammary gland. This result is supported by previous studies in the hematopoietic system (Asai et al., 2012; Liu et al., 2009). Moreover, previously it has been shown that a higher percentage of wild-type MaSCs reside in G2/M phase as compared to other compartments (Prater et al., 2014). In this study, we also observed this phenomenon not only in wild-type MaSCs but also in p53-null MaSCs and TICs, indicating that stem cells may be primed for rapid cell division to maintain tissue homeostasis.
DSB repair in hair follicle bulge stem cells, hematopoietic stem cells, and glioma stem cells has been shown to be more rapid than in other cell types in their respective tissues (Bao et al., 2006; Mohrin et al., 2010; Sotiropoulou et al., 2010). In the present study, however, we didn’t observe increased efficiency of DSB repair in MaSCs. It’s possible that, by 12 hr after IR, all three subpopulations have already repaired most of the IR-induced damage, as we observed similar levels of γH2AX positivity at the 12 and 24 hr post-IR time points. However, it is technically difficult to investigate shorter time points in these in vivo experiments. Nevertheless, we observed increased NHEJ activity in basal/MaSC compartment, which may help facilitate repair immediately after DNA damage, thereby ensuring genome integrity. On the other hand, p53 has been linked directly to DSB repair activity by regulating the HR or NHEJ pathways in in vitro studies (Marmorstein et al., 1998; Stürzbecher et al., 1996; Tang et al., 1999). Although in this study we didn’t observe a decrease in NHEJ activity in p53-null MECs, we showed that the repair of DSB was significantly slower in p53-null MECs as compared to wild-type MECs. This result suggests that p53-null cells may accumulate mutations more easily after DNA damage in vivo.
With respect to specific DSB repair mechanisms in the T7 p53-null tumor model, we demonstrated that TICs exhibited significantly higher NHEJ activity, contributing to their faster repair, as compared to non-TICs. However, the repair kinetics of TICs appeared much slower than normal MaSCs, which repaired most of the DSBs by 12 hr post-IR. In addition, a high percentage of p53-null tumor cells were actively cycling even following DNA damage. These observations suggest that, once damage occurs, p53-null tumor cells have an increase probability to accumulate mutations as compared to MECs. Finally, the increased activity of the NHEJ repair pathway in MaSCs and TICs might be a double-edged sword because NHEJ is an error-prone repair mechanism (Khanna and Jackson, 2001) and might, therefore, allow more mutations to accumulate over time (Mohrin et al., 2010).
In this study, we used three models to provide insight into the variations in the DDR from the normal mammary gland to breast cancer. First, we studied the DDR in normal and p53-null mammary glands from pubertal mice, which is the age when the mammary gland has a higher susceptibility to tumorigenesis after IR (Castiglioni et al., 2007; Land et al., 1980; Preston et al., 2002; Tang et al., 2014). We showed that loss of p53 in MECs interrupts DSB repair and cell-cycle regulation after IR, which potentially may increase the accumulation of mutations in mammary epithelium. Second, we used the T7 p53-null tumor model, which is histologically similar to human basal-like breast cancer (Zhang et al., 2008) and demonstrated that T7 TICs repaired DSB more rapidly with increased NHEJ activity and exhibited decreased apoptotic cell death, most likely contributing to their resistance to IR. These results may have important implications for designing targeting therapies to selectively sensitize TICs to radiation or chemotherapy by treating basal-like tumors with inhibitors of cell survival or NHEJ pathways.
Experimental Procedures
Mice and Treatment
Wild-type Balb/c female mice were used for the MEC study (5 to 6 weeks old) and tumor transplantation (3 to 4 weeks old). Balb/c p53-null mice were generously provided by Dr. Xiang Zhang at Baylor College of Medicine. All animal protocols were reviewed and approved by the Animal Protocol Review Committee at Baylor College of Medicine. For IR treatment, a 137Cs irradiator, unit GC-40 SN 192 (MDS Nordion), was used to irradiate the mice at room temperature. A single dose of 6 Gy was given.
Primary MEC Preparation
The primary MECs were isolated as previously described (Roarty et al., 2015). The third, fourth, and fifth pairs of mammary glands were harvested from 5- to 6-week-old wild-type or p53-null virgin female mice and minced into small pieces. DMEM/F12 containing 2 mg/ml collagenase A (Roche) was used to digest the glands for 1 hr at 37°C with constant rotation at 125 rpm. After digestion, organoids were centrifuged at 1,500 rpm for 5 min, followed by three short washes (1,500 rpm for 7 s) in PBS. The enriched organoids were then treated with 0.25% trypsin at 37°C for 5 min and filtered through 40-μm cell strainer, followed by one additional wash in PBS before labeling with antibodies.
Tumor Transplantation into the Cleared Mammary Fat Pads
Clearance of host mammary epithelial cells and transplantation procedures were performed as previously described (Medina, 1996). To generate fresh tumors, diced T7 tumor pieces (<1 mm3) were transplanted into cleared fat pad of the fourth mammary glands from 3-week-old Balb/c females. Tumors were allowed to grow for around 3 weeks and collected when the size reached 0.5–1 cm in diameter.
Primary Tumor Cell Preparation
The primary tumor cells were prepared as previous described (Zhang et al., 2008). Briefly, tumors were minced into small pieces and digested in DMEM/F12 containing collagenase type 3 (225 units/ml; Worthington) for 2 hr at 37°C with constant rotation at 125 rpm. After digestion, cells were first filtered through 70-μm cell strainer, centrifuged at 800 rpm and 1,500 rpm, respectively. The enriched cells were then filtered through 40-μm cell strainer to obtain single tumor cells and washed three times in HBSS before antibody labeling.
FACS Staining and Analysis
After primary MEC or tumor cell preparation, lineage-positive cells were depleted using either the EasySep Mouse Mammary Stem Cell Enrichment Kit (STEMCELL Technologies; 19758) or Biotin Mouse Lineage Panel (BD Biosciences; 559971) plus CD140a (eBioscience; 13-1401-82) and CD31 (BD Biosciences; 553371) antibodies. Cells were then resuspended at a concentration of 107 cells/ml in HBSS+ (HBSS containing 2% FBS with 10 mM HEPES buffer) for surface marker staining. All antibody incubations were performed either on ice for 30 min or at room temperature for 20 min. Anti-mouse antibodies used for surface marker staining include CD49f-FITC (1:100; BioLegend; 313606), CD49f-Pacific Blue (1:100; BioLegend; 313620), CD29-FITC (1:100; BioLegend; 102206), CD29-Pacific Blue (1:100; BioLegend; 102224), CD24-PE (1:100; BD Biosciences; 553262), CD24-Pacific Blue (1:100; BioLegend; 101820), and CD24-PE/Cy7 (1:100; BioLegend; 101821). For sorting, cells were also stained for Sytox Red (Life Technologies; S34859) for dead-cell depletion. For annexin V conjugation, cells with surface marker staining were resuspended in 100 μl of annexin-binding buffer and stained with 5 μl annexin V-Alexa Fluor 350 (Life Technologies; A23202) for 15 min, followed by Sytox Red staining. For intracellular γH2AX and Ki67 analysis, after surface marker staining, cells were stained with LIVE/DEAD Fixable Dead Cell Stain Kit (Life Technologies; L10120) before fixed and permeabilized with Transcription Factor (TF) Buffer Set (BD Biosciences; 562574). After fixation and permeabilization, cells were stained with anti-mouse H2AX (pS139)-Alexa Fluor 488 (5 μl/million cells; BD Biosciences; 560445) or Anti-Mouse Ki67-PE (1:200; eBioscience; 12-5698) at room temperature for 1 hr. For cell-cycle analysis, the cells were fixed with TF Buffer Set and then treated with PI (50 μg/ml; Sigma) and RNase A (100 μg/ml; Sigma) for 30 min at room temperature. All sorting experiments were done with BD AriaII sorter, and all flow cytometry analyses were done with BD LSRFortessa cell analyzer. Gating was based on isotype and single color controls. All FACS data were processed and analyzed using FlowJo.
Neutral Comet Assay
The neutral comet assay was performed by following the manufacturer’s protocol (Trevigen). Briefly, sorted cells were fixed in low-melting agarose gel, followed by lysis and electrophoresis. DNA was visualized with SYBR green staining, and images were analyzed with CometScore software.
High-Throughput NHEJ Assay
The pcDNA3-eGFP plasmid was digested with BamHI, which cleaved between promoter and open reading frame. The pCMV-tdtomato plasmid was linearized with NotI as an efficiency control. Two million freshly prepared MECs or tumor cells were co-electroporated with 5 μg of both linear plasmids using Nucleofector machine and Human Mammary Epithelial Cell Nucleofector Kit (Lonza). The cells were then cultured in PMEC medium (DMEM/F12 containing 5% FBS, 5 μg/ml insulin, and 10 ng/ml EGF) in suspension for 18–24 hr, followed by surface marker staining and flow cytometry analysis.
Statistical Analysis
The unpaired Student’s t test was utilized for statistical analysis. Plotted values represent means ± SE. n represents the numbers of independent experiments. A p value less than 0.05 was considered statistically significant (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
Author Contributions
C-H.C. conceived the project, designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. M.Z. designed experiments, input intellectual advice, created and maintained tumor models, and reviewed the manuscript. K.R. and C.C. analyzed and organized RPPA data. D.E. and S.H. helped perform RPPA analysis with intellectual input. J.M.R. conceived the project, designed experiments, input intellectual advice, and reviewed the manuscript.
Acknowledgments
This work was supported by NIH/NCI grant CA148761 (to J.M.R.) and NIH/NCI grant CA142898 (to M.Z.). We thank the outstanding technical support from the following core labs at Baylor College of Medicine: (1) the Cytometry and Cell Sorting Core with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574) and the expert assistance of Joel Sederstrom, Amanda White, Brandon Saxton, and Erin Cox; (2) the Proteomics Core with funding from the Cancer Prevention and Research Institute of Texas Proteomics and Metabolomics Core Facility Support Award (RP120092) and NCI Cancer Center Support Grant to Antibody-based Proteomics Core/Shared Resource (P30CA125123) and the assistance of Fuli Jia, Myra Grace Costello, and Dr. Kimberley Holloway; and (3) the Integrated Microscopy Core and the assistance of Dr. Fabio Stossi and Radhika Dandekar. We especially thank Dr. Cedric Blanpain for generously providing us with the pCMV-tdtomato plasmid, Dr. Xiang Zhang for providing us with the Balb/c p53 knockout mice, and Drs. Xiang Zhang and Amy Shore for critically reading and editing the manuscript. Finally, we would also like to thank Shirley Small and Alvenia Daniels for lab general management.
Published: August 20, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.07.009.
Supplemental Information
References
- Asai T., Liu Y., Di Giandomenico S., Bae N., Ndiaye-Lobry D., Deblasio A., Menendez S., Antipin Y., Reva B., Wevrick R., Nimer S.D. Necdin, a p53 target gene, regulates the quiescence and response to genotoxic stress of hematopoietic stem/progenitor cells. Blood. 2012;120:1601–1612. doi: 10.1182/blood-2011-11-393983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S., Wu Q., McLendon R.E., Hao Y., Shi Q., Hjelmeland A.B., Dewhirst M.W., Bigner D.D., Rich J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Barker N., van Es J.H., Kuipers J., Kujala P., van den Born M., Cozijnsen M., Haegebarth A., Korving J., Begthel H., Peters P.J., Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- Bergh J., Norberg T., Sjögren S., Lindgren A., Holmberg L. Complete sequencing of the p53 gene provides prognostic information in breast cancer patients, particularly in relation to adjuvant systemic therapy and radiotherapy. Nat. Med. 1995;1:1029–1034. doi: 10.1038/nm1095-1029. [DOI] [PubMed] [Google Scholar]
- Berns E.M., Foekens J.A., Vossen R., Look M.P., Devilee P., Henzen-Logmans S.C., van Staveren I.L., van Putten W.L., Inganäs M., Meijer-van Gelder M.E. Complete sequencing of TP53 predicts poor response to systemic therapy of advanced breast cancer. Cancer Res. 2000;60:2155–2162. [PubMed] [Google Scholar]
- Castiglioni F., Terenziani M., Carcangiu M.L., Miliano R., Aiello P., Bertola L., Triulzi T., Gasparini P., Camerini T., Sozzi G. Radiation effects on development of HER2-positive breast carcinomas. Clin. Cancer Res. 2007;13:46–51. doi: 10.1158/1078-0432.CCR-06-1490. [DOI] [PubMed] [Google Scholar]
- Creighton C.J., Li X., Landis M., Dixon J.M., Neumeister V.M., Sjolund A., Rimm D.L., Wong H., Rodriguez A., Herschkowitz J.I. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehn M., Cho R.W., Lobo N.A., Kalisky T., Dorie M.J., Kulp A.N., Qian D., Lam J.S., Ailles L.E., Wong M. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Díaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J.T., Tkáč J., Cook M.A., Rosebrock A.P., Munro M., Canny M.D. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Gasco M., Shami S., Crook T. The p53 pathway in breast cancer. Breast Cancer Res. 2002;4:70–76. doi: 10.1186/bcr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy G.J., Moulding H.A., Caldecott K.W., Rulten S.L. One ring to bring them all--the role of Ku in mammalian non-homologous end joining. DNA Repair (Amst.) 2014;17:30–38. doi: 10.1016/j.dnarep.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Gupta A., Hunt C.R., Chakraborty S., Pandita R.K., Yordy J., Ramnarain D.B., Horikoshi N., Pandita T.K. Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiat. Res. 2014;181:1–8. doi: 10.1667/RR13572.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J.W., Elledge S.J. The DNA damage response: ten years after. Mol. Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Herschkowitz J.I., Zhao W., Zhang M., Usary J., Murrow G., Edwards D., Knezevic J., Greene S.B., Darr D., Troester M.A. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc. Natl. Acad. Sci. USA. 2012;109:2778–2783. doi: 10.1073/pnas.1018862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009;361:1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- Humphreys R.C., Krajewska M., Krnacik S., Jaeger R., Weiher H., Krajewski S., Reed J.C., Rosen J.M. Apoptosis in the terminal endbud of the murine mammary gland: a mechanism of ductal morphogenesis. Development. 1996;122:4013–4022. doi: 10.1242/dev.122.12.4013. [DOI] [PubMed] [Google Scholar]
- Inomata K., Aoto T., Binh N.T., Okamoto N., Tanimura S., Wakayama T., Iseki S., Hara E., Masunaga T., Shimizu H., Nishimura E.K. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell. 2009;137:1088–1099. doi: 10.1016/j.cell.2009.03.037. [DOI] [PubMed] [Google Scholar]
- Insinga A., Cicalese A., Faretta M., Gallo B., Albano L., Ronzoni S., Furia L., Viale A., Pelicci P.G. DNA damage in stem cells activates p21, inhibits p53, and induces symmetric self-renewing divisions. Proc. Natl. Acad. Sci. USA. 2013;110:3931–3936. doi: 10.1073/pnas.1213394110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerry D.J., Kittrell F.S., Kuperwasser C., Laucirica R., Dickinson E.S., Bonilla P.J., Butel J.S., Medina D. A mammary-specific model demonstrates the role of the p53 tumor suppressor gene in tumor development. Oncogene. 2000;19:1052–1058. doi: 10.1038/sj.onc.1203270. [DOI] [PubMed] [Google Scholar]
- Khanna K.K., Jackson S.P. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- Kuerbitz S.J., Plunkett B.S., Walsh W.V., Kastan M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc. Natl. Acad. Sci. USA. 1992;89:7491–7495. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtova A.V., Xiao J., Mo Q., Pazhanisamy S., Krasnow R., Lerner S.P., Chen F., Roh T.T., Lay E., Ho P.L., Chan K.S. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517:209–213. doi: 10.1038/nature14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land C.E., Boice J.D., Jr., Shore R.E., Norman J.E., Tokunaga M. Breast cancer risk from low-dose exposures to ionizing radiation: results of parallel analysis of three exposed populations of women. J. Natl. Cancer Inst. 1980;65:353–376. [PubMed] [Google Scholar]
- Li X., Lewis M.T., Huang J., Gutierrez C., Osborne C.K., Wu M.F., Hilsenbeck S.G., Pavlick A., Zhang X., Chamness G.C. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- Liu Y., Elf S.E., Miyata Y., Sashida G., Liu Y., Huang G., Di Giandomenico S., Lee J.M., Deblasio A., Menendez S. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein L.Y., Ouchi T., Aaronson S.A. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc. Natl. Acad. Sci. USA. 1998;95:13869–13874. doi: 10.1073/pnas.95.23.13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascré G., Dekoninck S., Drogat B., Youssef K.K., Broheé S., Sotiropoulou P.A., Simons B.D., Blanpain C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature. 2012;489:257–262. doi: 10.1038/nature11393. [DOI] [PubMed] [Google Scholar]
- Medina D. The mammary gland: a unique organ for the study of development and tumorigenesis. J. Mammary Gland Biol. Neoplasia. 1996;1:5–19. doi: 10.1007/BF02096299. [DOI] [PubMed] [Google Scholar]
- Meek D.W. Tumour suppression by p53: a role for the DNA damage response? Nat. Rev. Cancer. 2009;9:714–723. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- Mohrin M., Bourke E., Alexander D., Warr M.R., Barry-Holson K., Le Beau M.M., Morrison C.G., Passegué E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7:174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potten C.S., Wilson J.W., Booth C. Regulation and significance of apoptosis in the stem cells of the gastrointestinal epithelium. Stem Cells. 1997;15:82–93. doi: 10.1002/stem.150082. [DOI] [PubMed] [Google Scholar]
- Pozarowski P., Grabarek J., Darzynkiewicz Z. Flow cytometry of apoptosis. Curr. Protoc. Cytom. 2003;25 doi: 10.1002/0471142956.cy0719s25. 7.19.1–7.19.33. [DOI] [PubMed] [Google Scholar]
- Prater M.D., Petit V., Alasdair Russell I., Giraddi R.R., Shehata M., Menon S., Schulte R., Kalajzic I., Rath N., Olson M.F. Mammary stem cells have myoepithelial cell properties. Nat. Cell Biol. 2014;16:942–950. doi: 10.1038/ncb3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston D.L., Mattsson A., Holmberg E., Shore R., Hildreth N.G., Boice J.D., Jr. Radiation effects on breast cancer risk: a pooled analysis of eight cohorts. Radiat. Res. 2002;158:220–235. doi: 10.1667/0033-7587(2002)158[0220:reobcr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Rios A.C., Fu N.Y., Lindeman G.J., Visvader J.E. In situ identification of bipotent stem cells in the mammary gland. Nature. 2014;506:322–327. doi: 10.1038/nature12948. [DOI] [PubMed] [Google Scholar]
- Roarty K., Shore A.N., Creighton C.J., Rosen J.M. Ror2 regulates branching, differentiation, and actin-cytoskeletal dynamics within the mammary epithelium. J. Cell Biol. 2015;208:351–366. doi: 10.1083/jcb.201408058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothkamm K., Krüger I., Thompson L.H., Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholzen T., Gerdes J. The Ki-67 protein: from the known and the unknown. J. Cell. Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Shackleton M., Vaillant F., Simpson K.J., Stingl J., Smyth G.K., Asselin-Labat M.L., Wu L., Lindeman G.J., Visvader J.E. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- Sørlie T., Perou C.M., Tibshirani R., Aas T., Geisler S., Johnsen H., Hastie T., Eisen M.B., van de Rijn M., Jeffrey S.S. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulou P.A., Candi A., Mascré G., De Clercq S., Youssef K.K., Lapouge G., Dahl E., Semeraro C., Denecker G., Marine J.C., Blanpain C. Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat. Cell Biol. 2010;12:572–582. doi: 10.1038/ncb2059. [DOI] [PubMed] [Google Scholar]
- Stingl J., Eirew P., Ricketson I., Shackleton M., Vaillant F., Choi D., Li H.I., Eaves C.J. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- Stürzbecher H.W., Donzelmann B., Henning W., Knippschild U., Buchhop S. p53 is linked directly to homologous recombination processes via RAD51/RecA protein interaction. EMBO J. 1996;15:1992–2002. [PMC free article] [PubMed] [Google Scholar]
- Tang W., Willers H., Powell S.N. p53 directly enhances rejoining of DNA double-strand breaks with cohesive ends in gamma-irradiated mouse fibroblasts. Cancer Res. 1999;59:2562–2565. [PubMed] [Google Scholar]
- Tang J., Fernandez-Garcia I., Vijayakumar S., Martinez-Ruis H., Illa-Bochaca I., Nguyen D.H., Mao J.H., Costes S.V., Barcellos-Hoff M.H. Irradiation of juvenile, but not adult, mammary gland increases stem cell self-renewal and estrogen receptor negative tumors. Stem Cells. 2014;32:649–661. doi: 10.1002/stem.1533. [DOI] [PubMed] [Google Scholar]
- Tomasetti C., Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78–81. doi: 10.1126/science.1260825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader J.E., Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014;28:1143–1158. doi: 10.1101/gad.242511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl A.F., Donaldson K.L., Fairchild C., Lee F.Y., Foster S.A., Demers G.W., Galloway D.A. Loss of normal p53 function confers sensitization to Taxol by increasing G2/M arrest and apoptosis. Nat. Med. 1996;2:72–79. doi: 10.1038/nm0196-72. [DOI] [PubMed] [Google Scholar]
- Woodward W.A., Chen M.S., Behbod F., Alfaro M.P., Buchholz T.A., Rosen J.M. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. USA. 2007;104:618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Behbod F., Atkinson R.L., Landis M.D., Kittrell F., Edwards D., Medina D., Tsimelzon A., Hilsenbeck S., Green J.E. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008;68:4674–4682. doi: 10.1158/0008-5472.CAN-07-6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Atkinson R.L., Rosen J.M. Selective targeting of radiation-resistant tumor-initiating cells. Proc. Natl. Acad. Sci. USA. 2010;107:3522–3527. doi: 10.1073/pnas.0910179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H., Chen L., Pledger W.J., Fang J., Chen J. p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene. 2014;33:734–744. doi: 10.1038/onc.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.B., Elledge S.J. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.