

Abstract
The yeast Saccharomyces cerevisiae is a model organism for replicative aging studies; however, conventional lifespan measurement platforms have several limitations. Here we present a microfluidics platform that facilitates simultaneous lifespan and gene expression measurements of aging yeast cells. Our multiplexed high-throughput platform offers the capability to perform independent lifespan experiments using different yeast strains or growth media. Using this platform in minimal media environments containing glucose, we measured the full lifespan of individual yeast cells in wild-type and canonical gene deletion backgrounds. Compared to glucose, in galactose we observed a 16.8% decrease in replicative lifespan accompanied by a ~2-fold increase in single-cell oxidative stress levels reported by PSOD1-mCherry. Using PGAL1-YFP to measure the activity of the bistable galactose network, we saw that OFF and ON cells are similar in their lifespan. Our work shows that aging cells are committed to a single phenotypic state throughout their lifespan.
Graphical Abstract
INTRODUCTION
Due to its short generation time and the abundance of genetic manipulation techniques, the yeast S. cerevisiae has been a commonly-used eukaryotic model organism for aging studies (Mortimer and Johnston, 1959; Müller et al., 1980; Kaeberlein et al., 2005; Steinkraus et al., 2008; Breitenbach et al., 2012; Longo et al., 1996; Fabrizio and Longo, 2003). Being a single-cell organism, yeast allows researchers to study in vivo organismal aspects of eukaryotic aging, as numerous genetic and cell biological processes are conserved between yeast and higher eukaryotes. Two different aging models can be studied by using yeast. The first model, replicative aging (Steinkraus et al., 2008; Breitenbach et al., 2012), is a measure of the number of daughter cells a mother cell mitotically produces before it senesces. The total number of daughter cells produced determines the replicative life span (RLS) of the mother cell. The second model, chronological aging (Breitenbach et al., 2012; Longo et al., 1996; Fabrizio and Longo, 2003), is a measure of how long a mother cell can live in a metabolically inactive state without losing the ability to revive itself when transferred to nutrient rich media. Here, we describe an automated platform to measure RLS in real time. Our platform can also be used for chronological aging measurements, which are relatively easier to perform due to their static nature.
For several decades, the conventional method to measure yeast RLS has required the use of micromanipulators (Steinkraus et al., 2008; Breitenbach et al., 2012). Mother cells are grown and followed on solid media environments, and to prevent crowding, each newborn daughter cell is physically separated from its mother using the micromanipulator. Typically, dozens of mother cells are processed to obtain sufficient statistics. This technique has several drawbacks. First, it is very labor-intensive and requires around-the-clock mother-daughter dissection. Since a mother cell can live dozens of generations, if performed uninterrupted, a single RLS experiment can take several days. This forces researchers to refrigerate the cells overnight and continue the micromanipulation process the next day. These unavoidable temperature fluctuations would complicate the interpretation of the results, as we do not comprehensively know how growth temperature dynamics affect the aging process. Second, the micromanipulation process can physically damage the mother cells and can lower the RLS depending on the level of damage. Third, cells growing on solid media environments can have cell-to-cell differences in their exposure to the two-dimensional plate surface. This is due to the fact that the contact surface area of large and small cells would be different, leading to differences in the transportation dynamics of the nutrients into the cells.
These drawbacks have recently forced researchers to use automated microfluidic devices (Ryley and Pereira-Smith, 2006; Lee et al., 2012; Zhang et al., 2012) for measuring RLS in liquid media environments. The first such study (Ryley and Pereira-Smith, 2006) reported the use of three different designs and compared their relative efficiencies in terms of measuring yeast RLS. However, even the best-performing design identified in this study could easily trap several cells, instead of just the original mother cell, making the mother-daughter identification process too challenging, as well as introducing problems in terms of having several cells getting stuck in the functional unit of the chip.
A different design introduced in a later study (Lee et al., 2012) used transparent pads on which cells were immobilized due to physical pressure. This design, too, had several issues. First, its functional unit was a flat surface that did not discriminate between mother and daughter cells. Second, the surface area of each unit could easily capture several yeast cells, instead of a single mother cell. These issues complicate the isolation of the mothers and therefore the tracking the mother-daughter pairs for RLS measurements. Also, when a daughter cell is separated from its mother with help from media flow, on its way out, it can attach to other pads, making it hard for the researcher to track the original mother cells.
Another study (Zhang et al., 2012) used a design that operated on the principle of randomly catching and holding a single yeast cell between a PDMS column and a glass slide. The column had a surface area that was similar to the cross-sectional area of a typical yeast cell. Despite offering improvements over the previous studies, this design did not allow the trapped mother cell to move freely between the PDMS column and the glass slide. Also, a daughter cell could not be born from the top or bottom portion of the immobilized mother cell without substantially squeezing the mother cell. The imposed physical constraints could negatively impact the RLS values of the mother cells studied. Yet other studies (Crane et al., 2014; Jo et al., 2015) used designs that were open from one side with a gap from the other side to allow the escape of the daughter cells. The maximal RLS values reported in these studies were only 40 (Crane et al., 2014) and ~45 (Jo et al., 2015) generations, while it is known that wild type mother cells can live more than 45 generations. This reduction in maximal RLS was most likely due to the loss of the old mother cells through the open side of the design (facilitated by mother-daughter attachment followed by the wash-out of both mother and daughter). Finally, neither these microfluidics designs nor the previously published ones could report the RLS values precisely, as the information on the exact age of the initially captured mother cell was missing. As one study (Zhang et al., 2012) reported by counting bud scars on WGA-stained (Wheat Germ Agglutinin) cells, the level of uncertainty on the starting age of the initially captured mother cells could be up to 8 generations (a significant fraction of the average RLS), corresponding to the level of maximal error on the ultimately measured RLS for a single cell.
Here we introduce a high-throughput microfluidics platform design that is free from the above issues, delivers high precision by starting the RLS measurement from the 0th generation, and is multiplexed for running independent experiments simultaneously. Using this platform, we performed single-cell aging experiments in different growth environments, and tracked cellular lifespan and gene expression levels in real time. As a validation of its functional performance, our platform was able to capture the effect of several canonical gene deletion mutants (fob1Δ, sgf73Δ, tor1Δ, sir2Δ, gpa2Δ, rpl31aΔ) on single cell RLS.
RESULTS
Design and fabrication of the multiplexed microfluidic chip
In order to automate replicative aging measurements for individual mother cells, we designed and fabricated a microfluidic platform (Fig. 1) that can be used to run multiple experiments in parallel and measure single-cell RLS values precisely by starting with virgin yeast cells.
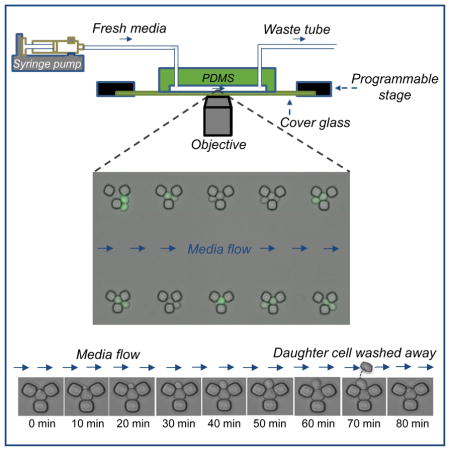
Figure 1. Design and fabrication of the multiplexed microfluidic platform.

A. Schematics of the experimental setup. B. Two identical PDMS platforms, each with 10 independent modules, drawn on a circular area representing the size of the silicon wafer. C. A single PDMS platform. D. One of the 10 independent modules of each PDMS platform. E. A zoomed-in area on a module. Each module has 32 rows and 332 columns of replicator units. Neighboring rows are separated by 45 microns, while neighboring columns are separated by 16 microns. F. Design details of a single replicator unit. G. Scanning Electron Microscope (SEM) picture of an area on an independent PDMS module. Scale bar is 100 microns. H. SEM picture of a single replicator unit. Scale bar is 5 microns. I. Time dynamic bright field images showing a trapped mother cell producing a daughter cell. Image sequence starts at top-left and ends at bottom-right.
Our multiplexed platform is composed of 10 independent modules allowing the researcher to run 10 independent experiments with different media or strains. The multiplexed PDMS structure fits onto a 43 x 50 mm coverslip. Each module has its own inlet and outlet connections to facilitate independent cell loading and media flow. Each module contains a two-dimensional array of 10,624 replicator units. The replicator is the smallest functional unit of our chip. It is composed of three PDMS bars of 5.3μm height, which allow the trapping of only one mother cell. Both the vertical height and the horizontal dimensions of each unit (including the size of the square bars and the corner-to-corner distance between them) have been the result of several rounds of optimization. If the trapping space inside each unit was too large, the unit did not function as intended due to the presence of multiple cells fitting into each unit, which clogs the unit and makes it too challenging to automate the separation of daughter cells. If the trapping space were too small, on the other hand, the trapped cell would be immobilized inside the unit and the spatial constraint would adversely affect cell growth.
In making the above optimization, we had to take into account how much volume increase was expected from each cell throughout its lifespan, as cell-volume is known to increase during the aging process. Unless accounted for, the increase in cell-volume would result in the squeezing of the cell with time. Our chip is optimized to accommodate the age-associated volume changes of a commonly used S. cerevisiae strain of ‘BY’ genetic background. Due to the availability of its genome sequence, this strain is one of the most commonly used yeast strain for a variety of yeast-based studies, including the majority of the previously published studies on aging. ‘W303’ is another S. cerevisiae strain background that is used by yeast researchers. While optimizing the size of our replicator unit for the BY background cells, we also tested it for cells of W303 genetic background. Compared to the BY background cells, yeast cells of W303 background are known to display a larger volume-increase during their aging (Zadrag-Tecza et al., 2009). Since the size of our replicator unit does not dynamically change during the aging process, a large difference in cell volume between when the cell is young vs. old means that the mother cell will be squeezed when it is old. When we used cells of W303 background in our chip, we indeed observed their relatively faster volume increase dynamics and physical squeezing, which was followed by premature cell death. The maximum replicative lifespan of the W303 cells measured using our platform was only 7 generations long.
Optimization of experimental parameters for initiating and running the RLS experiment
After optimizing the size of our replicator units as well as the overall dimensions of the multiplexed chip, next we fine-tuned the experimental parameters for cell loading and maintenance. For this, we characterized and optimized (i) flow rate during cell loading, (ii) density of the loaded cells, and (iii) rate of media ‘flush’ that is periodically applied in addition to the constant low-speed media flow. The flushing was necessary for two reasons: first, it facilitated the physical removal of the daughter cells from their mothers; second, it helped avoid clogging of the chip due to the accumulation of cells outside the traps.
We started our characterization by using cells grown to a density of 0.25 (OD600). Cells mixed in minimal media were pumped into the chip for 5 minutes. In three separate experiments, we applied the flow rates of 10, 30, and 50μL/min during cell loading (Fig. 2a), and then connected the inlet tube to a syringe that only contained fresh media without cells. After the loading process, the syringe pump was programmed to operate at two alternating flow rates during the entire aging experiment: a low-speed constant flow rate of 2μL/min applied for 18 min, followed by a flushing rate of 30μL/min applied for 2 min.
Figure 2. Characterization of experimental parameters for cell loading and maintenance.

A. The dependence of cell loading efficiency on the flow rate during cell loading. Cells with OD600 = 0.25 were loaded to separate modules of the multiplexed microfluidic platform, with each module experiencing a different culture flow rate for 5 minutes: 10μL/min, 30μL/min, or 50μL/min. The loading efficiency was calculated as the percentage of fillable units out of 1,120 replicator units investigated. A replicator unit was defined as fillable if it was not initially occupied by a cell, but it had a nearby cell touching one of its three gaps. For each flow rate, a rectangular region consisting of 1,120 replicator units provided the total number of replicator units investigated. B. The dependence of cell loading efficiency on the cell density during cell loading. Using the cell loading flow rate of 30μL/min for 5 minutes, three different cell densities (OD600 = 0.1, 0.25, 0.4) were characterized. As described in (A), the cell loading efficiency was calculated for each case. C. The dependence of cell loading efficiency on loading duration. Using the cell loading flow rate of 30μL/min and cell density of OD600 = 0.25, three different loading durations (3min, 5min, 10min) were characterized. As described in (A), the cell loading efficiency was calculated for each case. D. The effect of the media flush rate on the percentage of mother cells removed from their traps. The reduction in the percentage of the initially-trapped cells was separately measured for three different media flush rates: 10μL/min, 30μL/min, and 50μL/min. For each case, the media flush was applied for 5 minutes and the number of initially-trapped cells served as the denominator in calculating the percent reduction. In all panels, error bars are SEM (N=2).
The loading efficiencies plotted in Figure 2A correspond to the fraction of the inspected replicator units which have at least one cell right outside each unit. One of these cells eventually buds into the replicator unit and we measure the RLS starting from that unbudded virgin cell. For this efficiency quantification, we chose to inspect 1,120 replicator units positioned in a rectangular area which was at the left side of the outlet (with a distance of 10 replicator units between the inspection area and the outlet). Figure 2A shows that the cell loading efficiency increases as the cell loading flow rate increases. Despite the loading flow rate of 50μL/min gave rise to a slightly higher cell trapping efficiency compared to the efficiency obtained from 30μL/min, we also observed an increased chance of clogging during the multi-day aging experiment when we used the rate of 50μL/min. This was due to that the high loading speed also increased the number of cells that were not trapped, but were still present in the chip, and these loose cells caused clogging by exponentially growing in number. The flow rate of 30μL/min was optimal in terms of giving a sufficiently high trapping efficiency and minimal chance of clogging.
Next, to see the effect of cell density on cell loading efficiency, we characterized two more OD600 values (0.1 and 0.4) by keeping the rest of the experimental parameters the same as before (loading rate of 30μL/min and loading duration of 5 min). Compared to the efficiency observed when OD600 = 0.25, the efficiency observed at OD600 = 0.4 was slightly higher (Fig. 2B). However, compared to the case of OD600 = 0.25, we witnessed higher chances of chip clogging when the cell density was 0.4. Therefore, to avoid potential clogging issues, we picked OD600 = 0.25 as the ideal cell density among the densities characterized.
The next step of our characterization process involved testing the effect of loading duration on loading efficiency. At the previously optimized loading parameters (loading flow rate of 30μL/min and cell density of OD600 = 0.25), we measured the loading efficiency for the loading-durations of 3min, 5min, and 10min. We observed similar efficiency values among the three cases (Fig. 2C).
As the final step of the characterization process, we tested the effect of the periodic media flushing speed on the maintenance of the loaded cells throughout the entire lifespan measurement experiment. If the media flushing speed was too high (50μL/min), we saw that 72% of the trapped cells were lost within five minutes after the flush (Fig. 2D). On the other hand, using flushing speeds of 10μL/min and 30μL/min caused the loss of 1% and 24% of the trapped cells, respectively (Fig. 2D). Although choosing a flushing speed of 10μL/min or less minimized the fraction of lost cells, such a low speed also increased the chance of clogging the chip during the later stages of the RLS experiment due to the inefficient removal of daughter cells. We found that the media flushing speed of 30 μL/min balanced the two factors well.
In a single module of our chip, depending on the need, the total number of trapped cells can easily be increased by several folds. If a user uses several or all of the 10 independent modules of the chip, the time it takes for the objective to move around to take images needs to be taken into account. This is because the objective needs to come back to the same spot before the next bright field image is captured (in 10 minutes in our case).
Functional performance of the microfluidic chip
After the above optimizations, we ran a complete RLS measurement experiment with the goal of determining how our chip performs in terms of the average and maximal lifespan of wild type yeast cells. For this, overnight grown cells (16 hours in minimal media with 2% glucose) were loaded into the chip. The proper dilution of the cells made sure that, at the end of the overnight growth, the cell density was 0.25. Cell loading was performed at the loading speed of 30μL/min for 5 min. Throughout the 91 hours long RLS experiment, the media flow speed cycled between 2μL/min for 18min followed by 30μL/min of flushing speed for 2min. Every 10min, bright field images were acquired. Images were analyzed by counting the total number of daughter production events for 100 independent mother cells during their entire lifespan. The completion of lifespan for each mother cell was marked by cell bursting. We obtained an average lifespan of 29.3 generations (Fig. 3A–B). We also saw a maximal lifespan value of 67 generations, and to the best of our knowledge, this is the longest maximal lifespan value reported for wild type yeast cells grown in minimal media with 2% glucose.
Figure 3. Single-cell lifespan and daughter production duration analyses in the 2% glucose environment.

A. Distribution of replicative life spans (RLS) for 100 mother cells analyzed. The average RLS is 29.3 generations. B. Cellular viability as a function of the number of generations. Cellular viability was defined as the fraction of the initial cells that were still alive at a specific generation. Differently from the conventional ‘death curves’, the viability analysis performed here is based on single-cell level measurements. C. Separately for each mother cell, the relationship between the number of generations and time. 100 mother cells (specified by 100 different colors) were analyzed by marking the time points when a mother cell produced its daughters. Each color-coded trajectory corresponds to the entire lifespan of an individual mother cell. Each dot denotes the time point when a daughter cell was separated from its mother. Time t=0 corresponds to the completion of the birth event for each mother cell, after which its first daughter starts to be generated. D. The relationship between daughter production durations experienced by each mother cell and the specific number of the daughter cell it currently generates. 100 mother cells (specified by 100 different colors) were analyzed by measuring their daughter production durations for each passing generation. Each color-coded trajectory corresponds to the entire lifespan of an individual mother cell. Each dot denotes the time it takes for a mother cell (going through a specific generation) to complete generating a daughter cell. E. The single-cell level relationship between average daughter production duration and replicative lifespan. For each mother cell analyzed in (D), daughter production durations were averaged throughout its lifespan. F. The single-cell level relationship between the standard deviation of daughter production durations and replicative lifespan. See also Figure S1–S4.
Even though our device can trap, track, and report RLS from several fold more cells than 100 cells, it is desirable for the user to analyze 100 cells to save analysis time. To see how the RLS results obtained from the analysis of 100 cells would statistically compare to the results obtained from the analysis of 1,000 cells, we separately tracked and analyzed the RLS from 1,000 mother cells. Then, we randomly picked 100 cells out of these 1,000 cells and quantified their RLS statistics. We found that the mean RLS and its standard deviation were very similar between the two pools (Fig. S1).
To further validate the functional performance of our chip, we next measured the RLS of six different canonical gene deletion mutants that are expected to increase or decrease yeast lifespan based on previous work (Kaeberlein et al., 2005; McCormick et al., 2014; Kaeberlein et al., 2004). For this, we performed RLS experiments in our device by using minimal media supplemented with 2% glucose and quantified the single-cell RLS distributions for six strains: fob1Δ, sgf73Δ, tor1Δ, sir2Δ, gpa2Δ, and rpl31aΔ. As expected, we observed an RLS increase for fob1Δ, sgf73Δ, tor1Δ, gpa2Δ, and rpl31aΔ strains, while a decrease for the sir2Δ strain, compared to the wild type strain’s RLS value (Fig. S2–3). It is important to note that microfluidics-based lifespan measurements are free from potential non-cell autonomous effects while results obtained from the micromanipulator-based lifespan assay can be a combination of both cell autonomous and non-cell autonomous effects (Mei and Brenner, 2015), as during the application of the micromanipulator, cells can be exposed to small molecules that may be secreted to the solid growth surface during aging.
Finally, since the traditional micromanipulator-based RLS experiments are performed in rich media conditions (YPD with 2% glucose), we also tested our platform by running an RLS experiment using rich media conditions and compared the results to the one obtained from the minimal media environment. We saw similar RLS values between the two media conditions (Fig. S4).
Fluctuations in daughter production durations during aging
Next, we analyzed the acquired bright field images by recording the relationship between the number of generations and time (Fig. 3C), as well as the daughter production durations for each mother cell to produce and separate daughter cells until the completion of its lifespan (Fig. 3D). Despite being grown in the same environment, almost all mother cells showed significant fluctuations in their daughter production durations throughout their lifespan. Interestingly, some mothers consistently kept their daughter production durations around ~80min until their death (Fig. 3D). One possibility that could explain the elongated daughter production durations is that cells experience and repair DNA damage during aging. The production duration could be dependent on the level of damage, and if the repair is successful, the duration in the next generation could still be short.
To see whether or not there is a relationship between the lifespan of each mother cell and the range of daughter production durations it experiences throughout its lifespan, we plotted the mean (Fig. 3E) and standard deviation (Fig. 3F) of daughter production times as a function of RLS and calculated Pearson’s correlation coefficient. We saw a moderate degree of anti-correlation between the two phenotypes (r = −0.21 for Fig. 3E and r = −0.24 for Fig. 3F).
Galactose shortens the replicative lifespan of yeast cells
In order to understand which genetic or cell biological process contribute to the replicative lifespan of individual mother cells, it is often necessary to probe the relevant gene expression profiles in aging cells by using fluorescent reporter proteins, allowing one to combine the measurements of daughter production durations, maximal lifespan, and single cell gene expression levels. Using a haploid wild type yeast strain, we integrated a single copy of the PGAL1-YFP construct into its ho locus. To induce the GAL1 promoter activity (Suzuki-Fujimoto et al., 1996; Acar et al., 2005; Egriboz et al., 2011) in a bimodal fashion (Acar et al., 2005; Gardner et al., 2000; Becskei et al., 2001; Xiong and Ferrell Jr., 2003) so that OFF and ON cells’ aging profiles would be analyzed in the same environment as described in the next section, we supplemented the minimal media with 0.5% glucose and 2% galactose. Since high glucose concentrations repress the GAL network activity, using a glucose concentration lower than 2% was necessary to have an appreciable fraction of cells in the ON state. We loaded overnight grown cells into our chip and ran a ~4-days long RLS experiment as described in the previous sections. Bright field and fluorescence images of aging mother cells were acquired every 10min and 1hr, respectively. Performing analyses similar to the ones we performed for the cells grown in the 2% glucose environment, we observed a mean and maximal lifespan of 24.6 and 49 generations respectively (Fig. 4A–B), corresponding to a 16% decrease (from 29.3 to 24.6 generations) in the average RLS compared to the 2% glucose environment. If glucose was the only sugar in the media, lowering the glucose concentration from 2% to 0.5% would increase the average RLS by ~10% due to the caloric restriction23. Therefore, the 16% RLS reduction we observed indicates that it is the presence of galactose that lowers the lifespan.
Figure 4. Single-cell lifespan and daughter production duration analyses in the mixed sugar environment.

A. Distribution of replicative life spans (RLS) for 100 mother cells analyzed. The average RLS is 24.6 generations. B. Cellular viability as a function of the number of generations. Cellular viability was defined as the fraction of the initial cells that were still alive at a specific generation. C. Separately for each mother cell, the relationship between the number of generations and time. 100 mother cells (specified by 100 different colors) were analyzed by marking the time points when a mother cell produced its daughters. Each color-coded trajectory corresponds to the entire lifespan of an individual mother cell. Each dot denotes the time point when a daughter cell was separated from its mother. Time t=0 corresponds to the completion of the birth event for each mother cell, after which its first daughter starts to be generated. D. The relationship between daughter production durations experienced by each mother cell and the specific number of the daughter cell it currently generates. 100 mother cells (specified by 100 different colors) were analyzed by measuring their daughter production durations for each passing generation. Each color-coded trajectory corresponds to the entire lifespan of an individual mother cell. Each dot denotes the time it takes for a mother cell (going through a specific generation) to complete generating a daughter cell. E. The single-cell level relationship between average daughter production duration and replicative lifespan. For each mother cell analyzed in (D), daughter production durations were averaged throughout its lifespan. F. The single-cell level relationship between the standard deviation of daughter production durations and replicative lifespan.
As in the previous growth environment (Fig. 3C–D), we observed similar profiles for the mother cells in terms of their number of generations as a function of time (Fig. 4C) and the fluctuations in their daughter production durations throughout their lifespan (Fig. 4D). The average daughter production durations (Fig. 4E) (averaged over each mother cell’s lifespan) and their standard deviation (Fig. 4F) displayed anti-correlations with respect to the RLS values (r = −0.25 for Fig. 4E and r = −0.09 for Fig. 4F).
Phenotype-specific RLS analysis in the mixed sugar environment
To find out whether or not the 16% reduction in RLS was due to the galactose metabolism in individual mother cells, we measured the single-cell PGAL1-YFP level of each mother cell throughout its lifespan and plotted it as a function of the number of generations (Fig. 5A). Due to the bimodal nature (Acar et al., 2005; Acar et al., 2008; Acar et al., 2010; Peng et al., 2015) of the GAL network activity, 24% of the mother cells were in the ON state (or phenotype) with an ability to metabolize galactose, while the remaining 76% in the OFF state (Fig. 5A). The average RLS of the ON cells was 24.4 generations, while the OFF cells had an average RLS of 24.7 generations. Observing similar RLS values between the two phenotypes, we next analyzed the viability of the aging mother cells in terms of their displaying OFF or ON network activity profile (Fig. 5B). We saw that cells displaying each activity profile showed similar viability dynamics. Therefore, these results indicate that the 16% reduction in RLS was not due to the galactose metabolism in individual mother cells.
Figure 5. Phenotype-specific lifespan and daughter production duration analyses in the mixed sugar environment.

A. Tracking single-cell fluorescence levels in mother cells throughout their life span. Data from 100 mother cells are displayed using 100 different colors. The fluorescence level of 103 a.u. was used as the separation between the OFF and ON state of the GAL network activity. Lower inset: image of a trapped OFF cell. Upper inset: image of a trapped ON cell. The growth environment was minimal media containing 0.5% glucose and 2% galactose. B. Cellular viability curves for the ON and OFF cells. For the ON cells, the mean and standard deviation of their replicative lifespan were 24.4 and 10.9 generations, respectively. The OFF cells displayed a mean lifespan of 24.7 generations, with a standard deviation of 8.6 generations. Out of the 100 mother cells analyzed, 24 cells were in the ON-state of the GAL network, while 76 cells were in the OFF-state. C–D. Single-cell level relationships between average daughter production duration and replicative lifespan. For each mother cell analyzed in Figure 4d, daughter production durations were averaged throughout its lifespan separately for OFF (C) and ON (D) mother cells.
We also analyzed the OFF and ON cells in terms of their phenotype-specific relationships between their daughter production durations and lifespan (Fig. 5C–D). Using Pearson’s correlation coefficient, we quantified the degree of correlation between the lifespan of each mother cell and the average daughter production durations it experiences throughout its lifespan. For both OFF and ON phenotype cells, we saw anti-correlations (r = −0.24 for the OFF cells, and r = −0.30 for the ON cells) between their lifespan and averaged durations. Despite their moderate nature, the anti-correlations (Fig. 3E–F, Fig. 4E–F, Fig. 5C–D) we quantify in this work are significant. They suggest that cells that consistently spend less time to produce daughter cells would be expected to live longer. Whether or not there is indeed a causal relationship between a cell’s ability to divide quickly with similar durations and its extended lifespan will be explored in future studies.
Superoxide-dismutase activity is elevated at the presence of galactose
Our results in the previous section showed us that the 16% RLS reduction we observed in the mixed sugar environment was not due to the galactose metabolism or the expression of the GAL network proteins. Therefore, as an alternative explanation, we considered the possibility of increased oxidative damage levels when galactose is present in the growth environment. It is known that systemic exposure of mice, rats, and drosophila to galactose causes accelerated senescence (Cui et al., 2006). Despite that the molecular mechanism behind this phenotype is not fully understood, galactose-caused oxidant generation is hypothesized to make an important contribution (Cui et al., 2004).
To test whether or not yeast cells grown in the presence of galactose show increased levels of oxidative stress, we constructed a yeast strain in which the mCherry reporter protein was driven by the SOD1 promoter which is activated in the presence of oxidative stress. SOD1 encodes a superoxide-dismutase that catalyzes the breakdown of the superoxide radical O2− to dioxygen and hydrogen peroxide. Using minimal media supplemented with either 2% glucose or 2% galactose, we ran independent RLS experiments in our device and quantified single-cell mCherry expression levels during aging. Then, we analyzed the RLS difference between the two environments. We saw a 16.8% decrease in RLS in the 2% galactose environment compared to the 2% glucose environment (Fig. 6A), confirming our earlier prediction based on the mixed sugar environment (0.5% glucose + 2% galactose) that it is the presence of 2% galactose that lowers the lifespan. Next, we analyzed the single cell PSOD1-mCherry levels separately in the 2% glucose and 2% galactose environments and saw that there was indeed an increase (up to ~2-fold) in the reporter protein concentration in the galactose environment (Fig. 6B). These results further support the hypothesis that the RLS reduction observed in the galactose environment is due to the elevated oxidant levels in this environment.
Figure 6. Comparative analysis of lifespan and oxidative stress in glucose and galactose environments.
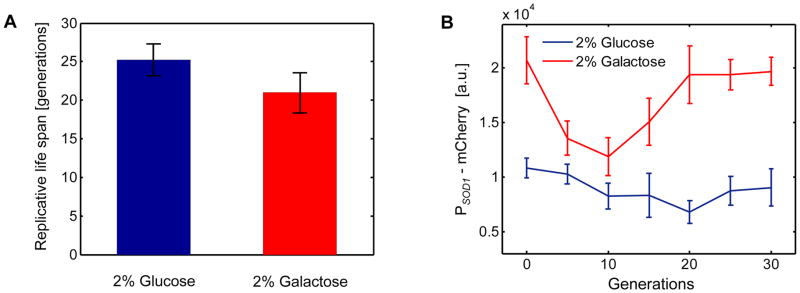
A. Average lifespan is 16.8% lower in the 2% galactose environment (RLSmean = 21.0) compared to the 2% glucose environment (RLSmean = 25.2). Error bars are SEM (N=3 experiments, glucose; N=5 experiments, galactose). B. PSOD1-mCherry levels quantified in aging mother cells in increments of 5 generations. 30 mother cells were tracked in each environment and single-cell mCherry levels from all living cells were averaged at the specific generation points plotted. Error bars are SEM (6 cells ≤ N ≤ 29 cells). For (A–B), the growth environment was minimal media.
In the galactose environment, we also observed an interesting dynamic behavior of the PSOD1-mCherry expression. During their aging, cells first lowered their SOD1 activity levels. This was followed by an increase at the later generations. This trend was also present in the glucose environment, however it was relatively weaker compared to the galactose environment. Our hypothesis into this dynamic behavior is that young cells perform better in detoxifying their intracellular environment compared to the old cells. Further, we attribute the raised mCherry levels in the newborn cells (0th generation in Fig. 6B) to the inheritance of their cytoplasmic content from their aged mothers. Future studies will elucidate the mechanistic details of this interesting observation.
Phenotypic commitment of single yeast cells during aging
As shown in Figure 5A, throughout their lifespan, ON and OFF cells display time-dynamic variations in their YFP content. These variations are due in part to the stochastic nature of the biochemical interactions in cells (Ozbudak et al., 2002; Elowitz et al., 2002; Blake et al., 2003; Raser and O’Shea, 2004), while age-induced genetic and cell-biological deformations and damage could also make contributions. If the level of age-induced deterioration of gene expression dynamics were high enough, one might expect to see frequent instances of an ON cell’s stopping expressing YFP, or an OFF cell’s starting expressing YFP. During the aging process, despite the time-dynamic variations observed at the single-cell YFP levels, for only 1 cell these variations were large enough to cause phenotypic switching between the OFF and ON expression state (Fig. 5A). Therefore, our work uncovers that single yeast cells commit to their gene expression states or phenotypes during their aging process.
Over shorter experimental durations, previous work (Zacharioudakis et al., 2007; Zhou and Zhou, 2011) showed that the transcriptional induction of GAL1 occurs with faster kinetics if it has been previously expressed (e.g. by altering the carbon sources between inducing galactose and repressing glucose), indicating transcriptional memory of the previous induction. In our work, we use the term phenotypic commitment to refer to the stability of cellular phenotype (cells’ tendency to stay in the ON state or OFF state), we elucidate this behavior over longer time periods in the context of aging, and we observe phenotypic commitment when cells are grown in the same environment (as opposed to altering the carbon sources). Despite these differences between our work and previous studies, the phenotypic commitment we observe here might still be facilitated by the same GAL1-mediated mechanism, as in the previous study (Zacharioudakis et al., 2007). In other words, the GAL1-facilitated phenotypic memory effect might help the ON state cells (which have high GAL1 expression) ‘stick’ to the ON state, despite there is no change in the environment. If this were true, then we would expect that cells would stick to the ON state better compared to their sticking to the OFF state (in which only a basal amount of GAL1 proteins are produced). As a result, the cellular switching frequency from ON-to-OFF state would be expected to be smaller than the cellular switching frequency from the OFF-to-ON state.
Our microscopic findings support these expectations despite the rare nature of the switching process: no ON cells switch to the opposite state while an OFF cell does switch to the ON state. To see if we could provide another layer of support to these expectations, we quantified the relative strengths of the OFF⇔ON phenotypic switching frequencies by running independent FACS experiments. The results further supported our expectations by showing that the OFF-to-ON phenotypic switching rate was higher than the opposite switching rate (Supplemental Experimental Procedures). It is worth noting that the FACS-based method for quantifying phenotypic switching rates is an average measure, but it can still be reliable for a relative comparison of switching rates between the two switching directions. In summary, here we directly showed that single cells could display commitment to their gene expression state throughout their entire lifespan. We hypothesized that the mechanism behind this behavior was the same as the mechanism that makes cells transcriptionally induce GAL1 with faster kinetics if it has been previously expressed. Based on this hypothesis, we expected the ON state cells to display relatively higher commitment compared to OFF state cells and our experimental findings supported this expectation.
DISCUSSION
What genetic and phenotypic changes accompany cellular aging? Are there a small set of master regulatory genes and dedicated gene networks that are primarily responsible for regulating the aging process? Despite the fundamental nature of these questions, our understanding into the mechanisms of cellular aging and how these mechanisms are coupled to the initiation of various disease states is very limited. For example, we know very little about how chromosome instabilities (McMurray and Gottschling, 2003; Miné-Hattab and Rothstein, 2012) occur in old cells and how cell size and gene expression dynamics change during aging (Tanouchi et al., 2015). This lack of understanding has been partly due to that cellular aging is a complex phenotype to measure and comprehensive studies on aging require the application of efficient experimental approaches and novel technological platforms.
Here we introduce a multiplexed high-throughput microfluidics platform that can automate quantitative single-cell measurements of cellular lifespan and gene expression in aging yeast cells with high precision by starting from virgin cells. Using this platform, we measured replicative lifespan of yeast cells in several different media conditions. We found that growing cells in galactose lowered their lifespan by 16.8% compared to glucose, cellular lifespan and daughter production durations were anti-correlated, and single cells displayed commitment to their gene expression states during aging.
For several decades, the labor-intensive nature of the conventional micromanipulator-based aging assays has limited the progress in the field of aging. The inability of the colony-based aging assays to quantify real-time gene expression profiles at the single cell level was also an important deficiency. High-precision multiplexed microfluidic platforms automating single-cell aging measurements will transform the aging field by overcoming the deficiencies and limitations of the conventional assays.
EXPERIMENTAL PROCEDURES
Design details of the multiplexed microfluidic platform
Using the software Layout Editor (http://www.layouteditor.net), two separate 10-module multiplexed platform was drawn in a circle of 100mm diameter. Each 10-module platform occupied a rectangular area of 38mm x 34mm. The horizontal and vertical distance between two neighboring modules were 5.5mm and 3.6mm, respectively. The size of each independent module is 10mm x 1.9mm, with a boundary thickness of 0.24mm. Each independent module contains 32 rows of the tri-square replicator units, symmetrically oriented with 16 rows facing up and the other 16 rows facing down. Row to row distance is 45 microns and in each row neighboring replicator units are separated by 16 microns. Each row consists of 332 replicator units, making a total number of 10,624 replicators inside each module. Each replicator unit consists of three squares, each with a size of 4.2 microns. The corner-to-corner distance between two neighboring squares is 3.2 microns.
Etching the design into silicon wafer in the cleanroom
Using a single-side polished silicon wafer (University Wafer) of 100mm diameter, we started the process by cleaning the wafer. The cleaning step consisted of two sub-steps: Piranha cleaning using a 3:1 mixture of H2SO4:H2O2, followed by the BOE (Buffered Oxide Etch) cleaning. After cleaning the wafer, the next step involved photoresist spinning and baking. Uniformly spreading a positive photoresist (S1805, Dow Chemical) onto the wafer, we used a designated spinner for positive photoresist and performed a 1 minute spin at 3500 rpm. After the spinning, the wafer was immediately transferred to a hot plate to bake at 110°C for 1 minute. Next, the pattern of the design was written on the wafer by using a laser writer. This was followed by the steps of photoresist development (for 1 minute in a glass container containing MF-319) and wafer inspection. Finally, 12 cycles of DRIE etching (Bosch Process) was performed by using Plasmalab System 100 manufactured by Oxford Instruments. Unnecessary photoresist and other chemicals were removed from the surface of the etched wafer by using oxygen plasma treatment.
Production and preparation of the PDMS platform
PDMS curing agent and PDMS elastomer were mixed (with a ratio of 1:10) thoroughly for 5 minutes. Once the PDMS mixture was prepared, it was poured onto the silicon wafer, followed by desiccation in a vacuum for about 4 hours until all air bubbles (caused by the mixing process) disappeared. Then, the container carrying both the wafer and the PDMS mixture was heated to solidify the PDMS mixture. After the cured PDMS cooled down, the area of the PDMS carrying the microfluidic design was cut using a razor blade. On each of the 10 independent modules, inlet and outlet holes were punched using Schmidt punch press (Syneo, LLC). Next, the punched PDMS platform was bonded onto a 43mm x 50mm glass coverslip. Tygon tubing was inserted into the inlet and outlet holes of the modules.
Setting up and priming the PDMS platform
The multiplexed platform was put onto the programmable stage of an inverted fluorescence microscope (Nikon Eclipse Ti). The platform was then connected to a syringe (NORM-JECT plastic syringe, 20 ml) by using connectors made from platinum-cured silicone tubing. A programmable syringe pump (Fusion 200, Chemyx, LLC) was used to control and program the flow rates of the media and cell cultures. To facilitate movement of cells inside each module, BSA (0.1 mg/ml) was pushed through the modules at 30 μL/min for 5 minutes using the syringe pump. After the BSA application, the modules were washed (at 30 μL/min for 5 minutes) with the prospective media which would be used in the RLS experiment.
Initiating and running the lifespan experiments
Cultures were grown in synthetic dropout media supplemented with the appropriate amino-acids and carbon sources as specified in the text. Overnight grown yeast cells (10ml culture volume in a shaker at 30°C) were diluted so that the cell den sity (OD600) was around 0.25. Approximately 3 ml cell culture was loaded into a syringe, which was then mounted to the syringe pump. The cells were pushed into the microfluidics platform at the flow rate of 30μL/min for 5 minutes. After loading the cells, using the software NIKON Elements, we recorded the x-y coordinates of 34 non-overlapping locations or fields of view for each independent module. A 60x oil objective was used and each field of view consisted of 10 replicator units (2 rows and 5 columns of replicator units), allowing to trap and track up to 10 mother cells for each location. Out of the 34 locations selected, 30 locations were selected for the RLS measurements, while the other 4 were selected for monitoring purposes to ensure that there were no clogging issues. For this, 4 locations surrounding the outlet (lower left, upper left, lower right, upper right) were chosen, as the chance of clogging (if any) was higher around the outlet. Next, the 30 positions for the RLS measurements were selected. The selection process started from the left hand side of the outlet (10 replicator units away from it). Each module was scanned from top to bottom and from right to left, one field of view at a time until 30 locations were chosen. At least 100 fillable replicator units were identified in these 30 locations, allowing us to perform replicative lifespan analysis for at least 100 mother cells. However, depending on a user’s need, by analyzing more than 30 locations, the total number of cells analyzed can easily be increased. To obtain the lifespan data from one thousand cells, we ran three independent experiments in three modules of our chip, and combined the single-cell RLS values into a single histogram (Fig. S1A).
The criterion for a fillable replicator unit was that the unit was not initially occupied by a cell, but it had at least one cell touching one of its three gaps. This way, the nearby cell would bud into the replicator unit and a newborn yeast cell would be trapped in the replicator unit, ensuring to start its RLS clock from zero. After the 30 locations were selected, the syringe pump was programmed to push fresh minimal media into the independent modules at two different media flow rates: the continuous rate at 2μL/min for 18 minutes, followed by the flushing flow rate of 30μL/min for 2 minutes. These flow rates cycled repeatedly until the end of the RLS experiment. The growth temperature during the lifespan experiments was ~30°C.
For each of the 30 locations selected, bright field and fluorescence images were captured every 10 minutes and 1 hour, respectively. The bright field images were used to quantify the number of budding events and daughter production durations throughout the lifespan of each trapped mother cell. The time series fluorescence images were analyzed using the NIKON Elements software. They were analyzed frame by frame, with each frame corresponding to the snapshot of one of the 30 recorded locations at each time point. In each frame, each trapped mother cell was circled in order to get the average YFP intensity inside the circled area at that time point. Then, for each frame, the background fluorescence intensity was measured and subtracted from the average YFP intensity reading of each trapped cell, giving rise to the final single-cell YFP intensity values plotted (Fig. 5A). To obtain the background fluorescence level of a frame, three square spots (with side length of 4μm) were identified at three locations (top left, center, bottom right) on each frame and the average YFP reading inside these locations were measured and averaged to obtain a single representative number for background subtraction. In the rare instances when a cell’s fluorescence level could potentially be affected by a nearby cell’s high fluorescence content, we interpolated the trapped cell’s YFP content at the nearest time points (one hour before and after) and used the resulting value for the middle time point.
The fluorescence measurements (Fig. 6B) for the strain carrying the PSOD1-mCherry reporter construct were similar to the measurements made for the strain carrying the PGAL1-YFP construct. Bright field and fluorescence images were captured every 10 minutes and 1 hour, respectively, in minimal media environments containing either 2% glucose or 2% galactose. For each environment, 30 mother cells were randomly picked from two independent runs and single-cell PSOD1-mCherry fluorescence values were extracted at each additional five generations until the 30th generation.
Construction of the yeast strains
All S. cerevisiae strains used in this study have the BY genetic background. The strains and their full genetic descriptions are listed in Table S1. The haploid gene-deletion strains (fob1Δ, sgf73Δ, tor1Δ, sir2Δ, gpa2Δ, rpl31aΔ) were obtained from Dharmacon, Inc. with the following catalog numbers, respectively: YSC6273-201935800, YSC6273-201936107, YSC6273-201937801, YSC6273-201935296, YSC6273-201921174, YSC6273-213589150. The strain with the YFP reporter was constructed in the following fashion. We constructed a haploid wild-type S. cerevisiae strain with the PGAL1-YFP reporter integrated in its ho locus. For this, KpnI–PGAL1–BamHI and BamHI–YFP–EcoRI fragments were cloned into a plasmid upstream of the CYC1 transcriptional terminator. The plasmid also carried the PTEF1-HIS5 marker positioned to the left of the PGAL1-YFP reporter. Using this plasmid as a template together with 5′-3′ primers having 60bp-long homology to the ho locus, the [PTEF1-HIS5 + PGAL1-YFP] region of the plasmid was PCR amplified and then transformed into a wild-type yeast strain. The PGAL1 promoter sequence corresponds to the 668 base-pair region directly upstream of the start codon of the S. cerevisiae GAL1 gene. Regarding the strain carrying the mCherry reporter, we constructed a haploid wild-type S. cerevisiae strain that carried the PSOD1-mCherry reporter construct in its ho locus. For this, KpnI–PSOD1–BamHI and BamHI–mCherry–EcoRI fragments were cloned into a plasmid upstream of the CYC1 transcriptional terminator. The plasmid also carried the PTEF1-HIS5 marker positioned to the left of the PSOD1-mCherry reporter. Using this plasmid as a template together with 5′-3′ primers having 60 bp-long homology to the yeast ho locus, the [PTEF1-HIS5 + PSOD1-mCherry] region of the plasmid was PCR amplified and then transformed into a wild-type yeast strain. The PSOD1 promoter sequence corresponds to the 566 base-pair region directly upstream of the start codon of the S. cerevisiae SOD1 gene.
Transformation protocol for the yeast strains
We used the standard Lithium Acetate (LiOAc) based yeast transformation technique. Yeast were grown overnight for ~15 hours in 5 ml YPD media in a 30°C shaker incubator. The overnight grown culture was diluted in 10mL YPD and grown for another ~2 hours so that the cells would be in mid log phase at the time of transformation. The mid log phase culture was washed with 10mM TrisCl and incubated in 100 mM LiOAc solution for 40 minutes in a 30°C shaker incubator. After the incubation, cells were spun down and resuspended in a small volume of 100mM LiOAc solution. The resuspended cells were added to a mixture containing 8μL of 10mg/ml salmon sperm DNA (Sigma Aldrich, D-9156), transforming PCR product, and 50μL of 100mM LiOAc solution. The cell-transformation mixture was vortexed with 300μL PEG solution (2g PEG3350 + 2ml 100mM LiOAc) and incubated for ~20 minutes. 50μL DMSO was then vortexed in, followed by a 15-minutes incubation at 42°C. Cells were spun down, resuspended in 900μL YPD, and grown for 2 hours in a rotator at 30°C. After this recovery, cells were spun down and re-suspended in 100μL of 10mM TrisCl. The resuspended cells were spread on CSM-ADE-HIS plates. Plates were grown 2 days at 30°C before colonies were picked for streaking and verification by colony PCR and sequencing.
Supplementary Material
Highlights.
Aging yeast cells are tracked to measure lifespan and real-time gene expression
Single-cell lifespan is measured precisely by starting from virgin cells
Oxidative stress levels are higher in galactose environment compared to glucose
Aging cells commit to a single phenotypic state throughout their lifespan
Acknowledgments
The authors thank M. Power, E. Karatekin, W. Vennekate, G. Kaufman, E. A. Sarnoski, M. Chatterjee, O. S. Koksaldi, and the rest of Acar Lab members for useful discussions. We thank M. Hochstrasser for providing us with the yeast strain AL002. MA acknowledges funding from a New Scholar in Aging Award (Ellison Medical Foundation, AG-NS-1015-13) and NIH Director’s New Innovator Award (NIH Director’s Office and National Institute on Aging, 1DP2AG050461-01). T.Z.Y. was partly funded by a Gruber Science Fellowship and the NIH Genetics Predoctoral Research Training Program (T32 GM007499).
Footnotes
AUTHOR CONTRIBUTIONS
MA conceived and guided the study, made the initial microfluidic design, planned the experiments and analyses, and wrote the manuscript. PL further developed the microfluidic design, fabricated the microfluidic platform, performed the experiments and data analyses, and made the figures. TZY constructed the strains carrying the PGAL1-YFP and PSOD1-mCherry reporters.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acar M, Becskei A, van Oudenaarden A. Enhancement of cellular memory by reducing stochastic transitions. Nature. 2005;435:228–232. doi: 10.1038/nature03524. [DOI] [PubMed] [Google Scholar]
- Acar M, Mettetal JT, van Oudenaarden A. Stochastic switching as a survival strategy in fluctuating environments. Nature Genetics. 2008;40:471–475. doi: 10.1038/ng.110. [DOI] [PubMed] [Google Scholar]
- Acar M, Pando BF, Arnold FH, Elowitz MB, van Oudenaarden A. A General Mechanism for Network-Dosage Compensation in Gene Circuits. Science. 2010;329:1656–1660. doi: 10.1126/science.1190544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becskei A, Seraphin B, Serrano L. Positive feedback in eukaryotic gene networks: cell differentiation by graded to binary response conversion. EMBO J. 2001;20:2528–2535. doi: 10.1093/emboj/20.10.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake WJ, Kaern M, Cantor CR, Collins JJ. Noise in eukaryotic gene expression. Nature. 2003;422:633–637. doi: 10.1038/nature01546. [DOI] [PubMed] [Google Scholar]
- Breitenbach M, Jazwinski SM, Laun P, editors. Aging research in yeast. Springer; New York: 2012. [Google Scholar]
- Crane MM, Clark IBN, Bakker E, Smith S, Swain PS. A microfluidic system for studying ageing and dynamic single-cell responses in budding yeast. PLoS ONE. 2014;9:e100042. doi: 10.1371/journal.pone.0100042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Wang L, Zuo P, Han Z, Fang Z, Li W, Liu J. D-galactose-caused life shortening in Drosophila melanogaster and Musca domestica is associated with oxidative stress. Biogerontology. 2004;5:317–326. doi: 10.1007/s10522-004-2570-3. [DOI] [PubMed] [Google Scholar]
- Cui X, Zuo P, Zhang Q, Li X, Hu Y, Long J, Packer L, Liu J. Chronic Systemic D-Galactose Exposure Induces Memory Loss, Neurodegeneration, and Oxidative Damage in Mice: Protective Effects of R-α-Lipoic Acid. J Neurosci Res. 2006;83:1584–1590. doi: 10.1002/jnr.20845. [DOI] [PubMed] [Google Scholar]
- Egriboz O, Jiang F, Hopper JE. Rapid GAL gene switch of Saccharomyces cerevisiae depends on nuclear Gal3, not nucleocytoplasmic trafficking of Gal3 and Gal80. Genetics. 2011;189:825–836. doi: 10.1534/genetics.111.131839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Longo VD. The chronological life span of Saccharomyces cerevisiae. Aging Cell. 2003;2:73–81. doi: 10.1046/j.1474-9728.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- Gardner TS, Cantor CR, Collins JJ. Construction of a genetic toggle switch in Escherichia coli. Nature. 2000;403:339–342. doi: 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- Jo MC, Liu W, Gu L, Dang W, Qin L. High-throughput analysis of yeast replicative aging using a microfluidic system. Proc Natl Acad Sci USA. 2015;112:9364–9369. doi: 10.1073/pnas.1510328112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Kirkland KT, Fields S, Kennedy BK. Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol. 2004;2:e296. doi: 10.1371/journal.pbio.0020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Lee SS, Vizcarraa IA, Hubertsc DHEW, Leeb LP, Heinemann M. Whole lifespan microscopic observation of budding yeast aging through a microfluidic dissection platform. Proc Natl Acad Sci USA. 2012;109:4916–4920. doi: 10.1073/pnas.1113505109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Gralla EB, Valentine JS. Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo. J Biol Chem. 1996;271:12275–12280. doi: 10.1074/jbc.271.21.12275. [DOI] [PubMed] [Google Scholar]
- McCormick MA, Mason AG, Guyenet SJ, Dang W, Garza RM, Ting MK, Moller RM, Berger SL, Kaeberlein M, Pillus L, La Spada AR, Kennedy BK. The SAGA histone deubiquitinase module controls yeast replicative lifespan via Sir2 interaction. Cell Rep. 2014;8:477–486. doi: 10.1016/j.celrep.2014.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray MA, Gottschling DE. An age-induced switch to a hyper-recombinational state. Science. 2003;301:1908–1911. doi: 10.1126/science.1087706. [DOI] [PubMed] [Google Scholar]
- Mei SC, Brenner C. Calorie Restriction-Mediated Replicative Lifespan Extension in Yeast Is Non-Cell Autonomous. PLoS Biol. 2015;13:e1002048. doi: 10.1371/journal.pbio.1002048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miné-Hattab J, Rothstein R. Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol. 2012;14:510–517. doi: 10.1038/ncb2472. [DOI] [PubMed] [Google Scholar]
- Mortimer RK, Johnston JR. Life span of individual yeast cells. Nature. 1959;183:1751–1752. doi: 10.1038/1831751a0. [DOI] [PubMed] [Google Scholar]
- Müller I, Zimmermann M, Becker D, Flomer M. Calendar life span versus budding life span of Saccharomyces cerevisiae. Mech Ageing Dev. 1980;12:47–52. doi: 10.1016/0047-6374(80)90028-7. [DOI] [PubMed] [Google Scholar]
- Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. Regulation of noise in the expression of a single gene. Nature Genetics. 2002;31:69–73. doi: 10.1038/ng869. [DOI] [PubMed] [Google Scholar]
- Peng W, Liu P, Xue Y, Acar M. Evolution of gene network activity by tuning the strength of negative-feedback regulation. Nat Commun. 2015;6:6226. doi: 10.1038/ncomms7226. [DOI] [PubMed] [Google Scholar]
- Raser JM, O’Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304:1811–1814. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryley J, Pereira-Smith OM. Microfluidics device for single cell gene expression analysis in Saccharomyces cerevisiae. Yeast. 2006;23:1065–1073. doi: 10.1002/yea.1412. [DOI] [PubMed] [Google Scholar]
- Schleita J, Waskoa BM, Kaeberlein M. Yeast as a model to understand the interaction between genotype and the response to calorie restriction. FEBS Lett. 2012;586:2868–2873. doi: 10.1016/j.febslet.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkraus KA, Kaeberlein M, Kennedy BK. Replicative aging in yeast: the means to the end. Annu Rev Cell Dev Biol. 2008;24:29–54. doi: 10.1146/annurev.cellbio.23.090506.123509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki-Fujimoto T, Fukuma M, Yano KI, Sakurai H, Vonika A, Johnston SA, Fukasawa T. Analysis of the galactose signal transduction pathway in Saccharomyces cerevisiae: interaction between Gal3p and Gal80p. Mol Cell Biol. 1996;16:2504–2508. doi: 10.1128/mcb.16.5.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanouchi Y, Pai A, Park H, Huang S, Stamatov R, Buchler NE, You L. A noisy linear map underlies oscillations in cell size and gene expression in bacteria. Nature. 2015;523:357–360. doi: 10.1038/nature14562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong W, Ferrell JE., Jr A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature. 2003;426:460–465. doi: 10.1038/nature02089. [DOI] [PubMed] [Google Scholar]
- Zacharioudakis I, Gligoris T, Tzamarias D. A yeast catabolic enzyme controls transcriptional memory. Curr Biol. 2007;17:2041–2046. doi: 10.1016/j.cub.2007.10.044. [DOI] [PubMed] [Google Scholar]
- Zadrag-Tecza R, Kwolek-Mirek M, Bartosz G, Bilinski T. Cell volume as a factor limiting the replicative lifespan of the yeast Saccharomyces cerevisiae. Biogerontology. 2009;10:481–488. doi: 10.1007/s10522-008-9192-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Luo C, Zou K, Xie Z, Brandman O, Ouyang Q, Li H. Single Cell Analysis of Yeast Replicative Aging Using a New Generation of Microfluidic Device. PLoS ONE. 2012;7:e48275. doi: 10.1371/journal.pone.0048275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BO, Zhou JQ. Recent transcription-induced histone H3 lysine 4 (H3K4) methylation inhibits gene reactivation. J Biol Chem. 2011;286:34770–34776. doi: 10.1074/jbc.M111.273128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.