

Summary
Secretory and membrane proteins from mammalian cells undergo post-translational modifications, including N-linked glycosylation, which can result in a large number of possible glycoforms. This sample heterogeneity can be problematic for structural studies, particularly X-ray crystallography. Thus, crystal structures of heavily glycosylated proteins such as the HIV-1 Env viral spike protein have been determined by removing the majority of glycans. This step is most frequently carried out using Endoglycosidase H (EndoH) and requires that all expressed glycans be in the high-mannose form, which is often not the native glycoform. With significantly improved technologies in single particle cryo-electron microscopy (cryoEM), we demonstrate that now it is possible to refine and build natively glycosylated HIV-1 Env structures in solution to 4.36 Å resolution. At this resolution we can now analyze the complete epitope of a broadly neutralizing antibody (bnAb), PGT128, in the context of the trimer expressed with native glycans.
Introduction
Many proteins, including those of viral and eukaryotic origin, often have post-translational modifications (PTMs) that make them difficult to study at high-resolution using X-ray crystallography due to intrinsic compositional or conformational heterogeneity. N- and O-linked glycosylation occurs in the majority of proteins that pass through the secretory pathway, and serves roles in both protein folding, and function. Glycosylated proteins have been especially difficult to study structurally due to the nature of the modification, which can be extremely diverse even at a single residue and may contain a large range of conformational motion. In X-ray crystallography, to overcome these challenges, glycosylation sites are either removed by mutagenesis, or the proteins are produced in expression systems that limit the glycosylation pathway. For example, GnTI-deficient HEK293S cells produce proteins with all high mannose glycans that can be deglycosylated with Endoglycosidase H (EndoH), leaving only the N-linked core N-Acetylglucosamine (N-GlcNAc) (Depetris et al., 2012; Julien et al., 2013a; Lee et al., 2008; Pancera et al., 2014). However, the major caveat of this approach is that the protein cannot be crystallized in its native form, which is especially disadvantageous when attempting to solve structures of glycoprotein complexes in which the binding partner recognizes complex glycans.
Human Immunodeficiency Virus-1 (HIV-1) envelope glycoprotein (Env), the fusion machine on the surface of HIV-1, is meta-stable and one of the most highly glycosylated protein complexes known, and has been subject to all of the above challenges. A soluble Env trimer construct termed BG505 SOSIP.664, derived from the sequence of clade A virus BG505, with an introduced gp120-gp41 heterodimer linking disulfide bond (SOS) (Binley et al., 2000), and pre-fusion conformation stabilizing I559P mutation (IP) (Sanders et al., 2002) was used to overcome the lack of trimer stability for multiple structural studies (Garces et al., 2014; Huang et al., 2014; Julien et al., 2013a; Julien et al., 2013b; Julien et al., 2013c; Khayat et al., 2013; Kong et al., 2013; Lyumkis et al., 2013; Pancera et al., 2014). Although the ability to generate a stable native-like soluble Env trimer was a major breakthrough, structural characterization of Env glycans remains an arduous task. Despite the fact that the outer domain of Env is heavily packed with glycans, they are highly flexible, and often not resolved unless directly bound, and thus stabilized, by an antibody that recognizes the particular glycan. Indeed, structures of the majority of glycans on Env larger than the core trisaccharide have been determined in this manner (Garces et al., 2014; Julien et al., 2013a; Julien et al., 2013c; Kong et al., 2013; McLellan et al., 2011; Pejchal et al., 2011), as the glycans not interacting with antibodies are disordered, or are accessible to EndoH in the deglycosylation step. As such, with current methods, antibodies that recognize complex glycans often cannot be co-crystallized as an antibody-antigen complex, and thus the untrimmed, large complex glycans predicted to be on trimeric Env have not been observed at high-resolution.
The advent of high-resolution single particle cryo-electron microscopy (cryoEM) overcomes the limitations of flexibility and PTMs, particularly glycosylation, because crystallization is not required. In fact, it has been shown previously that with cryoEM the natively glycosylated BG505 SOSIP.664 trimer with PGV04 Fragment antigen binding (Fab) can be determined to 5.8 Å resolution (Lyumkis et al., 2013), allowing a partial pseudo-atomic model of the peptide to be built. In this study, despite density corresponding to glycans being resolved in the EM map, the glycans were not built into the model (Lyumkis et al., 2013). Here we present a 4.36 Å cryoEM reconstruction of natively glycosylated BG505 SOSIP.664 in complex with a potent broadly neutralizing antibody (bnAb) PGT128, that binds the high-mannose patch of gp120 surrounding the N332 glycan (Pejchal et al., 2011; Sok et al., 2014; Walker et al., 2011). The resolution of this new model is similar to the resolutions of the two published X-ray structures of deglycosylated Env trimers in complex with different antibodies, at 4.7 Å and 3.5 Å resolution (Julien et al., 2013a; Pancera et al., 2014). Combining our high-resolution EM map and published X-ray data (Pancera et al., 2014; Pejchal et al., 2011) enabled us to build an atomic model of the HIV Env trimer, including many ordered glycans. This breakthrough enables resolution of the complete epitope of the bnAb PGT128 and provides a path forward for analyzing other bnAbs with complex, glycan-containing epitopes.
Results
CryoEM of BG505 SOSIP.664 Env trimer in complex with PGT128 Fab
Two datasets of the BG505 SOSIP.664-PGT128 Fab complex were collected, and initially independently processed (Figure S1). The reconstruction of the first dataset was resolved to 4.67 Å, while the second data set was resolved to 5.01 Å at a Fourier shell correlation (FSC) cut off of 0.143, both using the same gold standard refinement procedure (Figure S1E), with images recorded at similar dose and defocus range for both datasets (Table S1). Combining the two datasets improved the resolution to 4.47 Å (Figure S1E and Table S1). Throughout the 3D classification procedure and in the final refined models, it was apparent that the constant region of the Fab was highly flexible, and the greatest degree of variability among the 3D classes was in this region (Figure S1B). In order to improve resolution further, the model from the final refinement iteration was subjected to additional rounds of local refinement after imposing a mask shaped like the molecule but excluding the Fab constant region from particle alignment (Figure S1D). This resulted in a slight improvement in the resolution to 4.36 Å (Figures 1A, S1E and Table S1). Local resolution analysis of this reconstruction shows that the resolution of the molecule is mainly isotropic (Figure 1B). As expected, the Fab constant region exhibited the lowest resolution, ranging between ∼5-10 Å (Figure 1B).
Figure 1. CryoEM reconstruction of BG505 SOSIP.664 in complex with PGT128 Fab.
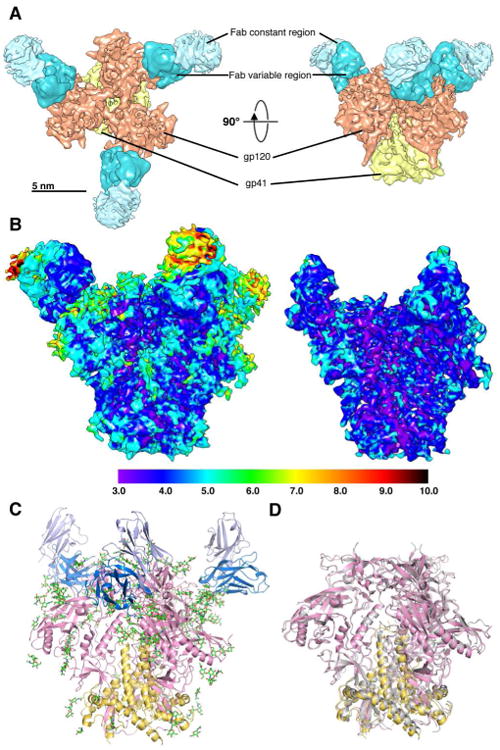
(A) Top (left) and side (right) views of the cryoEM reconstruction is segmented into gp120, gp41 in the trimer, and variable and constant regions in the Fab.
(B) Local resolution analysis of the 4.36 Å resolution reconstruction. Local resolution in Å is indicated by colors shown in the key at the bottom. The reconstruction shown at a higher threshold level (right) shows that the resolution is largely isotropic, especially at the core of the trimer.
(C) The refined PGT128 bound trimer model. gp120 and gp41 are shown in pink and yellow, respectively, and glycans are shown as green sticks. Only the Fab variable region was built. The heavy chain (HC) is shown in blue, and light chain (LC) in lavender. (D) Superposition of the PGT128 bound trimer (pink and yellow) with the PGT121 and 35O22 bound trimer solved by X-ray crystallography (gray) (Pancera et al., 2014).
Density map analysis and model building
The α-helical regions in both gp120 and gp41 were well resolved, including the helical pitch, with many side chains visible, particularly on internal helices (Figures 2A and S2A). Individual strands of β-sheets were also well resolved (Figures 2B and S2B). Docked coordinates of the 3.5 Å resolution X-ray structure of the SOSIP trimer (PDB ID: 4TVP) (Pancera et al., 2014), and the Fab variable region of the PGT128 Fab (PDB ID: 3TYG) (Pejchal et al., 2011) were used as the starting model for atomic model building and refinement into the cryoEM map, as the X-ray structures contained protein regions that was of the same construct as the used in the cryoEM complex, and each piece showed a good overall fit into the density. The majority of the structure did not have to be modified, with the exception of a change in V1, which is in a slightly different conformation relative to the conformation in the PGT122 bound state. The few inconsistencies were remodeled in Coot (Emsley and Cowtan, 2004), and then refined in RosettaRelax (DiMaio et al., 2015; DiMaio et al., 2009) (Figure S2C).
Figure 2. Analysis of high-resolution features and model validation.
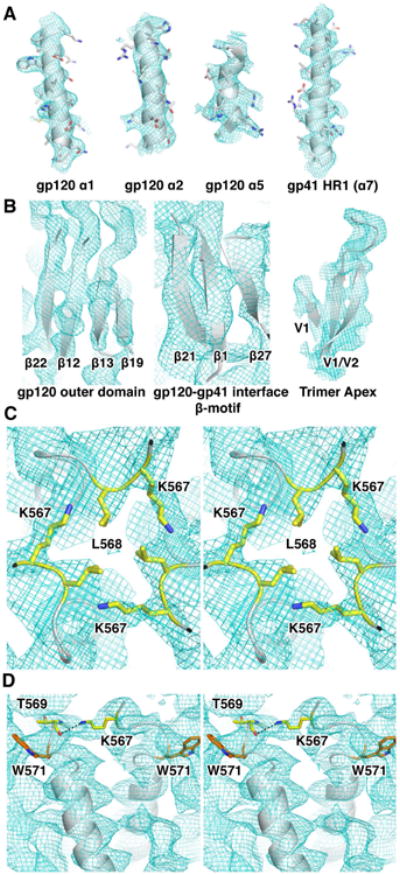
(A) Densities of some of the prominent α-helices in the Env trimer.
(B) Densities of some of the prominent β-sheets in the Env trimer.
(C) Stereo view of the N-terminal end of the α7 helix of gp41 following the unresolved FPPR region. L568 just prior to the start of the a7 helix of HR1 forms a hydrophobic interaction at the start of the trimeric coiled-coil at the center of the trimer, and K567 makes interactions with the adjacent HR1 helix. The two residues are shown in yellow sticks
(D) A stereo image of a different view of the K567 inter-HR1 interaction. The K567 side chain nitrogen could potentially interact with the backbone carbonyl oxygen of T569 (residues shown as yellow sticks, and interaction shown by the dashed line). We can be confident in the relative conformation due to the clear side chain density of W571 near by anchoring the peptide registration (orange).
A superimposition of the refined trimer structure to the initial model results in a Cα RMSD of 1.44 Å (Figure 1C), demonstrating that the majority of the structure is highly similar, in accordance with the low resolution EM observation that PGT128 does not cause a global conformational change in the trimer upon binding (Pejchal et al., 2011). Outside of the PGT128 epitope, two regions were structurally distinct from the crystal structure of the BG505 SOSIP trimer (PDB: 4TVP), the α0 helix of gp120 (residues 59-63) and the C-terminus of gp41. In the 4TVP X-ray structure (Pancera et al., 2014), the segment corresponding to residues 59-64 is modeled as a short α-helix, followed by a loop region in residues 64-74. This secondary structure configuration is reversed in the 4NCO X-ray structure (Julien et al., 2013a). In the cryoEM reconstruction, the density in both regions appears too disordered to be helical, and neither of the two structures fit the density corresponding to residues 59-67 well (Figure 3A). Helices are the first distinctive secondary structural elements seen in cryoEM maps once the resolution reaches <10 Å. Examination of α4, a helix of 6 residues (Figure 3B), suggests that this lack of helical definition is not due to limitations in resolving a short helix by cryoEM. Another indication of disorder in this region is the fact that the side chain density of Y61 is not visible, despite the majority of aromatic residue side chains being resolved in the EM map. Because we do not observe clear density in the cryoEM map between residues A58 to W68 we conclude that this region is natively disordered and removed this region from our model. This region may, however, become ordered upon binding to the receptor CD4, as suggested by the CD4 bound gp120 monomer structure (Pancera et al., 2010).
Figure 3. Regions of the map with missing density and the FPPR region.
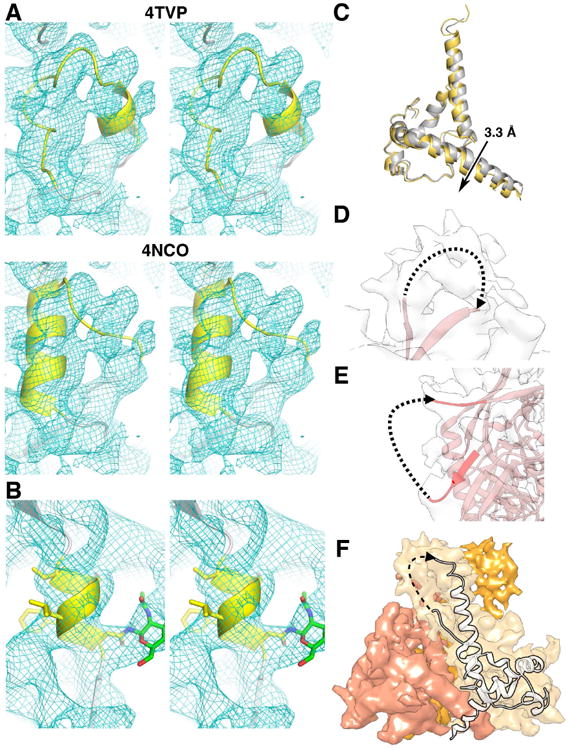
(A) Stereo view of the α0 region (yellow) of published trimer structures, 4TVP (top), 4NCO (bottom), and our model (blue mesh). The density of this region in our map does not fit neither 4NCO nor 4TVP well.
(B) The α4 helix of gp120 (residues T386-N392, yellow) is well resolved despite it being a small helix. The N-GlcNAc at N392 is shown in green.
(C) A close-up of the C-terminal region of HR2. Unlike HR1, there is a slight downward shift of ∼3.3 Å (measured between Q653 Cα) in the HR2 region of the PGT128 bound trimer (yellow) in comparison to the X-ray structure (gray).
(D) The density for the entire V2 loop is visible, although the lack of confidence in side-chain densities makes it difficult to build. The dotted line indicates the loop path of the 10 missing residues (E185a-S187).
(E) Density for the V4 loop is unresolved. The end and start of the V4 loop in the atomic model is shown in red, and the predicted path of missing regions (residues T399-S410) is shown by the dotted line.
(F) Three gp41 protomer densities are shown as shades of orange. As in (D), the entire FPPR density was visible but could not be built unambiguously. Residues Q551-L565 are missing from this atomic model.
The second structural difference between the cryoEM and X-ray models was localized to the C-terminal region of heptad repeat (HR) 2 of gp41. There is a small downward shift of ∼3 Å in HR2 helix of the cryoEM model compared to the 35O22 bound structure (Figure 3C), along with reduced helical integrity. Three possible explanations can be given for this observation. This region may be flexible as suggested by hydrogen-deuterium exchange mass spectrometry (HDXMS), showing rapid deuterium exchange in residues 648-664 of HR2 (Guttman et al., 2014). Secondly, 35O22, the antibody bound in the crystal structure, is a gp120-gp41 interface-binding antibody, and makes contact with the region surrounding N625 of gp41(Huang et al., 2014), which may cause a slight conformational shift. Lastly, and most likely, there are crystal-packing interactions between the constant domain of PGT122 Fab and the C-terminus of HR2 in 4TVP. Small differences in this region were also observed between the 5.8 Å EM structure (Lyumkis et al., 2013), and the 4.7 Å X-ray structure (Julien et al., 2013a). Although the latter two structures lack a full atomic model for gp41, the models provide additional support that the HR2 helix can shift slightly due to crystal contacts.
The gp120 regions that were unresolved in published X-ray structures of BG505 SOSIP.664 were similarly not resolved here. While density was visible in the V2 loop region, lack of definition in side chain densities made the Cα trace ambiguous (Figure 3D). The density corresponding to the V4 loop is almost entirely missing, as it is in the majority of gp120 structures (Figure 3E). These regions are consistently disordered in multiple X-ray and EM structures (Do Kwon et al., 2015; Julien et al., 2013a; Lyumkis et al., 2013; Pancera et al., 2010), and indicate that these regions are intrinsically disordered in the Env trimer.
The core of gp41 was one of the best-resolved regions of the trimer, and exhibited the highest local resolution in the overall structure (Figure 1B). The density corresponding to the fusion peptide proximal region (FPPR) was resolved as a continuous density (Figure 3F), but not ordered enough to build an atomic model. A few more residues (548-550 and 566-568) than what is visible in the 3.5 Å X-ray model could be built confidently from the visible side chain density. From this, new interactions potentially stabilizing the trimeric HR1 coiled-coil were resolved. Specifically L568 from the three protomers have hydrophobic interactions at the three-fold axis (Figure 2C), and there is also a potential hydrogen bonding interaction between K567 and the backbone carbonyl of T569 (Figure 2D).
Building and refinement of glycans
In X-ray crystallography, mercury labeling is often used as a means to confirm the Cα backbone trace of low-resolution structures. While N-linked glycans are flexible and often difficult to resolve fully, their occurrence is easy to predict (glycosylated at NXT/S), and the core N-GlcNAc is almost always visible as a small protrusion of density in EM and X-ray maps, even if the resolution is not high enough to resolve protein side chains or individual glycan residues. In this regard, N-GlcNAc densities can be utilized as a marker for confirming side chain registration in model building, allowing the model to be validated by glycan densities at potential N-glycosylation (PNG) sites, as it had been done for the previously published BG505 SOSIP.664 structure determined by cryoEM (Lyumkis et al., 2013). Indeed, in the current EM map, density corresponding to at least the first N-GlcNAc is visible at all PNG sites except for glycans in disordered loop regions, and N137 and N625 (Figures S3A and S3B). The N262 glycan was the only glycan not directly interacting with the Fab that had multiple visible branches due to its extensive contacts with gp120 (Figure 4A). A recent X-ray structure of a fully glycosylated gp120 core shows a large Man7 glycan at this position with a fully extended D1 arm (Kong et al., 2015). We also see large amounts of density for the D1 arm although the signal was not strong enough for us to build the entire D1 branch. This density extends toward the base of N301, and appears that the last D1 arm glycan residue would be able to interact with the core N-GlcNAc, and the D2 branch of N301 (Figure 4B). It has been previously noted that while glycosylation at N301 is not an absolute requirement for viral infectivity nor Env expression and folding, the deletion of this glycan renders the trimer more susceptible to neutralization by V3 loop binding antibodies (Binley et al., 2010). A similar but less pronounced effect was seen when all glycans were produced in high-mannose form (Binley et al., 2010). Because the N301 glycan is predicted to be complex (Cutalo et al., 2004; Leonard et al., 1990; Zhu et al., 2000), the authors concluded that the modification of the N301 glycan was linked to an increased V3 loop exposure (Binley et al., 2010). In this regard, N301 deletion induced V3 exposure may be due to the loss in glycan-glycan interaction between N262 and N301, which helps restrain the V3 loop in the closed configuration. While we have built the N301 glycan as high mannose due to the fact that we are using a published glycan model for N301, the D2 arm mannose residue is also likely to be a GlcNAc in complex glycans (Stanley, 2009). Thus it is possible that the N262-N301 glycan interactions are stronger when N301 is complex and has a GlcNAc at this position.
Figure 4. Glycan interactions resolved in the cryoEM structure.
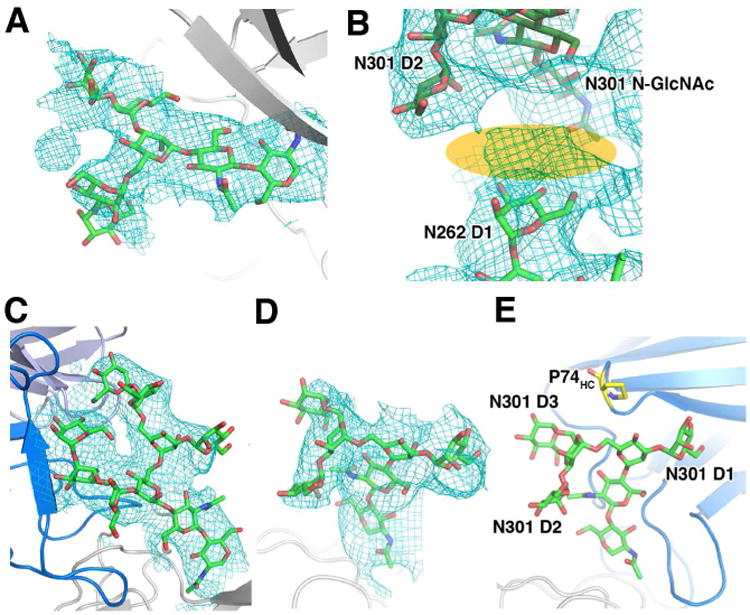
(A) Clear density for a Man5 glycan at N262 can be seen in the EM map.
(B) While the last glycan in the D1 am has not been built, we see a small amount of density in this region (orange) connecting the N262 D1 arm glycan to the base and D2 arm of the N301 glycan.
(C) The N332 glycan from 3TYG structure fits tightly into the EM density. In blue and lavender are PGT128 Fab HC and LC, respectively.
(D) The N301 glycan and its density.
(E) The D3 arm of N301 does not interact with P74 (yellow) in PGT128 (HC shown inblue) as was suggested by the X-ray structure.
Although most glycans were flexible and not visible beyond the first two GlcNAc residues, glycans interacting with PGT128 were well resolved, including the multiple branches of the glycans at N332 and N301. Rigid body docking of the glycan coordinates obtained from the PGT128 bound engineered mini gp120 outer domain (gp120 eOD) structure (PDB ID: 3TYG) localizes the glycans into the cryoEM density (Figures 4C and 4D), especially the Man8 N332 glycan (Figure 4C), which fits well into the EM map. We initially docked the N301 Man5 glycan into the EM density, which fit well overall, although the D3 arm torsions needed to be modified to fit into the density (Figures 4D and S3C). This D3 arm density seems to be slightly shifted relative to the X-ray coordinates, and the hydrogen bonding interaction that was proposed between the mannose O6 and the backbone carbonyl of P74 in frame work region heavy (FWRH) 3 of PGT128 (Pejchal et al., 2011), is lost (Figure 4E). Analysis of this linkage indicates that in the original structure, the torsion angle between this glycan and the previous mannose is highly unlikely, suggesting that perhaps the interaction seen is an artifact of the crystal structure (Figure S3C). Another possibility is that N301 in the native expression system is a complex glycan as discussed above, thus unlike the X-ray structure, the residues beyond the core pentasaccharide may not be mannoses and the branches adopt a slightly different structural configuration.
Defining the full PGT128 epitope
Many HIV-1 bnAbs have previously been solved in complex with either gp120 or peptide fragments that comprise a part of its epitope, due to difficulties in crystallizing the entire trimer. PGT128 binds the gp120 eOD with an ∼8-fold decrease in affinity relative to the gp120 core with the full V3 (Pejchal et al., 2011), likely due to lost interactions in the minimized epitope. Indeed, it has been shown that the extent of HIV-1 binding bnAb epitopes is often underestimated, as demonstrated by another N301 and N332 glycan dependent antibody, PGT122 (Julien et al., 2013c), which was confirmed to have a more expansive epitope involving V1/V2 glycans (Julien et al., 2013a). Other studies showed similar results for different antibodies that target the N332 site (Garces et al., 2014; Sok et al., 2014), and suggested that breadth of neutralization is derived from the ability of the bnAb to interact with other nearby glycans in a promiscuous manner when preferred glycans are not present (Sok et al., 2014). PGT128 and its clonal relatives were shown to have absolute dependence on the N301 glycan, but partial dependence on either N295 or N332. Both N295 and N332 must be knocked out simultaneously in order to eliminate the ability of the antibody to neutralize the virus (Sok et al., 2014). A model demonstrating how the N295 glycan would interact in place of the N332 glycan was previously calculated via computational methods (Sok et al., 2014). A comparison of this structure with our EM based model indicates that N295 glycan would have to move ∼60 degrees in order to interact with the PGT128 Fab in place of N332 (Figure 5A).
Figure 5. The PGT128 epitope in the context of the full trimer.
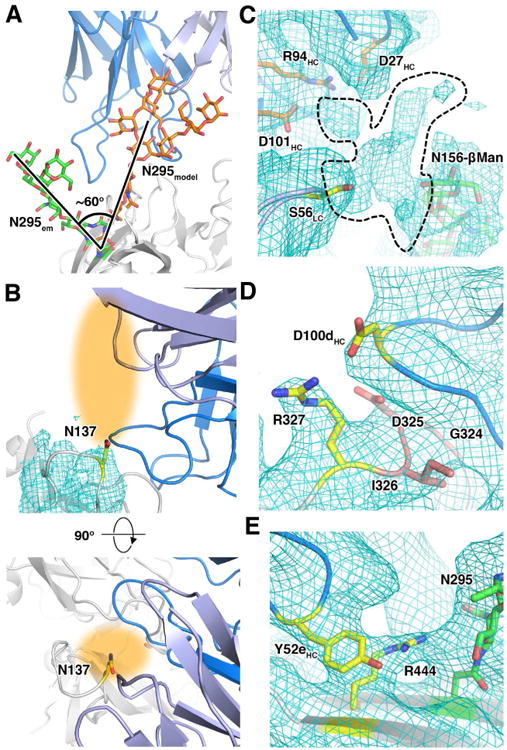
(A) The N295 glycan (green), which can act as a compensatory glycan when the N332 glycan is missing, must move ∼60 degrees (orange) in order to compensate for the loss of N332.
(B) Even though the glycan density cannot be seen, the position of N137 (yellow) indicates that a glycan projecting from this site (orange shading) could sterically affect PGT128 binding. The lower image is an orthogonal view of the upper image to show how the glycan may project out towards the Fab.
(C) Additional density branching off from N156 (green) can be seen, indicating the path of the higher-branch glycan residues (dotted line). Residues from both the HC (orange) and LC (yellow) of PGT128 may be contributing to interaction with atoms in this density.
(D) R327 in the GDIR motif (red) seems to be able to make electrostatic interactions with D100d in CDRH3, as opposed to the backbone interactions indicated in the X-ray structure. The two interacting residues are shown as yellow sticks.
(E) The density indicates an interaction between Y52e of CDRH2 and the hydrocarbon portion of R444 side chain in C5. An interaction between R444 and the base of N295 glycan (green) may also be present.
It was also shown that glycans at the trimer apex near the PGT128 binding site have minor effects on PGT128 binding. When the N137 glycan was knocked out in a BG505 virus lacking either the N332 or N334 glycan (the latter often being a compensatory glycosylation site when N332 is missing), virus neutralization by PGT128 was increased, while removal of the N156 glycan resulted in a slight reduction in neutralization potency (Sok et al., 2014). Because the gp120 eOD in the X-ray structure is missing most of the V1/V2 region including N137 and N156, which is present in our trimer construct, we are now able to examine potential interactions in this region more completely.
In the cryoEM map, there is very little density for N137 beyond the Asn side chain, suggesting that there is no specific interaction between N137 and the PGT128 Fab (Figures 5B and S3A). Still, in this V1 loop conformation, the N137 residue projects towards the outer face of PGT128 Fab light chain (LC) (Figure 5B). The proximity of N137 to the Fab LC suggests that a glycan in this position would sterically hinder PGT128 binding, corroborating the previous knockout study (Sok et al., 2014). On the contrary, density was visible for several sugar moieties of the N156 glycan at low map contour levels. Examination of this density suggests a possible interaction between S56 in the light chain complementarity determining region (CDRL) 2 of PGT128, and the D1 arm mannose of the N156 glycan (Figure 5C). Although density beyond the core trisaccharide was poorly defined, there are densities branching from the N156 glycan density that could indicate potential interactions between D27, R94, and D101 of the Fab heavy chain (HC) (Figure 5C).
In addition to new glycan contacts, we also observed protein-protein contacts not contained in the gp120 eOD bound structure (Pejchal et al., 2011). The GDIR motif is a highly conserved region in the gp120 V3 loop (residues G324-R327), and it has been predicted that this region is one of the most important peptide segments for binding of antibodies in the PGT128-like families (Garces et al., 2014). A comparison between the gp120 eOD-PGT128 Fab complex and gp120 monomer-PGT124 (another N332 dependent antibody) complex structures demonstrated that PGT124 interacts with the GDIR motif in a mostly side chain dependent manner, while PGT128 interacts with the backbone of the GDIR segment. Interestingly, the cryoEM density reveals a potential side chain interaction between D100d of PGT128 heavy chain complementarity determining region (CDRH) 3 and R327 of gp120 (Figure 5D). Previously published data showed only a small change in both neutralization IC50 and ELISA EC50 resulting from a D100dA mutation (Pejchal et al., 2011), indicating that this interaction may occur but not play a large role in epitope recognition. Another possible interaction was detected in β22 of gp120, which is engineered as a loop in the gp120 eOD, in which Y52e in CDRH2 packs against Cβ-Cδ of R444 (Figure 5E). Unlike the D100dA mutation, the Y52eA mutation resulted in a 21.1 and 12.9-fold increase in neutralization IC50 (for JR-FL and BaL viral isolates, respectively) relative to the wild type antibody (Pejchal et al., 2011). These additional interactions stress the importance of analyzing antibody-antigen contacts in the context of the full epitope, and the ability to resolve such interactions using cryoEM.
Discussion
Here we show that a 4.36 Å resolution cryoEM map, combined with refinement in Rosetta, enables model building of protein complexes with near atomic resolution details (DiMaio et al., 2015). PTMs, particularly glycans can serve as markers that help guide building and confirm registration of the amino acid sequence. The core N-GlcNAcs are almost always visible in the density map, while the higher branching is often too flexible to be resolved, unless constrained in some way. While similar to X-ray crystallography in this regard, the ability to express glycoproteins with native glycans, and not remove them prior to structural analysis is advantageous. Thus, with current EM and detector technology, there will be an increasing number of biological complexes of interest that require hybrid modeling and refinement, especially for glycans.
We have now shown that cryoEM is capable of generating rapid structural information for iterative, structure-based vaccine design (Correia et al., 2014; Jardine et al., 2013; McGuire et al., 2013; McLellan et al., 2013) as is being pursued for HIV-1. In X-ray crystallography, crystallization is highly dependent on stable protein-protein crystal contacts, and in fact there are only a few known combinations of trimer-Fab complexes that result in sufficiently well-ordered crystals that diffract to a reasonable resolution. CryoEM circumvents this problem and glycan and epitope binding analysis can be routinely achieved for any trimer-Fab complex. Even more promising, there is still room for improvement in resolution using newer refinement techniques, such as dose fractionation (Bartesaghi et al., 2014), and particle polishing (Scheres, 2014), which will better correct for imaging artifacts such as beam induced movement (Brilot et al., 2012; Campbell et al., 2012), and the loss of side chain resolution due to high-electron dose (Bartesaghi et al., 2014). Finally, we show that protein engineering, particularly deglycosylation, may result in structural information that does not fully recapitulate native interactions, as we have proposed here with possible complex glycan dependent interactions at N156, and the role of N301 for V3 stability. Studies of HIV-1 antibody lineages have shown that bnAbs undergo a dynamic shift in interaction with glycans proximal to the epitope during maturation. For example, N137 acts as an inhibitory glycan for early precursors of the PGT121 family of antibodies, whereas the mature antibody can utilize this glycan in the absence of N332 for neutralization (Garces et al., 2014). Similarly, the germline antibody of VRC01 does not bind gp120 that is glycosylated at N276 (Jardine et al., 2013; McGuire et al., 2013). Thus in order to guide the maturation of HIV bnAbs that require high levels of somatic mutation, it is crucial that we recognize all of the epitope contacts so we can develop the proper vaccine prime and boost immunogens that mimic the native Env trimer. CryoEM is now situated to make significant contributions in this area, complementing X-ray crystallographic studies that are often limited due to the nature of the technique.
Experimental Procedures
Complex formation and sample preparation
The purified BG505 SOSIP.664 trimers were mixed with PGT128 Fab using a 10-fold molar excess of the Fab. The mixture was incubated for 1 h at room temperature. This trimer-Fab complex was purified by size exclusion chromatography using a Superose 6 column (GE Healthcare) in 50 mM Tris pH 7.4, 150 mM NaCI. The fractions containing the complex were pooled and concentrated using a 100-kDa concentrator (Amicon Ultra, Millipore) to 2.5 mg/mL. 5 μL of the complex was incubated with 3 μL of fresh DDM solution at 1.8 mM, to prevent aggregation of the complex. At 4°C, 3 μL of this mixture was applied to a CF-2/2-4C C-Flat grid (Electron Microscopy Sciences, Protochips, Inc.) which had been plasma cleaned for 5 seconds using a mixture of Ar/O2 (Gatan Solarus 950 Plasma system), blotted off, and then immediately plunged into liquid ethane using a manual freeze plunger.
Data collection
Micrographs were collected on a FEI Titan Krios operating at 300 KeV coupled with Gatan K2 direct electron detector via the Leginon interface (Suloway et al., 2005). Each exposure image was collected at 22,500× mag resulting in a pixel size of 1.31 Å/pixel in the counting mode, using a dose rate of ∼10 e-/pix/sec, and 200 ms exposure per frame. The entire data were collected in 2 separate data collection sessions, resulting in a total of 2,111 micrographs collected in ∼85 hours. The total dose received for the first data collection accounting for 64% of the total collected micrographs was 32.88 e-/Å2, and 35.07 e-/Å2 the second data collection. The nominal defoci range used for both data sets were -1.50 to -3.50 μm (Table S1).
Data processing
All of the collected frames were aligned prior to processing (Li et al., 2013). CTF estimation was carried out using CTFFind3 (Mindell and Grigorieff, 2003), and particles were picked using an automated particle-picking program implemented in the Appion software package (Voss et al., 2009). Particles were stacked using a box size of 256×256 pixels at 1.31 Å/pix in EMAN boxer.py via Appion (Lander et al., 2009). Two rounds of reference-free 2D classification were carried out using MSA/MRA (Ogura et al., 2003) using a binning factor of 4, to remove amorphous particles that were clearly not the protein complex of interest. Particles in ambiguous classes were left in for sorting by 3D classification. To speed up data processing the two data sets were initially processed independently, using the same methods. After 2D sorting, the 4× binned particles were subject to 3D classification using RELION 1.3 (Scheres, 2012a, b), starting with an initial reference model of an unliganded trimer filtered to 60 Å resolution without imposing symmetry, and requesting 4 classes. Particles belonging to 3 fold symmetric, PGT128 bound models were then extracted, then were put through a second round of 3D classification. Classes with the best trimer density were selected for final, unbinned refinement. Refinements were carried out in RELION 1.3 with C3 symmetry imposed, independently for the 2 data sets and also with the two data sets combined (Table S1) (Scheres, 2012a, b; Scheres and Chen, 2012). Post-refinement, a shape-fitting mask excluding the Fab constant domain was generated. The final iteration of the previous refinement was then allowed to refine until convergence (3 additional iterations) with the mask applied. The final resolution was 4.36 Å at an FSC cutoff of 0.143. The FSC was calculated using a soft-edged mask with a Gaussian falloff, encompassing the entire structure, including the Fab constant regions, for all models.
Model building and refinement
The initial atomic model was generated by fitting the gp140 portion of the BG505 SOSIP.664 X-ray structure (4TVP) (Pancera et al., 2014) and the Fab variable region of gp120 eOD bound PGT128 Fab structure (3TYG) (Pejchal et al., 2011) into the 4.36 Å resolution EM structure. This model is the equivalent of a template model for phasing in X-ray crystal structure determination by molecular replacement. For this step, all glycans were temporarily removed, since RosettaRelax does not handle glycans. Building and refinement were carried out iteratively in Coot (Emsley and Cowtan, 2004) and RosettaRelax, a computational modeling program that uses cyroEM density as a constraint when refining a low resolution model by energy relaxation (DiMaio et al., 2015; DiMaio et al., 2009). After completion of the protein refinement, the glycans were modeled back into the structure. Large glycans were grafted from previously published PDB structures then edited (N301 and N332: 3TYG (Pejchal et al., 2011), N262: 4RQS (Kong et al., 2015)). All glycan torsions were manually adjusted and checked via pdb-care (Lutteke and von der Lieth, 2004). Few residues in the protein that Rosetta failed to place into the side chain densities were modified manually via rotamer flips in Coot. Model analysis and figure making were carried out in Coot, PyMOL and UCSF Chimera.
Supplementary Material
Highlights.
Natively glycosylated HIV-1 Env trimer solved to 4.36 Å resolution by cryoEM.
Complete description of the epitope of broadly neutralizing HIV-1 antibody PGT128.
Multi-branched oligomannose glycans can be visualized and modeled by cryoEM.
Acknowledgments
We thank A. Cupo and J.P. Moore for providing the BG505 SOSIP.664 trimers, J.-P. Julien for providing the PGT128 Fabs, T. Nieusma for technical assistance, J.-C. Ducom for help with computing cluster maintenance, and D. Sok, D. Culp, and S. Menis for sharing the computational model shown in Figure 4A. This work was supported by the California HIV/AIDS Research Program Dissertation Award D12-SRI-353 (to J.H.L), the Leona M. and Harry B. Helmsley Charitable Trust grant #2012-PG-MED002 (to D.L.), the Scripps CHAVI-ID (UM1 AI100663), P01 AI82362, International AIDS Vaccine Initiative, and the Bill and Melinda Gates Foundation (CAVD). This is manuscript #29088 from the Scripps Research Institute.
Footnotes
Accession Numbers: The EM reconstructions of BG505 SOSIP.664: PGT128 Fab complex at 4.47 Å and 4.36 Å resolution were deposited to the Electron Microscopy Data Bank under accession codes EMD-3120, and EMD-3121, respectively. The atomic model has been deposited to the PDB under accession code 5ACO.
Author Contributions: J.H.L, N.V., D.L., and A.B.W designed the research. J.H.L. and N.V. collected data. J.H.L. performed refinement and modeling. J.H.L. and A.B.W. analyzed the data and wrote the manuscript. All authors commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartesaghi A, Matthies D, Banerjee S, Merk A, Subramaniam S. Structure of beta-galactosidase at 3.2-A resolution obtained by cryo-electron microscopy. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:11709–11714. doi: 10.1073/pnas.1402809111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binley JM, Ban YE, Crooks ET, Eggink D, Osawa K, Schief WR, Sanders RW. Role of complex carbohydrates in human immunodeficiency virus type 1 infection and resistance to antibody neutralization. Journal of virology. 2010;84:5637–5655. doi: 10.1128/JVI.00105-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binley JM, Sanders RW, Clas B, Schuelke N, Master A, Guo Y, Kajumo F, Anselma DJ, Maddon PJ, Olson WC, et al. A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. Journal of virology. 2000;74:627–643. doi: 10.1128/jvi.74.2.627-643.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brilot AF, Chen JZ, Cheng A, Pan J, Harrison SC, Potter CS, Carragher B, Henderson R, Grigorieff N. Beam-induced motion of vitrified specimen on holey carbon film. Journal of structural biology. 2012;177:630–637. doi: 10.1016/j.jsb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MG, Cheng A, Brilot AF, Moeller A, Lyumkis D, Veesler D, Pan J, Harrison SC, Potter CS, Carragher B, et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 2012;20:1823–1828. doi: 10.1016/j.str.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia BE, Bates JT, Loomis RJ, Baneyx G, Carrico C, Jardine JG, Rupert P, Correnti C, Kalyuzhniy O, Vittal V, et al. Proof of principle for epitope-focused vaccine design. Nature. 2014;507:201–206. doi: 10.1038/nature12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutalo JM, Deterding LJ, Tomer KB. Characterization of glycopeptides from HIV-I(SF2) gp120 by liquid chromatography mass spectrometry. Journal of the American Society for Mass Spectrometry. 2004;15:1545–1555. doi: 10.1016/j.jasms.2004.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depetris RS, Julien JP, Khayat R, Lee JH, Pejchal R, Katpally U, Cocco N, Kachare M, Massi E, David KB, et al. Partial enzymatic deglycosylation preserves the structure of cleaved recombinant HIV-1 envelope glycoprotein trimers. The Journal of biological chemistry. 2012;287:24239–24254. doi: 10.1074/jbc.M112.371898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio F, Song Y, Li X, Brunner MJ, Xu C, Conticello V, Egelman E, Marlovits TC, Cheng Y, Baker D. Atomic-accuracy models from 4.5-A cryo-electron microscopy data with density-guided iterative local refinement. Nature methods. 2015;12:361–365. doi: 10.1038/nmeth.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio F, Tyka MD, Baker ML, Chiu W, Baker D. Refinement of protein structures into low-resolution density maps using rosetta. Journal of molecular biology. 2009;392:181–190. doi: 10.1016/j.jmb.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Kwon Y, Pancera M, Acharya P, Georgiev IS, Crooks ET, Gorman J, Joyce MG, Guttman M, Ma X, Narpala S, et al. Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nature structural & molecular biology. 2015;22:522–531. doi: 10.1038/nsmb.3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Garces F, Sok D, Kong L, McBride R, Kim HJ, Saye-Francisco KF, Julien JP, Hua Y, Cupo A, Moore JP, et al. Structural evolution of glycan recognition by a family of potent HIV antibodies. Cell. 2014;159:69–79. doi: 10.1016/j.cell.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Garcia NK, Cupo A, Matsui T, Julien JP, Sanders RW, Wilson IA, Moore JP, Lee KK. CD4-induced activation in a soluble HIV-1 Env trimer. Structure. 2014;22:974–984. doi: 10.1016/j.str.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kang BH, Pancera M, Lee JH, Tong T, Feng Y, Imamichi H, Georgiev IS, Chuang GY, Druz A, et al. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature. 2014;515:138–142. doi: 10.1038/nature13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine J, Julien JP, Menis S, Ota T, Kalyuzhniy O, McGuire A, Sok D, Huang PS, MacPherson S, Jones M, et al. Rational HIV immunogen design to target specific germline B cell receptors. Science. 2013;340:711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013a;342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien JP, Lee JH, Cupo A, Murin CD, Derking R, Hoffenberg S, Caulfield MJ, King CR, Marozsan AJ, Klasse PJ, et al. Asymmetric recognition of the HIV-1 trimer by broadly neutralizing antibody PG9. Proceedings of the National Academy of Sciences of the United States of America. 2013b;110:4351–4356. doi: 10.1073/pnas.1217537110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien JP, Sok D, Khayat R, Lee JH, Doores KJ, Walker LM, Ramos A, Diwanji DC, Pejchal R, Cupo A, et al. Broadly neutralizing antibody PGT121 allosterically modulates CD4 binding via recognition of the HIV-1 gp120 V3 base and multiple surrounding glycans. PLoS pathogens. 2013c;9:e1003342. doi: 10.1371/journal.ppat.1003342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khayat R, Lee JH, Julien JP, Cupo A, Klasse PJ, Sanders RW, Moore JP, Wilson IA, Ward AB. Structural characterization of cleaved, soluble HIV-1 envelope glycoprotein trimers. Journal of virology. 2013;87:9865–9872. doi: 10.1128/JVI.01222-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Lee JH, Doores KJ, Murin CD, Julien JP, McBride R, Liu Y, Marozsan A, Cupo A, Klasse PJ, et al. Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nature structural & molecular biology. 2013;20:796–803. doi: 10.1038/nsmb.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Wilson IA, Kwong PD. Crystal structure of a fully glycosylated HIV-1 gp120 core reveals a stabilizing role for the glycan at Asn262. Proteins. 2015;83:590–596. doi: 10.1002/prot.24747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of structural biology. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454:177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard CK, Spellman MW, Riddle L, Harris RJ, Thomas JN, Gregory TJ. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. The Journal of biological chemistry. 1990;265:10373–10382. [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nature methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutteke T, von der Lieth CW. pdb-care (PDB carbohydrate residue check): a program to support annotation of complex carbohydrate structures in PDB files. BMC bioinformatics. 2004;5:69. doi: 10.1186/1471-2105-5-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyumkis D, Julien JP, de Val N, Cupo A, Potter CS, Klasse PJ, Burton DR, Sanders RW, Moore JP, Carragher B, et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1484–1490. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire AT, Hoot S, Dreyer AM, Lippy A, Stuart A, Cohen KW, Jardine J, Menis S, Scheid JF, West AP, et al. Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4 binding site antibodies. The Journal of experimental medicine. 2013;210:655–663. doi: 10.1084/jem.20122824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GB, Yang Y, Zhang B, Chen L, Srivatsan S, Zheng A, et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science. 2013;342:592–598. doi: 10.1126/science.1243283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLellan JS, Pancera M, Carrico C, Gorman J, Julien JP, Khayat R, Louder R, Pejchal R, Sastry M, Dai K, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480:336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. Journal of structural biology. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Ogura T, Iwasaki K, Sato C. Topology representing network enables highly accurate classification of protein images taken by cryo electron-microscope without masking. Journal of structural biology. 2003;143:185–200. doi: 10.1016/j.jsb.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1166–1171. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature. 2014;514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejchal R, Doores KJ, Walker LM, Khayat R, Huang PS, Wang SK, Stanfield RL, Julien JP, Ramos A, Crispin M, et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science. 2011;334:1097–1103. doi: 10.1126/science.1213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RW, Vesanen M, Schuelke N, Master A, Schiffner L, Kalyanaraman R, Paluch M, Berkhout B, Maddon PJ, Olson WC, et al. Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. Journal of virology. 2002;76:8875–8889. doi: 10.1128/JVI.76.17.8875-8889.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. A Bayesian view on cryo-EM structure determination. Journal of molecular biology. 2012a;415:406–418. doi: 10.1016/j.jmb.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. Journal of structural biology. 2012b;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife. 2014;3:e03665. doi: 10.7554/eLife.03665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nature methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sok D, Doores KJ, Briney B, Le KM, Saye-Francisco KL, Ramos A, Kulp DW, Julien JP, Menis S, Wickramasinghe L, et al. Promiscuous glycan site recognition by antibodies to the high-mannose patch of gp120 broadens neutralization of HIV. Science translational medicine. 2014;6:236ra263. doi: 10.1126/scitranslmed.3008104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley PSHTN. N-Glycans. 2nd. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. Journal of structural biology. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. Journal of structural biology. 2009;166:205–213. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477:466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Borchers C, Bienstock RJ, Tomer KB. Mass spectrometric characterization of the glycosylation pattern of HIV-gp120 expressed in CHO cells. Biochemistry. 2000;39:11194–11204. doi: 10.1021/bi000432m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.