

Summary
CRISPR/Cas9 protein fused to transactivation domains can be used to control gene expression in human cells. In this study, we demonstrate that a dCas9 fusion with repeats of VP16 activator domains can efficiently activate human genes involved in pluripotency in various cell types. This activator in combination with guide RNAs targeted to the OCT4 promoter can be used to completely replace transgenic OCT4 in human cell reprogramming. Furthermore, we generated a chemically controllable dCas9 activator version by fusion with the dihydrofolate reductase (DHFR) destabilization domain. Finally, we show that the destabilized dCas9 activator can be used to control human pluripotent stem cell differentiation into endodermal lineages.
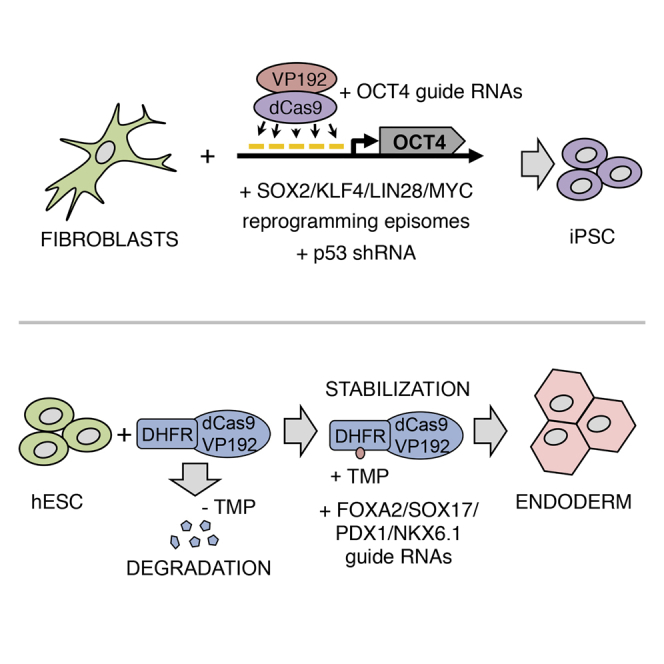
Graphical Abstract
Highlights
-
•
dCas9VP192 targeted to proximal promoters activates transcription in different loci
-
•
Transgenic OCT4 can be replaced by dCas9VP192
-
•
Destabilized dCas9VP192 enables temporal control of gene expression by TMP
-
•
TMP and DOX can be combined to control multiple genes in differentiation
Otonkoski and colleagues show transcription activation of multiple pluripotency and differentiation genes using a CRISPR/dCas9 system and functional validation by replacing transgenic OCT4 in the reprogramming of human fibroblasts to the pluripotent state. Additionally, a conditionally stabilized dCas9 activator is used to temporally control gene activation in the differentiation of pluripotent stem cells to the pancreatic lineage.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)-based systems have recently attracted a lot of attention due to their potential in biotechnological applications. CRISPRs originate from prokaryotic immune response systems and have been engineered to suit genome editing and transcriptional control purposes (Mali et al., 2013a). The most commonly utilized CRISPR system is derived from Streptococcus pyogenes and consists of a single CRISPR-associated (Cas) protein, Cas9, and small guide RNAs (gRNAs), either as two separate RNAs or fused into a single chimeric guide RNA. The complex formed by Cas9 and gRNA binds to DNA sequences complementary to the gRNA, upon which the Cas9 protein generates a double-strand break in the DNA. Modified versions of the Cas9 protein have been engineered by mutating important catalytical residues, generating Cas9 proteins that cut only one DNA strand (nickase Cas9) or that are completely inactive (Jinek et al., 2012). Catalytically inactivated Cas9 proteins (dead Cas9, dCas9) can be used to control gene expression, either by physically interfering with the transcription process (Gilbert et al., 2013; Qi et al., 2013) or as fusion proteins with factors that mediate transcriptional repression or activation (Bikard et al., 2013; Cheng et al., 2013; Gilbert et al., 2013; Hu et al., 2014; Maeder et al., 2013a; Mali et al., 2013b; Perez-Pinera et al., 2013). These systems have recently been utilized to mediate target gene activation and repression in mouse and human cells to promote differentiation of human cells (Chavez et al., 2015; Kearns et al., 2014) and transdifferentiation and reprogramming of mouse cells (Chakraborty et al., 2014; Gao et al., 2014). Moreover, various externally controllable Cas9 proteins have recently been described for gene editing using split Cas9 architecture with either rapamycin-inducible dimerization domains (Zetsche et al., 2015) or tamoxifen-dependent intein splicing (Davis et al., 2015).
In this study, we describe an efficient dCas9 activator with multimeric VP16 activation domain and a simplified method for guide RNA assembly and cloning. We demonstrate that the dCas9 activator can be fused to a dihydrofolate reductase (DHFR)-derived destabilization domain (DD) (Iwamoto et al., 2010) and used to control gene expression with addition of Trimethoprim (TMP) in multiple human cell types. Furthermore, we demonstrate that the dCas9 activator can be used to replace transgenic OCT4 in human cell reprogramming and that human pluripotent cell differentiation can be induced by the activator in a TMP-dependent manner.
Results
dCas9-Transactivator Optimization for Induction of OCT4 Expression
We introduced a second mutation to the previously published nicking version of humanized Streptococcus pyogenes Cas9 (Cong et al., 2013) to incorporate both D10A and H840A mutations that render it catalytically inactive (dCas9). This protein was then C-terminally fused with three repeats of the PADALDDFDLDML sequence of the Herpes simplex virus protein 16 (VP16) transactivation domain (Baron et al., 1997), generating the dCas9VP48 activator version. The function of the construct was validated in reporter HEK293 cells, where GFP expression is under the control of TetON promoter. After targeting dCas9VP48 to the TetON promoter with different gRNAs, we observed increased number of GFP-positive cells in the condition with TetOp2 gRNA, which binds four times to the TetON promoter sequence (Figure S1). Thus, dCas9VP48 showed transcriptional activation activity when targeted multiple times to a promoter sequence.
Most studies so far have used dCas9 activators with VP16 transactivation sequences repeated between three and ten times (Cheng et al., 2013; Maeder et al., 2013a; Mali et al., 2013b; Perez-Pinera et al., 2013). To test the effect of increasing the number of VP16 repeat domains, we cloned additional constructs with 6 and 12 repeats of the VP16 sequence (hereafter referred to as dCas9VP96 and dCas9VP192 accordingly) (Figure 1A) and targeted the endogenous OCT4 (POU5F1) promoter proximal area in HEK293 cells using five gRNAs. Efficient OCT4 upregulation was detected both at mRNA and protein level (Figures 1B and 1C). As dCas9VP192 caused the highest increase in OCT4 expression levels, up to 70-fold, it was used for further experiments.
Figure 1.

Strategy for dCas9-Mediated Activation of Pluripotency Genes
(A) Schematic representation of dCas9 activator versions tested.
(B) Upregulation of OCT4 in HEK293 cells by different dCas9 activator versions. Relative mRNA expression levels were measured by qRT-PCR 72 hr after transfection. VP96-only transfection was used as control. Data represent mean ± SEM, n = 5 independent transfections.
(C) Immunocytochemical analysis of OCT4 in HEK293 cells 72 hr after transfection with dCas9 activator versions and OCT4 targeting gRNAs. Scale bars represent 400 μm.
(D) Schematic representation of the gRNA cloning and testing strategy. gRNAs were first validated with PCR-amplified fragments before concatenating them into plasmids by Golden Gate assembly.
(E) Immunocytochemical detection of the products of the targeted genes in HEK293 cells 72 hr after transfection with PCR gRNA fragments and dCas9VP192 plasmid. Non-treated (NT) HEK293 cells were used as staining control. Scale bars represent 200 μm.
(F) Activation of targeted genes in HEK293 cells transfected with gRNA-concatenated plasmids and dCas9VP192 plasmid. Scale bars represent 200 μm.
(G) Upregulation of mRNA levels of targeted genes measured by qRT-PCR in HEK293 cells 72 hr after transfection with gRNA-concatenated plasmids and dCas9VP192 plasmid. Data represent mean ± SEM, n = 2–4 independent transfections.
dCas9VP192-Assisted Transcriptional Activation of Pluripotency Genes
We next tested the upregulation of additional endogenous pluripotency genes SOX2, NANOG, LIN28, KLF4, and CDH1 (E-Cadherin) in HEK293 cells by targeting dCas9VP192 to their promoters with five different gRNAs. Single gRNA transcriptional units were assembled by PCR and pooled and transfected together with dCas9VP192 encoding plasmid (Figure 1D). Expression of targeted endogenous factors could be detected by immunocytochemistry 72 hr after transfection (Figure 1E). PCR-gRNA fragments were then concatenated and cloned into plasmids using Golden Gate assembly (Figure 1D) and tested by transfection into HEK293 cells. Immunocytochemistry and qRT-PCR analysis showed activation of the endogenous-targeted genes (Figures 1F and 1G). OCT4, SOX2, NANOG, and CDH1 transcription was upregulated more than 20-fold over basal levels. LIN28 and KLF4 activation was more modest, possibly indicating either a less permissive chromatin state in the promoter of these genes or a high basal expression in HEK293 cells. These results showed that our method enables rapid testing of many gRNA candidates, which are easily generated with a single PCR. The subsequent concatenation of the successful ones in a single-step reaction allows for easy cloning of the guides into varying vectors.
To study the ability of the gRNA-encoding plasmids and the dCas9VP192 to activate gene expression in human skin fibroblasts, we cloned the dCas9VP192 activator and the concatenated guides into transiently replicating OriP-EBNA1-containing plasmids (Okita et al., 2011). Human skin fibroblasts were electroporated with dCas9VP192-EBNA and gRNA-encoding-EBNA plasmids. Activation of endogenous OCT4, SOX2, and LIN28A but not NANOG could be detected in cells 6 days after electroporation by immunocytochemistry and OCT4 and SOX2 activation by qRT-PCR (Figures 2A–2C).
Figure 2.

Functional Validation of dCas9 Activator in Reprogramming to Pluripotency
(A) Schematic representation of factors targeted in human skin fibroblasts with the dCasVP192 in transient electroporation.
(B and C) qRT-PCR and immunocytochemical detection of the targeted pluripotency factors 6 days after electroporation of human skin fibroblasts with dCas9VP192-EBNA and gRNA-encoding-EBNA. Scale bars represent 400 μm.
(D) Schematic representation of factors used in OCT4 replacement reprogramming protocol.
(E) Immunocytochemical detection of activation of endogenous OCT4 in human skin fibroblasts on day 6 of reprogramming. Cells were electroporated with pCXLE-GFP (-CTRL), episomal SOX2, KLF4, LIN28, and L-MYC plus dCas9VP192 and either OCT4 gRNAs plasmid or pCXLE-OCT4 (+CTRL). Scale bars represent 400 μm.
(F) Formation of AP+ colonies with dCas9VP192 replacing transgenic OCT4. Scale bars represent 200 μm.
(G) Comparison of episomal reprogramming efficiencies of adult human skin fibroblasts using either dCas9VP192 and OCT4 targeting guides or transgenic OCT4 together with transgenic SOX2, KLF4, LIN28, L-MYC, and p53 shRNA. Data represent mean ± SEM, n = 4 independent experiments, p = 0.177 using Student’s t test.
(H) Immunocytochemical detection of pluripotency markers TRA-1-81, NANOG, OCT4, and TRA-1-60 in passage 2 iPSCs derived from normal human fibroblasts with transgenic OCT4 replacement by dCas9VP192 and SKLM transgenes. Scale bars represent 400 μm.
(I) Immunocytochemical detection of markers of different germ layers in plated embryoid bodies of iPSCs derived from PNDM human fibroblasts by transgenic OCT4 replacement using dCas9VP192.FOXA2 (endoderm), VIMENTIN (mesoderm), and βIII-TUBULIN (ectoderm). Scale bars represent 200 μm for VIMENTIN.
See also Figure S2.
Reprogramming Factor OCT4 Replaced by dCas9VP192-Mediated Gene Activation
It has been previously demonstrated that dCas9 activators containing between four and ten VP16 repeats can be used to replace transgenic Oct4 in mouse fibroblast reprogramming by targeting the Oct4 distal enhancer (Gao et al., 2014). To determine whether dCas9VP192-mediated OCT4 activation would work in human cell reprogramming, we substituted the transgenic OCT4 plasmid in the episomal reprogramming protocol by dCas9VP192 and OCT4 promoter targeting guides (Okita et al., 2011; Yu et al., 2009) (Figure 2D). Activation of endogenous OCT4 could be detected in electroporated human skin fibroblasts expressing the dominant-negative form of p53 by day 6 of reprogramming (Figure 2E). Early colonies formed by day 9 of reprogramming and classical alkaline phosphatase positive (AP+) iPSC colonies were detected by day 18 (Figure 2F). Control conditions, where OCT4 guide plasmid had been replaced with GFP encoding plasmid, did not result in AP+ colony formation or endogenous OCT4 activation (Figure 2E).
We cloned short hairpin RNA for p53 into the dCas9VP192 plasmid to enable transient p53 inhibition in healthy fibroblasts during reprogramming. To compare the reprogramming efficiency of dCas9VP192-mediated endogenous OCT4 activation to transgenic OCT4 expression, we electroporated adult human skin fibroblasts with the reprogramming constructs and assessed the reprogramming efficiency by colony morphology and AP positivity. The colony formation efficiency was not found to differ significantly between the methods (0.0054% of plated cells for OCT4 activation versus 0.0021% for transgenic OCT4) (Figure 2G). Using sh-p53-dCas9VP192 construct and OCT4 targeting guides instead of exogenous OCT4 plasmid, we successfully derived iPSC from skin fibroblasts obtained from a 14-year-old donor with permanent neonatal diabetes mellitus (PNDM) and from a healthy 72-year-old donor (F72) (Figures S2A and S2B). The derived iPSC colonies were propagated for at least ten passages. All iPSC colonies expressed OCT4, NANOG, TRA-1-60, and TRA-1-81 (Figure 2H), and qRT-PCR analysis verified the expression of additional pluripotency markers (Figure S2A). iPSC lines were able to differentiate into the three germline derivatives positive for FOXA2 (endoderm), VIMENTIN (mesoderm), and βIII-TUBULIN (ectoderm) in embryoid bodies (Figure 2I). Clearance of the episomal reprogramming plasmids was confirmed in 12 of 17 iPSC clones in passage 2 (Figure S2B). Thus, human skin fibroblasts can be reprogrammed into iPSC by replacing OCT4 overexpression with dCas9 activator-mediated activation of endogenous OCT4.
Conditionally Stabilized dCas9 Activator
In order to control temporally the activity of the dCas9 activator, we fused the protein N-terminally to the E. coli-derived DHFR DD. This allowed us to control the stability of the activator in a TMP-dependent manner (Figure 3A). To test the construct in gene activation, we initially created a destabilized version of the dCas9VP48 activator (DDdCas9VP48) and generated HEK293 cells constitutively expressing DDdCas9VP48 with and without OCT4 targeting guides. In the absence of guides, OCT4 expression was not detected in the cells (Figure S3A). In the presence of guides, only some leaky OCT4 expression could be detected in a minor population of cells, which was drastically increased upon addition of TMP (Figure S3A).
Figure 3.

Gene Activation by Conditionally Stabilized dCas9VP192
(A) Schematic representation of DDdCas9VP192 stabilization. TMP, Trimethoprim.
(B) Immunocytochemical staining for OCT4 activation in selected HEK293 cells expressing DDdCas9VP192-T2A-GFP, TetON-dCas9VP192-T2A-GFP, or TetON-DDdCas9VP192-T2A-GFP activators and OCT4 targeting guides in the presence or absence of TMP and doxycycline. Scale bars represent 400 μm.
(C) Comparison of inducible DDdCas9, TetON-dCas9, and TetON-DDdCas9 activators in the upregulation of OCT4 levels in HEK293 cells. Cells were treated for up to 5 days with 1 μM TMP (for DDdCas9), 1 μg/ml of doxycycline (for TetON-dCas9), or both (for TetON-DDdCas9). Expression levels relative to H9 hESC are represented as mean ± SEM, n = 3 independently treated samples.
(D and E) Temporal activation of OCT4, SOX2 NANOG, and LIN28A induced with 1 μM TMP analyzed by qRT-PCR and OCT4 and LIN28A by immunocytochemistry in HEK293 cells expressing DDdCas9VP192 and gRNAs targeting these genes. Expression levels relative to H9 hESC are represented as mean ± SEM, n = 4 independently treated samples. Scale bars represent 400 μm.
(F and G) Temporal activation of OCT4, SOX2 NANOG, and LIN28A induced with 1 μM TMP analyzed by qRT-PCR and immunocytochemistry in primary human skin fibroblasts (F72) expressing DDdCas9VP192 and gRNAs targeting these genes. Expression levels relative to H9 hESC represented as mean ± SEM, n = 4 independently treated samples. Scale bars represent 400 μm.
See also Figure S3.
To compare the TMP-inducible dCas9 system with the more commonly used doxycycline inducible TetON system, we generated stable HEK293 cell lines with TetON-dCas9VP192, constitutive DDdCas9VP192, or TetON-DDdCas9VP192 and guides targeting the OCT4 promoter. Both TetON-dCas9VP192 and DDdCas9VP192 performed comparably (Figures 3B and 3C), presenting leaky expression at non-induced d0 samples, and upregulation of OCT4 after addition of doxycycline or TMP, to up to 5.2 and 6.6 times the expression levels of human embryonic stem cells (hESCs) accordingly. The combination of both TetON promoter and DHFR DD with the dCas9VP192 eliminated the nonspecific activation of OCT4 in the absence of doxycycline or TMP (Figures 3B and 3C).
We then generated HEK293 cells constitutively expressing the DDdCas9VP192 activator and guides targeting OCT4, SOX2, NANOG, and LIN28A promoters simultaneously. Some leaky activation of OCT4 and SOX2 but not NANOG or LIN28A could be detected in cells not treated with TMP (Figure 3D). After addition of TMP, the expression of OCT4, SOX2, and NANOG increased already by day 1, reaching 24%, 187%, and 41% of the expression levels measured in hESC (H9), respectively (Figure 3D). In contrast to the other factors, LIN28A upregulation was not detected by qRT-PCR (Figure 3D). Unlike the strongly activated OCT4, LIN28A expression could be detected in only a few cells by immunocytochemistry (Figure 3E).
To test the activator in primary human cells, we generated F72 human skin fibroblasts constitutively expressing the DDdCas9VP192 and OCT4, SOX2, NANOG, and LIN28A promoter targeting guides. Leaky activation was not detected in the fibroblasts in the absence of TMP (d0, Figures 3F and 3G). In the presence of TMP, OCT4, SOX2, and NANOG expression rose gradually reaching highest expression of 46%, 63%, and 7% of hESC levels, respectively, by the end of the 9-day period (Figure 3F). LIN28A upregulation was not robustly detected by qRT-PCR in fibroblasts (Figure 3F), but rare positive cells could be detected by immunocytochemistry (Figure 3G).
In an attempt to improve the activation efficiency of the target genes in the fibroblasts, we tested a set of inhibitors commonly used for iPSC induction to explore whether chemical compounds could improve dCas9VP192-mediated gene activation. However, treatment with these inhibitors did not significantly increase target gene activation efficiency (Figures S3B–S3D).
Activation of Endodermal and Pancreatic Transcription Factors by dCas9VP192
In order to examine the versatility of our method in activating different genes, we targeted a set of endodermal and pancreatic master transcription factors with dCas9VP192. We designed gRNAs targeting the proximal promoters of FOXA2, SOX17, GATA4, PDX1, and NKX6.1, PCR-amplified and tested them as described above. After preliminary confirmation of gene activation by immunocytochemistry, gRNAs were concatenated and validated in HEK293 cells. Analysis at 72 hr after transfection showed clear activation of the tested genes at the protein and mRNA level (Figure S4A).
Activation of endodermal transcription factor SOX17 using a dCas9 activator has been reported to work efficiently in hESCs (Kearns et al., 2014). To address whether our set of genes could be upregulated by dCas9VP192 in pluripotent stem cells, we transfected dCas9VP192 plus gRNA-encoding plasmids to human iPSCs. After 72 hr, activation of all the five tested genes was detected by immunocytochemistry and confirmed by qRT-PCR (Figures 4A, 4B, and S4C), demonstrating that dCas9VP192 can drive the expression of differentiation-relevant transcription factors also in pluripotent cells.
Figure 4.

Activation of Lineage-Specific Transcription Factors by dCas9VP192
(A) Immunocytochemical detection of dCas9VP192 targeted endodermal and pancreatic genes in hiPSC line HEL72.1. Scale bars represent 200 μm.
(B) qRT-PCR analysis of indicated targeted genes in hiPSC. Data represent mean ± SEM relative to non-transfected cells, n = 2 independent transfections. Analyses in (A) and (B) were performed 72 hr after delivery of gRNA-concatenated plasmids and dCas9VP192.
(C) Schematic representation of DDdCas9VP192-mediated activation of endodermal and pancreatic genes for differentiation of hiPSC.
(D) Immunocytochemistry of hESC H9 expressing DDdCas9VP192 and gRNAs for FOXA2, SOX17, PDX1, and NKX6.1 differentiated to definitive endoderm at day 3. hESC treated with 100 ng/ml Activin A (−TMP +AA), basal medium control (−TMP −AA) and basal medium with 1 μM TMP (+TMP −AA) stained for pancreatic progenitor markers PDX1, NKX6.1 (both targeted by DDdCas9VP192), and their downstream targets NKX2.2 and SOX9. Scale bars represent 200 μm.
(E) qRT-PCR analysis of cells treated as in (D) at day 3 of differentiation. Data represent mean ± SEM, n = 3 independently treated samples.
(F) Schematic representation of hPSC differentiation toward pancreatic progenitors using two inducible systems at different time points: TMP-inducible DDdCas9VP192 activating PDX1 and NKX6.1 endogenous expression (days 3 to 9) and DOX-inducible TetON-MAFA-ires-GFP overexpression system (days 7 to 9).
(G) qRT-PCR analysis of experiment depicted in (F). PDX1 and NKX6.1 expression levels are shown at days 7 and 9 in −/+ TMP-treated samples. MAFA and its downstream target GCK expression levels are shown at days 7 and 9 in −/+ DOX-treated samples. Data represent mean ± SEM, n = 3 independently treated samples.
(H) Immunocytochemistry for NKX6.1 and MAFA of hESC treated as depicted in (F). Scale bars represent 200 μm.
Next we generated a hESC line (H9) constitutively expressing DDdCas9VP192 activator and gRNAs targeting the endodermal transcription factors FOXA2 and SOX17 and the pancreatic factors PDX1 and NKX6.1 to test the applicability of this system in directed differentiation. The H9-DDdCas9VP192 cells were treated for 3 days following an endodermal differentiation protocol (Rezania et al., 2014) (Supplemental Experimental Procedures) either in the complete medium including Activin A (AA) (−TMP +AA condition), without Activin A as a control (−TMP −AA), or with 1 μM TMP and no Activin A (+TMP −AA) (Figure 4C). As expected, the cells treated for 3 days with AA differentiated into FOXA2+/SOX17+ definitive endoderm but did not express pancreatic progenitor marker genes (Figures 4D and S4D). In contrast, the simultaneous activation of endodermal and pancreatic transcription factors upon the addition of TMP upregulated pancreatic progenitor markers in the same time period. These cells expressed FOXA2, SOX17, PDX1, and NKX6.1 directly activated by DDdCas9VP192 (Figures 4D, 4E, and S4D). Importantly, also their downstream target transcription factors SOX9 and NKX2.2 were induced at both the mRNA and protein levels (Figures 4D and 4E).
To demonstrate the feasibility of using DDdCas9VP192 target gene activation in combination with other inducible expression systems, we generated a transgenic hESC line expressing DDdCas9VP192 activator, gRNAs targeting the pancreatic transcription factors PDX1 and NKX6.1, and a doxycycline-inducible overexpression cassette for mature beta cell transcription factor MAFA (TetON-MAFA-ires-GFP). The cells were treated with a pancreatic differentiation protocol for 9 days (Rezania et al., 2014). The experimental protocol is presented in Figure 4F. From days 3 to 9, the cells were treated with 1 μM TMP to induce PDX1 and NKX6.1 expression and from day 7 to 9 with 1 μg/ml doxycycline to induce MAFA (Figure 4F). At both days 7 and 9 of differentiation, PDX1 and NKX6.1 expression levels were increased in the +TMP condition, as shown by qRT-PCR and immunocytochemistry (Figures 4G and 4H). MAFA overexpression was robustly detected in the doxycycline-treated conditions at day 9, together with the upregulation of MAFA downstream target glucokinase (GCK) (Figures 4G and 4H) (Wang et al., 2007). These results demonstrate that the two inducible systems can be combined to control the expression of specific genes at distinct time points of the differentiation process.
Finally, we also tested these constructs in primary human foreskin fibroblasts. Endogenous gene activation was detected at protein and mRNA level (Figure S4B), albeit at low efficiency, indicating that dCas9VP192 can activate endodermal gene transcription even in somatic cells with a more restrictive epigenetic landscape.
Discussion
We report on the ability of dCas9 activators to upregulate the expression of endogenous human genes associated with reprogramming and maintenance of pluripotency as well as differentiation. We optimized the dCas9 activator efficiency and described a simplified workflow for testing and cloning the guide RNAs. The potent functional effect of the dCas9VP192 was demonstrated by replacing transgenic OCT4 with the activator in the reprogramming of primary human fibroblasts into iPSCs. Furthermore, we converted the activator into a conditionally stabilized version, which can be controlled by a small molecular compound, and demonstrated its function in controlling cell differentiation.
Our results show that increasing the number of VP16 transactivator repeats fused to dCas9 leads to improved activation of the endogenous OCT4. Using dCas9VP192 and five gRNAs, we obtained up to 70-fold OCT4 upregulation in HEK293 cells, in similar range of efficiency with previous studies (Cheng et al., 2013; Hu et al., 2014; Mali et al., 2013b). The fact that dCas9 activators can replace transgenic OCT4 as a reprogramming factor in human iPSC derivation speaks for the functional relevance of the gene activation and the potential for utilizing this approach for human cell type conversions. A similar approach has been previously demonstrated to work for mouse cell reprogramming (Chakraborty et al., 2014; Gao et al., 2014).
The potency of dCas9 activators varies between genes. We were not able to efficiently replace additional reprogramming factors, although their activation could be detected in fibroblasts. As NANOG and LIN28A activation was detected only in a minor population of cells, it is likely that the activation of these genes is stochastic by nature and limited by epigenetic barriers. Therefore, alternative approaches, for example, improved transactivation domains or catalytically active domains affecting the epigenetic state of the gene regions may be needed (Chavez et al., 2015; Hilton et al., 2015; Konermann et al., 2015; Maeder et al., 2013b). Additionally, the size of the construct could be reduced by incorporating multimerization sites for the activator domains, such as the recently reported SunTag peptide tail, which would implement an amplification step in the recruitment of transactivators to the proximal promoter that results in greatly enhanced gene activation (Tanenbaum et al., 2014). Targeting noncoding RNAs as well as genomic enhancer elements in addition to promoter proximal areas might also help in replacing transgenic factors with only dCas9 activators and gRNAs (Gao et al., 2014; Hilton et al., 2015).
The power of the dCas9 system for affecting gene expression lies in the multiplexing potential of the short gRNA sequences. Concatenation of gRNA expression cassettes using Golden Gate cloning has recently been described for multiplex genome editing (Sakuma et al., 2014) and gene activation (Kabadi et al., 2014). In contrast, our method avoids time-consuming traditional cloning for the initial gRNA-assembly step, being substituted by a simple PCR reaction. After gRNA efficiency validation, the gRNA-PCR fragments are concatenated with a single Golden Gate reaction, streamlining the generation of gRNA-encoding plasmids for different target genes. The small size of the gRNA construct compared with transgenic expression methods allows for efficient simultaneous targeting of multiple genomic loci and potentially full gene regulatory networks (GRNs). As dCas9 activators are targeting the endogenous genes for activation, activation of the proximal regulatory regions might benefit the establishment of proper transcriptional programs by affecting epigenetic barriers to the reprogramming process.
N-terminal fusion of the dCas9 activator with the DHFR DD leads to a functional TMP-dependent dCas9 activation. In terms of inducible gene transcription, DDdCas9 activator performs as efficiently as the TetON dCas9, with the advantage of not requiring an additional rtTA transactivator element. The DDdCas9 activator did, however, show a certain level of leakiness possibly due to incomplete protein degradation. This nonspecific activation can be controlled by combining the DDdCas9 activator with additional inducible systems like TetON, as shown in Figure 3B, or by controlling the expression level of the activator. Moreover, combining the S. pyogenes DDdCas9 activator with other orthogonal Cas9-derived conditional transcriptional modulators should allow for simultaneous temporal control over multiple gene expression programs (Davis et al., 2015; Esvelt et al., 2013; Ran et al., 2015; Zetsche et al., 2015).
Kearns et al. (2014) demonstrated that dCas9 transcriptional activators and repressors can be used to control human pluripotent stem cell differentiation by targeting endogenous SOX17 and OCT4. We have expanded the target gene list to additional key factors in the endodermal and pancreatic differentiation pathways. The induction of these genes may accelerate the differentiation of pluripotent stem cells to pancreatic progenitor-like cells to only 3 days, as compared with 10 or more days needed with current differentiation protocols (Rezania et al., 2014). The expression in these cells of genes downstream of FOXA2, SOX17, PDX1, and NKX6.1 indicates that simultaneous activation of a key gene set by DDdCas9 activator could help in establishing cell-type-specific GRNs (Arda et al., 2013). This constitutes a method to study and improve the differentiation of pluripotent stem cells to a particular lineage, for example, by activation of key regulators of lineage bifurcations or terminal differentiation. Moreover, DDdCas9-mediated transcriptional activation of genes can be utilized in combination with well-established overexpression methods, as we show here for doxycycline-inducible systems, adding an extra layer of tunable and temporally controlled gene expression.
In the future, combining the dCas9 activators with large-scale nucleic acid synthesis and high-throughput screening may enable the generation of gRNA arrays to activate simultaneously a determined set of genes (e.g., all the key components in a GRN), providing an unprecedented level of precision for controlling cellular reprogramming and differentiation (Gilbert et al., 2014; Shalem et al., 2014; Wang et al., 2014).
Experimental Procedures
Ethical Consent
The generation of the human induced pluripotent stem cell (hiPSC) lines used in this study was approved by the Coordinating Ethics Committee of the Helsinki and Uusimaa Hospital District (Nro 423/13/03/00/08).
Cell Culture
HEK293 cells, skin fibroblasts derived from a 14-year-old donor with PNDM, healthy 72-year-old skin fibroblast (F72), and human foreskin fibroblasts (HFFs, ATCC line CRL-2429) were cultured in fibroblast medium (DMEM; Life Technologies) containing 10% fetal bovine serum (FBS; Life Technologies), 2 mM GlutaMAX (Life Technologies), and 100 g/ml penicillin-streptomycin (Sigma). hESC and hiPSCs were cultured on Matrigel-coated dishes in E8 medium (Life Technologies, A1517001) in an incubator at 37°C and 5% CO2. The HEL72.1 hiPSC cell line was retrovirally derived from PNDM-patient fibroblasts, and in vitro differentiation through embryoid bodies was performed as described elsewhere (Vuoristo et al., 2013).
Guide RNA Design and Production
Guide RNA sequences were designed using the http://crispr.mit.edu web-based tool, targeting them to the proximal promoters (−400 to −50 base pairs from transcription start site) of the gene of interest. Possible guides were selected according to their off-target score and position. Guide RNA transcriptional units (gRNA-PCR) were prepared by PCR amplification and further concatenated using Golden Gate assembly. See Supplemental Experimental Procedures for further details.
Pluripotent Reprogramming
For human skin fibroblast reprogramming, 1 million cells were electroporated with Neon electroporator (Life Technologies), as described in Supplemental Experimental Procedures. A total of 6 μg of DNA was used per electroporation, containing 2 μg of pCXLE-dCas9VP192-GFP-shp53 and 1 μg of each other plasmids (guide encoding EBNA plasmids and reprogramming plasmids pCXLE-OCT4, pCXLE-hSK, and pCXLE-hUL). Afterward, electroporated cells were plated on gelatin-coated tissue culture dishes in fibroblast medium. On day 6, electroporated cells were split onto Matrigel-coated plates and cell culture medium was changed to a 3:1 mixture of hES and fibroblast-medium. After initial colony formation, the medium was changed to hES-medium (KnockOut DMEM; Life Technologies) supplemented with 20% knockout (KO) serum replacement (Life Technologies), 1% GlutaMAX, 0.1 mM beta-mercaptoethanol, 1% nonessential amino acids (Life Technologies), and 6 ng/ml basic fibroblast growth factor (bFGF, Life Technologies) supplemented with 0.25 mM sodium butyrate (NaB; Sigma). Medium was changed every other day. Emerging iPSC colonies were picked on Matrigel-coated dishes in E8 medium. P53 defective HFFs used in guide testing were infected with dominant-negative p53DD-encoding retrovirus (Addgene plasmid: 22729) and passaged twice before reprogramming. Clearance of episomal plasmids was demonstrated by PCR (Okita et al., 2011).
Author Contributions
D.B. and J.W. conceived the study and designed the experiments. D.B., J.W., and S.E. performed the experiments and analyzed the data. D.B., J.W., and T.O. wrote the manuscript with input from R.T. and K.W. T.O. provided funding for the work.
Acknowledgments
We thank Eila Korhonen and Heli Mononen for technical assistance. We are grateful to Alejandro Sarrión-Perdigones (Baylor College of Medicine) for advice on Golden Gate cloning. This study was supported by the Tekes Large Strategic Research Opening 3i Regeneration (project number 40395/13), the Academy of Finland, the Sigrid Juselius Foundation, the Novo Nordisk Foundation, and the Instrumentarium Science Foundation. D.B. and J.W. are supported by the Doctoral Program in Biomedicine, University of Helsinki.
Published: September 8, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2015.08.001.
Supplemental Information
References
- Arda H.E., Benitez C.M., Kim S.K. Gene regulatory networks governing pancreas development. Dev. Cell. 2013;25:5–13. doi: 10.1016/j.devcel.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron U., Gossen M., Bujard H. Tetracycline-controlled transcription in eukaryotes: novel transactivators with graded transactivation potential. Nucleic Acids Res. 1997;25:2723–2729. doi: 10.1093/nar/25.14.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikard D., Jiang W., Samai P., Hochschild A., Zhang F., Marraffini L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41:7429–7437. doi: 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S., Ji H., Kabadi A.M., Gersbach C.A., Christoforou N., Leong K.W. A CRISPR/Cas9-based system for reprogramming cell lineage specification. Stem Cell Reports. 2014;3:940–947. doi: 10.1016/j.stemcr.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A., Scheiman J., Vora S., Pruitt B.W., Tuttle M., P R Iyer E., Lin S., Kiani S., Guzman C.D., Wiegand D.J. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng A.W., Wang H., Yang H., Shi L., Katz Y., Theunissen T.W., Rangarajan S., Shivalila C.S., Dadon D.B., Jaenisch R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–1171. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis K.M., Pattanayak V., Thompson D.B., Zuris J.A., Liu D.R. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 2015;11:316–318. doi: 10.1038/nchembio.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvelt K.M., Mali P., Braff J.L., Moosburner M., Yaung S.J., Church G.M. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Tsang J.C.H., Gaba F., Wu D., Lu L., Liu P. Comparison of TALE designer transcription factors and the CRISPR/dCas9 in regulation of gene expression by targeting enhancers. Nucleic Acids Res. 2014;42:e155. doi: 10.1093/nar/gku836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton I.B., D’Ippolito A.M., Vockley C.M., Thakore P.I., Crawford G.E., Reddy T.E., Gersbach C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Lei Y., Wong W.-K., Liu S., Lee K.-C., He X., You W., Zhou R., Guo J.-T., Chen X. Direct activation of human and mouse Oct4 genes using engineered TALE and Cas9 transcription factors. Nucleic Acids Res. 2014;42:4375–4390. doi: 10.1093/nar/gku109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto M., Björklund T., Lundberg C., Kirik D., Wandless T.J. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem. Biol. 2010;17:981–988. doi: 10.1016/j.chembiol.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi A.M., Ousterout D.G., Hilton I.B., Gersbach C.A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014;42:e147. doi: 10.1093/nar/gku749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns N.A., Genga R.M., Enuameh M.S., Garber M., Wolfe S.A., Maehr R. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development. 2014;141:219–223. doi: 10.1242/dev.103341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder M.L., Linder S.J., Cascio V.M., Fu Y., Ho Q.H., Joung J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder M.L., Linder S.J., Reyon D., Angstman J.F., Fu Y., Sander J.D., Joung J.K. Robust, synergistic regulation of human gene expression using TALE activators. Nat. Methods. 2013;10:243–245. doi: 10.1038/nmeth.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P., Esvelt K.M., Church G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods. 2013;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K. A more efficient method to generate integration-free human iPS cells. Nat. Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Perez-Pinera P., Kocak D.D., Vockley C.M., Adler A.F., Kabadi A.M., Polstein L.R., Thakore P.I., Glass K.A., Ousterout D.G., Leong K.W. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezania A., Bruin J.E., Arora P., Rubin A., Batushansky I., Asadi A., O’Dwyer S., Quiskamp N., Mojibian M., Albrecht T. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014;32:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Sakuma T., Nishikawa A., Kume S., Chayama K., Yamamoto T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014;4:5400. doi: 10.1038/srep05400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G., Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum M.E.E., Gilbert L.A.A., Qi L.S.S., Weissman J.S.S., Vale R.D.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–646. doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuoristo S., Toivonen S., Weltner J., Mikkola M., Ustinov J., Trokovic R., Palgi J., Lund R., Tuuri T., Otonkoski T. A novel feeder-free culture system for human pluripotent stem cell culture and induced pluripotent stem cell derivation. PLoS ONE. 2013;8:e76205. doi: 10.1371/journal.pone.0076205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Brun T., Kataoka K., Sharma A.J., Wollheim C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia. 2007;50:348–358. doi: 10.1007/s00125-006-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Hu K., Smuga-Otto K., Tian S., Stewart R., Slukvin I.I., Thomson J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B., Volz S.E., Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015;33:139–142. doi: 10.1038/nbt.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.