

Summary
Ecteinascidin 743 (ET-743, Yondelis) is a clinically approved chemotherapeutic natural product isolated from the Caribbean mangrove tunicate Ecteinascidia turbinata. Researchers have long suspected that a microorganism may be the true producer of the anti-cancer drug, but its genome has remained elusive due to our inability to culture the bacterium in the laboratory using standard techniques. Here, we sequenced and assembled the complete genome of the ET-743 producer, Candidatus Endoecteinascidia frumentensis, directly from metagenomic DNA isolated from the tunicate. Analysis of the ~631 kb microbial genome revealed strong evidence of an endosymbiotic lifestyle and extreme genome reduction. Phylogenetic analysis suggested that the producer of the anti-cancer drug is taxonomically distinct from other sequenced microorganisms and could represent a new family of Gammaproteobacteria. The complete genome has also greatly expanded our understanding of ET-743 production and revealed new biosynthetic genes dispersed across more than 173 kb of the small genome. The gene cluster’s architecture and its preservation demonstrate that the drug is likely essential to the interactions of the microorganism with its mangrove tunicate host. Taken together, these studies elucidate the lifestyle of a unique, and pharmaceutically-important microorganism and highlight the wide diversity of bacteria capable of making potent natural products.
Introduction
Natural products are a critical source of pharmaceuticals and lead compounds in drug discovery efforts (Newman and Cragg, 2012). Over the last several decades, scientists have isolated thousands of biologically active metabolites from terrestrial and marine macroorganisms, including plants and animals.. Mounting evidence suggests that microbial symbionts may be the actual producers of many of these natural products (Piel, 2009).
Currently, the vast majority of drug-producing symbiotic microbes remain uncharacterized. Most fall into the > 99% of prokaryotic species currently incapable of being cultured in the laboratory, hindering their study (Staley and Konopka, 1985; Piel, 2009). Identifying these symbionts and understanding their genetic, biochemical, and metabolic characteristics is critical for advancing fundamental knowledge and potential applications. Many symbiont-derived secondary metabolites can only be isolated in low yields from their hosts, making large-scale production for pharmaceutical purposes unsustainable from both an economic and environmental perspective. Although total synthesis can sometimes solve the supply problem, it can be costly and fails to address our understanding of the unique biosynthetic processes that are mediated by these elusive microbes. Sequencing and analysis of symbiont genomes could provide insight into the lifestyles of these poorly understood bacteria, illuminate possible host-free cultivation methods, and provide a route to economical and sustainable large-scale production with the opportunity for genetic manipulation to produce novel drug analogs.
The chemotherapeutic compound ET-743 (1, Yondelis, Trabectedin) is one of the most important natural products suspected to be of symbiotic origin. Isolated directly from the mangrove tunicate Ecteinascidia turbinata (Fig. 1A and B), the biological activity of the drug against cancer cells has inspired over 40 years of research (Lichter et al., 1975; Rinehart et al., 1990). Currently, ET-743 is clinically approved in Europe against soft tissue sarcoma and relapsed ovarian cancer and is currently in phase III trials as an anticancer therapeutic in the United States (McLaughlin, 2015).
Figure 1.
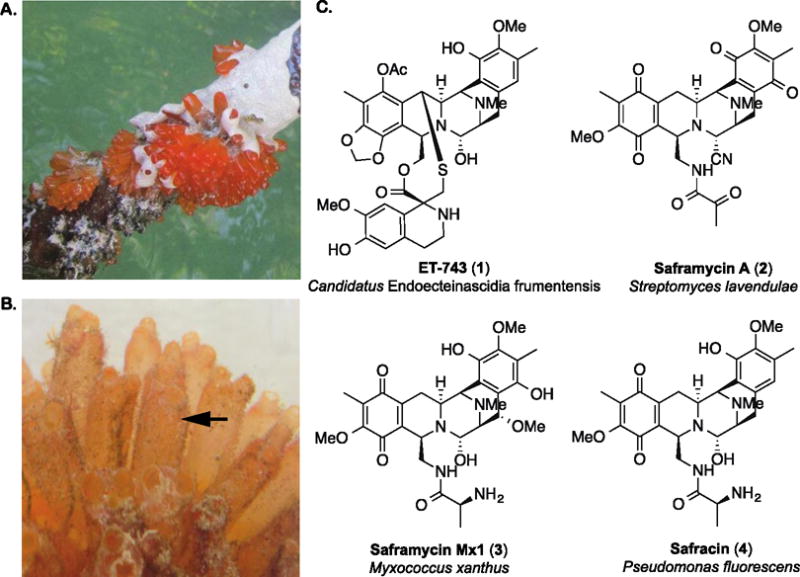
A. Tunicate colonies growing on the root of a mangrove tree in the Florida Keys. B. A tunicate colony composed of individual zooids (indicated by arrow). In this study, we sequenced the metagenomic DNA from four zooids. C. The chemotherapeutic compound ET-743 (1) and three natural products from cultivable bacteria that share a similar tetrahydroisoquinoline core.
The tetrahydroisoquinoline alkaloid natural products saframycin A (2), saframycin Mx1 (3) and safracin (4) are derived from three distinct cultivable bacteria and are structurally similar to ET-743, supporting a prokaryotic origin for the drug (Fig. 1C). Studies of the mangrove tunicate over a decade ago identified the potential intracellular Gammaproteobacterium Candidatus Endoecteinascida frumentensis to be the most prevalent member of the host microbial consortium (Moss et al., 2003; Pérez-Matos et al., 2007) and the only microorganism consistently associated with tunicates in both the Mediterranean and Caribbean seas (Pérez-Matos et al., 2007). A metagenomically-derived contig containing a partial ET-743 biosynthetic gene cluster was later indirectly linked to a separate contig bearing the 16S rRNA gene sequence for Ca. E. frumentensis through analysis of %G+C content and codon usage (Rath et al., 2011). Cultivation of the producing bacterium has so far been unsuccessful (Moss et al., 2003; Pérez-Matos et al., 2007), and aquaculture (Carballo et al., 2000) of the host tunicate and total synthesis (Corey et al., 1996) have also failed to provide sustainable access to the drug for clinical applications. ET-743 is therefore currently generated by a lengthy semisynthetic process starting from fermentation-derived cyanosafracin B (Cuevas and Francesch, 2009).
In this study, we utilized next generation sequencing technologies to expand our understanding of ET-743 biosynthesis and uncover the complete genome of the microorganism responsible the drug’s production. Analysis of phylogenetic markers and protein coding genes suggests that the microbe belongs to a novel family of Gammaproteobacteria. In-depth genomic analysis also provides initial insights into the endosymbiotic lifestyle of Ca. E. frumentensis, the ecological role of its sole secondary metabolic pathway, and key information that may provide access to host-cell free growth in the laboratory.
Results and Discussion
Overview of Samples and Dataset
The colonies of E. turbinata consist of thick bundles of individual zooids connected by a network of stolons that enable adherence of the animal to a stable surface. Our laboratory previously isolated metagenomic DNA from individual zooids and uncovered a 35 kb gene cluster responsible for ET-743 biosynthesis using 454 pyrosequencing (Rath et al., 2011). In the present study, we isolated additional metagenomic DNA from four zooids obtained from two colonies (Fig. S1). We shotgun sequenced the resulting DNA samples using Illumina HiSeq technology and assembled the data into contigs. The four zooids provided metagenome datasets each containing over 800 Mbp of sequence (Table S1).
We assigned the assembled contigs to taxonomic bins using tetranucleotide frequency with emergent self-organizing maps (tetra-ESOM) as previously described (Fig. S2) (Dick et al., 2009). Each of the four metagenomic samples possessed a single bin containing both the previously identified partial ET-743 biosynthetic gene cluster and the 16S rRNA gene for Ca. E. frumentensis (Table S1). The four bins containing the ET-743 producing microorganism were further assembled into a consensus genome containing three contigs. PCR amplification closed a 200 bp gap between two of the contigs to create a 630 kb scaffold. Additional PCR amplification closed a final 1.5 kb gap in the scaffold to create the closed genome for Ca. E. frumentensis (Fig. 2, Table 1, Fig. S1).
Figure 2.
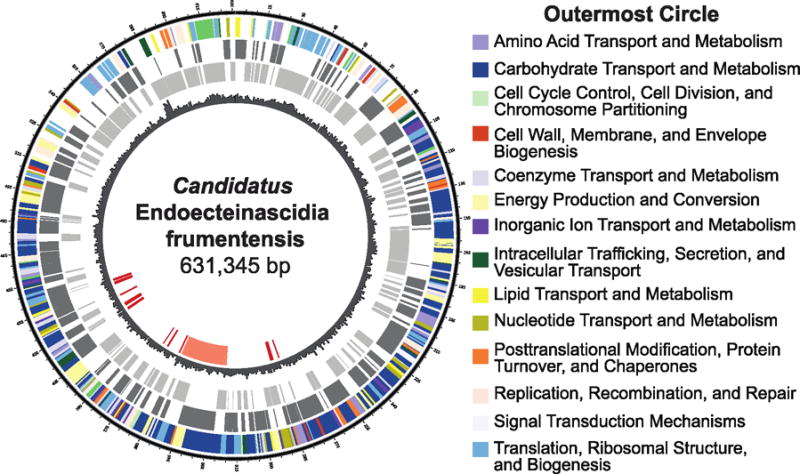
A circular map of the closed genome of Candidatus E. frumentensis. The outermost circle displays protein-coding genes assigned to Pfam categories (see key). The dark grey and light grey circles display protein-coding genes on the plus strand and minus strands, respectively. The fourth circle depicts a histogram of G+C content throughout the genome. The innermost circle represents ET-743 biosynthetic genes. Genes previously identified are depicted in light red while putative new genes are shown in dark red.
Table 1.
General features of the Candidatus E. frumentensis genome.
| Candidatus E. frumentensis | |
|---|---|
| Genome Size (bp) | 631,345 |
| Taxonomy | New family of Gammaproteobacteria |
| GC Content (%) | |
| Total | 23.3 |
| Coding Regions | 24.2 |
| Noncoding Regions | 12.7 |
| Coding Density (%) | 90.7 |
| Intergenic Pseudogenes | 10 |
| Protein-coding genes | 585 |
| With functional annotation | 556 (95.0%) |
| With ambiguous function | 29 (4.6%) |
| rRNA genes | 3 |
| tRNA genes | 32 |
The coverage depth for the endosymbiotic genome averaged 721x between the four samples (Table S2). However, one contig that consistently binned with the ET-743 producer and was retrieved in all four samples was not incorporated into the genome. This much smaller ~18 kb contig encodes a DNA primase and two protein-coding genes with ambiguous functions that repeat throughout the stretch of the sequence. Unlike the circular genome, the shorter contig has a coverage depth of only ~74x and reads could not be mapped to the sequence with confidence (Table S2). The excluded contig may be an extrachromosomal element that is present in only a subset of the Ca. E. frumentensis population or an artifact of the assembly and binning process. Given that the rest of the genome was closed and displayed even and deep coverage, we focused our analysis on the closed Ca. E. frumentensis genome in this study.
Very few other genomic bins were detected in the metagenomic datasets, despite prior evidence that the tunicate housed a complex microbial consortium (Table S1) (Moss et al., 2003; Rath et al., 2011). However, previous studies indicated Ca. E. frumentensis was one of the most abundant microorganisms in the consortium (Moss et al., 2003; Pérez-Matos et al., 2007; Rath et al., 2011) and the only microorganism found to be consistently associated with the tunicate host in both the Mediterranean and Caribbean marine habitats (Pérez-Matos et al., 2007). Further, metagenomic assembly of the symbiont population was likely facilitated due to its low genomic diversity compared to populations that are non-specifically associated. Thus, it is likely that the eukaryotic host and Ca. E. frumentensis monopolized the sequencing data, especially the large assembled contigs, despite the presence of a complex but lower abundance microbial community. The only other notable bin after tetra-ESOM was a cyanobacterium from the order Oscillatoriales that was present in two of the four metagenomic DNA samples (Table S1, Fig. S2).
Genome Reduction in the Symbiont
Previous in situ hybridization analysis provided an initial indication that Ca. E. frumentensis could be a bacterial endosymbiont (Shigenobu et al., 2000; Wernegreen, 2002). Assembly and analysis of the microbe’s complete genome provides further convincing evidence of an intracellular lifestyle and long-term evolution with the tunicate host, E. turbinata. Ca. E. frumentensis possesses many of the hallmarks of genome reduction, which is thought to be driven by a small bacterial population size and an inherent deletion bias (Moran, 1996; Moran et al., 2008; McCutcheon and Moran, 2012). The circular genome for Ca. E. frumentensis is quite small, totaling only 631,345 bp (Fig. 2). The small size of the genome rivals those of the model obligate endosymbionts Buchnera aphidicola in aphids and Wigglesworthia glossinidia in tsetse flies (Table S3). The functions maintained by Ca. E. frumentensis are also consistent with the minimal gene sets observed in these and other obligate symbionts (Figure S3). For example, Ca. E. frumentensis appears to have lost a number of genes involved in DNA replication and repair mechanisms (Figure S3). The loss of DNA repair mechanisms is thought to be a crucial turning point during the evolution of an endosymbiont (Moran et al., 2008; McCutcheon and Moran, 2012). Loss of these genes is frequently accompanied by increased mutation rates, an A+T DNA sequence bias, and the loss of additional nonessential genes.
Indeed, the exceptionally low total G+C content (23.3%) of Ca. E. frumentensis genomic DNA supports a mutational bias and an obligate endosymbiotic lifestyle. The G+C content disparity between the coding (24.2%) and noncoding (12.7%) regions of the genome (Table 1) further exemplifies this bias. Bacterial lineages that only recently became restricted to a host organism also often have higher numbers of pseudogenes within these noncoding regions and a consequently low overall coding density (Kuo et al., 2009). However, as bacteria continue to co-evolve with their hosts, pseudogenes gradually shrink and become unrecognizable through deletions while genomes become more compact (Moran, 1996; Kuo and Ochman, 2009). The noncoding regions of the Ca. E. frumentensis genome have only 10 pseudogenes whose predicted translation products show amino acid sequence similarity to known proteins (Table S4). The genome also has a higher overall coding density of 90.7% (Table 1), similar to B. aphidicola, W. glossinidia, and other obligate endosymbionts that co-evolved with their hosts along the order of millions of years (Moran and Munson, 1993; Moran et al., 2008). Taken together, these data provide strong support that Ca. E. frumentensis is an obligate endosymbiont that has undergone long-term co-evolution with the tunicate host, E. turbinata.
Phylogenetic analysis and novelty of Ca. E. frumentensis
The genome of Ca. E. frumentensis also appears to be remarkably distinct from other studied microorganisms. Analysis of conserved markers provided the first evidence that Ca. E. frumentensis may be phylogenetically distant from characterized bacterial species. The closest homologues for genes encoding the 16S rRNA gene, rpoB, and recA had 86.1%, 69.0%, and 74.8% sequence identities respectively (Figure S5).
Phylogenetic markers can be useful for microorganisms that have many well-studied and cultivable close relatives. However, in microorganisms with fewer obvious relatives, the average amino acid identity (AAI) of shared genes can be more revealing (Konstantinidis and Tiedje, 2005). To further explore the phylogenetic novelty of Ca. E. frumentensis, we compared the AAI and 16S rRNA gene of the microorganism to other bacterial species selected from a taxonomic profile of the Ca. E. frumentensis genome. This analysis confirmed that Ca. E. frumentensis is taxonomically distinct from many of its originally predicted relatives and likely represents a new family of Gammaproteobacteria (Fig. S5) (Yarza et al., 2014).
Primary Metabolism
Analysis of the endosymbiont’s primary metabolism provided further insight into the lifestyle of Ca. E. frumentensis (Fig. 3). The small genome appears to have portions of all three components of central metabolism, including the tricarboxylic acid cycle (TCA cycle), the non-oxidative branch of the pentose phosphate pathway, and most of the glycolytic pathway (Fig. 3). Although the genome is missing genes involved in early glucose catabolism, it does encode several sugar phosphate transporters. Sugar phosphates may therefore represent an important carbon source for the endosymbiont, similar to other microorganisms living in an intracellular environment (Munoz-Elias and McKinney, 2006).
Figure 3.
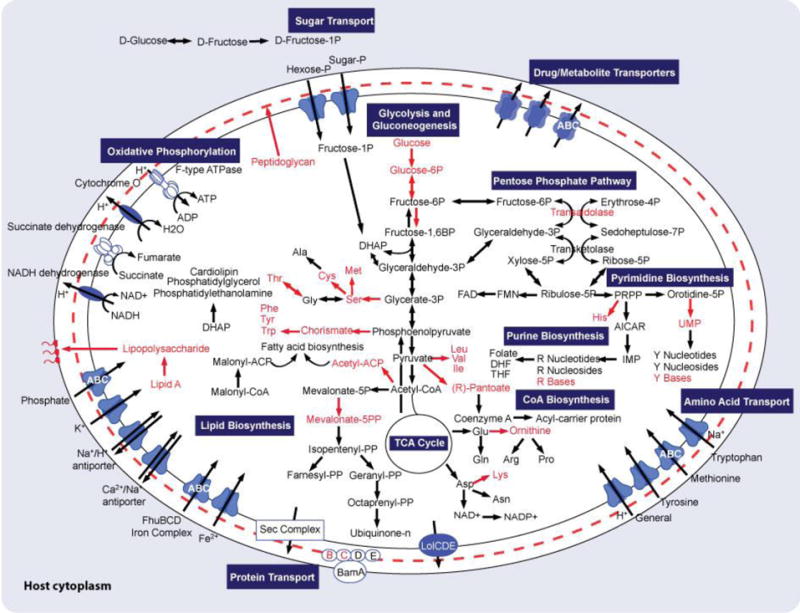
Overview of the metabolism of Ca. E. frumentensis deduced from genomic analysis. Reaction products depicted in red have either missing or partially missing biosynthetic pathways. ACP, acyl carrier protein; AICAR, 5-aminoimidazole carboxamide ribonucleotide; CoA, coenzyme A; DHAP, Dihydroxyacetone phosphate; DHF, dihydrofolate; DMAPP, dimethylallyl pyrophosphate; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; IMP, inosine monophosphate; NAD, nicotinamide adenine dinucleotide; PRPP, phosphoribosyl pyrophosphate; THF, tetrahydrofolate; UMP, uridine monophosphate.
Like most obligate endosymbionts and many intracellular pathogens, Ca. E. frumentensis is also missing a number of key amino acids and cofactors (Fig. 3). The genome only has the machinery to generate asparagine, aspartic acid, glutamate, and glutamic acid de novo. There are only partial gene sets for the remaining amino acids and several cofactors, including coenzyme A (CoA). It is likely that the endosymbiont acquires some of these essential metabolites or their precursors from the tunicate host. Indeed, the endosymbiont encodes 71 genes putatively linked to transporter function, including several involved in amino acid import (Fig. 3).
The Ca. E. frumentensis genome also has gene sets for the biosynthesis of lipids commonly incorporated into bacterial membranes, including phosphatidylethanolamine, cardiolipin, and phosphatidylglycerol (Fig. 3). However, the genome is missing a number of genes involved in the biosynthesis of peptidoglycan and lipid A biosynthesis. The vast majority of bacteria incorporate some level of peptidoglycan into their cell walls and most Gram-negative bacteria possess lipid A-containing lipopolysaccharides in their outer membrane. However, some microorganisms undergoing genome reduction have been known to lack both of these usually standard components (Pérez-Brocal et al., 2006; Wu et al., 2006; Moran et al., 2008; Nakabachi et al., 2013). The absence of the majority of these genes within Ca. E. frumentensis further highlights the extent of its genome reduction.
Secondary Metabolism
We previously identified a 35 kb contig containing many of the genes involved in the biosynthesis of the chemotherapeutic natural product ET-743 (Rath et al., 2011). However, close examination of ET-743, its previously isolated precursors (Rinehart et al., 1990), and other well-studied tetrahydroisoquinoline natural products (Pospiech et al., 1995; Velasco et al., 2005; Lei et al., 2008; Hiratsuka et al., 2013) led us to suspect that we were still missing a number of key biosynthetic genes (Rath et al., 2011). Expanding the 35 kb gene cluster to a complete genome for Ca. E. frumentensis has enabled us to identify many of these previously missing genes and improved our understanding of ET-743 biosynthesis. Key genes involved in production of the chemotherapeutic drug are dispersed over 173 kb of the small 631 kb genome (Fig. 2). Biosynthetic genes are split into three distinct regions within this expansive genomic range (Fig. 4A, Table S5). Newly detected gene products include the acetyltransferase EtuY and EtuM4, likely involved in acetylation and N-methylation to make 7 and ET-597 (9) respectively. We also identified three new flavoproteins in addition to the FAD-dependent monooxygenase (EtuO1) contained within the original ET-743 biosynthetic gene cluster (Rath et al., 2011).
Figure 4.
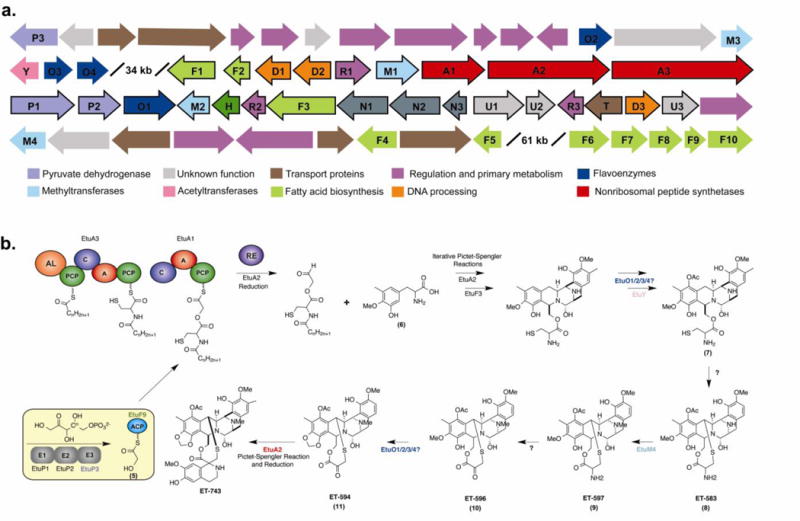
The identification of new genes with suspected involvement in ET-743 biosynthesis. The genes and their putative roles are also depicted in Table S5. A. New ET-743 biosynthetic genes were identified upstream and downstream of the original ET-743 biosynthetic gene cluster (outlined in black). Gene products are classified according to the corresponding color key. B. A condensed ET-743 biosynthetic pathway illustrating proposed new steps based on analysis of the complete genome. Colored steps represent new enzymes or new roles for previously identified enzymes. An updated proposal for the complete biosynthesis of ET-743 is depicted in Figure S6.
We additionally identified a gene encoding the E3 component of the pyruvate dehydrogenase complex (EtuP3, Fig. 4). The reactions catalyzed by this enzyme system typically provide the TCA cycle with acetyl-CoA (Patel et al., 2014). However, the primary metabolic enzymes were recently shown to also contribute to the biosynthesis of quinocarcin and naphtyridinomycin natural products (Peng et al., 2012). The enzyme complex can work with an acyl carrier protein (ACP) to provide a glycolicacyl-S-ACP extender unit (5) for a non-ribosomal peptide synthetase (NRPS). Both of these gene clusters in addition to SF-1739 (Hiratsuka et al., 2013) and the original ET-743 (Rath et al., 2011) biosynthetic gene cluster contain the E1 and E2 components for the enzyme complex. Although the E3 component has been absent in previously studied clusters, purified exogenous E3 does seem necessary for complete product conversion (Peng et al., 2012). The presence of the E3 component in Ca. E. frumentensis and its proximity to other ET-743 biosynthetic genes further exemplifies its importance in the biosynthesis of tetrahydroisoquinoline natural products.
Another genomic feature that may set the ET-743 biosynthesis apart from other natural products is the placement of the ACP that operates with the pyruvate dehydrogenase complex. The ACP is located in the main biosynthetic gene clusters for quinocarcin, naphtyridinomycin, and SF-1739. However, the only ACP in the entire Ca. E. frumentensis genome is located within a region containing fatty acid biosynthetic genes 61 kb downstream of the original ET-743 gene cluster (EtuF9, Fig. 4A). The location of the ACP and the presence of other fatty acid biosynthetic genes (EtuF1 and EtuF2) within the original ET-743 biosynthetic gene further supports potential interaction between primary and secondary metabolism during ET-743 biosynthesis. This ACP most likely functions in concert with EtuP1, EtuP2, and EtuP3 to provide the glycolicacyl-S-ACP extender unit (5) to EtuA1 (Fig. 4B).
Despite these new discoveries, we may still be missing some genes involved in ET-743 biosynthesis. For example, gene candidates for enzymes that catalyze formation of the thioether ring (8) and transamination to make ET-596 (10) remain to be identified. We cannot rule out that these genes may be located elsewhere in the Ca. E. frumentensis genome or that the microbe works together with its host to complete construction of the chemotherapeutic compound. Similar host-endosymbiont cooperation has been observed during the biosynthesis of parasitic plant fungus natural product rhizoxin (Lackner et al., 2011). Symbiotic bacteria have also been known to cooperatively biosynthesize compounds. However, previous findings suggesting that Ca. E. frumentensis is the only microorganism consistently associated with the tunicate {PerezMatos:2007ga} and make other symbionts a less likely source for additional biosynthetic genes.
The Ca. E. frumentensis genome also contains several widely dispersed genes found within the biosynthetic gene clusters of other tetrahydroisoquinoline natural products. For example, the gene encoding the excision nuclease subunit UvrA is found within the saframycin A, and SF-1739, and quinocarcin gene clusters, perhaps playing a role in repairing damage induced by these potent natural products. However, the gene in the Ca. E. frumentensis genome is located several hundred base pairs upstream from the original ET-743 gene cluster. The saframycin A gene cluster also contains a complete gene set for the recycling of S-adenosyl methionine (SAM), a coenzyme essential for methyltransferase activity during the biosynthesis of all tetrahydroisoquinoline natural products. The complete gene set for the recycling system is still present in the Ca. E. frumentensis genome, but the genes are located both upstream and downstream of the original 35 kb gene cluster.
The semi-dispersed nature of ET-743 biosynthetic genes is notable as microbial secondary metabolite systems are typically tightly clustered in bacteria with clearly identifiable boundaries (Walton, 2000; Chu et al., 2011). However, genes involved in ET-743 biosynthesis are located in different points throughout the genome, interspersed with genes involved in primary metabolism (Fig. 4). The fragmented nature of Ca. E. frumentensis secondary metabolism could be a consequence of horizontal gene transfer (Lawrence and Roth, 1996) and co-regulation of gene expression within operons (Price et al., 2005), which are two important forces thought to encourage selection and formation of gene clusters. However, the endosymbiont lifestyle provides few opportunities for horizontal gene transfer, and regulatory mechanisms are often among the first genetic elements lost during genome reduction (Moran et al., 2008; McCutcheon and Moran, 2012). The lack of selective pressure to retain clusters is thought to contribute to fragmentation of biosynthetic genes in other endosymbionts (Kwan et al., 2012), and likely also plays a role in the organization of genes involved in ET-743 production. The genome no longer possesses a canonical gene cluster, but instead contains scattered biosynthetic genes that may function in trans.
Analysis of the Ca. E. frumentensis genome has also improved our understanding of the importance of ET-743 biosynthesis in the relationship between the endosymbiont and the tunicate host, E. turbinata. In long-term co-evolution, bacterial genes that are useful to the host are retained despite ongoing genome erosion (Moran et al., 2008; McCutcheon and Moran, 2012). The survival of ET-743 biosynthetic genes despite clear evidence of extreme genome reduction is indicative of an important role for the secondary metabolite to the host. A query of the endosymbiont genome against the full complement of bioinformatics tools revealed that ET-743 was the only natural product gene cluster found within the genome, further exemplifying its ecological value to the tunicate. Adult ascidians such as E. turbinata are sessile marine invertebrates with soft-bodies, making them particularly vulnerable to predation. Their large larvae are released during daylight hours, making them similarly susceptible to predators. The secondary metabolite ET-743 could serve as a defense mechanism for the host. Many other ascidians and sponges are thought to produce secondary metabolites and inorganic acids that make them unpalatable (Lindquist et al., 1992). Indeed, ecological studies have already demonstrated that taste and orange coloring of larvae from E. turbinata protects the animal against predators (Young and Bingham, 1987). If ET-743 is the chemical deterrent responsible for protecting the host, it provides a driving force to assure the survival of ET-743 biosynthetic genes despite millions of years of genome reduction.
Conclusions
We have assembled a complete genome for Ca. E. frumentensis, an endosymbiont responsible for production of the chemotherapeutic drug ET-743. Microbial symbionts like Ca. E. frumentensis have long been thought to be the source of many natural products isolated from terrestrial and marine invertebrates. However, very little is known about the majority of these microbes due to our current inability to culture them in the laboratory.
The complete genome of Ca. E. frumentensis has enriched our understanding of ET-743 biosynthesis. The discovery of new ET-743 biosynthetic genes will enable future biochemical studies to confirm the roles of individual enzymes. A better understanding of its biosynthesis can facilitate future in vitro and heterologous expression efforts to engineer sustainable production of the drug and related analogs. Analysis of the complete genome has also highlighted the importance of ET-743 to the host-symbiont relationship. The lack of genomic evidence for other secondary metabolites, the survival of the gene cluster despite extreme genome reduction, and the dispersal of ET-743 genes across the small genome suggests the microbe has become specialized for production of the drug. The chemotherapeutic natural product is therefore likely crucial to the microorganism’s relationship with the tunicate host and its continued survival. This is intriguing since secondary metabolites are traditionally thought to be nonessential for microbial life (Williams et al., 1989) despite their prevalence in microbial genomes and ability to confer competitive advantages (Stone and Williams, 1992). However, improved sequencing technologies and metagenomic pipelines now permit more detailed studies of genomes undergoing reduction. Full genome studies on the endosymbionts found in macroorganisms like insects (Nakabachi et al., 2013), tunicates (Kwan et al., 2012; Kwan and Schmidt, 2013), or even fungi (Lackner et al., 2011) provide increasing evidence that natural products may sometimes play essential roles. When these secondary metabolites benefit a host organism, their preservation may ensure a microorganism’s survival and even facilitate co-evolution with a host. The drastically reduced genome of Ca. E. frumentensis presented here further supports this theory.
A better understanding of symbiont genomes along with their primary and secondary metabolism could provide new routes to economical and sustainable large-scale production of bioactive natural products. Analysis of the drastically reduced genome of Ca. E. frumentensis provides unique insight into the microorganism’s lifestyle and clues to possible host-free cultivation. Previous attempts to grow the microorganism in the laboratory were unsuccessful. However, our ability to culture elusive microorganisms is continually improving. Recent advances in host-cell free growth of Coxiella burnetii (Omsland et al., 2009) or the facultative symbionts Burkholderia spp., Rhodococcus rhodnii, and Wolbachia spp. (Kikuchi, 2009) motivates future efforts to develop suitable growing conditions and techniques to access the uncultivable majority of bacteria. Genome analysis in particular has proven a powerful method to pinpoint nutrient and oxygen requirements for microbial growth (Omsland et al., 2009; Kikuchi, 2009). The loss of key primary metabolic pathways in Ca. E. frumentensis suggests that the microorganism could not live independently of the host using standard media and cultivation techniques. The loss of genes involved in amino acid, coenzyme A, and glucose biosynthesis indicates that media enhanced with nutrients, cofactors, and alternative carbon sources may be necessary. However, genomic evidence for aerobic respiration and transporters for key metabolites indicates that the right environmental conditions might lead to host-cell free growth.
Experimental Procedures
Sample Collection and Isolation of Metagenomic DNA
Two tunicate colonies were collected off the coast of the Florida Keys. Animals were immediately frozen on dry ice after collection and stored at −80°C until processing. Metagenomic DNA was isolated from single zooids plucked from each colony (Figure S1) following the protocol outlined for mouse tails in the Wizard Genomic DNA Purification Kit (Promega).
Genome Sequencing, Assembly, Binning and Annotation
The four metagenomic samples were shipped on dry ice to the Joint Genome Institute (JGI) for immediate sequencing. Gene calling and annotation of the assembled metagenome was then completed through JGI IMG/M (Markowitz et al., 2013). Individual contigs from each assembly were assigned to taxonomic groups through binning with tetranucleotide frequency with ESOM as described previously (Dick et al., 2009). Since the metagenomes had an excess of sequences belonging to the eukaryotic host tunicate, iterative rounds of ESOM were required to hone in on microbial communities present in the sample.
Genes from the previously identified ET-743 biosynthetic gene cluster (Rath et al., 2011) and the 16S gene for E. frumentensis (Moss et al., 2003; Pérez-Matos et al., 2007; Rath et al., 2011) were used as BLAST queries to identify the bin containing the ET-743 producer in each of the four metagenomic samples. The four resulting bins were manually evaluated for completeness through the analysis of the distribution of conserved phylogenetic markers (Ciccarelli, 2006). Contigs from the four bins were assembled into a consensus genome with Geneious (v. 7.1.3).
Closing Genomic Gaps
We designed primers upstream of any suspected genomic gaps and carried out PCR using KOD Xtreme™ Hot Start DNA Polymerase (Novagen). Reactions contained 0.02 U/μL polymerase, 1X of the supplied buffer, 0.3 μM custom primers, 0.4 mM each dNTP, and 100 ng of metagenomic DNA. Reactions consisted of a hot start (94°C, 2 min), followed by 35 cycles of denaturing (98°C, 10 sec), annealing (variable temperatures for 30 sec), and extension (68°C for variable times). Since we were unsure about the size of genomic gaps, we began with a longer extension time of 5 minutes. If we saw a DNA band after running reactions on a 1% agarose gel, we repeated PCR and tailored the extension time to the size of the band (1 min/kbp) to limit any nonspecific amplification. Amplified DNA was then isolated from agarose gels using the standard protocol from the Wizard® SV Gel and PCR Clean-Up Kit (Promega).
Samples were submitted for Sanger sequencing with the primers used in the PCR reactions. Primer walking along the DNA strand then provided the missing sequence within both gaps. The complete consensus genome was submitted to JGI IMG (Markowitz et al., 2014) for gene calling and annotation. The final genome was reassessed for the completeness and accuracy through analysis of the distribution of conserved phylogenetic markers (Ciccarelli, 2006).
Genome Analysis
The common genes included in Figure S3 were compiled from other studies examining genome reduction in endosymbionts and intracellular pathogens (Moran et al., 2008; Kwan et al., 2014). Analysis of primary metabolic pathways was completed using the KEGG and MetaCyc annotations provided through JGI/IMG. To confirm the absence of any missing genes, protein sequences from a model organism (typically from E. coli E12) were used as queries in a BLASTP search against the Ca. E. frumentensis annotated genome.
To detect pseudogenes, all intergenic regions larger than 100 bp were used as BLASTX queries against the entire NR database using default settings. Any hits with e-values lower than 1×10−3 against nonhypothetical proteins were considered pseudogenes.
Visualization of the complete genome (Figure 2) was constructed using Circos (Krzywinski et al., 2009). Data for circles displaying Pfam categories for protein-coding genes, genes on the plus strands, and genes on the minus strands were provided directly through JGI IMG annotations and analysis.
To detect natural product gene clusters, the full genome was anlyazed with a host of previously described bioinformatics tools, including antiSMASH 2.0 (Blin et al., 2013), NP.searcher (Li et al., 2009), CLUSEAN (Weber et al., 2009), BAGEL3 (van Heel et al., 2013), and 2metdb (Bachmann and Ravel, 2009).
Phylogenetic Analysis
The gene sequences for conserved phylogenetic markers (16S rRNA, rpoB, and recA) were used as BLASTN queries against the NT database. Trees were constructed with Geneious (v. 7.1.3) after ClustalW multiple alignments with an IUB cost matrix (default settings). Neighbor-joining trees were constructed with the Jukes-Cantor genetic distance model (default settings). Top hits for cultivable or well-studied uncultivable microorganisms were included in the phylogenetic tree for 16S rRNA gene sequences. All unique hits for rpoB and recA were used in respective genetic trees.
To further explore taxonomic uniqueness (Figure S5), the complete or draft genomes of the top hits from phylogenetic analysis were used in a two-way BLAST against Ca. E. frumentensis to acquire average amino acid identity (AAI) as previously described (Konstantinidis and Tiedje, 2005). Thresholds for unique taxonomic rankings were based on 16S rRNA gene sequence identity as previously described (Yarza et al., 2014).
Acknowledgments
We thank Erich Bartels for use of facilities at Mote Marine Laboratories in the Florida Keys and for assistance with field collecting of E. turbinata. We also thank Tijana Glavina del Rio and Susannah Tringe at the Joint Genome Institute for their assistance. This research was supported by the International Cooperative Biodiversity Groups initiative (U01 TW007404) at the Fogarty International Center, the NSF under the CCI Center for Selective C–H Functionalization, CHE-1205646, and the Hans W. Vahlteich Professorship (D.H.S.). Support for M.M.S was provided by the NSF Graduate Research Fellowship Program (1256260). The work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
References
- Bachmann BO, Ravel J. Methods for In Silico Prediction of Microbial Polyketide and Nonribosomal Peptide Biosynthetic Pathways from DNA Sequence Data. Methods Enzymol. 2009;458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T. antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:W204–12. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo JL, Naranjo S, Kukurtzü B, Calle F, Hernández Zanuy A. Production of Ecteinascidia turbinata (Ascidiacea: Perophoridae) for Obtaining Anticancer Compounds. J World Aquac Soc. 2000;31:481–490. [Google Scholar]
- Chu HY, Wegel E, Osbourn A. From hormones to secondary metabolism: the emergence of metabolic gene clusters in plants. Plant J. 2011;66:66–79. doi: 10.1111/j.1365-313X.2011.04503.x. [DOI] [PubMed] [Google Scholar]
- Ciccarelli FD. Toward Automatic Reconstruction of a Highly Resolved Tree of Life. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- Corey EJ, Gin DY, Kania RS. Enantioselective total synthesis of ecteinascidin 743. J Am Chem Soc. 1996;118:9202–9203. [Google Scholar]
- Cuevas C, Francesch A. Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat Prod Rep. 2009;26:322–337. doi: 10.1039/b808331m. [DOI] [PubMed] [Google Scholar]
- Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP, Banfield JF. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009;10:R85. doi: 10.1186/gb-2009-10-8-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratsuka T, Koketsu K, Minami A, Kaneko S, Yamazaki C, Watanabe K, et al. Core assembly mechanism of quinocarcin/SF-1739: bimodular complex nonribosomal peptide synthetases for sequential mannich-type reactions. Chem Biol. 2013;20:1523–1535. doi: 10.1016/j.chembiol.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y. Endosymbiotic bacteria in insects: their diversity and culturability. Microbes Environ. 2009;24:195–204. doi: 10.1264/jsme2.me09140s. [DOI] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. Towards a genome-based taxonomy for prokaryotes. J Bacteriol. 2005;187:6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CH, Ochman H. Deletional bias across the three domains of life. Genome Biol Evol. 2009;1:145–152. doi: 10.1093/gbe/evp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CH, Moran NA, Ochman H. The consequences of genetic drift for bacterial genome complexity. Genome Res. 2009;19:1450–1454. doi: 10.1101/gr.091785.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan JC, Schmidt EW. Bacterial endosymbiosis in a chordate host: long-term co-evolution and conservation of secondary metabolism. PLoS ONE. 2013;8:e80822. doi: 10.1371/journal.pone.0080822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan JC, Donia MS, Han AW, Hirose E, Haygood MG, Schmidt EW. Genome streamlining and chemical defense in a coral reef symbiosis. Proc Natl Acad Sci USA. 2012;109:20655–20660. doi: 10.1073/pnas.1213820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan JC, Tianero MDB, Donia MS, Wyche TP, Bugni TS, Schmidt EW. Host control of symbiont natural product chemistry in cryptic populations of the tunicate Lissoclinum patella. PLoS ONE. 2014;9:e95850. doi: 10.1371/journal.pone.0095850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner G, Moebius N, Partida-Martinez LP, Boland S, Hertweck C. Evolution of an endofungal lifestyle: Deductions from the Burkholderia rhizoxinica genome. BMC Genomics. 2011;12:210. doi: 10.1186/1471-2164-12-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Roth JR. Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics. 1996;143:1843–1860. doi: 10.1093/genetics/143.4.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L, Deng W, Song J, Ding W, Zhao QF, Peng C, et al. Characterization of the saframycin A gene cluster from Streptomyces lavendulae NRRL 11002 revealing a nonribosomal peptide synthetase system for assembling the unusual tetrapeptidyl skeleton in an iterative manner. J Bacteriol. 2008;190:251–263. doi: 10.1128/JB.00826-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MH, Ung PMU, Zajkowski J, Garneau-Tsodikova S, Sherman DH. Automated genome mining for natural products. BMC Bioinformatics. 2009;10:185. doi: 10.1186/1471-2105-10-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter W, Lopez DM, Wellham L, Sigel MM. Ecteinascidia turbinata extracts inhibit DNA synthesis in lymphocytes after mitogenic stimulation by lectins. Exp Biol Med. 1975;150 doi: 10.3181/00379727-150-39059. [DOI] [PubMed] [Google Scholar]
- Lindquist N, Hay ME, Fenical W. Defense of ascidians and their conspicuous larvae: adult vs. larval chemical defenses. Ecol Monogr. 1992;62:547. [Google Scholar]
- Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Pillay M, et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 2014;42:D560–7. doi: 10.1093/nar/gkt963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Chen IMA, Chu K, Szeto E, Palaniappan K, Pillay M, et al. IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res. 2013;42:D568–D573. doi: 10.1093/nar/gkt919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- McLaughlin K. U.S. FDA Grants Priority Review for YONDELIS® (trabectedin) for the Treatment of Patients with Advan. 2015. [Google Scholar]
- Moran NA. Accelerated evolution and Muller’s rachet in endosymbiotic bacteria. Proc Natl Acad Sci USA. 1996;93:2873–2878. doi: 10.1073/pnas.93.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Munson MA. A molecular clock in endosymbiotic bacteria is calibrated using the insect hosts. Proc R Soc Lond B. 1993;253:167–171. [Google Scholar]
- Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–190. doi: 10.1146/annurev.genet.41.110306.130119. [DOI] [PubMed] [Google Scholar]
- Moss C, Green DH, Pérez B, Velasco A, Henríquez R. Intracellular bacteria associated with the ascidian Ecteinascidia turbinata: phylogenetic and in situ hybridisation analysis. Mar Biol. 2003;143:99–110. [Google Scholar]
- Munoz-Elias EJ, McKinney JD. Carbon metabolism of intracellular bacteria. Cell Microbiol. 2006;8:10–22. doi: 10.1111/j.1462-5822.2005.00648.x. [DOI] [PubMed] [Google Scholar]
- Nakabachi A, Ueoka R, Oshima K, Teta R, Mangoni A, Gurgui M, et al. Defensive bacteriome symbiont with a drastically reduced genome. Curr Biol. 2013;23:1478–1484. doi: 10.1016/j.cub.2013.06.027. [DOI] [PubMed] [Google Scholar]
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci USA. 2009;106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MS, Nemeria NS, Furey W, Jordan F. The pyruvate dehydrogenase complexes: structure-based function and regulation. J Biol Chem. 2014;289:16615–16623. doi: 10.1074/jbc.R114.563148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng C, Pu JY, Song LQ, Jian XH, Tang MC, Tang GL. Hijacking a hydroxyethyl unit from a central metabolic ketose into a nonribosomal peptide assembly line. Proc Natl Acad Sci USA. 2012;109:8540–8545. doi: 10.1073/pnas.1204232109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Brocal V, Gil R, Ramos S, Lamelas A, Postigo M, Michelena JM, et al. A small microbial genome: the end of a long symbiotic relationship? Science. 2006;314:312–313. doi: 10.1126/science.1130441. [DOI] [PubMed] [Google Scholar]
- Pérez-Matos AE, Rosado W, Govind NS. Bacterial diversity associated with the Caribbean tunicate Ecteinascidia turbinata. Antonie Van Leeuwenhoek. 2007;92:155–164. doi: 10.1007/s10482-007-9143-9. [DOI] [PubMed] [Google Scholar]
- Piel J. Metabolites from symbiotic bacteria. Nat Prod Rep. 2009;26:338–362. doi: 10.1039/b703499g. [DOI] [PubMed] [Google Scholar]
- Pospiech A, Cluzel B, Bietenhader J, Schupp T. A new Myxococcus xanthus gene cluster for the biosynthesis of the antibiotic saframycin Mx1 encoding a peptide synthetase. Microbiology. 1995;141:1793–1803. doi: 10.1099/13500872-141-8-1793. [DOI] [PubMed] [Google Scholar]
- Price MN, Huang KH, Arkin AP, Alm EJ. Operon formation is driven by co-regulation and not by horizontal gene transfer. Genome Res. 2005;15:809–819. doi: 10.1101/gr.3368805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath CM, Janto B, Earl J, Ahmed A, Hu FZ, Hiller L, et al. Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743. ACS Chem Biol. 2011;6:1244–1256. doi: 10.1021/cb200244t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinehart KL, Holt TG, Fregeau NL. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J Org Chem. 1990;55:4512–4515. [Google Scholar]
- Shigenobu S, Watanabe H, Hattori M, Sakaki Y, Ishikawa H. Genome sequence of the endocellular bacterial symbiont of aphids Buchnera sp. APS. Nature. 2000;407:81–86. doi: 10.1038/35024074. [DOI] [PubMed] [Google Scholar]
- Staley JT, Konopka A. Measurement of in situ activities of nonphotosynthetic microorganisms in aquatic and terrestrial habitats. Annu Rev Microbiol. 1985;39:321–346. doi: 10.1146/annurev.mi.39.100185.001541. [DOI] [PubMed] [Google Scholar]
- Stone MJ, Williams DH. On the evolution of functional secondary metabolites (natural products) Mol Microbiol. 1992;6:29–34. doi: 10.1111/j.1365-2958.1992.tb00834.x. [DOI] [PubMed] [Google Scholar]
- van Heel AJ, de Jong A, Montalbán-López M, Kok J, Kuipers OP. BAGEL3: automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013;41:W448–53. doi: 10.1093/nar/gkt391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco A, Acebo P, Gomez A, Schleissner C, Rodríguez P, Aparicio T, et al. Molecular characterization of the safracin biosynthetic pathway from Pseudomonas fluorescens A2-2: designing new cytotoxic compounds. Mol Microbiol. 2005;56:144–154. doi: 10.1111/j.1365-2958.2004.04433.x. [DOI] [PubMed] [Google Scholar]
- Walton JD. Horizontal gene transfer and the evolution of secondary metabolite gene clusters in fungi: an hypothesis. Fungal Genet Biol. 2000;30:167–171. doi: 10.1006/fgbi.2000.1224. [DOI] [PubMed] [Google Scholar]
- Weber T, Rausch C, Lopez P, Hoof I, Gaykova V. CLUSEAN: a computer-based framework for the automated analysis of bacterial secondary metabolite biosynthetic gene clusters. J Biotechnol. 2009;140:13–17. doi: 10.1016/j.jbiotec.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Wernegreen JJ. Genome evolution in bacterial endosymbionts of insects. Nat Rev Genet. 2002;3:850–861. doi: 10.1038/nrg931. [DOI] [PubMed] [Google Scholar]
- Williams DH, Stone MJ, Hauck PR, Rahman SK. Why are secondary metabolites (natural-products) biosynthesized? J Nat Prod. 1989;52:1189–1208. doi: 10.1021/np50066a001. [DOI] [PubMed] [Google Scholar]
- Wu D, Daugherty SC, Van Aken SE, Pai GH, Watkins KL, Khouri H, et al. Metabolic complementarity and genomics of the dual bacterial symbiosis of sharpshooters. PLoS Biol. 2006;4:e188. doi: 10.1371/journal.pbio.0040188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–645. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- Young CM, Bingham BL. Chemical defense and aposematic coloration in larvae of the ascidian Ecteinascidia turbinata. Mar Biol. 1987;96:539–544. [Google Scholar]