

Abstract
Dysregulation of the Notch1 receptor has been shown to facilitate the development and progression of colorectal cancer (CRC) and has been identified as an independent predictor of disease progression and worse survival. Although mutations in the NOTCH1 receptor have not been described in CRC, we have previously discovered a NOTCH1 gene copy number gain in a portion of CRC tumor samples. Here, we demonstrated that a NOTCH1 gene copy number gain is significantly associated with worse survival and a high percentage of gene duplication in a cohort of patients with advanced CRC. In our CRC patient-derived tumor xenograft (PDTX) model, tumors harboring a NOTCH1 gain exhibited significant elevation of the Notch1 receptor, JAG1 ligand and cleaved Notch1 activity. In addition, a significant association was identified between a gain in NOTCH1 gene copy number and sensitivity to a Notch1-targeting antibody. These findings suggest that patients with metastatic CRC that harbor a gain in NOTCH1 gene copy number have worse survival and that targeting this patient population with a Notch1 antibody may yield improved outcomes.
Keywords: colorectal cancer, Notch1, biomarker
Colorectal cancer (CRC) is the second leading cause of cancer-related death in the United States.1 Patients with metastatic disease have a poor 5-year survival rate, which is associated with the extent of metastatic lesions. Despite the increase in treatment options and newer therapies for this patient population, overall survival has only slightly improved. Accordingly, a concerted effort is underway to dissect the major pathways and genetic alterations that facilitate the development and progression of CRC that may ultimately translate into newer targeted therapies. Recently, the Cancer Genome Atlas Network (TCGA) has identified, in a large cohort of patients, several major pathways that are genetically altered in CRC including: WNT, phosphoinositide 3-kinase (PI3K), RAS, transforming growth factor-β (TGF-β) and p53.2 In addition to these major pathways, substantial evidence indicates that dysregulation of Notch signaling is an important factor in the development and progression of CRC.3–6 As such, developing agents targeting this pathway have a promising therapeutic utility.
The Notch signaling pathway plays a vital role in modulating various cellular functions. In cooperation with the WNT pathway, Notch ensures tissue homeostasis in the colon by regulating the self-renewal of the stem cell population and commitment to absorptive or secretory cell lineages.7–10 Notch pathway activation occurs through the cell–cell interaction of a ligand (DLL1, DLL3, DLL4, JAG1 and JAG2) with a Notch receptor (Notch1–4). This results in the proteolytic cleavage of the intracellular Notch (NICD) domain by the gamma-secretase complex, subsequent translocation into the nucleus and transcription of Notch target genes.11,12 In CRC, aberrant Notch activity has been reported to increase tumor burden by regulating a wide range of tumor cellular processes involved in enhancing cellular survival.6,13,14 In particular, overexpression of intracellular Notch (ICN) in an adenomatous polyposis colimin (Apcmin) model accelerated tumor growth that resulted in worse survival; treatment with a γ-secretase inhibitor (GSI) significantly reduced tumor burden in this model.6 In addition, in colon cancer-initiating cells (CCIC), the Notch pathway was significantly elevated; inhibition of the cleavage of ICN with a GSI resulted in an induction of apoptosis in these cells.14 In a CRC patient-derived tumor xenograft (PDTX) model, we demonstrated that tumors with elevated levels of the WNT and Notch pathways were sensitive to treatment with a clinical GSI.3 Together, these data as well as many others implicate the Notch signaling pathway as a critical contributor to tumorigenesis in CRC.
Recent studies suggest that the Notch1 receptor has a dominant role in the activation of the Notch pathway in CRC-mediating growth of colon cancer cells.15–18 Knockdown of the Notch1 receptor in colon cancer cells reduced proliferation in vitro and tumorigenic growth in a xenograft model.18 In contrast, overexpression of the Notch1 receptor enhanced cellular proliferation in vitro and the development of tumors in a xenograft model.18 In addition, tumors with elevated levels of the Notch1 receptor are associated with poor differentiation and more advanced stage of disease.17 Elevated Notch1 receptor protein expression has also been identified to be an independent predictor of prognosis and associated with poor survival in patients with CRC.16 We have discovered a NOTCH1 gene copy number gain in a subset of patients with CRC that may account for the increase in protein expression seen in patients with CRC.15
As the Notch1 receptor appears to be important in modulating tumor growth and an independent predictor of survival in CRC, we aimed to determine whether (i) a gain in the NOTCH1 gene was a prognostic indicator of survival in patients with metastatic CRC and (ii) targeting this receptor with a Notch1 targeting antibody will reduce tumor growth in our PDTX CRC model. We show that a gain in NOTCH1 gene copy number is a prognostic indicator of worse survival and a predictive biomarker to a Notch1-targeting antibody.
Material and Methods
Patients and specimens
Tumor specimens from 116 patients with metastatic CRC were obtained from consenting patients at MD Anderson in accordance with protocols approved by the Institutional Review Board. All available patients who received chemotherapy prior to tumor resection followed by adjuvant chemotherapy after liver resection were included in this retrospective cohort study. Formalin-fixed, paraffin-embedded (FFPE) samples of tumor tissue from archival specimens collected at the time of diagnosis were retrieved from storage at hospital pathology departments and a tissue microarray (TMA) was constructed. These tissue specimens were assembled onto TMAs with duplicate samples and both intraslide and interslide controls to control for edge effects and variation in slide staining. This TMA was stained with a Notch1 or CEP9 probe and subjected to FISH as described below. All patient samples were sequenced with respect to common mutations in CRC including: PIK3CA, KRAS, NRAS, CTNNB1 and BRAF genes. PTEN immunohistochemistry (IHC) was performed to determine PTEN status (loss or intact). There were no missing data in this data set with the exception of six patients where PTEN IHC failed. There were no patients who were lost to follow-up. The clinical endpoints evaluated in the study included relapse-free survival (RFS), defined as the period between surgery and tumor recurrence (death was not included as tumor recurrence in cases where patients were loss to follow-up) and overall survival, defined as the period between surgery and death. The influence of other clinical variables possibly related to survival, such as male vs. female; age at hepatectomy; tumor side (left vs. right); tumor diameter and number of lesions in the liver, was also considered in our study.
Immunohistochemistry
PTEN IHC staining was performed on 4-μm unstained TMA slides (as described above) from 116 patients with metastatic CRC. Sections were deparaffinized using standard histologic procedures, and an antigen retrieval method was used to ensure optimal antigen integrity and expression. A PTEN monoclonal antibody (6H2.1, Dako, Carpinteria, CA) was used at a concentration of 1:100 dilution. Diaminobenzidine was used as a chromogen and diaminobenzidine enhancer was applied, and hematoxylin was used for counterstaining. The IHC TMA-stained slides were examined by using standard light microscopy. The background stromal or non-neoplastic epithelial cells were used as an internal positive control. PTEN expression was categorized as loss of PTEN (no positive staining for PTEN, PTEN expression score =0) and as PTEN positive. Cleaved caspase 3 was assessed for overall staining of human cells in control and Notch1 antibody-treated CRC040 explant.
NOTCH1 FISH assay
A NOTCH1 FISH probe was developed by our group as previously described15 to determine NOTCH1 gene copy number variation. Formalin-fixed, paraffin-embedded sections from the CRC specimens were subjected to a dual-color FISH assay using the NOTCH1 (SpectrumRed) and the commercial probe CEP9 (labeled in SpectrumGreen, from Abbott Molecular, Des Plaines, Illinois) as a control for chromosome 9 aneusomy. Initially the slides were incubated for 4 hr at 56°C, deparaffinized in Citri-Solv (Fisher, Pittsburgh, PA) and washed in 100% ethanol for 10 min. The slides were sequentially incubated in 2× SSC at 75°C for 15–18 min, digested in 0.6 mg/ml Proteinase K/2× SSC at 45°C for 15–20 min, washed in 2× SSC for 5 min and dehydrated in ethanol. For a 113-mm2 hybridization area, a probe mixture was prepared using NOTCH1 (SR) (200 ng) and 1 μl of diluted CEP9 (SG). In each specimen, the probe was applied to the selected hybridization area, which was covered with a glass coverslip and sealed with rubber cement. DNA denaturation was performed for 15 min at 85°C and hybridization was allowed to occur at 37°C for 36–48 hr. Post-hybridization washes were performed sequentially with 2× SSC/0.3% NP40 (pH 7.0–7.5) at 74°C for 2 min and 2× SSC for 2 min, and dehydrated in ethanol. Chromatin was counterstained with DAPI (0.3 μg/ml in Vectashield mounting medium, Vector Laboratories, Burlingame, CA). Analysis was performed on epifluorescence microscope using single interference filter sets for green (FITC), red (Texas red), blue (DAPI), dual (red/green) and triple (blue, red, green) band pass filters. A minimum of 50 tumor cells per specimen was scored for NOTCH1 and CEP9 signals.
CRC explant xenograft model
Patient-derived colorectal adenocarcinoma tumor specimens were obtained from consenting patients at the University of Colorado Hospital in accordance with protocols approved by the Colorado Multiple Institutional Review Board. Four- to six-week-old female athymic (nu+/nu+) mice were obtained from Harlan Laboratories (Washington, DC) under an approved research protocol by the Institutional Animal Care and Use Committee. The tumor pieces were implanted in mice and expansion of the F1–F3 generations was carried out as previously described.19,20 Tumors were expanded in the left and right flanks of five to six mice (ten evaluable tumors per group). Mice were randomized into control antibody or PF-06293622 (Notch1-targeting antibody) groups when tumor volumes reached ~200 mm3. Mice were treated weekly with PF-06293622 (10 mg/kg) by intraperitoneal injections for 28 days. Mice were monitored daily for signs of toxicity and tumor size was evaluated twice per week by caliper measurements using the following formula: tumor volume =(length × width)2 × 0.52.
Immunoblotting
Tumor tissues (50–75 mg/mouse) were minced on ice and homogenized using a Dounce homogenizer and centrifuged at 16,000g at 4°C for 10 min. The total protein in samples was determined using the Pierce Protein Assay kit. Fifty micrograms of sample was electrophoresed on 4–12% Bis-Tris precast gels (Life Technologies, Carlsbad, CA). After electrotransfer to nitrocellulose, membranes were blocked at room temperature with TBST [10 mmol/L Tris-HCl (pH 7.5), 0.5 mol/L NaCl and 0.1% (v/v) Tween 20] containing 5% nonfat milk (BioRad, Hercules, CA) for 1 hr. Cleaved Notch, cleaved caspase 3 and actin primary antibodies (Cell Signaling Technologies, Danvers, MA) were diluted at 1:1,000 in TBST containing 5% protease-free bovine serum albumin (Sigma-Aldrich, St. Louis, MO), and the membranes were incubated overnight at 4°C with rocking. After washing three times with TBST, the membranes were incubated for 1 hr at room temperature with anti-rabbit IgG horseradish peroxidase-conjugated antibody at a final dilution of 1:50,000 in TBST. After washing three times with TBST, bound antibodies were detected by enhanced chemiluminescence (Millipore, Temecula, California).
Cleaved Notch1 ELISA
Normal colon and matching tumor tissue from nine CRC patients were evaluated for cleaved Notch1 activity. Normal colon and tumor tissue were lysed and protein levels were quantitated using the Pierce Protein Assay kit. Equal amounts of protein were plated on the ELISA plate and incubated for 2 hr according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). The ELISA plate was read at 450 nm on a Synergy 2 plate reader (Biotek, Winooski, VT). The detected absorbance for this developed color is relative to the amount of the activity of cleaved Notch1 (Val1744).
RNA isolation
Control and Notch1-treated CRC explants (end of study) were collected and immediately frozen in liquid nitrogen. RNA was isolated using an RNeasy kit (Qiagen, German-town, MD, Valencia, CA) according to the manufacturer’s protocol with an additional DNA digestion step. The RNA concentration and integrity were measured using a Nano-Drop (Thermo Fisher Scientific, Pittsburgh, PA) and a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA), respectively.
nCounterTM gene expression assay
nCounter probes were designed and obtained from Nano-String Technologies (Seattle, WA). The human probe sequences were screened against mouse RefSeq to eliminate potentially cross-hybridizing probes. Each assay was performed using at least two biological replicates. Total RNA (100 ng) was hybridized to nCounterTM capture and reporter probes at 65°C for 16 hr. The hybridized products were purified and processed using an automated sample prep station, and the images were prepared using the NanoString Digital Analyzer according to company’s standard gene expression assay protocol (http://www.NanoString.com).
nCounterTM data analysis
The counts were first normalized to six spiked-in positive controls to correct for experimental variability. A reference normalization factor was determined by first calculating the geometric mean of the positive controls for each sample and then computing the arithmetic mean across all 15 samples. The gene count for each sample was then normalized by dividing by the ratio of the geometric mean of the positive controls for the sample to the reference normalization factor. To account for the variability in RNA content, the normalized gene counts were further normalized against four endogenous control genes. This was performed by calculating the geometric mean of the endogenous controls for each sample, averaging across all samples and generating an endogenous normalization factor by computing the ratio of the geometric mean of the endogenous controls to the average value as described above. Each target gene count was divided by this endogenous normalization factor to compute the final normalized target gene count reflective of the transcript level. The detailed gene expression analysis guidelines can be found on the NanoString Technologies website (http://www.NanoString.com).
Statistical analysis
Kaplan–Meier survival curves were used to compare differences in overall survival and RFS between NOTCH1 gain and no gain. NOTCH1 signals were evaluated in a minimum of 50 cells and the number of signals per cell was averaged from the total of number of cells scored. Differences in survival were determined by a log-rank test. The influence of other clinical variables possibly related to survival such as male vs. female; age; tumor location (left vs. right); mutations in KRAS, NRAS, CTNNB1, PIK3CA and BRAF; and PTEN status (loss or intact) was examined by a univariate analysis. A p value <0.05 was considered significant. All analyses were performed using SAS statistical software (Cary, NC).
A paired t-test was used to compare the differences in cleaved Notch1 activity between normal and matching tumor tissue. An unpaired Student’s t-test was used to determine whether the means between control and Notch1 treated were significant at the end of treatment (~28 days) as well as differences in gene expression and protein expression between sensitive and resistant explants. The differences were considered significant when the p value was <0.05. All error bars are represented as the SEM. PDTX explants were considered to be responsive to treatment with the Notch1 antibody if the tumors treated with the Notch1 antibody have at least a 80% decrease in growth relative to the control tumors, where tumor growth is measured by the tumor growth inhibition index (TGII), a standardized measure of tumor growth, which is calculated using the following formula: TGII =(tumor volume of TX on Day 28 – tumor volume of TX on Day 0)/(tumor volume of Con on Day 28 – tumor volume of Con on Day 0) × 100, where TX is the Notch1 antibody-treated xenograft and CX is the control-treated xenograft.
Results
A NOTCH1 gene copy number gain is associated with worse RFS and a high percentage of gene duplication in patients with metastatic colorectal cancer
In a previous study, we identified a gain in NOTCH1 gene copy number in two of four CRC explants using FISH probes developed in our laboratory.15 These tumor specimens harbored more than three copies of the NOTCH1 gene. Thus, we explored whether a gain in the NOTCH1 gene was associated with clinical outcome. NOTCH1 gene copy number was examined in 116 patients (TMA) with metastatic CRC. All patients received chemotherapy before tumor resection followed by adjuvant chemotherapy after liver resection. As depicted in Figure 1a, patients with a NOTCH1 gain (≥3 copies) had worse RFS (log-rank p =0.025), defined as the period between surgery and tumor recurrence, when compared to patients with no gain (<3 copies). The median months to relapse were 12.0 months (95% CI: 8.3, 13.3) for NOTCH1 gain and 16.5 months (95% CI: 11.4, 21.4) for NOTCH1 no gain. No statistical difference was observed with NOTCH1 gene copy number and overall survival (data not shown), which is influenced by subsequent patient treatment(s). Interestingly, a NOTCH1 gene copy number dose effect was identified (Fig. 1b), evident by worse survival as NOTCH1 gene copy number increased. No significant differences with respect to common mutations (KRAS, NRAS, BRAF, CTNNB1 or PIK3CA), PTEN status (intact vs. loss) or other clinical variables and NOTCH1 gain vs. no gain were determined, suggesting that this genetic event may be an independent prognostic indicator of survival following resection (Supporting Information Table 1). The frequency of this genetic abnormality was 22% in this patient population. Although this gain in NOTCH1 gene copy number was accompanied with a gain in centromere 9, a duplication in the NOTCH1 gene was identified in 60% of tumors (Fig. 1c) with more than three copies of the NOTCH1 gene. A representative figure displaying three NOTCH1 signals (red probe) is shown in Figure 1d.
Figure 1.

Association between a gain in NOTCH1 gene copy number and worse RFS. (a) An association between a NOTCH1 gain and worse RFS was determined by Kaplan–Meier survival curve analysis (log-rank p =0.025). (b) A stepwise increase in NOTCH1 gene copy number gain was associated with worse RFS. (c) Percent gene duplication with respect to NOTCH1 gene copy number within each category (<2, 2.01–2.50, 2.51–2.99, >3). Percent duplication was determined by dividing the total number of patient samples with Notch1 doublets by the total number of patients within their respective category × 100. (d) Representative photograph of a patient with a NOTCH1 gene copy number gain (more than three copies) and gene duplication.
Next, we investigated NOTCH1 gene copy number and activation of the Notch pathway between normal colon and tumor tissue to determine if this genetic event is only present in tumor cells. As expected, Notch1 gene copy gain was tumor-specific (Figs. 2a and 2b). Consistent with these findings, Notch pathway activation measured by cleaved Notch1 activity was significantly elevated in tumor when compared to matched normal colon tissue in nine separate patients with CRC (Fig. 2c). These results were confirmed by Western blot showing an increase in cleaved Notch1 in tumor tissue when compared to matching normal tissue (Fig. 2d).
Figure 2.

A NOTCH1 gene copy number gain is a genetic evident in tumor cells. (a and b) A representative photograph of two CRC patients normal tissue and matched tumor tissue showing a gain in NOTCH1 gene copy number in tumor cells. The average number of NOTCH1 signals is shown above each representative figure for normal and tumor. (c) Tumors exhibit significantly elevated levels of cleaved Notch1 when compared to matched normal colon tissue (n =9). (d) A representative Western blot of cleaved Notch1 in CRC001 and CRC040 normal colon and tumor tissue.
Association between a NOTCH1 gain and sensitivity to a monoclonal Notch1 antibody
As we identified a gain in NOTCH1 gene copy as a prognostic indicator of worse RFS, we investigated the efficacy of a human anti-Notch1 monoclonal antibody PF-06293622 on tumor growth in a CRC PDTX model. Supporting Information Table 2 shows clinical and molecular features of each case. As displayed in Figure 3a, four CRC explants (CRC001, 034, 036 and 040) were sensitive (TGII: tumor growth rate relative to control ≤20%) and 11 were resistant (TGII >20%). A TGII of >20% (<80% tumor growth inhibition) was chosen as a cutoff for progressive disease to more closely reflect the Response Evaluation Criteria in Solid Tumors (RECIST) criteria used in the clinic. Representative graphs of the tumor growth rate of the four sensitive CRC explants are shown in Figures 3b–3e and 11 resistant CRC explants are displayed in Supporting Information Figure 1. A gain in NOTCH1 gene copy number (defined as NOTCH1 mean gene copy number ≥3) was found in five of the 15 CRC explants (Fig. 3a and Supporting Information Table 3 and Fig. 2). The four most sensitive CRC explants to the Notch1 antibody harbor a gain in NOTCH1 gene copy number. Using a Fisher’s exact test we identified a significant association (p <0.006) between a gain in NOTCH1 gene copy number and sensitivity to the Notch1 antibody. The positive predictive value was 80% and negative predictive value was 100%. Of note, the lone exception CRC125 harbors a FBXW7 mutation, which may account for lack of pathway inhibition.
Figure 3.

PF-06293622 (Notch1 monoclonal antibody) effects on tumor growth in CRC explants. (a) Fifteen CRC explants were treated with PF-06293622 10 mg/kg weekly for 28 days. Tumor size was evaluated twice per week by caliper measurements using the formula: tumor volume =(length × width)2 × 0.52. (a) A TGII, a standardized measure of tumor growth, which is calculated for each CRC explant using the following formula: TGII =(tumor volume of TX on Day 28 – tumor volume of TX on Day 0)/(tumor volume of Con on Day 28 – tumor volume of Con on Day 0) × 100. Cases with a TGII of ≤20% were considered sensitive, TGII of >20% were considered resistant to PF-06293622. Four xenografts (CRC001, CRC034, CRC036 and CRC040) were sensitive to PF-06293622 (TGI ≤20%) and 11 xenografts were resistant to PF-06293622 (TGI >20%). Columns, mean (n =10 tumors per group). (b–e) A representative figure of the growth curves from the CRC-sensitive explants (b) CRC040, (c) CRC036, (d) CRC034 and (e) CRC001. SEM. **Significant p <0.01.
Tumors sensitive to Notch1 therapy exhibit elevated levels of JAG1, Notch1 and cleaved Notch1 activation
On the basis of the observation that a gain in NOTCH1 gene copy is associated with sensitivity to Notch1 therapy, we explored baseline gene expression levels of the Notch ligands, receptors (RNA Seq) and Notch pathway activation (Western blot) between sensitive and resistant CRC explants to determine if sensitive tumors possess elevation of the pathway. As shown in Figure 4, the JAG1 ligand (A), Notch1 receptor (B) and Hey1 (C) and Hey2 (data not shown) were significantly elevated in tumors sensitive (NOTCH1 gain) to the Notch1-targeted antibody. These results were confirmed by nanostring (multiplexed measurement of gene expression); a significant increase in gene expression of JAG1 and the Notch1 receptor was observed in the sensitive CRC explants (data not shown). No differences were seen between sensitive and resistant tumors with respect to JAG2, DLL1, DLL3 and DLL4 and the Notch receptors 2, 3 or 4. Of note, comparison between NOTCH1 gene copy number gain (CRC001, CRC034, CRC036, CRC040 and CRC125) vs. no gain showed a trend (p =0.06) in Notch1 gene expression with tumors harboring a NOTCH1 gain displaying elevated gene expression of the Notch1 receptor (data not shown). Evaluation of activated cleaved Notch1 between three sensitive and three resistant tumors showed a 7.6-fold increase in cleaved Notch1 activity (Figs. 4d and 4e) in tumors that responded to Notch1. These findings imply that a gain in NOTCH1 gene copy may be functionally relevant as an increase in Notch1 receptor and JAG1 enhances activity of the Notch pathway.
Figure 4.

Notch pathway analysis between sensitive (CRC001, 0034, 036 and 040) vs. 11 resistant tumors. (A–C) RNA Seq analysis of baseline levels of Notch ligands, receptors, and the Notch dependent gene Hey1 showed a significant increase in (A) JAG1, (B) Notch1 and (C) Hey1 in sensitive tumors when compared to resistant tumors. (D) Baseline levels of cleaved Notch1 are elevated in sensitive tumors compared to resistant tumors. (E) Densitometry of cleaved Notch1/Actin ratio showed a significant increase in sensitive tumors compared to resistant tumors (p <0.01).
Notch1 treatment reduces Notch pathway activation and induces apoptosis
We examined the effects of Notch1 inhibition on activated cleaved Notch1 and the Notch1-dependent genes: Hes-1, Hes-3, Hes-4, Hes-5, Hey1, Hey2, HeyL, Myc and NRARP. Notch1 treatment resulted in potent inhibition of cleaved Notch1 activity in the sensitive tumors CRC001, 036, 040 and resistant tumor 042 (Fig. 5a). Notch1 treatment decreased activated Notch1 in CRC125 by ~50%. We also observed a significant reduction in gene expression of several Notch-dependent genes in CRC001, 036, 040 and 042 (Table 1). The specific Notch1-targeted gene that decreased varied among the CRC explants (Table 1); however, a decrease in Hes-1 was seen in CRC001, 036, 040 and 042. No decrease in Notch-dependent gene expression was seen in CRC125 following treatment, suggesting that a FBXW7 mutation in this tumor may be responsible for lack of pathway inhibition. Moreover, Notch1 blockade resulted in an increase in apoptosis evident by elevation of cleaved caspase 3 at Days 1, 3 and 7 in CRC001 and at Day 3 in CRC 036. This coincided with a reduction in cleaved Notch1 activity in these tumors (Fig. 5b). An increase in cleaved caspase 3 was also detected by IHC in the CRC040-sensitive explant (Fig. 5c). No significant elevations in cleaved caspase 3 were seen in the CRC027- and CRC042-resistant tumors (Supporting Information Fig. 3).
Figure 5.
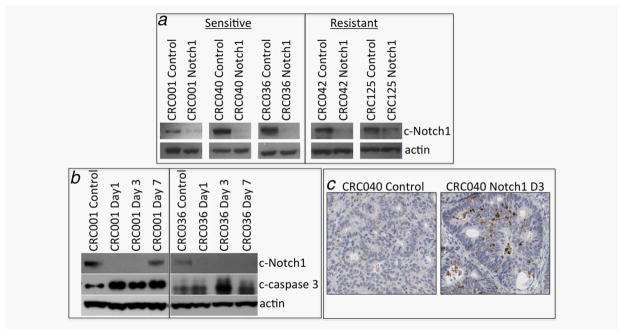
Notch1 inhibition abrogates cleaved Notch1 activity and increases apoptosis. (a) Treatment with the Notch1 antibody reduces cleaved Notch1 in sensitive and resistant tumors at the end of study. (b) Notch1 blockade increases apoptosis as shown by an increase in cleaved caspase 3 in the CRC001- and CRC036-sensitive CRC explants. (c) A representative picture showing elevations in cleaved caspase 3 (brown staining) in control and Notch1-treated after 3 days of treatment in CRC040.
Table 1.
The effects of Notch1 treatment on Notch-dependent transcription
| Gene | CRC001
|
CRC036
|
CRC040
|
CRC042
|
CRC125
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | Notch1 TX | TTEST | Control | Notch1 TX | TTEST | Control | Notch1 TX | TTEST | Control | Notch1 TX | TTEST | Control | Notch1 TX | TTEST | |
| Hes-1 | 665.8 | 414.7 | 0.001 | 753.0 | 413.1 | 0.016 | 1259.0 | 847.0 | 0.061 | 1083.9 | 671.6 | 0.017 | 2113.2 | 2136.5 | 0.869 |
|
| |||||||||||||||
| Hes-3 | 4.24 | 17.09 | 0.342 | 4.08 | 9.79 | 0.395 | 4.35 | 3.60 | 0.429 | 6.23 | 14.04 | 0.186 | 3.46 | 3.71 | 0.908 |
|
| |||||||||||||||
| Hes-4 | 117.7 | 84.17 | 0.129 | 129.9 | 51.96 | 0.003 | 427.5 | 168.2 | 0.007 | 188.3 | 224.0 | 0.182 | 47.1 | 49.5 | 0.831 |
|
| |||||||||||||||
| Hes-5 | 8.52 | 30.71 | 0.330 | 17.25 | 29.12 | 0.318 | 89.24 | 47.48 | 0.027 | 231.6 | 224.8 | 0.894 | 2.76 | 6.01 | 0.239 |
|
| |||||||||||||||
| Hey-1 | 15.0 | 22.11 | 0.286 | 14.39 | 16.15 | 0.793 | 177.8 | 168.0 | 0.815 | 7.87 | 15.41 | 0.115 | 6.83 | 13.74 | 0.315 |
|
| |||||||||||||||
| Hey-2 | 1.47 | 9.64 | 0.270 | 1.52 | 1.44 | 0.914 | 84.97 | 78.08 | 0.511 | 4.41 | 9.97 | 0.126 | 4.68 | 6.42 | 0.709 |
|
| |||||||||||||||
| HeyL | 4.45 | 15.12 | 0.235 | 8.15 | 9.71 | 0.678 | 6.25 | 10.96 | 0.410 | 19.21 | 24.6 | 0.403 | 6.57 | 10.2 | 0.434 |
|
| |||||||||||||||
| Myc | 45.74 | 49.84 | 0.634 | 704.9 | 251.1 | 0.004 | 905.5 | 672.7 | 0.058 | 225.5 | 251.5 | 0.479 | 2025.4 | 2263.9 | 0.541 |
|
| |||||||||||||||
| NRARP | 1914.8 | 1131.7 | 0.012 | 1371.2 | 867.7 | 0.002 | 1968.7 | 558.7 | 0.000 | 5555.8 | 4472.8 | 0.087 | 2831.7 | 2657.5 | 0.252 |
Discussion
The Notch signaling pathway is dysregulated in many cancers and shown to be an important driver of tumor growth and metastasis.3,4,6,14,21,22 In CRC, mounting evidence has implicated the Notch1 receptor to be essential in transducing signals that enhance tumorigenesis.15–18 In particular, increased protein levels of the Notch1 receptor have been shown in tumor tissue compared to normal tissue and is associated with a more poorly differentiated tumor and disease progression (Stages III and IV).17 In addition, elevated levels of the Notch1 receptor were associated with overall worse survival in CRC patients.16 We previously identified a Notch1 gene copy number gain (more than three copies of the NOTCH1 gene) in a small subset of patients with CRC.15 In contrast to other cancers where Notch1 is constitutively activated by a mutation or translocation, the genetic evidence in CRC linking NOTCH1 dysregulation to tumorigenesis remains elusive. We hypothesized that this gain in NOTCH1 gene copy may be the genetic abnormality responsible for increased activation of the Notch pathway.
We investigated whether a NOTCH1 gene copy number gain was associated with relapse in patients with resected metastatic CRC. Kaplan–Meier survival analysis showed a significantly worse RFS with respect to a gain in NOTCH1 gene copy number. This association was independent of known mutations in CRC such as p53, KRAS, NRAS, BRAF and CTNNB1 as well as PTEN status. In addition, we observed a gene copy number dose effect whereby an increase in NOTCH1 gene copy number was associated with worse RFS. These findings are consistent with Chu et al.16 demonstrating a stepwise increase in Notch1 protein expression was associated with worse survival. Although the gain in NOTCH1 gene copy number was accompanied with a gain in chromosome 9, we identified a significant amount of gene duplication (>60%) in tumors harboring more than three copies of the NOTCH1 gene. Further investigation is warranted to determine whether this gene duplication results in the NOTCH1 gene being regulated under a different promoter. Collectively, these results indicate that we may have identified the genetic abnormality responsible for increased activation of the Notch pathway and that a NOTCH1 gene copy number gain may identify patients at higher risk of relapse following liver resection for metastatic disease.
As the Notch1 receptor appears to be an important regulator of tumor growth18 and patient outcomes in CRC,16 we investigated the effects of an anti-Notch1 monoclonal antibody on tumor growth in our CRC PDTX model. We observed a range of sensitivity in our CRC patient-derived xenografts to the Notch1-targeting antibody. A cutoff of ≥80% tumor growth inhibition (TGII: tumor growth rate relative to control ≤20%) was chosen as cutoff for sensitivity to the Notch1 antibody. The “cutoffs” for determining sensitivity vs. resistance in PDTX models are controversial (ranging from 20 to 50%).23–27 In RECIST v1.0 and v1.128 a 20% growth from baseline is considered progressive disease, necessitating a change to treatment. Combined with the fact the 20% cutoff is related very well with the top quartile of sensitivity in our study and Figure 3 is akin to a waterfall plot in a clinical trial, we felt this was a reasonable approach. However, this remains an unresolved area of PDTX and patient avatars.
This particular Notch1 monoclonal antibody was also shown to reduce tumor growth in a breast cancer xenograft model.29 In comparison to a GSI we evaluated previously in our CRC explant model (0 of 16 reached a TGII >80%),3 the Notch1 antibody was more potent at reducing tumor growth. Moreover, we identified a significant association with a NOTCH1 gain and sensitivity to Notch1 inhibition with the four most sensitive explants (CRC001, CRC034, CRC036 and CRC040) having a gain in the NOTCH1 gene. Although CRC125 had a gain in NOTCH1 gene copy, a mutation in the FBXW7 gene may be in part responsible for lack of response to Notch1 inhibition in this CRC explant, as seen in leukemic cells.30 These results suggest that a NOTCH1 gene copy number gain may be used as a predictive biomarker for sensitivity to a Notch1-targeting antibody in CRC.
Given that tumors harboring a Notch1 gain exhibited the greatest sensitivity to the Notch1-targeting antibody, we compared baseline levels of Notch ligands, receptors and cleaved Notch1 activity in sensitive vs. resistant tumors. Sensitive Notch1 tumors displayed significantly higher levels of the Notch1 receptor and JAG1 ligand. This was accompanied with a 7.6-fold increase in activated cleaved Notch1. In addition to Notch1 playing an important role in CRC, other reports have implicated the Notch ligand JAG1 as a facilitator of tumor growth and metastasis.21,31–33 In particular, JAG1 is transcriptionally regulated by β-catenin32 and elevated JAG1 protein levels have been shown to be overexpressed in 50% of CRC tumors.32 In an Apcmin model, genetic knockout of the JAG1 gene significantly diminished the formation of intestinal tumors.32 Furthermore, JAG1 has been demonstrated to enhance the transition of epithelial cells to a mesenchymal phenotype through activation of the Notch1 receptor and upregulation of the Notch target gene Slug.31 The JAG1 gene is located on chromosome 20 and in a previous study we identified an increase in chromosome 20 in ~50% of CRC tumors (CRC cell lines and explants)34 and warrants further investigation. Of note, two of the sensitive explants (CRC034 and 036) had more than three copies of chromosome 20 indicating that JAG1 may be dysregulated in these tumors. These findings suggest that a gain in NOTCH1 gene copy number may be functionally relevant in transducing Notch1-dependent signals that enhance cell survival and tumorigenesis in CRC. Whether JAG1 is the main ligand that interacts with the Notch1 receptor remains to be determined.
In our study, we demonstrated that Notch1 blockade had consistent pharmacodynamics (PD) effects, with robust inhibition of cleaved Notch1 activity and a significant decrease in the Notch-dependent gene Hes-1 as well as other Notch target genes. This decrease in Notch1 activation resulted in enhanced cleavage of caspase 3 suggesting the mechanism of tumor cell death was by an induction of apoptosis. Similar results were found in breast cancer xenografts whereby Notch1 inhibition reduced tumor growth, decreased Notch-dependent transcription and increased the cleavage of capsase-3.29 These results indicate that Notch1 is playing an integral role in the growth of the sensitive tumors given that inhibition of the Notch1 receptor nearly eliminated the cleavage of the Notch1 receptor and enhanced cell death via apoptosis. Given that GSIs in the clinic are associated with gastrointestinal and other toxicities, targeting only the Notch1 receptor instead of all four receptors may reduce some of the side effects seen with typical GSIs.
In conclusion, our data provide strong evidence that a gain in NOTCH1 gene copy number may be a prognostic indicator of survival in patients with resected metastatic CRC. Tumors with a NOTCH1 gain also exhibited a high percentage of gene duplication, a significant increase in cleaved Notch1 activity and sensitivity to an anti-Notch1 monoclonal antibody in our CRC PDTX model. Therefore, targeting the Notch1 receptor with a novel and potent Notch1 antibody in patients with metastatic CRC who harbor a NOTCH1 gene copy number gain warrants clinical investigation.
Supplementary Material
What’s new?
There is mounting evidence that the Notch1 receptor is important in modulating tumor growth and an independent predictor of survival in colorectal cancer (CRC). While mutations in the NOTCH1 receptor have not yet been described in CRC, this study shows that a gain in NOTCH1 gene copy number is associated with worse survival. Targeting cells with a specific Notch1 antibody resulted in potent antitumor growth in a CRC patient-derived tumor xenograft model. A NOTCH1 gene copy number gain may thus be a prognostic marker for disease recurrence as well as a predictive biomarker of sensitivity to a Notch1 targeted therapy.
Acknowledgments
The authors thank Pfizer for providing the Notch1 monoclonal antibody. They acknowledge the University of Colorado Cancer Center pathology core for tissue analysis.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Conflict of interest: Peter Olson is an employee of Pfizer and owns stock in Pfizer; Xianxian Zheng is an employee of Pfizer and owns stock in Pfizer; Qin Zhang is an employee of Pfizer and owns stock in Pfizer.
References
- 1.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA: Cancer J Clin. 2014;64:104–17. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arcaroli JJ, Quackenbush KS, Purkey A, et al. Tumours with elevated levels of the Notch and Wnt pathways exhibit efficacy to PF-03084014, a gamma-secretase inhibitor, in a preclinical colorectal explant model. Br J Cancer. 2013;109:667–75. doi: 10.1038/bjc.2013.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fischer M, Yen WC, Kapoun AM, et al. Anti-DLL4 inhibits growth and reduces tumor-initiating cell frequency in colorectal tumors with oncogenic KRAS mutations. Cancer Res. 2011;71:1520–5. doi: 10.1158/0008-5472.CAN-10-2817. [DOI] [PubMed] [Google Scholar]
- 5.Hoey T, Yen WC, Axelrod F, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009;5:168–77. doi: 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 6.van Es JH, van Gijn ME, Riccio O, et al. Notch/ gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–63. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura T, Tsuchiya K, Watanabe M. Crosstalk between Wnt and Notch signaling in intestinal epithelial cell fate decision. J Gastroenterol. 2007;42:705–10. doi: 10.1007/s00535-007-2087-z. [DOI] [PubMed] [Google Scholar]
- 8.Noah TK, Shroyer NF. Notch in the intestine: regulation of homeostasis and pathogenesis. Annu Rev Physiol. 2013;75:263–88. doi: 10.1146/annurev-physiol-030212-183741. [DOI] [PubMed] [Google Scholar]
- 9.Pannuti A, Foreman K, Rizzo P, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010;16:3141–52. doi: 10.1158/1078-0432.CCR-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–15. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 11.van Es JH, Clevers H. Notch and Wnt inhibitors as potential new drugs for intestinal neoplastic disease. Trends Mol Med. 2005;11:496–502. doi: 10.1016/j.molmed.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Zolkiewska A. ADAM proteases: ligand processing and modulation of the Notch pathway. Cell Mol Life Sci. 2008;65:2056–68. doi: 10.1007/s00018-008-7586-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fre S, Huyghe M, Mourikis P, et al. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964–8. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 14.Sikandar SS, Pate KT, Anderson S, et al. NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res. 2010;70:1469–78. doi: 10.1158/0008-5472.CAN-09-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arcaroli JJ, Powell RW, Varella-Garcia M, et al. ALDH+ tumor-initiating cells exhibiting gain in NOTCH1 gene copy number have enhanced regrowth sensitivity to a gamma-secretase inhibitor and irinotecan in colorectal cancer. Mol Oncol. 2012;6:370–81. doi: 10.1016/j.molonc.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu D, Li Y, Wang W, et al. High level of Notch1 protein is associated with poor overall survival in colorectal cancer. Ann Surg Oncol. 2010;17:1337–42. doi: 10.1245/s10434-009-0893-7. [DOI] [PubMed] [Google Scholar]
- 17.Chu D, Wang W, Xie H, et al. Notch1 expression in colorectal carcinoma determines tumor differentiation status. J Gastrointest Surg. 2009;13:253–60. doi: 10.1007/s11605-008-0689-2. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Li B, Ji ZZ, et al. Notch1 regulates the growth of human colon cancers. Cancer. 2010;116:5207–18. doi: 10.1002/cncr.25449. [DOI] [PubMed] [Google Scholar]
- 19.Rubio-Viqueira B, Jimeno A, Cusatis G, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–61. doi: 10.1158/1078-0432.CCR-06-0113. [DOI] [PubMed] [Google Scholar]
- 20.Dangles-Marie V, Pocard M, Richon S, et al. Establishment of human colon cancer cell lines from fresh tumors versus xenografts: comparison of success rate and cell line features. Cancer Res. 2007;67:398–407. doi: 10.1158/0008-5472.CAN-06-0594. [DOI] [PubMed] [Google Scholar]
- 21.Kim MH, Kim HB, Yoon SP, et al. Colon cancer progression is driven by APEX1-mediated upregulation of Jagged. J Clin Invest. 2013;123:3211–3230. doi: 10.1172/JCI65521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonoshita M, Aoki M, Fuwa H, et al. Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell. 2011;19:125–37. doi: 10.1016/j.ccr.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Bertotti A, Migliardi G, Galimi F, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–23. doi: 10.1158/2159-8290.CD-11-0109. [DOI] [PubMed] [Google Scholar]
- 24.Song EK, Tai WM, Messersmith WA, et al. Potent antitumor activity of cabozantinib, a c-MET and VEGFR2 inhibitor, in a colorectal cancer patient-derived tumor explant model. Int J Cancer. 2015;136:1967–75. doi: 10.1002/ijc.29225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Migliardi G, Sassi F, Torti D, et al. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res. 2012;18:2515–25. doi: 10.1158/1078-0432.CCR-11-2683. [DOI] [PubMed] [Google Scholar]
- 26.Anderson RT, Keysar SB, Bowles DW, et al. The dual pathway inhibitor rigosertib is effective in direct patient tumor xenografts of head and neck squamous cell carcinomas. Mol Cancer Ther. 2013;12:1994–2005. doi: 10.1158/1535-7163.MCT-13-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Micel LN, Tentler JJ, Tan AC, et al. Antitumor activity of the MEK inhibitor TAK-733 against melanoma cell lines and patient-derived tumor explants. Mol Cancer Ther. 2015;14:317–25. doi: 10.1158/1535-7163.MCT-13-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 29.Qiu M, Peng Q, Jiang I, et al. Specific inhibition of Notch1 signaling enhances the antitumor efficacy of chemotherapy in triple negative breast cancer through reduction of cancer stem cells. Cancer Lett. 2013;328:261–70. doi: 10.1016/j.canlet.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 30.O’Neil J, Grim J, Strack P, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204:1813–24. doi: 10.1084/jem.20070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leong KG, Niessen K, Kulic I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007;204:2935–48. doi: 10.1084/jem.20071082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodilla V, Villanueva A, Obrador-Hevia A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci USA. 2009;106:6315–20. doi: 10.1073/pnas.0813221106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santagata S, Demichelis F, Riva A, et al. JAGGED1 expression is associated with prostate cancer metastasis and recurrence. Cancer Res. 2004;64:6854–7. doi: 10.1158/0008-5472.CAN-04-2500. [DOI] [PubMed] [Google Scholar]
- 34.Arcaroli JJ, Touban BM, Tan AC, et al. Gene array and fluorescence in situ hybridization biomarkers of activity of saracatinib (AZD0530), a Src inhibitor, in a preclinical model of colorectal cancer. Clin Cancer Res. 2010;16:4165–77. doi: 10.1158/1078-0432.CCR-10-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.