

Abstract
Functionally significant polymorphisms in endothelial nitric oxide synthase (eNOS) and reduced vascular eNOS activity have been associated with increased human diabetic nephropathy (DN), but the pathogenic role of eNOS deficiency in the development of DN has not yet been confirmed. This study characterizes the severity of DN in eNOS−/− mice that were backcrossed to C57BLKS/J db/db mice. Although the severity of hyperglycemia was similar to C57BLKS/J db/db mice, by 26 wk, eNOS−/− C57BLKS/J db/db mice exhibited dramatic albuminuria, arteriolar hyalinosis, increased glomerular basement membrane thickness, mesangial expansion, mesangiolysis, and focal segmental and early nodular glomerulosclerosis. Even more remarkable, eNOS−/− C57BLKS db/db exhibited decreases in GFR to levels <50% of that in eNOS+/+ C57BLKS db/db, as confirmed by increased serum creatinine. In summary, eNOS−/− db/db mice provide the most robust model of type II DN that has been described to date and support a role for deficient eNOS-derived NO production in the pathogenesis of DN.
Diabetic hyperglycemia causes microvascular dysfunction, which contributes to the development of ESRD (1–3). In recent years, studies have focused first on the mesangial cell and more recently on the podocyte as both initiators and targets of diabetic nephropathy (DN) (4–6). Although these cells undoubtedly are involved in the development of DN, there is equally compelling reason to consider a role for endothelial cell dysfunction in the initiation and propagation of the characteristic glomerular lesions that are seen in DN.
Endothelial cell–derived vasodilators, such as nitric oxide (NO), seem to be important modulators of permeability in the vasculature. Inhibition or genetic deletion of endothelial NO synthase (eNOS, NOS III) induces opening of the interendothelial junctions and increases vascular permeability in microvascular beds, suggesting that NO production may play an important role in regulating the endothelial barrier function (7,8).
Functionally significant polymorphisms in eNOS have been recognized in human DN (9). In addition, vascular eNOS activity is altered in diabetes. NO products have been reported to be increased early after the onset of diabetes and may be involved in mediating renal vasodilation and hyperfiltration; however, with more prolonged diabetes, renal eNOS production decreases (10). The exact function of eNOS in the development of DN remains undetermined. These studies were designed not only to understand better the role of eNOS-mediated events in the development and progression of DN but also to determine whether eNOS deficiency results in a mouse model of DN that more closely approximates the functional and structural changes that are seen in human DN.
Materials and Methods
Animals
eNOS+/− mice initially on the C57/B6 background and db heterozygous mice on the C57BLKS/J (BKS) background were purchased from The Jackson Laboratory (Bar Harbor, ME). The eNOS+/− mice were backcrossed for 10 generations to the C57BLKS/J background and then crossed with db heterozygous mice. Genotyping was performed by PCR. Whenever possible, the same mice underwent all physiologic assessments (BP measurement, GFR, and albumin/creatinine ratio [ACR] measurement) and procurement of kidney tissue for histologic analysis.
BP Measurement
Systolic BP (SBP) was measured in conscious, trained mice at room temperature using a tail-cuff monitor (BP-2000 BP Analysis system, Visitech System, NC).
Determination of Blood Glucose and Creatinine
Blood glucose was determined using the OneTouch glucometer and test strips (LifeScan, Milpitas, CA). Serum creatinine was measured by a previously described HPLC method (11).
Urine Albumin and Creatinine
Spot urine was collected from individually caged mice using polycarbonate metabolic cages. Urinary albumin and creatinine excretion was determined using Albuwell-M kits (Exocell Inc., Philadelphia, PA).
Measurement of GFR
GFR was measured by a single-bolus FITC-inulin injection method, as described previously (12).
Histologic Analysis
Renal histology was assessed in mice that were killed at 24 to 26 wk of age. The perfused kidneys were removed and fixed overnight in 10% formalin at 4°C, and 3-μm-thick sections were stained with periodic acid-Schiff. Histologic evaluation was performed without knowledge of the identity of the various groups. A semiquantitative index was used to evaluate the degree of glomerular mesangial expansion and sclerosis. Each glomerulus on a single section was graded from 0 to 4, where 0 represents no lesion, and 1, 2, 3, and 4 represent mesangial matrix expansion or sclerosis, involving ≤25, 25 to 50, 50 to 75, or >75% of the glomerular tuft area, respectively.
Immunohistochemistry
Immunohistochemical detection of fibronectin staining was performed using an anti-fibronectin antibody (Sigma, St. Louis, MO). The sections then were incubated using the avidin-biotin-horseradish peroxidase technique (Elite Vectastain ABC kit; Vector Laboratories, Burlingame, CA), and staining was visualized using 3,3′-diaminobenzidine.
Statistical Analyses
All values are presented as means ± SEM. Bonferroni t test corrected for multiple comparisons was used for statistical analysis, and differences were considered significant at P < 0.05.
Results
Development of Type 2 Diabetes in eNOS−/− C57BLKS db/db Mice
We first investigated the timing of development of diabetes in this model. Hyperglycemia was evident by approximately 6 to 8 wk of age. The fasting blood glucose level in db/db mice was significantly higher than that in the lean controls at 26 wk of age (P < 0.05; Figure 1A). eNOS deletion did not alter fasting blood glucose in either db/db or control mice. As was described previously (13,14), moderate hypertension was observed in lean mice with genetic deletion of eNOS by age 24 to 28 wk (Figure 1B). SBP was significantly greater in eNOS−/− C57BLKS/J db/db mice (eNOS−/−db/db) than in db/db C57BLKS/J mice (db/db; 158 ± 10 versus 110 ± 3; n = 4; P < 0.05) but was not significantly higher than in nondiabetic eNOS−/− C57BLKS/J mice (eNOS−/−; 143 ± 7 mmHg; n = 5; NS).
Figure 1.

Metabolic and physiologic parameters. (A) Fasting blood glucose at 24 to 26 wk in db/db mice and age-matched controls ± eNOS−/−. *P < 0.05 versus control and eNOS−/−. Values are means ± SE of at least eight mice. (B) Systolic BP. *P < 0.05 eNOS−/− or eNOS−/− db/db versus control or db/db. Values are means ± SE of at least four mice.
Albuminuria in eNOS−/− C57BLKS/J db/db Mice
Albuminuria is a hallmark of DN. At 26 wk of age, moderate albuminuria was observed in db/db (262 ± 24 μg albumin/mg creatinine; n = 10) and eNOS−/− (387 ± 27; n = 19), compared with control mice (43 ± 9; n = 8; P < 0.05; Figure 2A). In contrast, eNOS−/−db/db exhibited a marked increase in spot urine ACR (1503 ± 176; n = 12; P < 0.001 compared with their age-matched controls, db/db, or eNOS−/−). Urine ACR was elevated by age 8 wk in eNOS−/−db/db (data not shown).
Figure 2.

Functional renal parameters. (A) Urinary albumin/creatinine ratio (ACR) at 24 to 26 wk of age. Values are means ± SE of at least eight mice. *P < 0.05 versus respective controls, db/db, and eNOS−/−. (B) GFR in diabetic mice. *P < 0.05 versus control, db/db, and eNOS−/−. Values are means ± SE of at least eight mice. (C) Correlation of serum creatinine with GFR.
GFR in eNOS−/− C57BLKS/J db/db Mice
HPLC serum creatinine determined at 26 wk was 0.10 ± 0.001 (n = 5) in the control lean mice, 0.11 ± 0.01 (n = 7) in db/db, 0.115 ± 0.001 (n = 9) in eNOS−/−, and 0.17 ± 0.02 (n = 5) in eNOS−/−db/db (P < 0.05). GFR was examined in diabetic mice and age-matched controls using FITC-inulin clearance. Given the disparity in body weights between the control (lean) and diabetic (obese) mice, GFR was calculated per mouse as well as per gram of body weight. At 26 wk of age, GFR determined per mouse was numerically increased in db/db (366 ± 39 μl/min per mouse; n = 8) but was not statistically significant compared with age-matched controls (331 ± 25; n = 10). In contrast, the GFR of eNOS−/−db/db (164 ± 27; n = 10) was significantly decreased compared with db/db (P < 0.001) and eNOS−/− (265 ± 21; n = 12; P < 0.05; Figure 2B). At 26 wk, the body weights of the obese mice were more than double those of the aged-matched controls (lean controls 23.4 ± 0.9 g; db/db 59.4 ± 2.4; eNOS−/− 26.5 ± 1.5; eNOS−/−db/db 58.1 ± 2.3). GFR per gram of body weight were as follows: lean controls 14.1 ± 1.1 μl/min per g; db/db 6.2 ± 0.6; eNOS−/− 9.9 ± 0.6; and eNOS−/−db/db 2.5 ± 0.4. Given that body fat represents approximately 50% of total body weight in db/db mice (15), determination of GFR/body weight in the obese mice will underestimate the true GFR. Serum creatinines correlated with the GFR measurements in mice from all groups in which both measurements were obtained (n = 23; r2 = 0.699, P < 0.0001; Figure 2C).
Histologic Analysis
Kidneys were assessed by light and electron microscopy (Figure 3). Compared with control (Figure 3A, a), moderate mesangial expansion was observed in glomeruli of db/db (Figure 3A, b and e) and eNOS−/− (Figure 3A, c) at 26 wk of age. In contrast, marked mesangial expansion, focal nodular sclerosis, and mesangiolysis (Figure 3A, d and f) as well as arteriolar hyalinosis (Figure 3A, f) were noted in eNOS−/−db/db glomeruli at 26 wk of age. There was only minimal tubulointerstitial fibrosis in eNOS−/− db/db kidneys. Immunohistochemical examination also revealed minimal accumulation of fibronectin in the mesangial regions of control (Figure 3B, a), db/db (Figure 3B, b), and eNOS−/− (Figure 3B, c), compared with the striking fibronectin accumulation that was observed in the eNOS−/−db/db glomeruli (Figure 3B, d).
Figure 3.
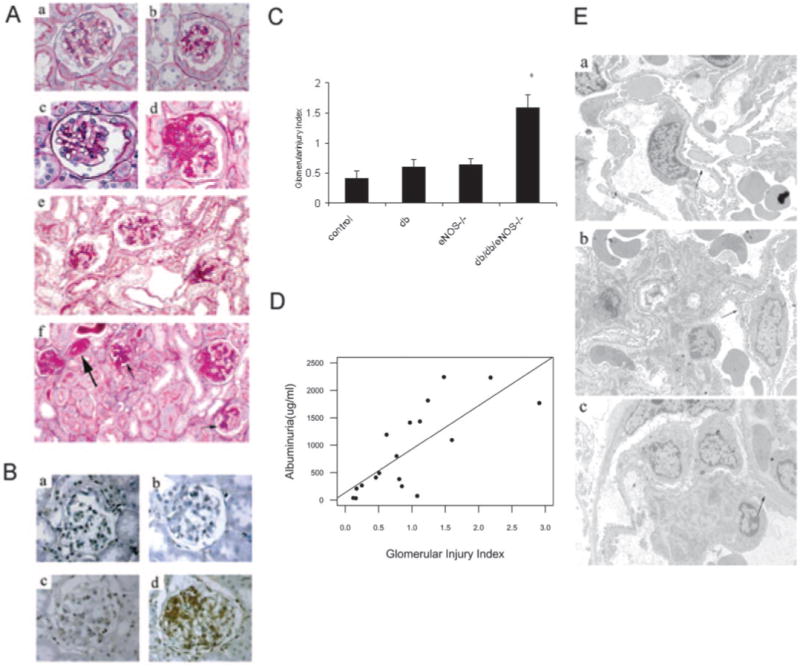
Glomerular histopathology. (A) Representative glomerular lesions of diabetic mouse kidneys at 24 to 26 wk: control (a); db/db (b and e); eNOS−/− (c); eNOS−/−db/db (d and f). (f) Arteriolar hyalinosis (big arrow) and early nodular glomerulosclerosis (small arrow) in eNOS−/− db/db mice (periodic acid-Schiff). (B) Glomerular fibronectin expression in control (a), db/db (b), eNOS−/− (c), and eNOS−/− db/db mice (d). (C) Glomerular injury scores in diabetic mice. Mesangial expansion and sclerosis were scored on a scale from 0 to 4+ as described in The Materials and Methods section. *P < 0.05, eNOS−/− db/db (24 to 26 wk) mice versus age-matched controls, db/db, and eNOS−/− mice. (D) Correlation of glomerular injury index with albuminuria. (E) Electron micrographs of glomeruli (16 wk). Opposed arrows indicate GBM. GBM thickening was notable in eNOS−/−db/db mice (c) compared with control (a) and db/db mice (b). Magnifications: ×400 in A, a through d, and B; ×200 in A, e and f.
Glomerular injury that was assessed semiquantitatively was significantly increased in eNOS−/−db/db at 24 to 26 wk of age, as compared with other groups (n = 6 to 9/group; P < 0.05; Figure 3C). The amount of albuminuria correlated with the glomerular injury index in mice from all groups in which both measures were obtained (n = 19; r2 = 0.58, P < 0.0002; Figure 3D).
Glomerular ultrastructure was examined by electron microscopy at age 16 wk (Figure 3E). Compared with control mice (270 ± 47 nm; Figure 3E, a), at this age, there was no thickening of glomerular basement membrane (GBM) in db/db mice (240 ± 76 nm; Figure 3E, b), in contrast to the markedly thickened GBM that was seen in eNOS−/−db/db mice (322 ± 57 nm; Figure 3E, c). No electron-dense deposits were present.
Discussion
Human DN is a characteristic clinical syndrome that consists of albuminuria, progressively declining GFR, and defined histopathologic features that include thickening of the GBM and mesangial expansion, often with nodular glomerulosclerosis, arteriolar hyalinosis, and tubulointerstitial fibrosis (16,17). These features would be important features of a robust animal model of DN (18).
Previous animal models of diabetic kidney disease have manifested albuminuria and early renal pathologic changes such as GBM thickening and mesangial expansion but with only minimal or inconsistent expression of other characteristic histopathologic features such as arteriolar hyalinosis and nodular glomerulosclerosis. Furthermore, the failure of previous models to manifest a decline in renal function has called into question their utility as analogues of human DN (16,18,19).
eNOS−/− C57BLKS db/db mice not only developed striking albuminuria and characteristic pathologic changes of DN but also exhibited remarkably decreased GFR on the basis of inulin clearance and serum creatinine. Furthermore, the onset and the progression of the pathologic features in eNOS−/−db/db mice were rapid, which makes this DN model attractive as a potential platform for testing efficacy of new therapeutic agents.
The eNOS−/− db/db mice had significant obesity and hypertension, which typically is seen in humans with type 2 diabetes (20,21). The mutated leptin receptor in db/db mice leads to defective signaling of leptin in the hypothalamus and results in persistent hyperphagia and obesity and development of peripheral insulin resistance (19). In addition, when the db/db mutation is on the C57BLKS background, mice develop a late (>4 mo) insulitis so that insulin levels are inappropriately low for the level of hyperglycemia (22). eNOS−/− mice were reported previously to develop moderate systemic hypertension (13,14). This increased SBP was slightly greater in diabetic eNOS−/−db/db mice, although the results did not reach statistical significance.
There is growing evidence that endothelial cell dysfunction contributes to hypertension and microvascular complications of diabetes (2,23). A major defense of endothelial cells against vascular injury is eNOS, which generates NO in the presence of the substrate l-arginine, and the co-factor (6R)-5,6,7,8-tetrahydro-l-biopterin (BH4). NADPH oxidases are major sources of reactive oxygen species in endothelium and are activated in animal models of hypertension and diabetes. Superoxide reacts avidly with vascular NO· to form peroxynitrite, leading to BH4 oxidation and subsequent promotion of superoxide production by eNOS itself, so-called “eNOS uncoupling” (24,25). Uncoupled eNOS is detected in conditions that are associated with oxidant stress, hypertension, and diabetes (23).
Human DN progresses through several pathophysiologic stages, initially characterized by early hyperfiltration and hypertrophy followed by microalbuminuria and mesangial expansion, and then overt proteinuria, sclerosis, and a progressive decline of GFR (16,17). Early in diabetes, increased endothelial NO production may contribute to the observed hyperfiltration and microalbuminuria (10), but with advancing nephropathy, there is progressive NO deficiency. Hyperglycemia, advanced glycosylation end products, increased oxidant stress, endogenous inhibitors of NO such as asymmetric dimethylarginine, activation of protein kinase C, and TGF-β all are potential contributors to decreased NO production and/or eNOS uncoupling (26,27).
In humans, the eNOS gene is found on chromosome 7q. Genome-wide scans have indicated that regions on 7q, 18q, and 22q may influence proteinuria and/or development of DN in type 2 diabetes (27). Of note, evidence for a region on 7q overlapped all studies (28). Recent studies have reported an association between eNOS polymorphisms that lead to de creased eNOS expression and the development of advanced DN in both patients with type 1 (29-31) and with type 2 diabetes (32,33). Other studies also have found an association of these polymorphisms with nondiabetic causes of ESRD (9,34). However, not all studies have detected an association of disease with these eNOS polymorphisms (35-39).
Although short-term studies have examined the effect of administration of NOS inhibitors in experimental models of diabetes (10,40), we are not aware of any published studies to date that have examined long-term chronic administration (4 to 6 mo). Whereas administration of NOS inhibitors alone produces renal lesions that are consistent with ischemic nephropathy with minimal glomerulosclerosis (41), chronic administration of nonspecific NOS inhibitors does accelerate glomerulosclerosis in the remnant model of rat glomerulopathy (42), although similar studies are yet to be performed in mouse models. Therefore, it is possible that NOS inhibitors may impart a similar acceleration of injury in experimental diabetic models, although a potential advantage of the current genetic model in dissecting pathophysiologic mechanisms of DN is that eNOS is selectively deleted while other NOS isoforms remain active.
Conclusion
Our results demonstrate that eNOS−/−db/db mice exhibit significant albuminuria and glomerular pathology that parallel the later phase of DN in patients with type 2 diabetes and include arteriolar hyalinosis, mesangial expansion, thickening of GBM, and focal segmental and early nodular glomerulosclerosis. This model should prove useful for studying the role of endothelial dysfunction in development of DN and in facilitating the development of new diagnostic and therapeutic interventions.
Acknowledgments
This work was supported by National Institutes of Health grants DK061018 (Mouse Models of Diabetic Complications Consortium), DK39261 (Vanderbilt George O'Brien Kidney and Urologic Diseases Center), and DK59637 (Vanderbilt Mice Metabolic Phenotype Center) and funds from the Department of Veterans Affairs.
We thank Dr. Stephen Dunn for measuring plasma creatinine levels and Xiaofeng Fan for technical support.
References
- 1.Futrakul N, Butthep P, Vongthavarawat V, Futrakul P, Sirisalipoch S, Chaivatanarat T, Suwanwalaikorn S. Early detection of endothelial injury and dysfunction in conjunction with correction of hemodynamic maladjustment can effectively restore renal function in type 2 diabetic nephropathy. Clin Hemorheol Microcirc. 2006;34:373–381. [PubMed] [Google Scholar]
- 2.Jawa A, Nachimuthu S, Pendergrass M, Asnani S, Fonseca V. Impaired vascular reactivity in African-American patients with type 2 diabetes mellitus and microalbuminuria or proteinuria despite angiotensin-converting enzyme inhibitor therapy. J Clin Endocrinol Metab. 2006;91:31–35. doi: 10.1210/jc.2005-1632. [DOI] [PubMed] [Google Scholar]
- 3.Wautier JL, Schmidt AM. Protein glycation: A firm link to endothelial cell dysfunction. Circ Res. 2004;95:233–238. doi: 10.1161/01.RES.0000137876.28454.64. [DOI] [PubMed] [Google Scholar]
- 4.Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol. 2003;14:1358–1373. doi: 10.1097/01.asn.0000065640.77499.d7. [DOI] [PubMed] [Google Scholar]
- 5.Wendt TM, Tanji N, Guo J, Kislinger TR, Qu W, Lu Y, Bucciarelli LG, Rong LL, Moser B, Markowitz GS, Stein G, Bierhaus A, Liliensiek B, Arnold B, Nawroth PP, Stern DM, D'Agati VD, Schmidt AM. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White KE, Bilous RW. Structural alterations to the podocyte are related to proteinuria in type 2 diabetic patients. Nephrol Dial Transplant. 2004;19:1437–1440. doi: 10.1093/ndt/gfh129. [DOI] [PubMed] [Google Scholar]
- 7.Predescu D, Predescu S, Shimizu J, Miyawaki-Shimizu K, Malik AB. Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am J Physiol Lung Cell Mol Physiol. 2005;289:L371–L381. doi: 10.1152/ajplung.00175.2004. [DOI] [PubMed] [Google Scholar]
- 8.Sessa WC. eNOS at a glance. J Cell Sci. 2004;117:2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- 9.Noiri E, Satoh H, Taguchi J, Brodsky SV, Nakao A, Ogawa Y, Nishijima S, Yokomizo T, Tokunaga K, Fujita T. Association of eNOS Glu298Asp polymorphism with end-stage renal disease. Hypertension. 2002;40:535–540. doi: 10.1161/01.hyp.0000033974.57407.82. [DOI] [PubMed] [Google Scholar]
- 10.Prabhakar SS. Role of nitric oxide in diabetic nephropathy. Semin Nephrol. 2004;24:333–344. doi: 10.1016/j.semnephrol.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 11.Yuen PST, Dunn SR, Miyaji T, Yasuda H, Sharma K, Star RA. A simplified method for HPLC determination of creatinine in mouse serum. Am J Physiol Renal Physiol. 2004;286:F1116–F1119. doi: 10.1152/ajprenal.00366.2003. [DOI] [PubMed] [Google Scholar]
- 12.Qi Z, Whitt I, Mehta A, Jin J, Zhao M, Harris RC, Fogo AB, Breyer MD. Serial determination of glomerular filtration rate in conscious mice using FITC-inulin clearance. Am J Physiol Renal Physiol. 2004;286:F590–F596. doi: 10.1152/ajprenal.00324.2003. [DOI] [PubMed] [Google Scholar]
- 13.Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Vliet BN, Chafe LL, Montani JP. Characteristics of 24 h telemetered blood pressure in eNOS-knockout and C57Bl/6J control mice. J Physiol. 2003;549:313–325. doi: 10.1113/jphysiol.2003.041897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cox JE, Powley TL. Development of obesity in diabetic mice pair-fed with lean siblings. J Comp Physiol Psychol. 1977;91:347–358. doi: 10.1037/h0077322. [DOI] [PubMed] [Google Scholar]
- 16.Breyer MD, Bottinger E, Brosius FC, 3rd, Coffman TM, Harris RC, Heilig CW, Sharma K. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. doi: 10.1681/ASN.2004080648. [DOI] [PubMed] [Google Scholar]
- 17.Parving HH, Mauer M, Ritz E. Diabetic nephropathy. In: Brenner BM, editor. The Kidney. 7th. Philadelphia: WB Saunders; 2004. pp. 1777–1818. [Google Scholar]
- 18.Breyer MD, Bottinger E, Brosius FC, Coffman TM, Fogo A, Harris RC, Heilig CW, Sharma K. Diabetic nephropathy: Of mice and men. Adv Chronic Kidney Dis. 2005;12:128–145. doi: 10.1053/j.ackd.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284:F1138–F1144. doi: 10.1152/ajprenal.00315.2002. [DOI] [PubMed] [Google Scholar]
- 20.Sarafidis PA, Ruilope LM. Insulin resistance, hyperinsulinemia, and renal injury: Mechanisms and implications. Am J Nephrol. 2006;26:232–244. doi: 10.1159/000093632. [DOI] [PubMed] [Google Scholar]
- 21.Dobesh PP. Managing hypertension in patients with type 2 diabetes mellitus. Am J Health Syst Pharm. 2006;63:1140–1149. doi: 10.2146/ajhp050385. [DOI] [PubMed] [Google Scholar]
- 22.Coleman DL, Leiter EH, Schwizer RW. Therapeutic effects of dehydroepiandrosterone (DHEA) in diabetic mice. Diabetes. 1982;31:830–833. doi: 10.2337/diab.31.9.830. [DOI] [PubMed] [Google Scholar]
- 23.Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 24.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: Critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2005;102:9056–9061. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prabhakar SS. Tetrahydrobiopterin reverses the inhibition of nitric oxide by high glucose in cultured murine mesangial cells. Am J Physiol Renal Physiol. 2001;281:F179–F188. doi: 10.1152/ajprenal.2001.281.1.F179. [DOI] [PubMed] [Google Scholar]
- 26.Saran R, Novak JE, Desai A, Abdulhayoglu E, Warren JS, Bustami R, Handelman GJ, Barbato D, Weitzel W, D'Alecy LG, Rajagopalan S. Impact of vitamin E on plasma asymmetric dimethylarginine (ADMA) in chronic kidney disease (CKD): A pilot study. Nephrol Dial Transplant. 2003;18:2415–2420. doi: 10.1093/ndt/gfg406. [DOI] [PubMed] [Google Scholar]
- 27.Tarnow L, Hovind P, Teerlink T, Stehouwer CD, Parving HH. Elevated plasma asymmetric dimethylarginine as a marker of cardiovascular morbidity in early diabetic nephropathy in type 1 diabetes. Diabetes Care. 2004;27:765–769. doi: 10.2337/diacare.27.3.765. [DOI] [PubMed] [Google Scholar]
- 28.Ng DP, Krolewski AS. Molecular genetic approaches for studying the etiology of diabetic nephropathy. Curr Mol Med. 2005;5:509–525. doi: 10.2174/1566524054553504. [DOI] [PubMed] [Google Scholar]
- 29.Frost D, Chitu J, Meyer M, Beischer W, Pfohl M. Endothelial nitric oxide synthase (ecNOS) 4 a/b gene polymorphism and carotid artery intima-media thickness in type-1 diabetic patients. Exp Clin Endocrinol Diabetes. 2003;111:12–15. doi: 10.1055/s-2003-37494. [DOI] [PubMed] [Google Scholar]
- 30.Ksiazek P, Wojewoda P, Muc K, Buraczynska M. Endothelial nitric oxide synthase gene intron 4 polymorphism in type 2 diabetes mellitus. Mol Diagn. 2003;7:119–123. doi: 10.1007/BF03260027. [DOI] [PubMed] [Google Scholar]
- 31.Zanchi A, Moczulski DK, Hanna LS, Wantman M, Warram JH, Krolewski AS. Risk of advanced diabetic nephropathy in type 1 diabetes is associated with endothelial nitric oxide synthase gene polymorphism. Kidney Int. 2000;57:405–413. doi: 10.1046/j.1523-1755.2000.00860.x. [DOI] [PubMed] [Google Scholar]
- 32.Li C, Dong Y, Lu W. The association between polymorphism of endothelial nitric oxide synthase gene and diabetic nephropathy [in Chinese] Zhonghua Nei Ke Za Zhi. 2001;40:729–732. [PubMed] [Google Scholar]
- 33.Neugebauer S, Baba T, Watanabe T. Association of the nitric oxide synthase gene polymorphism with an increased risk for progression to diabetic nephropathy in type 2 diabetes. Diabetes. 2000;49:500–503. doi: 10.2337/diabetes.49.3.500. [DOI] [PubMed] [Google Scholar]
- 34.Asakimori Y, Yorioka N, Yamamoto I, Okumoto S, Doi S, Hirai T, Taniguchi Y. Endothelial nitric oxide synthase intron 4 polymorphism influences the progression of renal disease. Nephron. 2001;89:219–223. doi: 10.1159/000046071. [DOI] [PubMed] [Google Scholar]
- 35.Degen B, Schmidt S, Ritz E. A polymorphism in the gene for the endothelial nitric oxide synthase and diabetic nephropathy. Nephrol Dial Transplant. 2001;16:185. doi: 10.1093/ndt/16.1.185. [DOI] [PubMed] [Google Scholar]
- 36.Freedman BI, Yu H, Anderson PJ, Roh BH, Rich SS, Bowden DW. Genetic analysis of nitric oxide and endothelin in end-stage renal disease. Nephrol Dial Transplant. 2000;15:1794–1800. doi: 10.1093/ndt/15.11.1794. [DOI] [PubMed] [Google Scholar]
- 37.Lin S, Qu H, Qiu M. Allele A in intron 4 of ecNOS gene will not increase the risk of diabetic nephropathy in type 2 diabetes of Chinese population. Nephron. 2002;91:768. doi: 10.1159/000065048. [DOI] [PubMed] [Google Scholar]
- 38.Rippin JD, Patel A, Belyaev ND, Gill GV, Barnett AH, Bain SC. Nitric oxide synthase gene polymorphisms and diabetic nephropathy. Diabetologia. 2003;46:426–428. doi: 10.1007/s00125-003-1046-3. [DOI] [PubMed] [Google Scholar]
- 39.Shimizu T, Onuma T, Kawamori R, Makita Y, Tomino Y. Endothelial nitric oxide synthase gene and the development of diabetic nephropathy. Diabetes Res Clin Pract. 2002;58:179–185. doi: 10.1016/s0168-8227(02)00156-0. [DOI] [PubMed] [Google Scholar]
- 40.Komers R, Anderson S. Paradoxes of nitric oxide in the diabetic kidney. Am J Renal Physiol. 2003;284:F1121–F1137. doi: 10.1152/ajprenal.00265.2002. [DOI] [PubMed] [Google Scholar]
- 41.Ribeiro MO, Antunes E, de Nucci G, Lovisolo SM, Zatz R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension. 1992;20:298–303. doi: 10.1161/01.hyp.20.3.298. [DOI] [PubMed] [Google Scholar]
- 42.Fujihara CK, De Nucci G, Zatz R. Chronic nitric oxide synthase inhibition aggravates glomerular injury in rats with subtotal nephrectomy. J Am Nephrol. 1995;5:1498–1507. doi: 10.1681/ASN.V571498. [DOI] [PubMed] [Google Scholar]