

Abstract
Mexiletine is a sodium channel blocker, primarily used in the treatment of ventricular arrhythmias. Moreover, recent studies have demonstrated its therapeutic value to treat myotonic syndromes and to relieve neuropathic pain. The present study aims at investigating the direct blockade of hERG potassium channel by mexiletine and its metabolite m-hydroxymexiletine (MHM). Our data show that mexiletine inhibits hERG in a time- and voltage-dependent manner, with an IC50 of 3.7 ± 0.7 μmol/L. Analysis of the initial onset of current inhibition during a depolarizing test pulse indicates mexiletine binds preferentially to the open state of the hERG channel. Looking for a possible mexiletine alternative, we show that m-hydroxymexiletine (MHM), a minor mexiletine metabolite recently reported to be as active as the parent compound in an arrhythmia animal model, is a weaker hERG channel blocker, compared to mexiletine (IC50 = 22.4 ± 1.2 μmol/L). The hERG aromatic residues located in the S6 helix (Tyr652 and Phe656) are crucial in the binding of mexiletine and the different affinities of mexiletine and MHM with hERG channel are interpreted by modeling their corresponding binding interactions through ab initio calculations. The simulations demonstrate that the introduction of a hydroxyl group on the meta-position of the aromatic portion of mexiletine weakens the interaction of the drug xylyloxy moiety with Tyr652. These results provide further insights into the molecular basis of drug/hERG interactions and, in agreement with previously reported results on clofilium and ibutilide analogs, support the possibility of reducing hERG potency and related toxicity by modifying the aromatic pattern of substitution of clinically relevant compounds.
Keywords: Ab initio calculations, antiarrhythmic drug, hERG channel, metabolite switch, mexiletine
Introduction
Potassium channels have a central role in the repolarization phase of the action potential and the control of the cellular resting membrane potential in the heart (Tamargo et al. 2004). The delayed rectifier K+ current IK can be separated (on the basis of biophysical properties, pharmacological modulation, and molecular biology) into a rapidly activating IKr and a slowly activating IKs component (Sanguinetti and Jurkiewicz 1990).
The “human Ether-à-go-go-Related Gene” (hERG) encodes the α-subunit of the IKr potassium channel, whereas the KCNQ1 (potassium voltage-gated channel, KQT-like subfamily, member 1) with KCNE1 (potassium voltage-gated channel, Isk-related family, member 1) regulatory subunits encodes the α-subunit of the IKs potassium channel (Barhanin et al. 1996; Sanguinetti et al. 1996b). Polymorphisms in both hERG and KCNQ1 genes can impair channel function and thereby lead to long QT syndrome (LQTS), which consists of an abnormal prolongation of the time between the Q wave and the T wave of the heart's electrical cycle (Sanguinetti et al. 1996a; Hancox et al. 2008). hERG channel may also mediate the “acquired” (drug-induced) form of the LQTS syndrome (Sanguinetti and Tristani-Firouzi 2006). Indeed, it is now known that structurally different drugs (e.g., Class III antiarrhythmics, antibiotics, antihistamines, antidepressants, and antipsychotic agents) are able to block the hERG channel, causing a concomitant risk of sudden death, as a side effect; for this reason hERG inhibition is an important antitarget during the drug discovery process. Recent evidence suggests that the pharmacological sensitivity of the hERG channel is associated with (1) the larger inner cavity, compared to other voltage-gated K+ channels, that allows many molecules to enter and block the channel; (2) the presence of particular aromatic amino acid residues in the pore and in the S6 helix which are critical for high-affinity binding of drugs that contain aromatic rings (Sanguinetti and Mitcheson 2005).
Aside from the well-known QT-prolonging effects of class III antiarrhythmic agents (Witchel and Hancox 2000), class I antiarrhythmic agents are also known to prolong repolarization and QT interval, carrying a risk of LQTS and associated pro-arrhythmia (Vaughan Williams 1984). Indeed, IKr blockade contributes to the actions of the class Ia antiarrhythmics quinidine, procainamide, and disopyramide (Paul et al. 2002; Ridley et al. 2003; El Harchi et al. 2012) and class Ib antiarrhythmic agents aprindine (Horie and Yoshida 1999) and phenytoin (Danielsson et al. 2003). IKr blockade has also been reported for class Ic drugs such as propafenone (Windisch et al. 2011) and flecainide (Paul et al. 2002).
Mexiletine is a class Ib antiarrhythmic drug which is used predominantly in the treatment of ventricular arrhythmias. Moreover, recent studies have demonstrated its therapeutic value to treat many disorders associated with voltage-gated sodium channel dysfunction, for example, neuropathic pain (Challapalli et al. 2005), erythromelalgia (Cregg et al. 2014), myotonic syndromes (Conte Camerino et al. 2007; Logigian et al. 2010; Statland et al. 2012), and Timothy syndrome (Gao et al. 2013). Unfortunately, mexiletine has been withdrawn from the market in many countries (Matthews and Hanna 2014) and attempts are made to develop a “mexiletine alternative.”
While effective in treatment and prophylaxis of ventricular tachycardias, there is some controversial evidence that mexiletine, especially at high doses, can cause adverse effects such as hypotension and atrioventricular heart block (Brochu et al. 2006; Akinci et al. 2011; Eijkelkamp et al. 2012).
A study on guinea pig ventricular myocytes reported no effect of mexiletine on IKr at 30 μmol/L (Wang et al. 1996), whereas a significant block at this concentration was observed in a later study (Mitcheson and Hancox 1997). Furthermore, a more recent article reported, on the basis of a predictive model for hERG/drug interaction, an estimation of IC50 ≫ 10 μmol/L for mexiletine (Roche et al. 2002), suggesting a very low or even null effect of the drug on the hERG channel. Taking these controversial data into account, we decided to investigate in detail the interaction of mexiletine with hERG channel, heterologously expressed in HEK cells.
Recently (Catalano et al. 2012), it has been shown that a minor metabolite of mexiletine, meta-hydroxymexiletine (MHM), has the same antiarrhythmic activity on guinea pig isolated left atria and a more favorable toxicological profile, when compared to mexiletine. This is the prerequisite property for the so-called “metabolite switch,” namely the selection of an active metabolite as the substitute for the parent compound, as long as the former has more favorable properties compared with the latter. This work was also intended to perform a comparative study of the direct blockade of hERG channel by mexiletine and its metabolite MHM.
Materials and Methods
Chemistry
m-hydroxymexiletine was prepared as previously reported (Catalano et al. 2012). Mexiletine was synthesized modifying a procedure previously applied to the preparation of mexiletine analogs (Franchini et al. 2003). Briefly, 2,6-xylyloxyacetone was obtained by Williamson etherification of 2,6-dimethylphenol with chloroacetone under either conventional conditions or microwave-assisted heating. The so-obtained ketone underwent either conventional (Bruno et al. 2006) or microwave assisted (Cavalluzzi et al. 2013) reductive amination to give the target compound (see Supporting Information for details).
Maintenance of mammalian cell lines and cell transfection
Patch-clamp studies were carried out in human embryonic kidney 293 (HEK) cells stably expressing hERG channel and Chinese Hamster Ovary (CHO) cells transiently expressing KCNQ1/KCNE1 channels or hERG-F656A and Y652A mutants. For heterologous protein expression, cells were plated in 6-well cell culture dishes with 2-mL growth medium, 24 h before transfection. Cells were transiently transfected using X-tremeGENE 9 transfection reagent (Roche, Roche Diagnostics, Mannheim, Germany), according to the protocol supplied by the manufacturer. EGFP fluorescence was used as marker of successful transfection. Electrophysiology studies were performed 48–72 h after transfection.
Electrophysiological recordings
Electrophysiological recordings were performed using the whole-cell mode of the patch-clamp technique. The extracellular recording solution used for patch-clamp recordings had the following composition: 140 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 2 mmol/L CaCl2, 10 mmol/L Glucose, 10 mmol/L HEPES, pH 7.4 with NaOH. The following intracellular solution was used: 130 mmol/L KCl, 1 mmol/L MgCl2, 10 mmol/L HEPES, 10 mmol/L EGTA, 5 MgATP pH 7.2 with KOH. Solutions were applied to the cell via a gravity-fed perfusion system (VC-6 Six Channel Valve Controller, Warner Instruments, Hamden, CT, USA). Patch-clamp electrode was pulled from Sutter capillary glass (Novato, CA) on a Flaming/Brown type puller (Sutter P-87), and fire polished to 3–4 MΩ resistance, using a microforge (Narishige). Patch-clamp recordings of cell cultures were carried out at room temperature 48 h after transfection. For recordings, a Multiclamp 200B amplifier (Molecular Devices, Inc., Sunnyvale, CA) and Digidata 1440 data acquisition board (Molecular Devices, Inc.) with pCLAMP 10 software (Molecular Devices, Inc.) were used. Series resistance was compensated by ∼60–80%. The data analysis was performed using Origin 8.0 (OriginLab Corporation, Northampton, MA). Further information for data and statistical analysis is available in the online supplementary information.
Quantum mechanical calculations
The models of 3-methoxy-2,4-dimethylphenol (MHM moiety, MHMm), 2,6-dimethylphenyl methyl ether (mexiletine moiety, MEXm), p-cresol (Tyr moiety, TYRm), and toluene (Phe moiety, PHEm) were generated from atomic fragments incorporated into Spartan'14 (Wavefunction Inc., Irvine, CA) inner fragment library and assuming the suggested default starting geometries. The generated geometries were optimized by the molecular mechanics MMFF routine offered by the software (Halgren 1996) and then submitted to a systematic conformational distribution analysis using the default step sizes. All conformers in a window of 10 Kcal/mol above the global minimum conformer were retained. When two conformers differed by dihedral values lower than 10˚, the less stable conformer was left out. Conformers were then classified according to their ab initio gas phase energy content calculated at the RHF/3-21G*level. All conformers falling within a window of 5 kcal/mol above global minimum were retained and submitted to RHF/3-21G* geometry optimization. After removal of redundant conformers (i.e., each conformer differing from a more stable one by less than 5° in their corresponding dihedral values), the single point energy content for all the remaining conformers were calculated at the RHF/6-31G** level. The optimized structures were confirmed as real minima by IR frequency calculation. The most stable conformer of either MHMm or MEXm was then arbitrarily coupled with the most stable conformer of either TYRm or PHEm, and the so-obtained putative complexes underwent geometry optimization by density function theory (DFT) implemented in Spartan'14 with B3LYP functional (Becke 1988) and several basis sets [6-31G*, 6-31G**, 6-31+G*, 6-311G*, 6-311+G**, 6-311++G**, and 6-311++G(2df,2p)] (Davidson and Feller 1986) in the gas phase. The same geometry optimization procedures were applied to the separated guest and host species. Where solution calculations are concerned, water contribution to the total energy was computed by means of the continuum solvation model SM8 implemented for 6-31G*, 6-31G**, and 6-31+G* basis sets, the only ones proposed as reliable in Spartan'14 (Marenich et al. 2007). The highest occupied and lowest unoccupied molecular orbital (HOMO and LUMO, respectively) energies (EHOMO and ELUMO, respectively) of the complexes were used to calculate the corresponding electrophilicity indexes (ω) (Maynard et al. 1998; Parr et al. 1999) in agreement with the following equations (Parthasarathi et al. 2004):
where I and A denote the ionization potential (IP) and the electron affinity (EA), respectively. The interaction energy involved in complex formation (ΔE) was simply obtained by the energy of the complex (Ecomplex) subtracted by the sum of energies of isolated constituents (i.e., MEXm, MHMm, TYRm, and PHEm):
each energy being calculated at the above reported levels (see Table S2 in the Supporting Material for details).
Results
Concentration dependence of hERG channel block by mexiletine and MHM
To investigate the effect of mexiletine and MHM (Fig.1A) on hERG potassium channels, patch-clamp experiments were performed on HEK cells heterologously expressing the hERG channel. To elicit the activating outward current, a depolarizing step from a holding potential of −80 mV to −10 mV (3 sec in duration) was applied. Tail current was evoked by repolarizing to −50 mV for 3 sec (Fig.1B). Successive command pulses were applied at 10-sec intervals. In the presence of 50 μmol/L mexiletine, the tail current was reduced almost completely and the IhERG block induced by mexiletine was reversible (Fig.1C). Fitting the concentration–response relationship for block of hERG tail currents with a Hill equation yielded an IC50 of 3.7 ± 0.7 and 22.4 ± 1.2 μmol/L and a Hill slope of 0.93 ± 0.09 and 1.3 ± 0.08, for mexiletine and MHM, respectively (Fig.1D).
Figure 1.
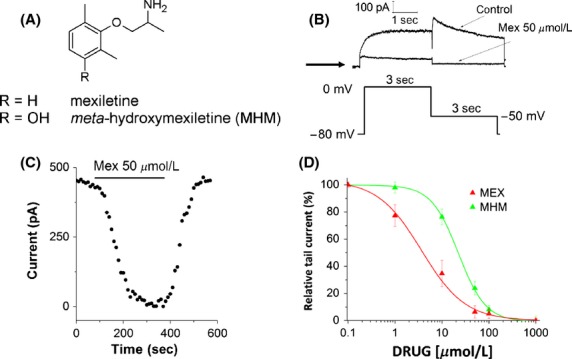
(A) Structures of mexiletine and its hydroxylated metabolite m-hydroxymexiletine (MHM). (B) Representative hERG currents obtained before (control) and after addition of 50 μmol/L mexiletine, recorded using the pulse protocol shown. The arrow indicates zero current level. (C) Time course of mexiletine-induced hERG tail current inhibition from the same cell shown in (B). (D) Concentration–response relationship (mean ± SEM) for block of hERG tail current by mexiletine and MHM. A Hill equation fit yielded an IC50 of 3.7 ± 0.7 and 22.4 ± 1.2 μmol/L and a Hill slope of 0.93 ± 0.09 and 1.3 ± 0.08, for mexiletine and MHM, respectively (n ≥ 5 per data point).
In order to evaluate the binding selectivity of mexiletine and MHM, we tested their modulatory effect on two different cardiac targets: KCNQ1/KCNE1 channel and Na+,K+-ATPase. In both cases, we observed little or no effect (Fig. S2, Supporting Information) on the current signals generated by the two transport proteins. Finally, we studied the interaction of mexiletine and MHM with two ATPase proteins highly expressed in biological membranes, such as P-glycoprotein (P-gp) and MultiDrug Resistance-associated protein 1 (MRP1), which affect pharmacokinetics, efficacy, safety, or tissue levels of drugs. Each compound has been tested in a cell line overexpressing the ATPase proteins P-gp and MRP1 (MDCK-MDR1 and MDCK-MRP1 cells) in order to investigate the interaction of mexiletine and MHM with the selected proteins. Both compounds, at 100 μmol/L concentration, did not interfere with the activity of the pumps as they did not influence the P-gp and MRP1-mediated efflux of the probe (CalceinAM), in cell line overexpressing MDCK-MDR1 and MDCK-MRP1 proteins. Moreover, the consumption of ATP cell level has been also measured to confirm the absence of P-gp and MRP1-mediated efflux (data not shown; for details see Supporting Information).
Voltage- and time-dependent block of hERG channel by mexiletine and MHM
The voltage dependence of mexiletine- and MHM-induced inhibition was measured on hERG K+ current amplitudes, applying the protocol shown in Figure2A. Figure2B shows the current–voltage relationships (I–V curve) for currents measured at the end of the test pulse. The hERG current has an activation threshold of −40 mV, increases progressively with potentials up to 0 mV, but then declines at more positive voltages, as a consequence of the inward rectification. Tail currents were elicited by repolarization to −60 mV. Individual tail current amplitudes, recorded before (control) or after 10 min incubation with 10 μmol/L mexiletine and MHM, were normalized to the maximal control amplitude and fitted with a Boltzmann function (Fig.2C). The voltage required for half-maximal activation (V1/2) was slightly shifted from −17.4 ± 0.6 mV (control), to −19.6 ± 1.3 mV (MHM) and −21.2 ± 0.9 mV (mexiletine). This shift to more negative potentials might be explained by mexiletine and MHM binding preferentially to the channel in an open state, thus limiting a hERG conductance increase at greater depolarized test potentials (Smith et al. 1996).
Figure 2.
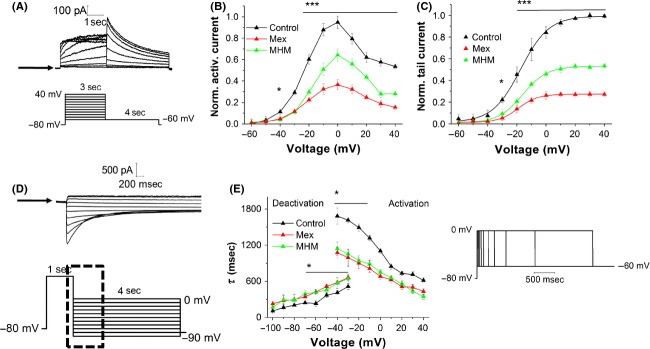
(A) Representative hERG current traces elicited by the voltage protocol shown in the lower panel, testing steady-state activation currents. The arrow indicates zero current level. (B, C) Normalized (with respect to the control currents) I–V relationships for current measured at the end of the test pulse (B) and at the tail current peaks (C), before (control) and after addition (time of incubation ≥ 10 min) of 10 μmol/L mexiletine and MHM. Error bars for some points are masked by symbols (n ≥ 5 cells per data-point). I–V relationships for tail current amplitudes were fitted with a Boltzmann function. V1/2 shifted from −17.4 ± 0.6 mV (control), to −19.6 ± 1.3 mV (MHM; P < 0.05 paired Student's t-test; n = 6 cells) and −21.2 ± 0.9 mV (mexiletine; P < 0.001, paired t-test; n = 6 cells). *P < 0.05, two-way ANOVA followed by a Bonferroni posttest; ***P < 0.001 two-way ANOVA followed by a Bonferroni posttest. (D) Representative hERG current traces elicited by the voltage protocol shown in the lower panel, testing deactivation currents. The arrow indicates zero current level. (E) Activation time constants were derived from currents acquired using the envelope of tails voltage protocol shown on the right-hand side of the figure, fitted with a single exponential function. Deactivation time course was fitted by a double exponential function and derived from currents acquired using the protocol shown in (D). *P < 0.05, two-way ANOVA followed by a Bonferroni posttest; n ≥ 5 cells.
Drug block of ion channels may affect protein kinetics. For this reason we investigated a possible effect of mexiletine and MHM on activation or deactivation kinetics of hERG channel. To evaluate the time course of hERG channel activation, ionic currents were recorded using the envelop tail protocol shown in Figure2E (Liu et al. 1996; Vandenberg et al. 2012). The time constants, determined by fitting with a single exponential function the latter part of the activation time course, were plotted against the membrane voltage (Fig.2E). We observed that in the presence of 10 μmol/L mexiletine or MHM there was a significant decrease in the activation time constant. Moreover, deactivation time constants were obtained by fitting with a double exponential function the currents recorded using the protocol shown in Figure2D. In this case, the inhibitory effect of mexiletine or MHM caused a slight increase in the fast component of the deactivation current (Fig.2E), whereas the slow component (τslow) was not affected by the presence of both compounds (Fig. S1, Supporting Information).
Effect of mexiletine and MHM on hERG channel inactivation
Steady-state inactivation currents were measured following the protocol shown in Figure3A: channels were inactivated at +60 mV, before short test pulses from −100 mV to +20 mV were applied, to recover the channels from inactivation. Depolarization to +60 mV after these test pulses evoked a large outward inactivating current. Current amplitudes measured after the return to +60 mV were normalized, plotted against the voltage and fitted by a Boltzmann function. Our data show that mexiletine and MHM do not cause any significant change in steady-state inactivation, as shown by the V1/2 values: −90.6 ± 4.7 mV in the absence of drugs (control), −89.1 ± 3.5 mV for mexiletine and −90.4 ± 4.2 mV for MHM (Fig.3C). This indicates that both drugs have no effect on the inactivation gating of the hERG channel. Finally, the effect of mexiletine and MHM on the inactivation time course was investigated using the protocol shown in Figure3B. From a holding potential of −80 mV, a 200 ms test pulse to +60 mV was applied to inactivate the channel. Moreover, a short pulse to −100 mV and voltage steps from −20 mV to +60 mV (300 ms, 10 mV-increments) were applied to elicit a large outward inactivating current. Inactivation currents were fitted by a single exponential function to extrapolate time constant values. Figure3D shows that the inactivation time course was not affected by the presence of mexiletine and MHM.
Figure 3.
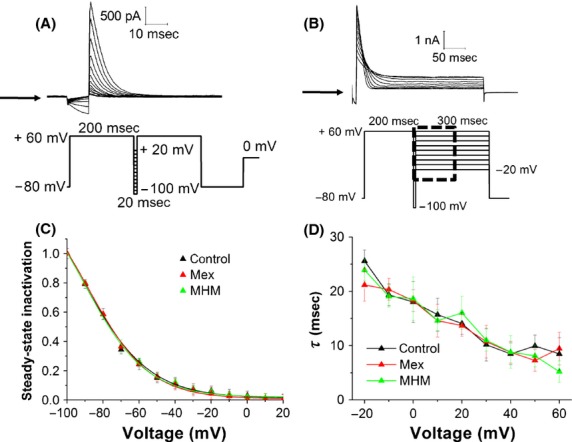
(A) Representative hERG current traces elicited by the voltage protocol shown in the lower panel, testing steady-state inactivation currents. The arrow indicates zero current level. (B) Representative onset of inactivation currents, elicited by a test pulse from +60 mV, to potentials ranging from −20 to +60 mV, in 10 mV-increments. The arrow indicates zero current level. (C) Normalized steady-state I–V inactivation curves before (control) and after application of 10 μmol/L mexiletine and MHM. Data are expressed as mean ± SEM; error bars for some points are masked by symbols (n ≥ 5 per data point). Solid lines represent fits with Boltzmann function. The V1/2 obtained are −90.6 ± 4.7 mV (control), −90.4 ± 2.1 mV (mexiletine), −89.1 ± 3.5 mV (MHM). (D) Inactivation time constants were derived from currents acquired using the protocol shown in (B), fitted with a single exponential function and plotted against the membrane potential. Data are expressed as mean ± SEM (n ≥ 5 per data point).
State dependence of hERG channel block by mexiletine
To investigate the state dependence of hERG inhibition, we evaluated the effect of mexiletine on hERG current amplitude during long depolarizing steps. Current was activated by a 100 ms depolarization step from −80 mV (to keep the channels in the closed state) to +80 mV, then the cell was held at 0 mV for 1 sec (the protocol is shown in Fig.4A, B). This protocol is a useful strategy to study in details the possibility of hERG close-state block by drugs (Kiehn et al. 1996; Thomas et al. 2001). After performing the control measurements, 10 μmol/L mexiletine was applied for 10 min without pulsing. After the pulse-free incubation period, the pulse protocol was repeated. Figure4A shows that the initial current during the step to +80 mV and the current activation peak, recorded in the absence (control, trace “a”) or presence of mexiletine (trace “b”), overlap perfectly, suggesting minimal closed-state block by mexiletine. Moreover, in trace “b” we observe a time-dependent increase in blockage, which can be fitted by a single exponential function with a time constant of 1.18 ± 0.11 sec. This is shown more clearly in Figure4B, reporting the “mexiletine-sensitive” current, obtained by subtracting trace “b” from trace “a.” The trace in Figure4B shows an exponential development with a time constant of 0.88 ± 0.08 sec, supporting the hypothesis that the closed state of hERG channel is not targeted by mexiletine.
Figure 4.
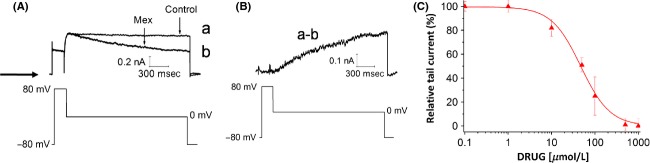
(A) Representative hERG current traces recorded before (control) or after addition of 10 μmol/L mexiletine (10 min incubation), using the protocol shown in the lower panel. To verify the reproducibility of the experiments, each measurement was repeated n ≥ 5 times. (B) “Mexiletine sensitive” current obtained by subtracting traces “a” and “b” shown in (A). (C) Concentration–response relationship (mean ± SEM) for block by mexiletine of hERG tail current, induced by repolarization from +80 mV to −50 mV. A Hill equation fit yielded an IC50 of 47.2 ± 5.3 μmol/L and a Hill slope of 1.13 ± 0.20 (n ≥ 5 per data point).
Finally we evaluated the possibility of block induced by mexiletine when the hERG channel is in the inactivated state. To this end, we observed that the value of maxiletine IC50, calculated at +80 mV (at which all channels are inactivated), is 47.2 ± 5.3 μmol/L (Fig.4C), compared to −10 mV (at which most channels are open) is almost 15-fold higher than the corresponding value at −10 mV (Fig.1D), suggesting mexiletine preferential binding to the open state of hERG channel, rather than the inactivated state.
Role of S6 inner helix aromatic residues on mexiletine binding
Most hERG channel blockers bind within the channel inner cavity, interacting with one or two aromatic residues of the S6 domain (Tyr652 and Phe656). To evaluate if mexiletine binds to sites within the inner cavity of hERG channel, we recorded ionic currents through CHO cells expressing Y652A or F656A hERG mutants, in the presence of mexiletine. Due to the known low expression and altered kinetic properties of F656A hERG clone, we measured F656A hERG inward tail current using a high [K+] external solution (94 mmol/L, Fig.5A) (Mitcheson et al. 2000a; Du et al. 2014); on the contrary, the Y652A hERG outward tail current (Fig.5B) was acquired in the “normal” [K+] solution (5 mmol/L).
Figure 5.

(A) Representative IhERG traces of WT hERG and F656A hERG in the absence (black line) and presence (gray line) of 50 μmol/L mexiletine. The tail current was evoked by repolarization from +20 to −120 mV (protocol shown in the lower panel) and was recorded using “high” [K+] solution (94 mmol/L). The arrows indicate zero current level. (B) Representative IhERG traces of WT hERG and Y652A hERG mutant in the absence (black line) and presence (gray line) of 50 μmol/L mexiletine. The tail current was evoked by repolarization from 0 to −50 mV (same protocol shown in the lower panel of Fig.1B) and was recorded under the “normal” external solution ([K+] = 5 mmol/L). The arrows indicate zero current level. (C, D) Concentration–response relationships for block of hERG channel currents by mexiletine (C) and MHM (D). Each value represents mean ± SEM of n ≥ 5 cells. (E) Summary of fold changes in IC50 values for Y652A and F656A hERG mutants compared with WT-hERG.
Figure5C, D show the inhibition curves of mexiletine and MHM for wild-type and mutant hERG channels. The extrapolated IC50 values calculated in the presence of mexiletine are 3.7 ± 0.7 μmol/L, 41.6 ± 6.6 μmol/L, and 111.5 ± 1.8 μmol/L for WT, Y652A, and F656A, respectively. Moreover, the extrapolated IC50 values calculated in the presence of MHM are 22.4 ± 1.2 μmol/L, 42.6 ± 4.6 μmol/L, and 436 ± 130 μmol/L for WT, Y652A and F656A, respectively. Thus, both mutant channels Tyr652 and Phe656 exhibit decreased sensitivity to block by mexiletine and MHM, particularly in the case of Phe656. Moreover, our results suggest that Y652A mutation affects MHM binding less than mexiletine binding. In fact the greatest fold shift in IC50 of the mutant Y652A is observed in the presence of mexiletine (∼11), compared to MHM (∼2) (Fig.5E).
Ab initio calculations
It has been suggested that both Tyr652 and Phe656 may give π–π stacking interactions with the same aromatic portion of hERG blockers (Gemma et al. 2012) while Tyr652 should preferentially be involved in π-cation interactions with the protonated basic nitrogen of the ligand (Pearlstein et al. 2003). Since mexiletine and MHM share the same basic aliphatic moieties, we focused our attention on the aromatic portions of the ligands, the ones that should elicit the differences observed when comparing their corresponding pharmacological profiles. In particular, (1) we assumed the moieties drawn in bold in Figure6 (panels A and B) as representative of the whole MHM and mexiletine molecules, (2) modelled the corresponding truncated compounds – 3-methoxy-2,4-dimethylphenol (MHM moiety, MHMm) and 2,6-dimethylphenyl methyl ether (mexiletine moiety, MEXm), respectively –, and (3) optimized them at the RHF/3-21G* level (Fig.6, panel C). The same reductionist process (1–3) was used to represent the aromatic rings of the two amino acids supposed to be involved in direct interactions with the aromatic portions of the guest molecules. Thus, p-cresol and toluene were assumed as models of Tyr652 (Tyr moiety, TYRm) and Phe656 (Phe moiety, PHEm), respectively (Fig.6, panels A–C).
Figure 6.
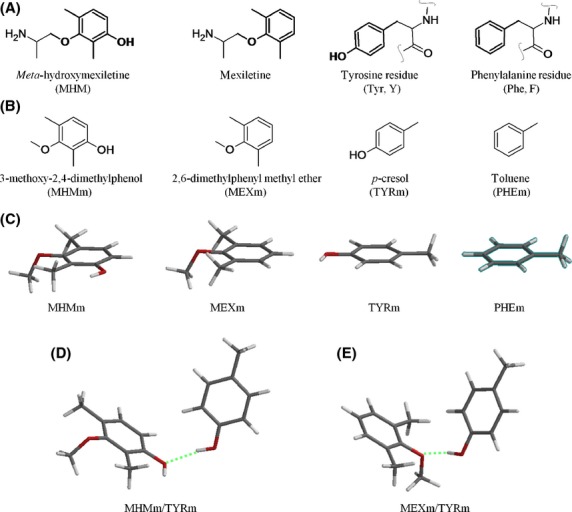
Structures of the model compounds used to predict the interaction energies between mexiletine and MHM with their putative binding aromatic residues – Tyr652 and Phe656. (A) guest and host molecules presenting in bold the moieties studied by ab initio calculations; (B) corresponding truncated molecules assumed as representative of the whole guest compounds and host aromatic residues; (C) tube representation of the most stable conformer [B3LYP/6-311++G(2df,2p)//B3LYP/6-311++G(2df,2p)] of each model compound undergoing complex formation; (D) complex formed by MHMm with TYRm, optimized at the above level of calculation (hydrogen bond shown as a green broken line); (E) complex formed by MEXm with TYRm, optimized at the above level of calculation (hydrogen bond shown as a green broken line).
To evaluate the possibility that the aromatic rings of the ligands would display different tendencies to form sandwich-like stacked complexes with both Tyr652 and Phe656 (Gemma et al. 2012), the geometries of the complexes MHMm/TYRm, MEXm/TYRm, MHMm/PHEm, and MEXm/PHEm were optimized at several levels of DFT calculation (see Materials and Methods). Since the binding site of hERG blockers is supposed to be located into a restricted space lined by four residues each of Phe656 and Tyr652, calculations were performed in vacuum (i.e., ε = 0) to simulate a highly lipophilic environment. Generally, complexes were T shaped denoting the contribution of both hydrogen bonding and dispersion interactions (Paton and Goodman 2009). In particular, TYRm behaved as a hydrogen bond donor while the phenolic hydroxy and methoxy groups of MHMm and MEXm, respectively, performed as hydrogen bond acceptors (Fig.6, panels D and E). MHMm and MEXm interact rotated of 120° with respect to TYRm. This apparently dramatic difference may be accepted as possible, given the reduced size of both mexiletine and MHM compared with the relatively large inner cavity of the channel. Furthermore, the symmetry of the latter and the presence of four equally accessible Tyr residues let hypothesize the possible flipping of ligand/Tyr interactions within the pore. However, given the limitations of the model, the above considerations should be considered as mere speculations and should be assumed with caution when predicting the actual binding orientation of the whole binding partners. For each optimized complex, the corresponding electrophilicity index (ω) value was assumed as a measure of the residual tendency to “soak” electrons from a further aromatic ring as an electron donor. Regardless of the level of calculation used, the complexes formed by MHM showed slightly higher ω values (see Table S1 in the Supporting Information); however, no significant difference was observed when the corresponding mean values were compared. When the energies of formation of the complexes were considered, the complexes involving TYRm resulted significantly more stable than the corresponding complexes formed with PHEm (mean energies of formation of about 6–7 and 1–3 Kcal/mol, respectively; see Table S2 in the Supporting Information), MHMm/TYRm being significantly less stable than MEXm/TYRm (mean difference = 1.2 ± 0.3 Kcal/mol, P = 0.01). When the interaction with PHEm is concerned, the complex formed with MHM resulted more stable than the corresponding complex formed with MEXm of about the same difference previously observed between the two complexes formed with TYRm. However, compensation should be ruled out considering the significantly higher levering interactions with Tyr652. Thus, the interaction of the aromatic moieties of mexiletine and MHM with Tyr652 might explain the difference observed in their relative potency of hERG block. Since the same interaction was proposed as a pivotal contribution to the blocking activity of both compounds on cardiac sodium channels (Desaphy et al. 2012) where MHM was at least as potent as mexiletine, we hypothesized that this apparently contradictory result might be explained assuming that the binding site for the two blocking agents is located in a relatively less lipophilic environment in the sodium channel in comparison with the one present in the hERG vestibule. Running our calculations in the presence of water, we were gratified by the results indicating comparable stability for MHMm/TYRm (E ± SE = −6.2 ± 1.8 Kcal/mol) in respect of MEXm/TYRm (E ± SE = −6.7 ± 1.8 Kcal/mol) (Table S3 in the Supporting Information). Once again, the complexes involving TYRm resulted significantly more stable than the corresponding complexes formed with PHEm (mean energies of formation of about 6–7 and 1–2 Kcal/mol, respectively; see Table S4 in the Supporting Information), thus supporting the suggestion that, when both hydrogen bonding and dispersion interactions contribute to binding, the Tyr aromatic ring should display a higher levering interaction in comparison with the one involving Phe residues. MHMm/PHEm and MEX/PHEm also presented comparable stability in water (E ± SE = −0.6 ± 0.7 Kcal/mol and E ± SE = −1.6 ± 1.1 Kcal/mol, respectively).
Discussion
This is the first report of direct blockade of heterologously expressed hERG potassium channel by the antiarrhythmic agent mexiletine and one of its metabolite m-hydroxymexiletine (MHM).
We found that mexiletine is a moderate-potency inhibitor of hERG channel, with an IC50 of 3.7 ± 0.7 μmol/L, a value close to its therapeutic concentration (4–11 μmol/L) (Campbell et al. 1978). Interestingly, MHM, a minor metabolite of mexiletine, performed as a weaker hERG blocker, compared to mexiletine, with an IC50 of 22.4 ± 1.2 μmol/L.
Target specificity is critical to limit toxicity. To assess the specific relationship between the risk of mexiletine-induced cardiotoxicity and other potential targets for cardiac adverse effects, we investigated the effect of mexiletine and MHM on the following proteins: (1) the Na+,K+-ATPase, an ion pump of cardiac cells, which is blocked by the action of cardiac glycosides like digoxin used to treat heart disease; (2) the KCNQ1/KCNE1 channel, which is responsible for the slow component of the action potential repolarization phase (IKs); (3) two ATPase proteins highly expressed in biological membranes such as P-glycoprotein (P-gp) and MultiDrug Resistance-associated protein 1 (MRP1), which affect pharmacokinetics, efficacy, safety, and tissue distribution of drugs. The absence of interaction with these antitargets (see Supporting Information), together with the previously reported absence of CNS side effects and null cytotoxicity (Catalano et al. 2012), reinforce the role of MHM as a possible metabolite switch candidate.
As most open-channel blockers, mexiletine and MHM accelerate significantly the rate of activation and cause a small deceleration of the deactivation time-course (Fig.2E). This kinetic effect is due to the reopening of channels caused by charged drug unbinding (Wang et al. 1999; Mitcheson et al. 2000b; Tie et al. 2000). Given these observations, we conclude that mexiletine blocks the hERG channel preferentially in an open state, with features similar to those of other open-channel blockers, for example, miconazole, mesoridazine, and ketanserin (Su et al. 2004; Kikuchi et al. 2005; Tang et al. 2008).
The molecular mechanism of many hERG inhibitors is an open-state-dependent trapping model of drugs into the inner cavity of the channel (Vandenberg et al. 2012). In this model, aromatic residues on the S6 helix (Tyr652 and Phe656) and polar residues at the base of the pore helix (Thr623 and Ser624) line the inner cavity of the channel (Sanguinetti and Mitcheson 2005; Perry et al. 2006). Mexiletine has two pharmacophoric points: the aromatic portion that could interact with Tyr652, and a charged group, which could form π-cationic interaction with Phe656. Thus, considering also the analogy between Nav1.5- and hERG-binding sites, we hypothesized that both strictly related compounds Mex and MHM should preferentially reside in the bottom corner of the pore, the one lined by Tyr652 and Phe656. To evaluate this hypothesis, we recorded ionic currents through Y652A or F656A hERG mutants. Our data show that mutation of Tyr652 and Phe656 residues dramatically reduces the blocking effect by mexiletine (Fig.5); in particular the Phe656 residue seems to play a prominent role in the binding of mexiletine. Moreover, we observed that Y652A mutation affects MHM binding less than mexiletine binding (Fig.5C and D), in accordance to ab initio calculations.
According to the modulated receptor theory for channel block (Hille 1977) and in agreement with previous results on both sodium (Desaphy et al. 2012) and hERG (Mitcheson et al. 2000a,b) channels, it may be assumed that block by mexiletine and MHM occurs from the cytoplasmic side of the membrane. Thus, the lower potency found for the less lipophilic analog – MHM (log D = 0.02) – in comparison with the one found for mexiletine (log D = 0.53) (Desaphy et al. 2012) might be explained as a reflex of reduced access of the former to its binding site when compared with an easier access of the latter. However, lipophilicity might also play a critical role as a determinant of the intimate interaction between the compounds and their binding site. Our ab initio investigations on possible subtle differences in MHM and mexiletine relative binding modes suggest that the two compounds should display similar tendencies to attract electrons from aromatic ring residues as indicated by the mean ω values found for the complexes formed by their corresponding truncated aromatic moieties with the ones corresponding to Tyr652 (0.90 ± 0.13 and 0.88 ± 0.10, respectively) and Phe656 (0.94 ± 0.13 and 0.89 ± 0.12, respectively). However, in high lipophilic environments MHM should give relatively less stable complexes with Tyr residues with respect to those formed by mexiletine: a significant mean difference of 1.2 ± 0.3 Kcal/mol (P = 0.01) was found between the interaction energy of the truncated aromatic moiety of MHM with the moiety representative of Tyr (TYRm), and that of the truncated moiety representative of mexiletine with the same TYRm. Indeed, mexiletine was about six times more potent as a hERG blocker than MHM, in good quantitative agreement with the prediction. In more hydrophilic environments MHM interactions are predicted to be at least as strong as the ones of mexiletine. This seems to be the case for MHM and mexiletine when their corresponding sodium channel blocking activities are considered: MHM was only twice as potent as mexiletine on cardiac sodium channels (Catalano et al. 2012).
The possibility to reduce hERG potency by modifying the pattern of aromatic substitution of hERG blocking compounds has been previously explored, and results from elegant mutagenesis and molecular modeling studies on both clofilium and ibutilide analogs suggested that phenyl ring para-substituents strongly affect Tyr binding, with polar and electronegative substituents reinforcing the binding interaction (Perry et al. 2006). Perry et al. concluded that modifying the para-substituent could be a useful strategy for reducing hERG potency. Our results are in general agreement with the latter statement. However, when the relatively less explored meta-position was substituted with a polar group such as the hydroxyl one in MHM, a reduction in hERG potency was observed, thus opposing to what previously observed with para-substituents in both clofilium and ibutilide series (Perry et al. 2006). Puzzling as it may appear, this observations provide further insights into the molecular basis of drug/hERG interactions.
Of course, our model is tainted with a number of limitations (current inhibition measurements as indirect evidence of binding energies; truncated moieties assumed as representative of the whole partner entities with consequent overlooking of protonated amine group contributions and conformation issues; Manichean assumptions about dielectric constants; relative limited calculation levels used for energy predictions in water solution; allosteric perturbations as consequences of mutations). However, some of the above were attenuated by the fact that two strictly related compounds were considered where the only structural difference was an OH group in the meta-position of the xylyloxy moiety.
Finally, in the light of our results, it is important to observe that, even though our data show that mexiletine inhibits hERG at physiologically relevant concentration, mexiletine has not been documented to significantly affect cardiac repolarization (normal QT-interval in the ECG) and is generally a drug well tolerated in man at low doses. Conversely, mexiletine has been shown to cause a significant reduction in action potential duration (APD) mainly through its inhibition of the sodium current (Matsuo et al. 1985; Roden et al. 1987); this mechanism has been considered as an advantage for the treatment of patients with LQT3 (Wang et al. 1997) and LQT8 (Gao et al. 2013) syndromes, suggesting the possibility of gene-specific therapy for the two distinct forms of the congenital LQTS. More specifically, mexiletine has been shown to preferentially block at therapeutic concentrations the late sodium current (INaL) compared to the peak sodium current (Wang et al. 1997) and this selectivity contributes to the mechanism of reverse use dependence of select APD/QT-prolonging agents (Guo et al. 2010). With regard to our data and in accordance to the literature, we can speculate that the low documented cardiotoxicity of mexiletine could be due to the fact that mexiletine inhibition of INaL may counteract any simultaneous prolongation of the QT interval induced by the block of IKr/hERG channel. Noteworthy, clinical relevant combined effects on INa and IKr have been also reported for the antianginal agent ranolazine, which has been shown to be of potential interest (Hancox and Doggrell 2010; Du et al. 2014) for the treatment of atrial fibrillation; in the case of ranolazine the inhibition of IKr/hERG appears even as a “positive” side effect.
From the point of view of synthetic chemist, the beneficial effect offered by the introduction of a meta-phenol function as a structural feature to reduce hERG affinity might be of general applicability. If validated through the study of other well-known hERG blockers opportunely hydroxylated on their phenyl rings, this chemical modification might provide the key to rescue useful drugs previously withdrawn from clinical use because of their hERG-related toxicity, and orientate the design of safer new drugs. Studies in this respect have been undertaken.
Acknowledgments
We thank M. D'Amico and A. Arcangeli (University of Florence) for the kind gift of HEK-hERG trasfected cells; J. Barhanin (Université de Nice-Sophia Antipolis, Nice, France) for PCI-hKCNQ1 and pIRES-CD8-hKCNE1 plasmids; D. Golenbock (University of Massachusetts Medical School, Boston, USA) for providing pcDNA3-EGFP (Addgene plasmid 13031); X. Koenig for providing F656A and Y652A hERG plasmids.
Glossary
- CHO
Chinese Hamster Ovary
- HEK
human embryonic kidney 293
- hERG
human Ether-à-go-go-related gene
- MHM
m-hydroxymexiletine
- V1/2
voltage required for half-maximal activation
Author Contributions
R. G., G. L. designed the research study; R. G., G. L., F. T.-B., M. R., I. D., M. C. performed the experiments; R. G., G. L., F. T.-B., N. A. C. analyzed the data; R. G., G. L., F. T.-B. wrote or contribute to the writing of the article.
Disclosure
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1. Materials and methods.
Figure S1. Slow time constants (τslow) of hERG deactivation elicited before and after application of mexiletine and MHM.
Figure S2. Effect of mexiletine and MHM on Na+,K+-ATPase and KCNQ1/KCNE1 channels.
Table S1. Calculated electrophilicity index ω (Kcal/mol) for complexes formed by either MHMm or MEXm with either TYRm or PHEm (in vacuum).
Table S2. Calculated interaction energies (Kcal/mol) for complexes formed by either MHMm or MEXm with either TYRm or PHEm (in vacuum).
Table S3. Calculated interaction energies (Kcal/mol) for complexes formed by either MHMm or MEXm with TYRm (in water solution).
Table S4. Calculated interaction energies (Kcal/mol) for complexes formed by either MHMm or MEXm with PHEm (in water solution).
References
- Akinci E, Yüzbaşioglu Y, Coşkun F. Hemodialysis as an alternative treatment of mexiletine intoxication. Am J Emerg Med. 2011;29:1235–1236. doi: 10.1016/j.ajem.2010.08.027. [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- Brochu RM, Dick IE, Tarpley JW, McGowan E, Gunner D, Herrington J. Block of peripheral nerve sodium channels selectively inhibits features of neuropathic pain in rats. Mol Pharmacol. 2006;69:823–832. doi: 10.1124/mol.105.018127. , et al. ( [DOI] [PubMed] [Google Scholar]
- Bruno C, Carocci A, Catalano A, Cavalluzzi MM, Corbo F, Franchini C. Facile, alternative route to lubeluzole, its enantiomer, and the racemate. Chirality. 2006;18:227–231. doi: 10.1002/chir.20240. , et al. ( [DOI] [PubMed] [Google Scholar]
- Campbell NP, Kelly JG, Adgey AA, Shanks RG. The clinical pharmacology of mexiletine. Br J Clin Pharmacol. 1978;6:103–108. doi: 10.1111/j.1365-2125.1978.tb00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano A, Desaphy JF, Lentini G, Carocci A, Di Mola A, Bruno C. Synthesis and toxicopharmacological evaluation of m-hydroxymexiletine, the first metabolite of mexiletine more potent than the parent compound on voltage-gated sodium channels. J Med Chem. 2012;55:1418–1422. doi: 10.1021/jm201197z. , et al. ( [DOI] [PubMed] [Google Scholar]
- Cavalluzzi MM, Viale M, Bruno C, Carocci A, Catalano A, Carrieri A. A convenient synthesis of lubeluzole and its enantiomer: evaluation as chemosensitizing agents on human ovarian adenocarcinoma and lung carcinoma cells. Bioorg Med Chem Lett. 2013;23:4820–4823. doi: 10.1016/j.bmcl.2013.06.077. , et al. ( [DOI] [PubMed] [Google Scholar]
- Challapalli V, Tremont-Lukats IW, McNicol ED, Lau J, Carr DB. Systemic administration of local anesthetic agents to relieve neuropathic pain. Cochrane Database Syst Rev. 2005;4:CD003345. doi: 10.1002/14651858.CD003345.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte Camerino D, Tricarico D, Desaphy J-F. Ion channel pharmacology. Neurotherapeutics. 2007;4:184–198. doi: 10.1016/j.nurt.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Cregg R, Cox JJ, Bennett DL, Wood JN, Werdehausen R. Mexiletine as a treatment for primary erythromelalgia: normalization of biophysical properties of mutant L858F NaV1.7 sodium channels. Br J Pharmacol. 2014;171:4455–4463. doi: 10.1111/bph.12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsson BR, Lansdell K, Patmore L, Tomson T. Phenytoin and phenobarbital inhibit human hERG potassium channels. Epilepsy Res. 2003;55:147–157. doi: 10.1016/s0920-1211(03)00119-0. [DOI] [PubMed] [Google Scholar]
- Davidson ER, Feller D. Basis set selection for molecular calculations. Chem Rev. 1986;86:681–696. [Google Scholar]
- Desaphy JF, Dipalma A, Costanza T, Carbonara R, Dinardo MM, Catalano A. Molecular insights into the local anesthetic receptor within voltage-gated sodium channels using hydroxylated analogs of mexiletine. Front Pharmacol. 2012;3:17. doi: 10.3389/fphar.2012.00017. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Zhang Y, El Harchi A, Dempsey CE, Hancox JC. Ranolazine inhibition of hERG potassium channels: drug-pore interactions and reduced potency against inactivation mutants. J Mol Cell Cardiol. 2014;74:220–230. doi: 10.1016/j.yjmcc.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N, Linley JE, Baker MD, Minett MS, Cregg R, Werdehausen R. Neurological perspectives on voltage-gated sodium channels. Brain. 2012;135:2585–2612. doi: 10.1093/brain/aws225. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Harchi A, Zhang YH, Hussein L, Dempsey CE, Hancox JC. Molecular determinants of hERG potassium channel inhibition by disopyramide. J Mol Cell Cardiol. 2012;52:185–195. doi: 10.1016/j.yjmcc.2011.09.021. [DOI] [PubMed] [Google Scholar]
- Franchini C, Carocci A, Catalano A, Cavalluzzi MM, Corbo F, Lentini G. Optically active mexiletine analogues as stereoselective blockers of voltage-gated Na+ channels. J Med Chem. 2003;46:5238–5248. doi: 10.1021/jm030865y. , et al. ( [DOI] [PubMed] [Google Scholar]
- Gao Y, Xue X, Hu D, Liu W, Yuan Y, Sun H. Inhibition of late sodium current by mexiletine: a novel pharmotherapeutical approach in timothy syndrome. Circ Arrhythm Electrophysiol. 2013;6:614–622. doi: 10.1161/CIRCEP.113.000092. , et al. ( [DOI] [PubMed] [Google Scholar]
- Gemma S, Camodeca C, Brindisi M, Brogi S, Kukreja G, Kunjir S. Mimicking the intramolecular hydrogen bond: synthesis, biological evaluation, and molecular modeling of benzoxazines and quinazolines as potential antimalarial agents. J Med Chem. 2012;55:10387–10404. doi: 10.1021/jm300831b. , et al. ( [DOI] [PubMed] [Google Scholar]
- Guo D, Lian J, Liu T, Cox R, Margulies KB, Kowey PR. Contribution of late sodium current (I(Na-L)) to rate adaptation of ventricular repolarization and reverse use-dependence of QT-prolonging agents. Heart Rhythm. 2010;8:762–769. doi: 10.1016/j.hrthm.2010.12.026. , et al. ( [DOI] [PubMed] [Google Scholar]
- Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Computational Chem. 1996;17:490–519. [Google Scholar]
- Hancox JC, Doggrell SA. Perspective: does ranolazine have potential for the treatment of atrial fibrillation? Expert Opin Investig Drugs. 2010;19:1465–1474. doi: 10.1517/13543784.2010.535517. [DOI] [PubMed] [Google Scholar]
- Hancox JC, McPate MJ, El Harchi A, Zhang YH. The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol Ther. 2008;119:118–132. doi: 10.1016/j.pharmthera.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J Gen Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie M, Yoshida H. An antiarrhythmic agent with class Ib action, aprinidine, inhibits human IKr but not IKs heterologously expressed in COS7 cells. Circulation. 1999;100:I–280. [Google Scholar]
- Kiehn J, Lacerda AE, Wible I, Brown AM. Molecular physiology and pharmacology of hERG. Single-channel currents and block by dofetilide. Circulation. 1996;94:2572–2579. doi: 10.1161/01.cir.94.10.2572. [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Nagatomo T, Abe H, Kawakami K, Duff HJ, Makielski JC. Blockade of hERG cardiac K+ current by antifungal drug miconazole. Br J Pharmacol. 2005;144:840–848. doi: 10.1038/sj.bjp.0706095. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Rasmusson RL, Campbell DL, Wang S, Strauss HC. Activation and inactivation kinetics of an E-4031-sensitive current from single ferret atrial myocytes. Biophys J. 1996;70:2704–2715. doi: 10.1016/S0006-3495(96)79840-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logigian EL, Martens WB, Moxley RT, IV, McDermott MP, Dilek N, Wiegner AW. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology. 2010;74:1441–1448. doi: 10.1212/WNL.0b013e3181dc1a3a. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marenich AV, Olsen RM, Kelly CP, Cramer CJ, Truhlar DG. Self-consistent reaction field model for aqueous and nonaqueous solutions based on accurate polarized partial charges. J Chem Theory Comput. 2007;3:2011–2033. doi: 10.1021/ct7001418. [DOI] [PubMed] [Google Scholar]
- Matsuo S, Kishida H, Munakata K, Atarashi H. The effects of mexiletine on action potential duration and its restitution in guinea pig ventricular muscles. Jpn Heart J. 1985;26:271–287. doi: 10.1536/ihj.26.271. [DOI] [PubMed] [Google Scholar]
- Matthews E, Hanna MG. Repurposing of sodium channel antagonists as potential new anti-myotonic drugs. Exp Neurol. 2014;261:812–815. doi: 10.1016/j.expneurol.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Maynard AT, Huang M, Rice WG, Covell DG. Reactivity of the HIV-1 nucleocapsid protein p7 zinc finger domains from the perspective of density-functional theory. Proc Natl Acad Sci USA. 1998;95:11578–11583. doi: 10.1073/pnas.95.20.11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Hancox JC. Modulation by mexiletine of action potentials, L-type Ca current and delayed rectifier K+ current recorded from isolated rabbit atrioventricular nodal myocytes. Pflügers Arch. 1997;434:855–858. doi: 10.1007/s004240050476. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA. 2000a;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Sanguinetti MC. Trapping of a methanesulfonanilide by closure of the hERG potassium channel activation gate. J Gen Physiol. 2000b;115:229–240. doi: 10.1085/jgp.115.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr RG, Szentpály LV, Liu S. Electrophilicity Index. J Am Chem Soc. 1999;121:1922–1924. [Google Scholar]
- Parthasarathi R, Subramanian V, Roy DR, Chattaraj PK. Electrophilicity index as a possible descriptor of biological activity. Bioorg Med Chem. 2004;12:5533–5543. doi: 10.1016/j.bmc.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Paton RS, Goodman JM. Hydrogen bonding and pi-stacking: how reliable are force fields? A critical evaluation of force field descriptions of nonbonded interactions. J Chem Inf Model. 2009;49:944–955. doi: 10.1021/ci900009f. [DOI] [PubMed] [Google Scholar]
- Paul AA, Witchel HJ, Hancox JC. Inhibition of the current of heterologously expressed hERG potassium channels by flecainide and comparison with quinidine, propafenone and lignocaine. Br J Pharmacol. 2002;136:717–729. doi: 10.1038/sj.bjp.0704784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlstein R, Vaz R, Rampe D. Understanding the structure-activity relationship of the human ether-a-go-go-related gene cardiac K+ channel. A model for bad behavior. J Med Chem. 2003;46:2017–2022. doi: 10.1021/jm0205651. [DOI] [PubMed] [Google Scholar]
- Perry M, Stansfeld PJ, Leaney J, Wood C, de Groot MJ, Leishman D. Drug binding interactions in the inner cavity of HERG channels: molecular insights from structure-activity relationships of clofilium and ibutilide analogs. Mol Pharmacol. 2006;69:509–519. doi: 10.1124/mol.105.016741. , et al. ( [DOI] [PubMed] [Google Scholar]
- Ridley JM, Milnes JT, Benest AV, Masters JD, Witchel HJ, Hancox JC. Characterization of recombinant hERG K+ channel blockade by the Class Ia antiarrhythmic drug procainamide. Biochem Biophys Res Commun. 2003;306:388–393. doi: 10.1016/s0006-291x(03)00980-x. [DOI] [PubMed] [Google Scholar]
- Roche O, Trube G, Zuegge J, Pflimlin P, Alanine A, Schneider G. A virtual screening method for prediction of the hERG potassium channel liability of compound libraries. ChemBioChem. 2002;3:455–459. doi: 10.1002/1439-7633(20020503)3:5<455::AID-CBIC455>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Roden DM, Iansmith DH, Woosley RL. Frequency-dependent interactions of mexiletine and quinidine on depolarization and repolarization in canine Purkinje fibers. J Pharmacol Exp Ther. 1987;243:1218–1224. [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of cardiac delayed rectifier K+ current. Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Mitcheson JS. Predicting drug-hERG channel interactions that cause acquired long QT syndrome. Trends Pharmacol Sci. 2005;26:119–124. doi: 10.1016/j.tips.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–469. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Spector PS, Keating MS. Spectrum of hERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc Natl Acad Sci USA. 1996a;93:2208–2212. doi: 10.1073/pnas.93.5.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996b;384:80–83. doi: 10.1038/384080a0. , et al. ( [DOI] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the hERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Statland JM, Bundy BN, Wang Y, Rayan DR, Trivedi JR, Sansone VA. Consortium for Clinical Investigation of Neurologic Channelopathies. Mexiletine for symptoms and signs of myotonia in nondystrophic myotonia: a randomized controlled trial. JAMA. 2012;308:1357–1365. doi: 10.1001/jama.2012.12607. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Martin R, Cox BF, Gintant G. Mesoridazine: an open-channel blocker of human ether-a-go-go-related gene K+ channel. J Mol Cell Cardiol. 2004;36:151–160. doi: 10.1016/j.yjmcc.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Tamargo J, Caballero R, Gomez R, Valenzuela C, Delpon E. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004;62:9–33. doi: 10.1016/j.cardiores.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Tang Q, Li Z-Q, Li W, Guo J, Sun H-Y, Lau C-P. The 5-HT2 antagonist ketanserin is an open channel blocker of human cardiac ether-à-go-go-related gene (hERG) potassium channels. Br J Pharmacol. 2008;155:365–373. doi: 10.1038/bjp.2008.261. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Wendt-Nordahl G, Rockl K, Ficker E, Brown AM, Kiehn J. High-affinity blockade of human ether-a-go-go-related gene human cardiac potassium channels by the novel antiarrhythmic drug BRL-32872. J Pharmacol Exp Ther. 2001;297:753–761. [PubMed] [Google Scholar]
- Tie H, Walker BD, Singleton CB, Valenzuela SM, Bursill JA, Wyse KR. Inhibition of hERG potassium channels by the antimalarial agent halofantrine. Br J Pharmacol. 2000;130:1967–1975. doi: 10.1038/sj.bjp.0703470. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K(+) channels: structure, function, and clinical significance. Physiol Rev. 2012;92:1393–1478. doi: 10.1152/physrev.00036.2011. [DOI] [PubMed] [Google Scholar]
- Vaughan Williams EM. A classification of antiarrhythmic actions reassessed after a decade of new drugs. J Clin Pharmacol. 1984;24:129–147. doi: 10.1002/j.1552-4604.1984.tb01822.x. [DOI] [PubMed] [Google Scholar]
- Wang DW, Kiyosue T, Sato T, Arita M. Comparison of the effects of class I anti-arrhythmic drugs, cibenzoline, mexiletine and flecainide, on the delayed rectifier K+ current of guinea-pig ventricular myocytes. J Mol Cell Cardiol. 1996;28:893–903. doi: 10.1006/jmcc.1996.0084. [DOI] [PubMed] [Google Scholar]
- Wang DW, Yazawa K, Makita N, George AL, Jr, Bennett PB. Pharmacological targeting of long QT mutant sodium channels. J Clin Invest. 1997;99:1714–1720. doi: 10.1172/JCI119335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HZ, Shi H, Liao SJ, Wang Z. Inactivation gating determines nicotine blockade of human hERG channels. Am J Physiol. 1999;277:H1081–H1088. doi: 10.1152/ajpheart.1999.277.3.H1081. [DOI] [PubMed] [Google Scholar]
- Windisch A, Timin E, Schwarz T, Stork-Riedler D, Erker T, Ecker G. Trapping and dissociation of propafenone derivatives in hERG channels. Br J Pharmacol. 2011;162:1542–1552. doi: 10.1111/j.1476-5381.2010.01159.x. , et al. ( [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witchel HJ, Hancox JC. Familial and acquired long QT syndrome and the cardiac rapid delayed rectifier potassium current. Clin Exp Pharmacol Physiol. 2000;27:753–766. doi: 10.1046/j.1440-1681.2000.03337.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Materials and methods.
Figure S1. Slow time constants (τslow) of hERG deactivation elicited before and after application of mexiletine and MHM.
Figure S2. Effect of mexiletine and MHM on Na+,K+-ATPase and KCNQ1/KCNE1 channels.
Table S1. Calculated electrophilicity index ω (Kcal/mol) for complexes formed by either MHMm or MEXm with either TYRm or PHEm (in vacuum).
Table S2. Calculated interaction energies (Kcal/mol) for complexes formed by either MHMm or MEXm with either TYRm or PHEm (in vacuum).
Table S3. Calculated interaction energies (Kcal/mol) for complexes formed by either MHMm or MEXm with TYRm (in water solution).
Table S4. Calculated interaction energies (Kcal/mol) for complexes formed by either MHMm or MEXm with PHEm (in water solution).