

Abstract
In this study, we demonstrate that treatment of T lymphoblastic leukemic Molt4 cells with farnesol activates the apoptosome via the intrinsic pathway of apoptosis. This induction was associated with changes in the level of intracellular potassium and calcium, the dissipation of the mitochondrial and plasma membrane potential, release of cytochrome c, activation of several caspases, and PARP cleavage. The induction of apoptosis by farnesol was inhibited by the addition of the pan-caspase inhibitor Z-VAD-fmk and by the exogenous expression of the anti-apoptotic protein Bcl2. Analysis of the gene expression profiles by microarray analysis revealed that farnesol increased the expression of several genes related to the unfolded protein response (UPR), including CHOP and CHAC1. This induction was associated with the activation of the PERK-eIF2α-ATF3/4 cascade, but not the XBP-1 branch of the UPR. Although farnesol induced activation of the ERK1/2, p38, and JNK pathways, inhibition of these MAPKs had little effect on farnesol-induced apoptosis or the induction of UPR-related genes. Our data indicate that the induction of apoptosis in leukemic cells by farnesol is mediated through a pathway that involves activation of the apoptosome via the intrinsic pathway and induction of the PERK-eIF2α-ATF3/4 cascade in a manner that is independent of the farnesol-induced activation of MAPKs.
Keywords: Leukemia, Molt4, ER stress, Apoptosis, Gene expression
1. Introduction
High intake of fruit and vegetables has been found to reduce cancer risk in several studies [1–4] and this has been in part attributed to increased consumption of pharmacologically active agents, including isoprenoids. Farnesol is naturally found with several related isoprenoids, including perillyl alcohol and geraniol, in many fruits and essential oils of many plants [1]. Isoprenoids are implicated in many biochemical and physiological processes, including lipid biosynthesis and the regulation of cell differentiation [5]. These isoprenoid alcohols also inhibit cell proliferation and induce apoptosis in a number of malignant cell lines [5–11] and exhibit anti-tumor and anti-carcinogenic effects in vivo [1,12–19]. Isoprenoid alcohols have been reported to act as chemopreventative agents in colon carcinogenesis and pancreatic cancer models in animals and anti-tumorigenic effects have also been observed in an initiation-promotion hepatocarcinogenesis model in rats [14]. A recent study showed that farnesol inhibited tumor growth and enhanced the anti-cancer effects of bortezomib in multiple myeloma xenografts in mice [20]. Administration of farnesol also significantly suppressed azoxymethane-induced formation of aberrant crypt foci and crypt multiplicity in the rat colon [15] and exhibited anti-genotoxic effects against benzo(a) pyrene by reducing DNA strand breaks and the formation of DNA adducts in vivo [13]. In addition to its anti-cancer effects, farnesol exhibits immunomodulatory properties. Recent studies showed that farnesol exhibited anti-inflammatory and anti-allergic effects in a mouse model of allergic asthma and suppressed cellular adaptive immunity [21,22]. A different study reported that farnesol ameliorated cigarette smoke extract-induced inflammation and lung injury [23] and inhibited lipopolysaccharide-induced neuro-degeneration [24]. Together, the anti-cancer, anti-inflammatory and anti-allergic properties of farnesol have raised the potential of farnesol as a chemopreventative pharmacological agent as well as a putative therapeutic drug in the treatment of certain cancers and inflammatory diseases.
The sensitivity to farnesol-induced apoptosis varies widely among cancer cells. The mechanisms by which farnesol induces apoptosis varies greatlyamong cell types [5,11,25–28]. Leukemia and lymphoma cells are particularly sensitive to farnesol-induced apoptosis [7–9,11]; however, little is know about its mechanism. To study this further, we examined the signaling pathways by which farnesol induces apoptosis in human T lymphoblastic leukemia Molt4 cells. We demonstrated that farnesol treatment results in the activation of the apoptosome as indicated by changes in the intracellular level of several cations, the dissipation of the mitochondrial membrane potential (ΔΨm) and plasma membrane potential, release of cytochrome c from the mitochondria into the cytoplasm, increased annexin V binding and poly(ADP-ribose) polymerase (PARP) cleavage, and induction of caspase activity. Comparison of the gene expression profiles of Molt4 cells treated with or without farnesol by microarray analysis revealed that in Molt4 cells farnesol induces a partial unfolded protein response (UPR). We further show that this increase is independent of the farnesol-induced activation of several mitogen-activated protein kinases (MAPKs). Our study indicates that farnesol-induced apoptosis in Molt4 cells is mediated through activation of the apoptosome via the intrinsic pathway and induction of a partial UPR. The greater sensitivity of leukemic and lymphoma cells to farnesol-induced apoptosis suggests that these cells might be particularly good chemotherapeutic targets for cancer therapy using farnesol.
2. Materials and methods
2.1. Materials
Farnesol ([2E, 6E]-3,7,11-trimethyl-2,6,10-dodecatrien-1-ol) was obtained from Sigma (St. Louis, MO). Z-VAD-fmk was purchased from Kamiya Biomed. (Seattle, WA). The MEK1/ 2 inhibitor U0126 was obtained from Promega (Madison, WI), and the p38 MAPK and JNK inhibitors, SB203580 and SP600125, respectively, were purchased from Calbiochem (La Jolla, CA).
2.2. Cell culture
The human T cell leukemia cell line Jurkat (clone E6-1) cells were obtained from the American Typing Culture Collection (ATCC, Manassas, VA). Human T lymphoblastic leukemia Molt4-Bcl2 and Molt4-hyg, derived through stable transfection of the expression vector pMEP with and without full length murine Bcl2, respectively, were provided by Dr. Y.A. Hannun (Department of Medicine, Duke University Medical Center, Durham, NC) [29]. DEL, an ALK-positive anaplastic large-cell lymphoma, was obtained from Dr. S. Mathas (Medical University, Berlin, Germany). Fibrosarcoma Ht1080 and Ht1080mut containing wild type p53 and mutant p53 were obtained from Dr. A. Merrick, NIEHS. All cells were cultured in RPMI-1640 medium (Gibco, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Atlanta, GA), 2 mM L-glutamine, penicillin, and streptomycin (Gibco). The T-cell hybridoma cell line DO11.10 was provided by Dr. B.A. Osborne (University of Massachusetts, Amherst, MA) and grown in RPMI-1640 medium supplemented with 10% horse serum (Hyclone Laboratories, Logan, UT). Farnesol was dissolved in DMSO and cells were treated with farnesol at the concentrations indicated or with vehicle control (0.1% DMSO final concentration).
2.3. Flow cytometry
For cell cycle analysis, cells were fixed in 70% ethanol and resuspended in PBS containing 50 μg/ml of propidium iodide (PI; Sigma) and 0.1 mg/ml of DNase-free RNase A. The analysis of Annexin V binding was carried out using the ApoAlert Annexin V-FITC kit from Clontech (Mountain View, CA) according to the manufacturer’s protocol. Cells (106) were resuspended in 200 μl of binding buffer containing Annexin V-FITC and PI. Changes in mitochondrial membrane potential (ΔΨm) were measured by flow cytometry using 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylben-zimidazolcarbocyanine iodide (JC-1; Thermo Fisher Sci., Carlsbad, CA). Cells (5 × 105) were incubated in 1 ml of culture media containing 10 μM JC-1 for 30 min at 37 °C and analyzed as described [30]. Changes in the plasma membrane potential were measured by using DiBAC4 (Thermo Fisher Sci.). Cells (5 × 105) were resuspended in 1 ml of culture media containing 150 nM DiBAC4. For measuring the changes of intracellular K+, Na+, and Ca2 + level, PBFI, SBFI, and Fluo-3 (Thermo Fisher Sci.) were used, respectively. Cells (5 × 105) were pretreated with 75 μM farnesol for 4 h and then loaded with PBFI (10 μM), SBFI (10 μM), or Fluo-3 (5 μM) for 1 h. Flow cytometric analyses of labeled cells were performed using a FACSort (Becton Dickinson, San Jose, CA) flow cytometer with CellQuest software (DNA, Annexin, JC-1, DiBAC4), or an LSRII (Becton Dickinson) flow cytometer with FACSDiVa software (PBFI, SBFI, Fluo-3).
2.4. Caspase activity assay and apoptosis detection ELISA
Cells treated with 75 μM farnesol and vehicle-treated cells (2 × 106 cells) were harvested at the times indicated and resus-pended in 50 μl of hypotonic buffer containing 25 mM HEPES (pH 7.4), 5 mM MgCl2, 5 mM EDTA, 5 mM DTT, and protease inhibitor cocktail (Sigma). Cells were then lysed by four cycles of freezing and thawing. Caspase activity was assayed using the CaspACETM Assay System (Promega, Madison, WI) in a Fluoroskan Ascent FL (Lab Systems, Kennett Square, PA) according to the manufacturer’s instructions. Apoptosis was measured with a Cell Death Detection ELISA Kit (Roche, Indianapolis, IN).
2.5. Western blot analysis
Cells were treated with 75 or 100 μM farnesol or vehicle at the times indicated and then washed twice with ice-cold PBS, pH 7.4, followed by centrifugation at 500 × g for 5 min. The cell pellet was resuspended in 500 μl of extraction buffer, containing 220 μM mannitol, 68 mM sucrose, 50 mM PIPES-KOH (pH 7.4), 50 mM KCl, 5 mM EGTA, 2 mM MgCl2, 1 mM dithiothreitol (DTT) and protease inhibitors (Cocktail, Sigma). After 30 min incubation on ice, cells were homogenized using a Sonifier (Branson). Cell homogenates were centrifuged at 14,000 × g for 15 min. The supernatants were removed and stored at −70 °C. Cytosolic proteins were examined by Western blot analysis with antibodies specific for cytochrome c (7H8.2C12, Pharmingen, San Diego, CA), caspase-9, -3, PARP, p-p38, p38, p-ERK, ERK, p-JNK, JNK, eIF2α (Cell Signaling Technology, Danvers, MA) and secondary antibodies conjugated to horseradish peroxidase (EDM Millipore, Billerica, MA) followed by visualization by enhanced chemiluminescence (Pierce) following the manufacturer’s protocol. To examine autophagy, Western blot analysis was performed using an antibody against LC3 (Cell Signaling Technology). Proteins were quantified using ImageQuant TL software analysis (GE Healthcare, Piscataway, NJ). The intensities of the experimental bands minus the background were normalized against the intensity of β-actin bands minus the background.
2.6. Quantitative real-time PCR (QRT-PCR)
Cells treated with farnesol or vehicle at the concentration and time indicated, were collected and RNA was isolated using TriReagent (Sigma) following the manufacturer’s protocol and was reversed-transcribed using a high capacity cDNA archive kit according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA). QRT-PCR reactions were performed as described previously using the POWER SYBER® Green PCR master mix (Applied Biosystems) [11]. The forward and reverse oligonucleotide primers for ATF3 (5′-CTGCAGAAAGAGTCGGAG, 5′-TGAGCCCG-GACAATACAC), GRP78 (5′-CCAGAATCGCCTGACACCTG, 5′-AGCACTAGCAGATCAGTGTC), CHAC1 (5′-CCTGAAGTACCTGAATGT-GC-GAGA, 5′-GCAGCAAGTATTCAAGGTTGTGGC), and CHOP (5′-GAAACGG-AAACAGAGTGGTCATTCCCC, 5′-GTGGGATTGAGGGTCA-CATCATTGGCA) were purchased from Sigma. PCR assays were performed using the 7300 Real Time PCR System (Applied Biosystems). All results were normalized relatively to the 18S rRNA or GAPDH transcripts and are presented as mean ± SD of three independent experiments. No significant differences were observed in the relative expression pattern when data were normalized against 18S or GAPDH. The nonconventional splicing of X-box binding protein 1 (XBP1) mRNA was examined by reverse transcription-PCR (RT-PCR) using 5′-CCTTGTAGTTGAGAACCAGG and 5′-GGGGCTTGGTATATATGTGG as primers. This will amplify both unspliced (XBP1u) and spliced (XBP1s) XBP1 mRNAs. The siRNAs to knockdown CHAC1 and CHOP expression were obtained from Dharmacon (Lafayette, CO).
2.7. Microarray analysis
Microarray analyses were carried out by the NIEHS Microarray Group (NMG) using Agilent whole human genome oligo arrays (14850) (Agilent Technologies, Palo Alto, CA) following the Agilent 1-color microarray-based gene expression analysis protocol as described previously [11]. Total RNA was isolated from Molt4 cells treated with vehicle or 75 μM farnesol for 4 h using Qiagen (Germantown, MD) RNeasy Mini Kit and subsequently amplified using the Agilent Low RNA Input Fluorescent Linear Amplification Kit protocol. RNA from 3 independent experiments was analyzed in duplicate. Hybridizations were performed as described previously [11]. The Agilent Feature Extraction Software performed error modeling, adjusting for additive and multiplicative noise. The resulting data were processed using the Rosetta Resolver® system (version 7.2) (Rosetta Biosoftware, Kirkland, WA). In order to identify differentially expressed probes, analysis of variance (ANOVA) was used to determine if there was a statistical difference between the means of groups. In addition, we used a multiple test correction to reduce the number of false positives. Specifically, an error-weighted ANOVA and Benjamini–Hochberg multiple test correction with a p value of p <0.01was performed using Rosetta Resolver (www.rosettabio.com). The microarray data discussed in this study have been deposited in the NCBI’s Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) as GSE46670.
2.8. Measurement of intracellular reactive oxygen species (ROS)
The generation of intracellular ROS was measured by using the oxidation-sensitive fluorescent dye 5-(and-6)-carboxy-2′,-7′-dichlorofluorescein diacetate (DCFH-DA; Thermo Fisher Sci.). Molt4 cells were incubated with 20 μM DCFH-DA for 30 min. The DCFH-DA loaded cells were then seeded in 96-well plate (20,000 cells/well) and subsequently treated with 100 μM farnesol in the presence or absence of 300 μM vitamin C or 100 μM butylated hydroxyanisol (BHA). After excitation at 480 nm, fluorescence intensity at 530 nm was measured at 30 or 60 min intervals using a CytoFluor 2350 Fluorescence Measurement system (Millipore, Bedford, MA).
3. Results
3.1. Farnesol inhibits the proliferation of human T lymphoblastic leukemia cells
Previous studies have shown that farnesol inhibits the proliferation of a number of different carcinoma cell lines [5–7,11]. As shown in Fig. 1, farnesol treatment greatly inhibited the growth of and induced cell death in the lymphoblastic leukemia cell lines, Molt4-hyg and Jurkat, which contain mutant p53, as well as the anaplastic large-cell lymphoma cell line DEL, the murine T cell lymphoma S49, and the T cell hybridoma DO11.10 cells. The anaplastic lymphoma kinase (ALK)-positive DEL cells were as sensitive as the ALK-negative Molt4-hyg and Jurkat cells. The inhibition of cell proliferation by farnesol was concentration-dependent and observed at concentrations as low as 10 μM (Fig. 1A). All lymphoma cell lines showed a much greater sensitivity to the growth–inhibitory effects of farnesol than the fibrosarcoma Ht1080 cells (Fig. 1A) and several lung carcinoma cell lines, as reported previously [11]. No significant difference in farnesol sensitivity was observed between Ht1080 cells containing wild type or mutant p53. Of the more sensitive cell lines, Molt4-hyg was selected to further investigate the induction of apoptosis by farnesol.
Fig. 1.

Inhibition of proliferation and induction of apoptosis in T lymphoblastic Molt4-hyg and Molt4-Bcl2 cells. (A) Molt4-hyg, Jurkat, DEL, DO11.10, Ht1080, and Ht1080mut cells were treated for 24 h with farnesol at the concentrations indicated before cell number was determined. Each error bar represents mean ± SE; *indicates statistically different from vehicle-treated control (p <0.01). (B) Effect of Bcl-2 expression on the farnesol-induced inhibition of Molt4 cell proliferation. Molt4-hyg and Molt4-Bcl2 cells, that overexpress Bcl-2, were treated with farnesol at the concentrations indicated and 16 h later the relative cell number determined and plotted. *Indicates statistically different from vehicle-treated control and from farnesol-treated Molt4-Bcl2 cells (p <0.01). (C and D) Cell cycle analysis. Molt4-hyg and Molt4-Bcl2 cells were treated with 75 μM farnesol or vehicle in the presence or absence of Z-VAD-fmk as indicated. At 0 (control), 8 and 16 h cell cycle distribution was performed as described in Section 2. The percentage of the cells in sub-G1/G0 is indicated.
To determine the effect of farnesol on cell cycle progression, propidium iodide stained cells were analyzed by flow cytometry. As shown in Fig. 1C, treatment of Molt4-hyg cells with farnesol for 8 h and 16 h caused a substantial increase in the sub-G1/G0 population constituting dead cells. At 8 h and 16 h, this percentage was 21.4% and 55.6%, respectively.
To determine whether this sub-G1/G0 population constituted apoptotic cells, Molt4-hyg cells were treated with 75 μM farnesol and at different time points cells were analyzed for the induction of several apoptotic markers, including the cysteine proteases, caspase-9 and caspase-3, and the caspase-3 target PARP-1. Western blot analysis showed that farnesol treatment of Molt4-hyg cells induced proteolytic cleavage of caspase-9, caspase-3, and PARP-1 (Fig. 2A and B). Cleavage of these proteins was observed as early as 2 h after the addition of farnesol and increased further at 4 and 6 h of treatment. The activation of caspases was supported by the increased cleavage of the fluorogenic substrate Ac-YVAD-AMC by cellular extracts from farnesol-treated cells. The activation of caspases by farnesol was dose-dependent (Fig. 2C), while the addition of the pan-caspase inhibitor Z-VAD-fmk totally blocked farnesol-induced caspase-3-like activity (Fig. 2D). The induction of apoptosis was supported by the increase in the percentage of the annexin-Vhigh cell population (Fig. 2E). Together these results indicated that the increase in the sub-G1/G0 population in farnesol-treated Molt4-hyg cells is due to the induction of apoptosis. No changes in the autophagy markers, LC3I and LC3 II, was observed suggesting that farnesol did not induce autophagy in Molt4 cells (not shown).
Fig. 2.

Induction of apoptosis in farnesol-treated Molt4-hyg cells. (A) Molt4-hyg cells were treated with 75 μM farnesol for the times indicated. Total cell lysates were isolated and examined by Western blot analysis with antibodies recognizing full-length and cleaved caspase-3, caspase-9, and PARP. β-Actin was used as a control for equal loading. Shown are representative images of two independent experiments. (B) The levels of cleaved PARP, caspase 9 and 3 shown under A were quantitated as described in Section 2. Intensities were normalized against β-actin. The relative levels were plotted. (C) Dose-dependent induction of caspase activity by farnesol. (D) Molt4-hyg cells were pretreated with pan-caspase inhibitor Z-VAD-fmk (10 μM) for 30 min before the addition of farnesol (75 μM) and 8 h later analyzed for caspase-3-like activity. (E and F) Bcl2 inhibits farnesol-induced apoptosis in Molt4 cells. Molt4-hyg and Molt4-Bcl2 cells were treated with 75 μM farnesol and at the time intervals indicated cells were collected and assayed for annexin V binding (E) and caspase 3-like activity (F). Each error bar represents mean ± SE; *indicates statistically different from vehicle-treated Molt4-hyg control and farnesol-treated Molt4-Bcl2 cells (p <0.01).
3.2. Bcl-2 inhibits farnesol-induced cellular apoptosis
Bcl-2 functions as an anti-apoptotic protein and with other members of the family play a critical role in regulating apoptosis in a number of cell systems [29,31]. To determine whether Bcl-2 was able to inhibit farnesol-induced apoptosis, we examined the effect of farnesol in Molt4-Bcl2 cells, a stable cell line ectopically expressing Bcl-2. Fig. 1B shows that Molt4-Bcl2 cells were significantly less sensitive to the growth–inhibitory effects of farnesol than control Molt4-hyg cells. Cell cycle analysis demon-strated that the apoptotic sub-G1/G0 population was substantially smaller in farnesol-treated Molt4-Bcl2 cells than Molt4-hyg cells (Fig. 1C and D); at 16 h 15% of the Molt4-Bcl2 cells were in the sub-G1/G0 population, compared to 55.6% of the Molt4-hyg cells. The decreased sensitivity of Molt-Bcl2 cells to farnesol-induced apoptosis was further supported by the lower percentage of annexin Vhigh cells (Fig. 2E) and the reduced induction of caspase activity (Fig. 2F).
The mitochondrial membrane potential (ΔΨm) plays a critical role in the early phase of apoptosis [32]. To determine the effect of farnesol on ΔΨm, the ΔΨm was examined by monitoring the fluorescence of the cationic lipophilic cationic probe JC-1 that has the unique property of forming J-aggregates (red fluorescence) under high mitochondrial ΔΨm conditions and the monomeric form (green fluorescence) when ΔΨm is low [30]. Fig. 3A shows that treatment of Molt4-hyg with 75 μM farnesol resulted in increased formation of JC-1 monomers suggesting that farnesol-induced apoptosis in Molt4-hyg cells is accompanied by a reduction in ΔΨm. To determine whether this collapse of ΔΨm was dependent on caspase activation, cells were treated with the pan-caspase inhibitor Z-VAD-fmk. Z-VAD-fmk did not block the formation of JC-1 monomers suggesting that the collapse of ΔΨm was not dependent on caspase activation (Fig. 3C). As shown in Fig. 3B and C, the farnesol-induced dissipation of ΔΨm was significantly reduced in Molt4-Bcl2 cells.
Fig. 3.

Farnesol induces dissipation of the mitochondrial membrane potential and release of cytochrome c in Molt4 cells. (A and B) Molt4-hyg and Molt4-Bcl2 cells were treated with 75 μM farnesol or vehicle (DMSO) for 8 h before JC-1 aggregation, a measurement of mitochondrial membrane potential, was examined by flow cytometry. The percent monomers and aggregates are indicated. (C) In contrast to expression of Bcl-2, the pan-caspase inhibitor Z-VAD-fmk does not affect the dissipation of the mitochondrial potential in farnesol-treated Molt4-hyg cells. Cells were treated with farnesol in the presence or absence of the Z-VAD-fmk and at the times indicated the percent JC-1 monomers determined. *Indicates statistically different from vehicle-treated Molt4-hyg control and farnesol-treated Molt4-Bcl2 cells (p <0.01). (D) Release of cytochrome c (Cytos. Cyt. C) in the cytoplasm. Cells were collected at the times indicated and cytosolic fractions analyzed by Western blot analysis using a cytochrome c-specific antibody. Shown are representative images of two independent experiments. (E) The levels of Cytos. Cyt. C shown under A were quantitated and normalized against β-actin and plotted. (For interpretation of the references to color in text, the reader is referred to the web version of this article.)
Dissipation of the ΔΨm can lead to a release of cytochrome c from mitochondria into the cytoplasm and subsequently result in the formation of the apoptosome and activation of caspase-9 [33,34]. As shown in Fig. 3D and E, treatment of Molt4-hyg cells with farnesol induced the release of significant amounts of cytochrome c into the cytoplasm. This release was greatly inhibited in Molt4-Bcl2 cells.
3.3. Farnesol changes intracellular cation levels and induces plasma membrane depolarization
While externalization of membrane phosphatidylserine, depolarization of the mitochondrial membrane potential, and activation of caspases are characteristics of the apoptotic process, plasma membrane depolarization and loss of intracellular potassium are also critical early steps during apoptosis [35,36]. Analysis of changes in plasma membrane potential showed that farnesol induced depolarization of the plasma membrane in Molt4-hyg cells by approximately 15%, that was partially inhibited by the addition of the pan-caspase inhibitor ZVAD (Fig. 4). In contrast, overexpression of Bcl-2 largely prevented plasma membrane depolarization upon farnesol treatment. Interestingly, farnesol treatment resulted in an increase in cell size (increased forward scattered light), regardless of the presence or absence of Bcl-2. However, a loss of cell size is observed for the depolarized population of cells, characteristic of apoptosis.
Fig. 4.

Farnesol induced depolarization of plasma membrane potential in Molt4 cells. (A) Molt4-hyg and Molt4-Bcl2 cells were treated with 75 μM farnesol for 8 h with or without Z-VAD-fmk. Then changes in the plasma membrane potential were measured using DiBAC4 as described in Section 2. (B) Time course of plasma membrane depolarization.
Cationic fluxes have been shown to play a critical role in regulating cell shrinkage, plasma membrane depolarization, as well as activation of caspases and apoptotic nucleases [35–37]. As shown in Table 1 and Fig. 5, farnesol induced a loss of intracellular potassium, along with an increase in intracellular sodium in Molt4-hyg cells. In contrast, Molt4-Bcl-2 cells showed very little change in these intracellular ions (Table 1 and Fig. 5). Moreover, only Molt4-hyg cells capable of undergoing apoptosis upon farnesol treatment, but not Molt4-Bcl2 cells, showed an increase in intracellular calcium, characteristic of this mode of cell death (Table 1 and Fig. 5). Together, these data suggest that the loss of intracellular potassium along with the increase in intracellular sodium and calcium correlates with cells undergoing apoptosis.
Table 1.
Percent change in the intracellular K+, Na+, and Ca2+ in farnesol-treated Molt4-hyg and Molt4-Bcl2 cells.
| K+ | Control
|
Farnesol
|
||
|---|---|---|---|---|
| High K+ | Low K+ | High K+ | Low K+ | |
| Molt4-hyg | 97.4% ± 0.1 | 2.6% ± 0.1 | 83.7% ± 2.7 | 16.3% ± 2.7 |
| Molt4-Bcl2 | 98.5% ± 0.3 | 1.5% ± 0.3 | 95.3% ± 1.5 | 4.7% ± 1.5 |
|
| ||||
| Na+ | Control
|
Farnesol
|
||
| High Na+ | Low Na+ | High Na+ | Low Na+ | |
|
| ||||
| Molt4-hyg | 2.3% ± 0.1 | 97.7% ± 0.1 | 19.6% ± 2.9 | 80.4% ± 2.9 |
| Molt4-Bcl2 | 4.0% ± 2.7 | 96.0% ± 2.7 | 5.3% ± 3.8 | 94.7% ± 3.8 |
|
| ||||
| Ca2+ | Control
|
Farnesol
|
||
| High Ca2+ | Low Ca2+ | High Ca2+ | Low Ca2+ | |
|
| ||||
| Molt4-hyg | 2.5% ± 0.2 | 97.5% ± 0.2 | 39.1% ± 13.9 | 60.9% ± 13.9 |
| Molt4-Bcl2 | 3.8% ± 0.8 | 96.2% ± 0.8 | 4.0% ± 0.2 | 96.0% ± 0.2 |
Fig. 5.

Effect of farnesol on the intracellular level of potassium, sodium, and calcium of Molt4-hyg and Molt-Bcl2 cells. Cells were treated with 75 μM farnesol or vehicle for 4 h and then loaded with PBFI, SBFI, and Fluo-3 to measure the intracellular level of K+, Na+, and Ca2+, respectively, as described in Section 2.
3.4. Farnesol-induced changes in gene expression in Molt4 cells
To obtain greater insight into the mechanism by which farnesol induced apoptosis in Molt4 cells, we compared the gene expression profiles between Molt4-hyg cells treated for 4 h with vehicle (DMSO) or with 75 μM farnesol by microarray analysis. We treated at suboptimal conditions to focus on early changes in apoptosis-related gene expression. This analysis revealed that 185 and 280 genes were, respectively, induced or repressed more than 1.5 fold in farnesol-treated Molt4-hyg cells. The full data set is available at NCBI’s Gene Expression Omnibus as GSE46670. Based on their roles in ER stress, cell death and proliferation, a selective list of differentially regulated genes is shown in Table 2. In Molt4-hyg cells, farnesol induced the expression of several UPR-related genes, including the activating transcription factor 3 (ATF3), DNA damage-inducible transcript 3 (DDIT3 or CHOP/ GADD153), and homocysteine-inducible, ER stress-inducible, ubiquitin-like domain member 2 (HERPUD2); however, the expression of another UPR-related gene, the chaperone GRP78 (BIP/HSPA5), was down regulated. In addition, farnesol altered the expression of several other apoptosis and cell proliferation related genes, including the cation transport regulator-like protein 1 (CHAC1) and chloride intracellular channel 4 (CLIC4) (Table 2).
Table 2.
Farnesol-induced changes in gene expression in Molt4-hyg cells.
| Gene name | Description | GeneBank # | Fold change |
|---|---|---|---|
| ER stress | |||
| ATF3 | Activating transcription factor 3 | NM_004024 | 2.8 |
| DDIT3 (CHOP/GADD153) | DNA-damage-inducible transcript 3 | NM_004083 | 1.8 |
| HERPUD2 | HERPUD family member 2 | BC035153 | 1.6 |
| HERPUD1 | HERPUD family member 1 | NM_014685 | 1.5 |
| ATF4 | Activating transcription factor 4 | NM_001675 | 1.3 |
| GRP78 | 78 kD glucose-regulated protein (BIP/ HSPA5) | NM_005347 | −2.5 |
| Cell death | |||
| SESN2 | Sestrin 2 | NM_031459 | 4.3 |
| CHAC1 | ChaC, cation transport regulator homolog 1 | NM_024111 | 4.0 |
| SARS | Seryl-tRNA synthetase | AK022339 | 2.7 |
| CLIC4 | Chloride intracellular channel 4 | NM_013943 | 2.4 |
| FOXA3 | Forkhead box A3 | NM_004497 | 2.3 |
| PTK2B (PYK2) | PTK2B protein tyrosine kinase 2 beta | NM_173174 | 2.0 |
| SLC3a2 | Solute carrier family 3, member 2 | NM_002394 | 1.9 |
| PDCD6IP | Programmed cell death 6 interacting protein | NM_013374 | 1.5 |
| Bcl2L11 | Bcl2-like 11 | BF675199 | −1.3 |
| NAIP (BIRC1) | NRL family, apoptosis inhibitory protein | BF675199 | −1.3 |
| PDCD7 | Programmed cell death 7 | NM_005707 | −1.3 |
| BAX | BCL2-associated X protein | NM_004324 | −1.3 |
| NAIF1 | Nuclear apoptosis inducing factor 1 | BC021580 | −1.3 |
| MLL | Myeloid/lymphoid or mixed-lineage leukemia | NM_005933 | −1.3 |
| DRAM | Damage-regulated autophagy modulator | NM_018370 | −1.4 |
| XAF1 | XIAP associated factor 1 | NM_017523 | −1.4 |
| PRKDC | Protein kinase, DNA-activated, catalytic polypeptide | NM_006904 | −1.4 |
| MAGI3 | Membrane associated guanylate kinase, WW and PDZ domain containing 3 | NM_020965 | −1.4 |
| P53AIP1 | p53-regulated apoptosis-inducing protein 1 | AB045831 | −1.5 |
| PPP1R13B | Protein phosphatase 1, regulatory (inhibitor) subunit 13B | NM_015316 | −1.5 |
| EP300 | E1A binding protein p300 | NM_001429 | −1.5 |
| FANCG | Fanconi anemia, complementation group G | NM_007126 | −1.6 |
| DHCR24 | 24-dehydrocholesterol reductase | NM_014762 | −1.6 |
| ACIN1 | Apoptotic chromatin condensation inducer 1 | NM_014977 | −1.6 |
| PDCD1 | Programmed cell death 1 | NM_005018 | −1.7 |
| RAG1 | Recombination activating gene 1 | NM_000448 | −2.1 |
| AATK | Apoptosis-associated tyrosine kinase | AB014541 | −2.6 |
| Proliferation | |||
| JDP2 | Jun dimerization protein 2 | NM_130469 | 5.3 |
| NANOG | Nanog homeobox | NM_024865 | 2.0 |
| FGF7 | Fibroblast growth factor 7 | NM_002009 | 2.0 |
| CDK11B | Cyclin-dependent kinase 11B | NM_005605 | 1.5 |
| CDC25A | Cell division cycle 25 homolog A | NM_001789 | −1.3 |
| CDC2L1 | Cell division cycle 2-like 1 (PITSLRE proteins) | NM_033487 | −1.3 |
| APPL1 | Adaptor protein, phosphotyrosine interaction | NM_012096 | −1.4 |
| MKI67 | PH domain and leucine zipper containing 1 antigen identified by monoclonal antibody Ki-67 | NM_002417 | −1.4 |
| CBFA2T2 | Core-binding factor, runt domain, alpha subunit 2, translocated to 2 | AK021595 | −1.4 |
| ZEB1 | Zinc finger E-box binding homeobox 1 | NM_030751 | −1.4 |
| PDAP1 | PDGFA associated protein 1 | NM_014891 | −1.5 |
| PA2G4 | Proliferation-associated 2G4 | NM_006191 | −1.5 |
| AR | Androgen receptor | NM_000044 | −1.5 |
| MTCP1 | Mature T-cell proliferation 1 | AK095886 | −1.5 |
| CDCA2 | Cell division cycle associated 2 | NM_152562 | −1.8 |
| MPHOSH8 | M-phase phosphoprotein 8 | NM_017520 | −1.8 |
| Others | |||
| SLC7A11 | Solute carrier family 7 | NM_014331 | 5.1 |
| PCK2 | Phosphoenolpyruvate carboxykinase 2 | NM_004563 | 3.8 |
| CEBPB | CCAAT/enhancer binding protein, beta | NM_005194 | 3.3 |
| LMO2 | LIM domain only 2 (rhombotin-like 1) | NM_005574 | 2.8 |
| CEACAM21 | Carcinoembryonic antigen-related cell adhesion molecule 21 | NM_033543 | 2.8 |
| FOXA3 | Forkhead box A3 | NM_004497 | 2.3 |
| MMP14 | Matrix metallopeptidase 14 | NM_004995 | 2.2 |
| EGR1 | Early growth response 1 | NM_001964 | 2.2 |
| HDAC5 | Histone deacetylase 5 | NM_005474 | 1.8 |
| KLF13 | Kruppel-like factor 13 | NM_015995 | −1.7 |
| MPHOSPH8 | M-phase phosphoprotein | NM_017520 | −1.8 |
| GDF10 | Growth differentiation factor 10 | NM_004962 | −1.8 |
| CEBPA | CCAAT/enhancer binding protein, alpha | NM_004364 | −2.1 |
The differential expression of several of these genes was confirmed by QRT-PCR analysis. Farnesol induced ATF3 and CHAC1 expression in a concentration-dependent manner (Fig. 6A). Moreover, ATF3, CHAC1, and CHOP mRNA expression was increased in a time-dependent manner upon the addition of farnesol, whereas the expression of GRP78 was decreased (Fig. 6B). The induction of CHAC1 mRNA was the most dramatic and increased about 200-fold after 4 h of farnesol treatment. Although Bcl-2 inhibited farnesol-induced cell death in Molt4 cells (Figs. 1 and 2), it did not block the changes in ATF3, GRP78, and CHAC1 mRNA expression suggesting that this induction was independent of Bcl-2 (Fig. 6C). Thus, Bcl-2 was able to inhibit apoptosis despite the increase in UPR-related gene expression suggesting this induction was not sufficient to induce apoptosis in Molt4 cells. Both CHOP or CHAC1 play critical roles in apoptosis in some cell types; however, knockdown of CHOP or CHAC1 by siRNAs had little effect on farnesol-induced apoptosis (not shown) suggesting that they are not an absolutely requirement for the induction of apoptosis in Molt4 cells.
Fig. 6.

Farnesol enhances the expression of ATF3, GRP78, CHOP, and CHAC1 in Molt4-hyg cells. (A) Molt4-hyg cells were treated with farnesol at the concentrations indicated. 6 h later, RNA was collected and analyzed for ATF3 and CHAC1 mRNA expression by QRT-PCR as described in Section 2. (B) Molt4-hyg cells were treated with 100 μM farnesol and at the times indicated cells were collected and analyzed for ATF3, GRP78, CHOP, and CHAC1 mRNA expression by QRT-PCR. Each value is the mean ± SD of three separate experiments. *Indicates statistically different from vehicle-treated Molt4-hyg control (p <0.01). (C) Bcl-2 does not block the farnesol-induced changes in ATF3, CHAC1, and GRP78 mRNA expression in Molt4 cells. Molt4-hyg and Molt4-Bcl2 cells were treated with farnesol or vehicle and analyzed as described under A. (D) Farnesol (100 μM) increased phosphorylation of eIF2αconsistent with activation of the PERK-eIF2αbranch of the UPR. (E) The levels of p-eIF2αshown under A were quantitated and normalized against β-actin as described in Section 2. (F) Farnesol does not induce alternative splicing of XBP1 mRNA in Molt4 cells. Molt4-hyg cells (1–3) were treated with 100 μM farnesol for 4 h before RNA was isolated and the presence of unspliced and spliced forms of XBP1 (XBP1u and XBP1s, respectively) were analyzed by RT-PCR. H460 cells were used as a positive control.
ER stress triggers the UPR and activation of the PERK-eIF2α and IRE1-XBP1 pathways, which subsequently leads to induction of UPR-related genes, such as CHOP and CHAC1. Fig. 6D and E shows that farnesol caused an increase in phosphorylated eIF-2α; however, farnesol did not induce alternative splicing of XBP1 in Molt4-hyg cells (Fig. 6F). These results indicate that farnesol-induced ER stress in Molt4-hyg cells activates the PERK-eIF2α, but not the IRE1-XBP1 branch of the UPR, suggesting that only a partial activation of the UPR.
3.5. Activation of MEK/ERK1/2, p38, and JNK pathways
Several mitogen-activated protein kinases (MAPK), including MEK/ERK1/2, p38, and JNK, have been implicated in the regulation of the ER stress response and apoptosis. We, therefore, analyzed whether farnesol had any effect on the activation of these MAPKs in Molt4-hyg cells. As shown in Fig. 7A and B, activation of ERK1/2 and p38 occurred as early as 15–30 min after the addition of farnesol, while activation of JNK was observed 3–4 h later. However, inhibition of MEK/ERK1/2, p38, and JNK activation by U0126, SB203580, and SP600125, respectively, had little effect on farnesol-induced cell death in Molt4-hyg cells (Fig. 7C). These results suggested that activation of these MAPKs did not play a critical role in farnesol-induced apoptosis in Molt4-hyg cells. Study of the effect of MAPK inhibitors on the induction of ER-stress-related genes showed that neither U0126 nor SB203580 blocked farnesol-induced expression of ATF3, CHAC1, and CHOP mRNA (Fig. 7D). These results are in contrast to data obtained with H460 cells in which the MEK inhibitor U0126, but not the p38 inhibitor, effectively inhibited the induction of ATF3 and CHAC1 mRNA in H460 cells (Fig. 7D, right panel). These results indicate that mechanism of the farnesol-induced activation of the UPR is independent of MEK/ERK1/2, p38, and JNK activation.
Fig. 7.

Farnesol-induced activation of p38, ERK1/2, and JNK in Molt4-hyg cells. (A) Molt4-hyg cells were treated with 75 μM farnesol. At the time intervals indicated cell lysates were examined by Western blot analysis with antibodies against phosphorylated p38 MAPK (p-p38 MAPK), total p38 MAPK, phosphorylated ERK1/2 (p-ERK), total ERK1/2, phosphorylated JNK (p-JNK), and total JNK. (B) The levels of p-p38, p-ERK, and p-JNK shown under A was quantitated and normalized against β-actin as described in Section 2. (C) Molt4-hyg cells were pretreated with vehicle C, p38 MAPK inhibitor SB203580 (SB, 10 μM), JNK inhibitor SP600125 (SP, 10 μM), or MEK inhibitor U0126 (U, 10 μM) for 30 min and then treated with 75 μM farnesol for 6 h before apoptotic index was determined using a cell death detection ELISA kit. (D) Effect of MEK1/2 and p38 inhibitors on the induction of ATF3, GRP78, CHOP, and CHAC1 in Molt4-hyg and H460 cells. Cells were pretreated with MEK inhibitor U0126 (10 μM) or p38 inhibitor SB203580 (10 μM) for 30 min and then treated with farnesol. After 4 h cells ATF3, GRP78, CHOP, and CHAC1 mRNA expression were evaluated by QRT-PCR as described in Section 2. Each value is the mean ± SD of three separate experiments. *Indicates statistically different from vehicle-treated Molt4-hyg or H460 control (p <0.01).
Farnesol also enhanced the generation of reactive oxygen species (ROS) in Molt4 cells, which has been implicated in the growth–inhibitory effects of farnesol. And although vitamin C and butylated hydroxyanisol (BHA) blocked the generation of ROS, they did not greatly affect PARP cleavage (Fig. 8).
Fig. 8.
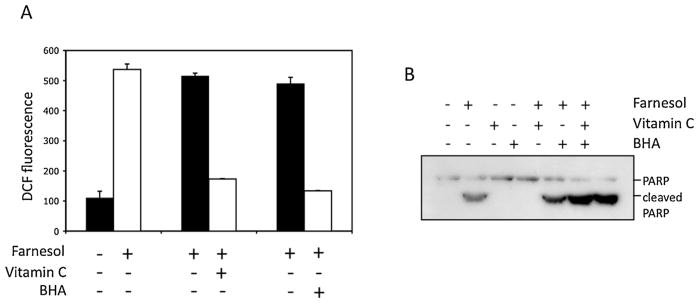
Farnesol-induced apoptosis in Molt4-hyg cells is independent of ROS generation. Cells were treated with 100 μM farnesol or vehicle for 4 h in the presence or absence of vitamin C or BHA. ROS and PARP cleavage were analyzed as described in Section 2. Results show that vitamin C and BHA inhibited ROS, but did not block PARP cleavage suggesting that farnesol-induced apoptosis was independent of ROS generation.
4. Discussion
Alcohol isoprenoids, including farnesol, perillyl alcohol, and geranylgeraniol, exhibit anti-tumor and anti-carcinogenic effects as well anti-inflammatory and anti-allergic properties in vivo [1,12–17]. These studies have suggested that these agents might be useful in chemoprevention as well as in the treatment of certain cancers and inflammatory diseases. High intake of fruit and vegetables has been found to reduce leukemia risk [1–4], which might be in part attributed to increased consumption of farnesol and related isoprenoids. In this study, we demonstrate that farnesol effectively inhibits the growth of human T lymphoblastic leukemia cells by inducing apoptosis as indicated by increased caspase activation, annexin V binding, and PARP cleavage. Permeabilization of the outer mitochondrial membrane and the subsequent release of cytochrome c have been reported to play a critical role in the intrinsic pathway of apoptosis [32–34]. The released cytochrome c interacts with Apaf1, which subsequently undergoes oligomerization forming a complex referred to as the apoptosome. The apoptosome then activates the initiator caspase 9, which subsequently leads to the activation of downstream executioner caspases, such as caspase 3. In this study, we demonstrate that treatment of Molt4 cells with farnesol causes a reduction in mitochondrial membrane potential (ΔΨm), suggesting opening of the permeability transition pores (PTP), that subsequently results in the release of cytochrome c from the mitochondria into the cytoplasm, and activation of caspases 9 and 3. Additionally, farnesol-induced apoptosis in Molt4 cells was associated with externalization of phosphatidylserine, plasma membrane depolarization, a loss of intracellular potassium, and an increase in intracellular sodium and calcium. In total, these observations support the concept that the induction of cell death by farnesol in Molt4 is mediated by the activation of the apoptosome through the intrinsic apoptotic pathway.
The intrinsic pathway is tightly regulated by members of the Bcl-2 family, which among other things control mitochondrial membrane integrity [38,39]. Pro-apoptotic members, such as Bax, oligomerize and form pores in the outer mitochondrial membrane that cause release of cytochrome c, while anti-apoptotic proteins, such as Bcl-2, inhibit this process. We show that expression of Bcl-2 in Molt4 cells inhibit depolarization of the mitochondrial membrane potential, the release of cytochrome c and caspase activation consistent with the conclusion that farnesol activates the intrinsic pathway of apoptosis. Furthermore, we show that expression of Bcl-2 in Molt4 cells results in the absence of many hallmarks of apoptosis including a subdiploid peak of DNA, externalization of phosphatidylserine, plasma membrane depolarization, along with the changes the intracellular ions. Bcl-2 has been reported to regulate the expression of several ion channels and might control K+ levels by regulating the expression and/or activity of potassium channels [36,40–43]. Interestingly, farnesol treatment of Molt4 cells induced cell swelling (Fig. 4; forward scatter) that occurred independently of Bcl-2 expression. The varying degree of change in intracellular sodium coupled with the increase in cell size upon farnesol treatment, regardless of the presence of absence of Bcl-2, supports the hypothesis that sodium may play a more vital role in changes in cell size, while the loss of potassium controls the progression of the apoptotic process [35,36]. Finally, Bcl-2 family members have also been reported to play a critical role in the control of calcium homeostasis and regulate ER as well as mitochondrial calcium dynamics during apoptosis [37,41,43]. For example, high Ca2+ can cause loss of mitochondrial potential thereby initiating the intrinsic apoptotic pathway. Our observed increase in intracellular Ca2+ levels that is prevented by the expression of Bcl-2 is consistent with a role of calcium dynamics in the induction of farnesol-induced apoptosis in Molt4 cells.
The signaling pathways involved in farnesol-induced apoptosis are not yet fully understood and appear to greatly depend on cell type [5]. The generation of reactive oxygen species (ROS) has implicated in the growth–inhibitory effects of farnesol in mammalian cells, fungi, and yeast [5,44,45]. Although we showed that farnesol also enhanced the generation of ROS in Molt4 cells, co-treatment with vitamin C or butylated hydroxyanisol (BHA) blocked the generation of ROS, but did not significantly affect PARP cleavage suggesting that the generation of ROS is not the direct cause of farnesol-induced cell death in Molt4 cells.
To obtain greater insights into the mechanism involved in the induction of apoptosis in lymphoblastic leukemia Molt4 cells by farnesol, we carried out microarray analysis to compare the gene expression profiles between human Molt4-hyg cells treated with farnesol or vehicle. This analysis showed that farnesol treatment affected the expression of a large number of genes and revealed that farnesol induced the expression of a select group of genes implicated in UPR signaling [26,46]. Interestingly, farnesol induced the expression of the UPR-related genes, ATF3, CHOP, HERPUD1 and 2, and ATF4, whereas the expression of several other UPR-related genes was either not significantly changed or down-regulated, such as GRP78. Interestingly, GRP78 functions as an anti-apoptotic protein and plays a role in the recovery from ER stress [47]. Its down-regulation might inhibit the rescue from ER stress-induced apoptosis and part of the mechanism by which Molt4 cells are more sensitive to farnesol-induced apoptosis than other cells, such as H460, in which GRP78 is induced [11]. We further show that farnesol enhances eIF2α phosphorylation (Fig. 6D and E), but has no effect on the alternative splicing of XBP1 (Fig. 6F), suggesting that farnesol activates the PERK-eIF2α, but not the IRE1-XBP1 branch of the UPR indicating that in Molt4 cells farnesol appears to induce only a partial UPR response. This is in contrast to the farnesol-induced ER stress response observed in human lung carcinoma H460 cells, which is associated with the induction of many UPR-related genes, including, ATF3, CHOP, XBP1, GRP78, GRP94, PDIA4, and HERPUD1, as well as by the alternative splicing of XBP1 [5,11]. And although farnesol activates several MAPK pathways, as indicated by the increased phosphory-lation of ERK1/2, JNK and p38, in contrast to H460 cells [11], inhibition of these MAPK pathways by chemical inhibitors did not affect the apoptotic index or the induction of the UPR-related genes, ATF4 and CHOP. Recent studies have revealed that ATF4 and/or CHOP directly regulate the transcription of ATF3, SARS, HERPUD1, and SLC3a2 [46], all of which are also increased in farnesol-treated Molt4 cells (Table 2). These observations suggest that the increased expression of these genes in Molt4 cells is related to the increased expression and activation of ATF4 and CHOP. Together, these results are consistent with the concept that activation of the PERK-eIF2α pathways leads to activation of the ATFs and the subsequent induction of UPR genes, such as CHOP and CHAC1 in a manner that is independent of the activation of MEK/ERK or the JNK and p38 pathways. Down-regulation of CHOP or CHAC1 expression had little effect on farnesol-induced apoptosis suggesting that these functionally different proteins act independently from each other in inducing apoptosis in Molt4 cells and have redundant roles in farnesol-induced apoptosis. Although Molt4-Bcl2 cells are greatly resistant to farnesol-induced apoptosis, UPR-related genes are induced to the same extent as in Molt4-hyg cells suggesting that induction of the UPR is not sufficient to induced apoptosis. However, the activation of a partial UPR response might contribute to increased sensitivity of Molt4 cells to farnesol-induced apoptosis.
Interestingly, CHAC1 was one of the genes most dramatically induced by farnesol in Molt4 cells. Although not well-studied, a recent report demonstrated that CHAC1 functions as a pro-apoptotic protein in endothelial cells and its expression was found to be directly regulated by ATF4 and ATF3 through a bipartite ATF/CRE site in its promoter [48–50]. Thus, the induction of the ATF4-ATF3-CHOP cascade and increased CHAC1 expression in farnesol-treated Molt4 cells is consistent with a link between the regulation of CHAC-1 and ATF4/ATF3 pathway. As ATF3 and CHOP, the induction of CHAC1 was independent of the activation of MAPKs. Recently, CHAC1 was found to function as a γ-glutamyl cyclotransferase in yeast [50,51]. Increased CHAC1 expression depletes yeast cells from glutathione and reduces the cell’s protection against ROS-induced apoptosis, which could be reversed by the addition of glutathione. However, the addition of glutathione had little effect on farnesol-induced apoptosis in Molt4 cells (data not shown) consistent with our conclusion that ROS is not the immediate cause of farnesol-induced apoptosis in these cells.
In summary, in this study we show that farnesol effectively induces apoptosis in human T lymphoblastic leukemia Molt4 cells by activating the apoptosome through the intrinsic pathway of apoptosis. Increased expression of Bcl-2 inhibits this induction. Comparison of gene expression profiles indicated that this induction is accompanied by activation of the PERK-eIF2 α-ATF3/4, but not the IRE1-XBP-1 branch of the UPR. Although farnesol activates the JNK, MEK/ERK1/2, and p38 MAPK pathways, the induction of this cascade is independent of the activation of these MAPK pathways. A recent study indicated that the sensitivity to farnesol is independent of drug resistance genes and the expression of p53, the EGF receptor and several genes involved in apoptosis [3]. It was suggested that farnesol may possess a novel mechanism of action that may bypass drug resistance to established chemotherapeutics. These observations together with those in this study suggest that farnesol might be particularly effective in the prevention or treatment of leukemic and lymphomas cells.
Acknowledgments
The authors would like to thank Drs. Gary ZeRuth and Hong Soon Kang for their comments on the manuscript and Dr. Kevin Gerrish from the Microarray Core for his assistance. This research was supported by the Intramural Research Program of the National Institute of Environmental Health Sciences, the National Institutes of Health (Z01-ES-101586).
Abbreviations
- ATF
activating transcription factor
- ALK
anaplasic lymphoma kinase
- CHAC1
cation transport regulator like protein 1
- ER
endoplasmic reticulum
- JC-1
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenz-imidazolcarbocyanine iodide
- MAPK
mitogen-activated protein kinase
- PARP-1
poly(ADP-ribose) polymerase-1
- eIF2α
eukaryotic translation initiation factor 2A
- PI
propidium iodide
- UPR
unfolded protein response
- GRP78
78 kD glucose-regulated protein
- XBP1
X-box binding protein 1
- Z-VAD-fmk
benzyloxycarbonyl-Val-Ala-Asp fluoromethylketone
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Crowell PL. Prevention and therapy of cancer by dietary monoterpenes. J Nutr. 1999;129:775S–778S. doi: 10.1093/jn/129.3.775S. [DOI] [PubMed] [Google Scholar]
- 2.Jensen CD, Block G, Buffer P, Ma X, Selvin S, Month S. Maternal dietary risk factors in childhood acute lymphoblastic leukemia (United States) Cancer Causes Control. 2004;15:559–570. doi: 10.1023/B:CACO.0000036161.98734.17. [DOI] [PubMed] [Google Scholar]
- 3.Kuete V, Efferth T. Molecular determinants of cancer cell sensitivity and resistance towards the sesquiterpene farnesol. Pharmazie. 2013;68:608–615. [PubMed] [Google Scholar]
- 4.Saberi H, Peeters P, Romieu I, Kelly R, Riboli E, Olsen A, et al. Dietary intakes and risk of lymphoid and myeloid leukemia in the European Prospective Investigation into Cancer and Nutrition (EPIC) Nutr Cancer. 2014;66:14–28. doi: 10.1080/01635581.2014.847471. [DOI] [PubMed] [Google Scholar]
- 5.Joo JH, Jetten AM. Molecular mechanisms involved in farnesol-induced apoptosis. Cancer Lett. 2010;287:123–135. doi: 10.1016/j.canlet.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haug JS, Goldner CM, Yazlovitskaya EM, Voziyan PA, Melnykovych G. Directed cell killing (apoptosis) in human lymphoblastoid cells incubated in the presence of farnesol: effect of phosphatidylcholine. Biochim Biophys Acta. 1994;1223:133–140. doi: 10.1016/0167-4889(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 7.Rioja A, Pizzey AR, Marson CM, Thomas NS. Preferential induction of apoptosis of leukaemic cells by farnesol. FEBS Lett. 2000;467:291–295. doi: 10.1016/s0014-5793(00)01168-6. [DOI] [PubMed] [Google Scholar]
- 8.Wiseman DA, Werner SR, Crowell PL. Cell cycle arrest by the isoprenoids perillyl alcohol, geraniol, and farnesol is mediated by p21(Cip1) and p27(Kip1) in human pancreatic adenocarcinoma cells. J Pharmacol Exp Ther. 2007;320:1163–1170. doi: 10.1124/jpet.106.111666. [DOI] [PubMed] [Google Scholar]
- 9.Melnykovych G, Haug JS, Goldner CM. Growth inhibition of leukemia cell line CEM-C1 by farnesol: effects of phosphatidylcholine and diacylglycerol. Biochem Biophys Res Commun. 1992;186:543–548. doi: 10.1016/s0006-291x(05)80842-3. [DOI] [PubMed] [Google Scholar]
- 10.Lagace TA, Miller JR, Ridgway ND. Caspase processing and nuclear export of CTP: phosphocholine cytidylyltransferase alpha during farnesol-induced apoptosis. Mol Cell Biol. 2002;22:4851–4862. doi: 10.1128/MCB.22.13.4851-4862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joo JH, Liao G, Collins JB, Grissom SF, Jetten AM. Farnesol-induced apoptosis in human lung carcinoma cells is coupled to the endoplasmic reticulum stress response. Cancer Res. 2007;67:7929–7936. doi: 10.1158/0008-5472.CAN-07-0931. [DOI] [PubMed] [Google Scholar]
- 12.Burke YD, Stark MJ, Roach SL, Sen SE, Crowell PL. Inhibition of pancreatic cancer growth by the dietary isoprenoids farnesol and geraniol. Lipids. 1997;32:151–156. doi: 10.1007/s11745-997-0019-y. [DOI] [PubMed] [Google Scholar]
- 13.Jahangir T, Sultana S. Benzo(a) pyrene-induced genotoxicity: attenuation by farnesol in a mouse model. J Enzyme Inhib Med Chem. 2008;23:888–894. doi: 10.1080/14756360701448768. [DOI] [PubMed] [Google Scholar]
- 14.Ong TP, Heidor R, deAonti C, Dagli ML, Moreno FS. Farnesol and geraniol chemopreventive activities during the initial phases of hepatocarcinogenesis involve similar actions on cell proliferation and DNA damage, but distinct actions on apoptosis, plasma cholesterol and HMGCoA reductase. Carcinogenesis. 2006;27:1194–1203. doi: 10.1093/carcin/bgi291. [DOI] [PubMed] [Google Scholar]
- 15.Rao CV, Newmark HL, Reddy BS. Chemopreventive effect of farnesol and lanosterol on colon carcinogenesis. Cancer Detect Prev. 2002;26:419–425. doi: 10.1016/s0361-090x(02)00119-8. [DOI] [PubMed] [Google Scholar]
- 16.Stayrook KR, McKinzie JH, Burke YD, Burke YA, Crowell PL. Induction of the apoptosis-promoting protein Bak by perillyl alcohol in pancreatic ductal adenocarcinoma relative to untransformed ductal epithelial cells. Carcinogenesis. 1997;18:1655–1658. doi: 10.1093/carcin/18.8.1655. [DOI] [PubMed] [Google Scholar]
- 17.Wargovich MJ, Jimenez A, McKee K, Steele VE, Velasco M, Woods J, et al. Efficacy of potential chemopreventive agents on rat colon aberrant crypt formation and progression. Carcinogenesis. 2000;21:1149–1155. [PubMed] [Google Scholar]
- 18.Chen M, Knifley T, Subramanian T, Spielmann HP, O’Connor KL. Use of synthetic isoprenoids to target protein prenylation and Rho GTPases in breast cancer invasion. PLoS One. 2014;9:e89892. doi: 10.1371/journal.pone.0089892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfister C, Pfrommer H, Tatagiba MS, Roser F. Detection and quantification of farnesol-induced apoptosis in difficult primary cell cultures by TaqMan protein assay. Apoptosis: Int J Program Cell Death. 2013 doi: 10.1007/s10495-012-0796-0. [DOI] [PubMed] [Google Scholar]
- 20.Lee JH, Kim C, Kim SH, Sethi G, Ahn KS. Farnesol inhibits tumor growth and enhances the anticancer effects of bortezomib in multiple myeloma xenograft mouse model through the modulation of STAT3 signaling pathway. Cancer Lett. 2015;360:280–293. doi: 10.1016/j.canlet.2015.02.024. [DOI] [PubMed] [Google Scholar]
- 21.Ku CM, Lin JY. Farnesol sesquiterpene alcohol in herbal plants, exerts anti-inflammatory and antiallergic effects on ovalbumin-sensitized and -challenged asthmatic mice. Evid Based Complement Altern Med. 2015;2015:1. doi: 10.1155/2015/387357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leonhardt I, Spielberg S, Weber M, Albrecht-Eckardt D, Blass M, Claus R, et al. The fungal quorum-sensing molecule farnesol activates innate immune cells but suppresses cellular adaptive immunity. MBio. 2015;6:e00143. doi: 10.1128/mBio.00143-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qamar W, Sultana S. Farnesol ameliorates massive inflammation, oxidative stress and lung injury induced by intratracheal instillation of cigarette smoke extract in rats: an initial step in lung chemoprevention. Chem Biol Interact. 2008;176:79–87. doi: 10.1016/j.cbi.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 24.Santhanasabapathy R, Sudhandiran G. Farnesol attenuates lipopolysaccharide-induced neurodegeneration in Swiss albino mice by regulating intrinsic apoptotic cascade. Brain Res. 2015 doi: 10.1016/j.brainres.2015.04.043. [DOI] [PubMed] [Google Scholar]
- 25.Joo JH, Jetten AM. NF-kappaB-dependent transcriptional activation in lung carcinoma cells by farnesol involves p65/RelA(Ser276) phosphorylation via the MEK-MSK1 signaling pathway. J Biol Chem. 2008;283:16391–16399. doi: 10.1074/jbc.M800945200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Logue SE, Cleary P, Saveljeva S, Samali A. New directions in ER stress-induced cell death. Apoptosis: Int J Program Cell Death. 2013;18:537–546. doi: 10.1007/s10495-013-0818-6. [DOI] [PubMed] [Google Scholar]
- 27.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 28.Wu J, Kaufman RJ. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006;13:374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Alter N, Reed JC, Borner C, Obeid LM, Hannun YA. Bcl-2 interrupts the ceramide-mediated pathway of cell death. Proc Natl Acad Sci U S A. 1996;93:5325–5328. doi: 10.1073/pnas.93.11.5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (JC-1) Biochem Biophys Res Commun. 1993;197:40–45. doi: 10.1006/bbrc.1993.2438. [DOI] [PubMed] [Google Scholar]
- 31.Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, et al. Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc Natl Acad Sci U S A. 1998;95:1455–1459. doi: 10.1073/pnas.95.4.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ow YP, Green DR, Hao Z, Mak TW. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008;9:532–542. doi: 10.1038/nrm2434. [DOI] [PubMed] [Google Scholar]
- 33.Adams JM, Cory S. Engines for caspase activation. Curr Opin Cell Biol. 2002;14:715–720. doi: 10.1016/s0955-0674(02)00381-2. [DOI] [PubMed] [Google Scholar]
- 34.Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–413. doi: 10.1038/nrm2153. [DOI] [PubMed] [Google Scholar]
- 35.Bortner CD, Cidlowski JA. Uncoupling cell shrinkage from apoptosis reveals that Na+ influx is required for volume loss during programmed cell death. J Biol Chem. 2003;278:39176–39184. doi: 10.1074/jbc.M303516200. [DOI] [PubMed] [Google Scholar]
- 36.Bortner CD, Cidlowski JA. Cell shrinkage and monovalent cation fluxes: role in apoptosis. Arch Biochem Biophys. 2007;462:176–188. doi: 10.1016/j.abb.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smaili SS, Pereira GJ, Costa MM, Rocha KK, Rodrigues L, do Carmo LG, et al. The role of calcium stores in apoptosis and autophagy. Curr Mol Med. 2013;13:252–265. doi: 10.2174/156652413804810772. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, et al. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. (see comments) [DOI] [PubMed] [Google Scholar]
- 39.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. (see comments) [DOI] [PubMed] [Google Scholar]
- 40.Storey NM, Gomez-Angelats M, Bortner CD, Armstrong DL, Cidlowski JA. Stimulation of Kv1. 3 potassium channels by death receptors during apoptosis in Jurkat T lymphocytes. J Biol Chem. 2003;278:33319–33326. doi: 10.1074/jbc.M300443200. [DOI] [PubMed] [Google Scholar]
- 41.Bonneau B, Prudent J, Popgeorgiev N, Gillet G. Non-apoptotic roles of Bcl-2 family: the calcium connection. Biochim Biophys Acta. 2013;1833:1755–1765. doi: 10.1016/j.bbamcr.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 42.Ekhterae D, Platoshyn O, Krick S, Yu Y, McDaniel SS, Yuan JX. Bcl-2 decreases voltage-gated K+ channel activity and enhances survival in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2001;281:C157–C165. doi: 10.1152/ajpcell.2001.281.1.C157. [DOI] [PubMed] [Google Scholar]
- 43.Yamamura Y, Oum R, Gbito KY, Garcia-Manero G, Strom SS. Dietary intake of vegetables, fruits, and meats/beans as potential risk factors of acute myeloid leukemia: a Texas case-control study. Nutr Cancer. 2013;65:1132–1140. doi: 10.1080/01635581.2013.834946. [DOI] [PubMed] [Google Scholar]
- 44.Fairn GD, Macdonald K, McMaster CR. A chemogenomic screen in Saccharomyces cerevisiae uncovers a primary role for the mitochondria in farnesol toxicity and its regulation by the Pkc1 pathway. J Biol Chem. 2007;282:4868–4874. doi: 10.1074/jbc.M610575200. [DOI] [PubMed] [Google Scholar]
- 45.Machida K, Tanaka T, Fujita K, Taniguchi M. Farnesol-induced generation of reactive oxygen species via indirect inhibition of the mitochondrial electron transport chain in the yeast Saccharomyces cerevisiae. J Bacteriol. 1998;180:4460–4465. doi: 10.1128/jb.180.17.4460-4465.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013 doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Curr Opin Cell Biol. 2011;23:150–156. doi: 10.1016/j.ceb.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mungrue IN, Pagnon J, Kohannim O, Gargalovic PS, Lusis AJ. CHAC1/ MGC4504 is a novel proapoptotic component of the unfolded protein response, downstream of the ATF4-ATF3-CHOP cascade. J Immunol. 2009;182:466–476. doi: 10.4049/jimmunol.182.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romanoski CE, Che N, Yin F, Mai N, Pouldar D, Civelek M, et al. Network for activation of human endothelial cells by oxidized phospholipids: a critical role of heme oxygenase 1. Circ Res. 2011;109:e27–e41. doi: 10.1161/CIRCRESAHA.111.241869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crawford RR, Prescott ET, Sylvester CF, Higdon AN, Shan J, Kilberg MS, et al. Human CHAC1 protein degrades glutathione and mRNA induction is regulated by the transcription factors ATF4 and ATF3 and the bipartite ATF/CRE element. J Biol Chem. 2015;290:15878–15891. doi: 10.1074/jbc.M114.635144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar A, Tikoo S, Maity S, Sengupta S, Kaur A, Bachhawat AK. Mammalian proapoptotic factor ChaC1 and its homologues function as gamma-glutamyl cyclotransferases acting specifically on glutathione. EMBO Rep. 2012;13:1095–1101. doi: 10.1038/embor.2012.156. [DOI] [PMC free article] [PubMed] [Google Scholar]