

Abstract
Excess glutamatergic neurotransmission may contribute to excitotoxic loss of nigrostriatal neurons in Parkinson’s disease (PD). Here, we determined if increasing glutamate uptake could reduce the extent of tyrosine hydroxylase (TH) loss in PD progression. The beta-lactam antibiotic, ceftriaxone, increases the expression of glutamate transporter 1 (GLT-1), a glutamate transporter that plays a major role in glutamate clearance in central nervous system and may attenuate adverse behavioral or neurobiological function in other neurodegenerative disease models. In association with >80 % TH loss, we observed a significant decrease in glutamate uptake in the established 6-hydroxydopamine (6-OHDA) PD model. Ceftriaxone (200 mg/kg, i.p.) increased striatal glutamate uptake with ≥ 5 consecutive days of injection in nonlesioned rats and lasted out to 14 days postinjection, a time beyond that required for 6-OHDA to produce >70 % TH loss (~9 days). When ceftriaxone was given at the time of 6-OHDA, TH loss was ~57 % compared to ~85 % in temporally matched vehicle-injected controls and amphetamine-induced rotation was reduced about 2-fold. This attenuation of TH loss was associated with increased glutamate uptake, increased GLT-1 expression, and reduced Serine 19 TH phosphorylation, a calcium-dependent target specific for nigrostriatal neurons. These results reveal that glutamate uptake can be targeted in a PD model, decrease the rate of TH loss in a calcium-dependent manner, and attenuate locomotor behavior associated with 6-OHDA lesion. Given that detection of reliable PD markers will eventually be employed in susceptible populations, our results give credence to the possibility that increasing glutamate uptake may prolong the time period before locomotor impairment occurs.
Keywords: Parkinson’s disease, Ceftriaxone, GLT-1, Tyrosine hydroxylase, 6-OHDA, Neuroprotection
Introduction
Glutamate mediates excitatory signal transduction, playing a primary role in brain physiology [1]. However, excessive glutamatergic neurotransmission (excitotoxicity) has been implicated in neurodegenerative disease processes such as Huntington’s disease [2–4], amyotrophic lateral sclerosis [5], and Parkinson’s disease [6]. Although Parkinson’s disease is a disease characterized by the progressive degeneration of dopaminergic neurons [7, 8], concomitant increases in glutamatergic tone have also been observed [9–11]. Indeed, converging literature has shown increased levels of extracellular glutamate [12–16] and increased glutamate receptor expression in Parkinson’s disease models or patients alike [17–19].
Despite evidence that excess glutamatergic excitotoxicity is associated with Parkinson’s disease, it is not clear at what point (if at all) in Parkinson’s disease progression excitotoxicity could contribute to the loss of nigrostriatal neurons. Glutamate increases Ca2+ influx through the binding of N-methyl-D-aspartate (NMDA) receptors. However, in excess, increased Ca2+ influx can trigger the activation of calcium-dependent proteases that promote cell injury and death [20–25]. Calpain is one such protease, and, indeed, there is evidence of increased calpain-related proteolytic activity in postmortem tissue of Parkinson’s disease patients contributing to the loss of dopaminergic neurons [26, 27], while calpain inhibitors can alleviate motor loss in Parkinson’s disease models [26]. There is also evidence that diminishing glutamate receptor activation can mitigate nigrostriatal damage in Parkinson’s disease models [28–30]. Unfortunately, however, the use of glutamate receptor antagonists in the Parkinson’s disease patient have had mixed clinical outcomes [31–33] with some NMDAR antagonists, although showing promise in its ability to block excitotoxicity, can also inhibit normal excitatory synaptic activity leading to untoward side effects [34, 35]. Therefore, an alternate intervention targeting elevated glutamatergic signaling could prove useful for mitigating nigrostriatal neuron loss, without compromising the essential components of glutamate signaling in neuronal functioning. However, demonstration of evidence that reducing extracellular glutamate could protect loss of nigrostriatal DA neurons is still a necessary first step for advancing prospects that mitigating excitotoxicity is a viable strategy to diminish, if not entirely halt, Parkinson’s disease progression.
One possible approach is to reduce excess glutamatergic tone and test the impact upon nigrostriatal loss induction in in vivo Parkinson’s disease models. Glutamate transporters, expressed primarily in glial cells and secondarily in neurons, aid in the removal of glutamate from the extracellular space, thereby minimizing its synaptic accumulation [36–39]. The glutamate transporter-1 (GLT-1), predominantly expressed by the glia, may be responsible for the majority of glutamate uptake [39, 40], although GLAST, also expressed in glia, can upregulate in expression when GLT-1 function is compromised [41, 42]. The importance of GLT-1, however, is highlighted by findings indicating GLT-1 dysfunction in amyotrophic lateral sclerosis [43] as well as traumatic brain injury [44] patients. Experimental reduction of the glial transporters increases synaptic glutamate levels, triggering excitotoxic neurodegeneration and paralysis [37, 45].
In Parkinson’s disease models, evidence suggests that increasing GLT-1 expression could be a strategy to diminish nigrostriatal loss. Glial cells themselves seem to play a protective role in PD models [46, 47]. Ceftriaxone, a beta-lactam antibiotic, increases GLT-1 expression [43] and could be thus be a feasible venue for augmenting glutamate uptake to reduce excitotixicy. A recent proof-of-concept study indicates that ceftriaxone, when given prior to lesion induction, decreased the degree of nigrostriatal neuron loss [48]. Ceftriaxone may attenuate the Huntington’s disease phenotype in the R6/2 mouse [4, 49] and alleviate motor neuron degeneration in an animal model of amyotrophic lateral sclerosis [50]. Obviously, the next step forward is to determine if ceftriaxone could mitigate damage to the nigrostriatal pathway once lesion progression is underway. Thus, once reliable biomarkers for Parkinson’s disease are revealed, increasing glutamate uptake could delay the onset of locomotor symptoms that emerge with >70 % striatal DA loss. Given that ceftriaxone requires ≥5 daily injections to increase GLT-1 [43], we determined if tyrosine hydroxylase (TH) loss, already underway in the 6-hydroxydopamine (6-OHDA) Parkinson’s disease model, could be mitigated by initiating ceftriaxone treatment at the same time of the lesion induction.
Methods
Animals
Male Sprague–Dawley rats, purchased from Charles River, were used in all experiments. All rats were 4–8 months old, and were housed under controlled lighting conditions (12:12 light/dark cycle) with standard animal chow and water available ad libitum. All animals were used in compliance with federal guidelines and the institutional Animal Care and Use Committee guidelines at LSU Health Sciences Center–Shreveport.
Ceftriaxone Administration
Ceftriaxone (Hospira, Lake Forest, IL, USA) was dissolved in sterile saline (0.9 %) and given by intraperitoneal injection (i.p) at 200 mg/kg. To determine the impact of ceftriaxone on GLT-1 expression and glutamate uptake in naive (not 6-OHDA-lesioned) animals, ceftriaxone was given for seven consecutive days at 200 mg/kg. Control animals were given 0.9 % saline for the same time frame. In order to determine longevity of ceftriaxone effect, 4–8-month-old naive Sprague–Dawley rats were treated with 200 mg/kg of ceftriaxone or vehicle (0.9 % saline) for seven consecutive days. Rats were euthanized, and glutamate uptake was assessed in both groups at either 1 or 4 days and 1 or 2 weeks post last injection (Fig. 1a). In rats receiving 6-OHDA lesion, ceftriaxone was given on the day of lesion and for seven consecutive days thereafter (Fig. 1b). As a control to ceftriaxone injection, the 6-OHDA lesion only group had consecutive daily injections of vehicle.
Fig. 1.
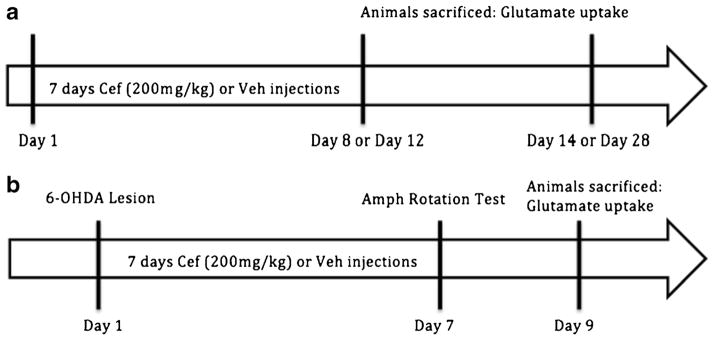
Timeline of ceftriaxone regimen. a Ceftriaxone longevity experiment: 7 days of 200 mg/kg ceftriaxone or vehicle was injected i.p. in naive male Sprague–Dawley rats. Rats were then killed at various time points after the last (seventh) cef injection: 1 or 4 days after, 1 or 2 weeks after. b Ceftriaxone lesion experiment: all male Sprague–Dawley rats were lesioned on day 1. On the seventh consecutive day of ceftriaxone or vehicle (200 mg/kg), amphetamine (Amph) induced rotation was assessed. On day 9, rats were killed for glutamate reuptake and biochemical analysis (TH, GLT-1, and GLAST)
6-OHDA Lesions
Each animal underwent survival surgery to deliver the neurotoxin 6-OHDA to the medial forebrain bundle as previously described [51]. Rats were anesthetized with 40 mg/kg Nembutal intraperitoneal (i.p.) (pentobarbital Lundbeck Inc, Deerfield, IL, USA) with supplement of 9.0, 0.6, and 0.3 mg/kg ketamine, xylazine, and acepromazine, respectively. Animals were immobilized in a stereotaxic frame to target the medial forebrain bundle at coordinates ML +1.5, AP −3.8, DV −8.0, relative to Bregma, according to Paxinos and Watson rat brain atlas [52]. A total of 16 μg of 6-OHDA in 4 μL in 0.02 % ascorbic acid (concentration of 4 mg/mL) was infused unilaterally at a rate of 1 μL/min. To control for any effects of the lesions surgery alone, the contralateral striatum was also infused with vehicle (0.02 % ascorbic acid) at a rate of 1 μL/min. The syringe was left in place for 10 min before removal to allow for maximal diffusion and to minimize mechanical damage to the tissue. Body temperature was maintained at 37 °C during surgery using a temperature monitor with probe and heating pad (FHC, Bowdoin, ME, USA).
Amphetamine Testing for Lesion Verification
Rotational behavior was assessed for 60 min following amphetamine (2 mg/kg i.p) administration. Rotations ipsilateral to the lesioned side were quantified at 7 days post-6-OHDA infusion (Fig. 1b). Amphetamine-induced rotations increase in response to a lesion and can detect at least a 50 % loss of dopamine [53]. Rats were killed 9 days following 6-OHDA for glutamate uptake analysis, GLT-1, TH, TH ser19 phosphorylation, a Ca2+-dependent process in DA neurons [42, 54], and calpain analyses, 2 days after the amphetamine test (allowing for practically complete clearance of amphetamine).
Preparation of Synaptosomes
Crude synaptosomes were prepared from striatal tissue was homogenized in 5 mL of 0.32 M sucrose solution using 10 up and down strokes of a Teflon/glass homogenizing wand (Glas-Col, Terre Haute, IN, USA) then spun at 1,000×g for 10 min. The resulting pellet was stored as the P1 fraction, from which the analysis of total and phosphorylated TH was later conducted by sonicating the pellet in sodium dodecyl sulfate and performing Western blot analysis (we have previously reported the utility of using this fraction in determining the expression level of cytosolic proteins such as TH Chotibut et al. [51]). The resulting supernatant was spun further at 17,500×g for 30 min yielding the P2 fraction. The P2 fraction was used to determine glutamate uptake on the day of preparation, and aliquots were frozen to later analyze GLT-1 and GLAST protein expression. The supernatant was aspirated and resuspended in 1 mL of Kreb’s buffer. Protein concentration was determined using a BCA colormetric assay (Thermo Scientific, Rockford, IL, USA). This protocol has been used to determine the reuptake of glutamate [42] and other neurotransmitters endogenous to striatum [55].
Glutamate Uptake Protocol
Synaptosomal P2 fraction contain glial components [56], and ~70 % of the levels of glial fibrillary acid protein are recovered in purified glial plasmalemmal vesicles [57] and thus are adequate for assessment of glutamate reuptake [42]. Synaptosomes were distributed in test tubes at equal protein quantity to prepare for glutamate reuptake, with an aliquot saved for later determination of the protein quantities of GLT-1 TH, ser19 TH phosphorylation, and calpain activity (spectrin breakdown products) [58].
Synaptosomes were used in a quantity of 30 μg of total protein in a 200-μL final volume for glutamate reuptake. In 100 μL, the combination of the synaptosome prep to constitute 30 μg synaptosomal protein and oxygenated Kreb’s buffer was prepared at 4°C. The synaptosomes were then placed in a water bath at 35 °C for 5 min, followed by the addition of 100 μL of 10 μM 14C(U)-L-glutamic acid (Perkin-Elmer, specific activity 260 mCi/mmol, catalogue no. NEC290E050UC) to the synaptosome preparations (giving a 5 μM final [glutamate]), allowed to incubate for reuptake for 90 s. The reaction was then terminated with 1 mL of ice-cold Kreb’s buffer, and the tubes were reimmersed the tubes into an ice bath. The reuptake time was chosen to be as close as technically and practically possible to the reuptake time of glutamate observed in vivo, which occurs within 10 s [59, 60]. Synaptosomes were washed multiple times in order to remove excess labeled glutamate with equal-osmolarity phosphate-buffered saline through a Brandel M24-TI (Gaithersburg, MD, USA) cell harvester with Brandel GF/C filter paper pretreated with a 2 % polyethylenimine solution to reduce nonspecific binding of label. The filter paper containing the rinsed synaptosomes were then transferred into scintillation vials containing 5 mL of biodegradable scintillation cocktail (Research Products International, Mount Prospect, IL, USA) and counted with a Beckman Coulter LS6500 scintillation counter (Brea, CA, USA).
Quantifying [14C]Glu Uptake into Synaptosomes
To determine the quantity of glutamate reuptake, the percent of glutamate (as the label) recovered in the synaptosomes against the total amount of glutamate (as the label) in the reuptake experiment was first determined. This percentage of reuptake averaged 1.50±0.28 % (mean±SEM, n =7 experiments) using 5 μM glutamate (final concentration) for reuptake studies. In pilot experiments, the percent reuptake was not significantly different using 1 or 2 μM glutamate (n =5 experiments). The picomole of glutamate was then determined based upon this percent of label recovery in the synaptosomes and normalized to mg protein per minute based upon the allotted 90 s uptake time allowed.
The determination of [14C]Glu uptake in the crude synaptosomes from dorsal striatum harvested from the contralateral vehicle-infused and 6-OHDA-infused hemispheres was conducted simultaneously. Each determination was done in triplicate for each assay condition.
Tissue Preparation and Western Immunoblotting
Synaptosome fraction (P1) and the processed preparatory sample (P2) were sonicated in a 1 % sodium dodecyl sulfate solution (pH ~8) using a Branson Sonifier 150 (Danbury, CT, USA). Protein concentration was determined using the bichinchoninic acid colometric assay. Following gel electrophoresis, proteins were transferred for 500 Vh in a Tris/glycine/methanol buffer onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA).
The nitrocellulose membrane was stained with Ponceau S to further normalize staining in each sample lane [61]. These lanes were scanned and quantified by Image J to normalize protein in each sample. This relative total level then served as an additional normalizing value to determine the quantity of each protein assayed [61]. To continue processing, the membranes were blocked in PVP buffer (1 % polyvinylpyrrolidone and 0.05 % Tween 20) for a minimum of 2 h to reduce nonspecific antibody binding. The membrane was soaked in primary antibody for 1–3 h. Specific primary antibodies were as follows: Specific primary antibodies were as follows; GLT-1 (Santa Cruz, Santa Cruz, CA, catalogue no. 15317), TH (Millipore, catalogue no. AB152), GLAST (Novus Biologicals, Littleton, CO, catalogue no. NB100-1869), and serine 19 phosphorylation (PhosphoSolutions, catalogue no. p1580-19). P2 fraction was used to assess GLT-1 expression and P1 fraction used for TH expression Western blot analysis. Nominal protein loads for linear detection of GLT-1 was 20–30 μg total protein 8 μg for TH protein, and ~5–10 μg for serine 19 TH phosphorylation. After primary treatment, blots were exposed to secondary antibody (swine anti-rabbit IgG) for signal enhancement, followed by 1 h incubation with [125I] protein A (PerkinElmer, Waltham, MA, USA).
Statistics
All glutamate uptake studies were done in conjunction with assessment of GLAST and GLT-1, as assessed in aliquots of synaptosomes that were used to determine glutamate uptake. Tissue harvested from the striatum contralateral to 6-OHDA-lesion served as the inherent control to the lesioned striatum for each rat/test subject. We excluded vehicle and ceftriaxone pairs where the vehicle did not have >65 % TH loss (4 of 13 surgery pairs). Surgeries were performed in pairs with the assumption that any irregularities in efficacy of the 6-OHDA lesion may occur in either the vehicle- and ceftriaxone-treated animals. Specifically, surgeries were performed in multiples of two each day to accommodate for the two treatment (Cef and Veh) groups. For instance, on one day, two surgeries were performed and treatment was assigned (Cef, saline). On another day, two additional surgeries were performed and treatment was assigned (Saline, Cef). Thus, any variances that may be encountered on each day of surgery that may affect lesion severity/experiment could be accounted for by analyzing the data using a paired t test due to the cef/saline-matched pairing. As a result, a Student’s paired two-tailed t test was also used to compare glutamate uptake differences between the two groups and for the lesioned and contralateral striatum, to ascertain differences in TH protein loss between lesioned and contra-lateral striatum, differences in ser19 TH phosphorylation, calpain activity, and GLT-1 expression associated with 6-OHDA lesion in the vehicle-injected control and the ceftriaxone-treatment group. These measures were all conducted from the same tissue sources, thereby obtaining all dependent measures, including rotational behavior, in operationally matched manner. As such, Student’s paired two-tailed t test was also used for analysis of rotational behavior.
Results
Glutamate Uptake in 6-OHDA Lesioned Rats
We found evidence of decreased striatal glutamate uptake in the 6-OHDA lesioned striatum compared with its contralateral (vehicle-infused) striatum by ~35 % (Fig. 2). To verify the degree of lesion in association with this impairment in glutamate uptake, we determined TH loss in this set of studies to be ~84 % (data not shown), the threshold above which clinical Parkinson’s disease symptoms are seen [62].
Fig. 2.

Impaired glutamate reuptake in the 6-OHDA lesioned striatum. In association with >80 % TH protein loss (data not shown), after 6-OHDA (16 μg) infusion into the medial forebrain bundle, striatal glutamate reuptake was decreased ~45 % in 4–6-month-old male Sprague–Dawley rats (n =5, Student’s paired t test, **p <0.01, t =7.499)
The decrease in glutamate reuptake capacity, at least when TH loss is at PD levels, suggests the possibility that this impairment could contribute to nigrostriatal neuron loss once 6-OHDA initiates damage to the neuron.
Ceftriaxone Effect on Glutamate Uptake in Naive Rats
Treatment with 200 mg/kg (i.p.) ceftriaxone for 5–7 days has been shown to increase glutamate reuptake capacity [43, 49]. Consistent with these previous observations, we also observed increased GLT-1 expression with a similar regimen 4 days after the last day of administration (Fig. 3).
Fig. 3.
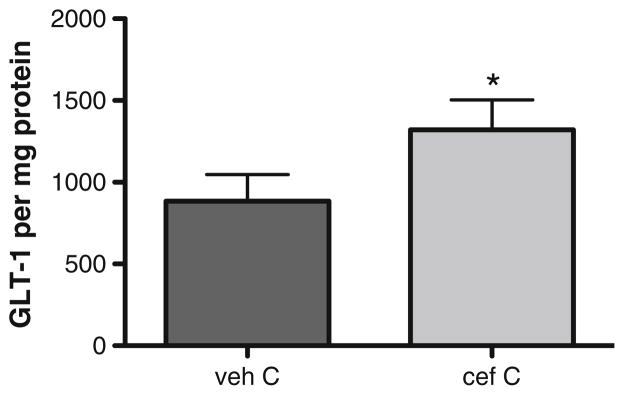
Ceftriaxone increases GLT-1 expression in naive rats. Four- to six-month-old male Sprague–Dawley rats were treated with 200 mg/kg ceftriaxone (cef) or saline (veh) for seven consecutive days. GLT-1 expression was determined 4 days later (n =5, Student’s paired t test, *p <0.05, t =3.567)
Longevity of the Ceftriaxone Effect on Glutamate Uptake in Naive Rats
In order to ascertain the longevity of the ceftriaxone effect, we examined glutamate uptake dynamics at 1, 4, 7, and 14 days after five or seven consecutive days of ceftriaxone treatment. We first determined the duration of ceftriaxone-mediated enhancement of glutamate reuptake 1 and 4 days after final (seventh) consecutive day of ceftriaxone administration, representing a time period encompassing the length of time (~9 days) necessary for the 6-OHDA lesions to produce ~70 % loss of TH. Glutamate reuptake was increased ~50 % 1 and 4 days (pooled) after the final injection (Fig. 4a). This increase in glutamate reuptake was also observed between 7 and 14 days after last injection, increasing ~20 % overall (Fig. 4b).
Fig. 4.

Longevity of ceftriaxone enhancement of striatal glutamate reuptake. Four to eight-month-old male Sprague–Dawley rats were treated with 200 mg/kg ceftriaxone (cef) or saline (veh) for five or seven consecutive days. Glutamate reuptake was assessed and data collapsed for analysis in two time periods post-cef: a 1 and 4 days after last cef administration (seven consecutive days). Glutamate reuptake increased ~1.5-fold (154 % of veh) in cef group relative to reuptake in veh group (Student’s paired t test, *p <0.05, t =2.99, n =6). Mean±SEM increase: after day 1: 176±20 %; after day 4: 131±27 %. Data presented as percent difference in uptake in cef group vs. veh group. b 7 and 14 days after last cef administration (five consecutive days). Glutamate reuptake increased ~1.2-fold (122 % of veh) in cef group relative to reuptake in veh group (Student’s paired t test, *p <0.05, t =3.04, n =5). Mean±SEM increase: after day 7: 118±6 %; after day 14: 127±22 %. Data presented as total glutamate reuptake (picomole glutamate per minute per milligram protein) cef group vs. veh group
Ceftriaxone Treatment in a 6-OHDA PD Rat Model: Tyrosine Hydroxylase, GLT-1, and GLAST Expression and Glutamate Uptake
With 6-OHDA lesion and at 2 days after seven consecutive injections of ceftriaxone (200 mg/kg), the increase in GLT-1 expression observed in naive rats (Fig. 3) was also observed with 6-OHDA lesion (Fig. 5a). Predictably, this was associated with increased glutamate uptake, with an increase of ~30 % compared to that in the lesioned striatum of the vehicle-injected control group (Fig. 5b). A similar increase in GLT-1 was seen in the unlesioned striatum of ceftriaxone-treated rats compared to the unlesioned striatum vehicle-treated rats (data not shown).
Fig. 5.

Ceftriaxone increases GLT-1 expression and glutamate reuptake in lesioned striatum. a GLT-1 per microgram protein (n =5, paired t test, *p <0.05, t =2.954) and b glutamate reuptake (n =5, paired t test, t = 4.987, p <0.01) in 6-OHDA-lesioned striatum is increased by ~30 % with seven consecutive injections of 200 mg/kg ceftriaxone (cef) in 4–8-month-old male Sprague Dawely rats
There were no significant differences in striatal glutamate–aspartate transporter (GLAST) expression observed between these same 6-OHDA lesioned rats treated with vehicle versus ceftriaxone (data not shown).
In comparison to contralateral control striatum, this increased ceftriaxone-mediated glutamate reuptake was associated with a significant attenuation of TH protein loss, with ~3-fold more striatal TH remaining in cef-treated rats compared to that in the vehicle-injected control group (Fig. 6a). A Western blot representing the difference in TH protein loss produced by 6-OHDA between the ceftriaxone-versus vehicle-treated groups is shown,with ~55 % loss seen with ceftriaxone and ~90 % loss seen with vehicle treatment (Fig. 6b).
Fig. 6.

Ceftriaxone attenuates tyrosine hydroxylase (TH) loss in 6-OHDA lesioned rats. a Seven consecutive daily injections intraperitoneally of (200 mg/kg) ceftriaxone starting on the day of 6-OHDA infusion attenuated TH loss by ~30 % in 4–8-month-old male Sprague–Dawley rats, as compared to an average TH loss of vehicle Treated rats was 86 % (n =9, paired t test, t =5.24, p <0.05). b Representative Western blot of a (left to right): ceftriaxone-treated rat [lesion (L), unlesioned (UL)] and a vehicle treated rat. Standards of TH shown in nanograms (0.5, 2, 3, and 4)
Rotational Behavior in 6-OHDA PD Rat Model with Ceftriaxone Treatment
Behavioral testing was performed after amphetamine (2 mg/kg) i.p. injection on day 7 after lesion. In addition to attenuating TH loss due to 6-OHDA, the ceftriaxone regimen significantly reduced amphetamine-induced rotation (ipsilateral to the lesion) compared to vehicle treatment by ~50 % in these same rats (Fig. 7). Thus, ceftriaxone treatment also reduced 6-OHDA lesion impact on rotational behavior, an index of nigrostriatal neuron loss.
Fig. 7.
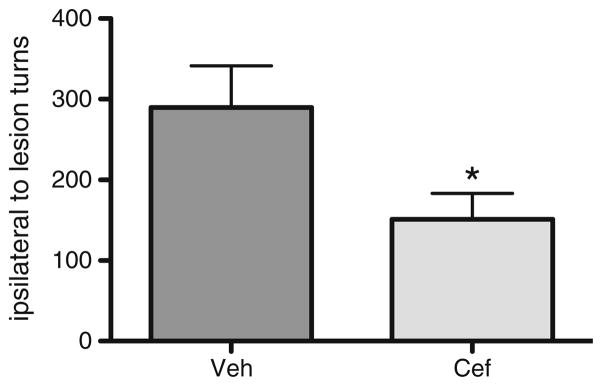
Ceftriaxone decreases amphetamine-induced rotational turns in 6-OHDA lesioned rats. Seven consecutive daily injections of ceftriaxone (200 mg/kg, i.p.) starting on the day of 6-OHDA infusion ellicited a reduction in amphetamine-induced rotation compared to vehicle treatment by ~50 % (n =7, paired t test, t =3.552, p <0.05)
Mechanism of Ceftriaxone Protection: Evidence for Reduced Ca2+-Influx Serine 19 TH Phosphorylation
Ser19 phosphorylation, while not directly affecting TH activity, may alter its confirmation increasing the likelihood for TH degradation [63]. Ser19 phosphorylation is also Ca2+ dependent [54], and the phosphorylation stoichiometry decreases in association with increased striatal glutamate uptake [42]. Therefore, ser19 TH phosphorylation was used as an indirect index of Ca2+ influx into DA terminals. The stoichiometry of ser19 TH phosphorylation in lesioned striatum was significantly reduced in the ceftriaxone group compared with that in the vehicle group (Fig. 8a, c), without any significant difference between the two groups in the unlesioned contralateral control striatum (Fig. 8b, c).
Fig. 8.

Ceftriaxone affects serine19 TH phosphorylation in 6-OHDA lesioned rats. Seven consecutive daily injections of ceftriaxone (200 mg/kg i.p.) starting on the day of 6-hydroxydopamine infusion reduced Ser19 TH phosphorylation in the lesioned striatum (a) but did not affect the unlesioned striatum (b) (a n =7, paired t test, t =5.869, p < 0.01). c Representative Western blot of a (left to right): ceftriaxone-treated rat [unlesion (UL), lesion (L)] and a vehicle-treated rat (UL, L). Calibrated standards of phosphoSer19 shown in nanograms (0.5, 1, 2.5, and 4), used as previously described [42]
Discussion
Glutamate excitotoxicty has long since been thought to be a component of various neurodegenerative disorders, as seen in models of Huntington’s disease [1, 64, 65] and, more recently, Parkinson’s disease [6, 16, 29, 30]. Dopamine and glutamate have reciprocal interaction within the striatum and affect each others’ release, thereby playing a role in basal ganglia function and dysfunction [12, 66–71], with both neurotransmitters cooperatively modulating the activity of striatal output (medium spiny) neurons. Indeed, the onset of motor symptoms in Parkinson’s disease animal models has been shown to be closely tied with the increase of glutamate levels within the basal ganglia [12–14, 69, 72, 73]. These changes in corticostriatal glutamate release due to dopaminergic denervation imply circuitry imbalance and have been speculated to contribute to further loss of nigrostriatal neurons. The expression of both ionotropic and metabotropic glutamate receptors on nigrostriatal neurons increases its sensitivity to glutamate excitotoxicity [68]. Increased expression of NMDA and mGluR5 glutamate receptors have also been observed in PD patients [17, 18]. Taken together, these scenarios may not only facilitate glutamate excitotoxicty but also play an underlying role in Parkinson’s disease progression. The ability to minimize this increase in glutamate activation in Parkinson’s disease pharmacologically remains an elusive goal given the high incidence of side effects from glutamate receptor antagonists [31–35]. Our work here suggests that through ceftriaxone-induced glutamate reuptake, the progression of nigrostriatal neuron loss may be mitigated when administered early in PD pathogenesis.
There is evidence that glutamate can contribute to the demise of nigrostriatal neurons via activation of TH, the rate-limiting enzyme in dopamine biosynthesis [74], further supporting the role that excitotoxicity may be playing in the initiation of Parkinson’s disease progression. Additionally, protection against 6-OHDA lesion in mGluR5 receptor knockout mice has also been observed indicating the importance glutamate activation may play in DA denervation [75]. As such, preclinical studies and postmortem analysis of central nervous system (CNS) tissue show evidence that glutamate transporters can provide some measure of protection against excitotoxicity in the CNS, as the main transporter responsible for glutamate reuptake (GLT-1), can increase in expression in response to injury or excitotoxic insult [76, 77]. In line with this, DA denervation may trigger modulations in GLT-1 [9, 78], indicating that GLT-1 dysfunction likely plays a role in Parkinson’s disease progression.
In this line, we first demonstrate that there is impaired glutamate uptake, presumably through GLT-1, in the striatum of the 6-OHDA-lesioned rat, supporting the idea of glutamatergic dysfunction in Parkinson’s disease, as suggested by others [10–12, 78]. Secondly, when a 7-day treatment period of ceftriaxone is initiated at the time of 6-hydroxydopamine infusion, not only is glutamate uptake increased with increased GLT-1 expression, but TH loss is attenuated by ~3 times compared to vehicle treatment. Amphetamine-induced rotational behavior after lesion also was attenuated by ~50 % after a 7-day regimen of ceftriaxone compared to vehicle, suggesting the potential for ceftriaxone to minimize Parkinson’s disease symptoms after the lesion has progressed.
TH loss attenuation at 9 days postlesion
Given that the onset of motor symptoms in Parkinson’s disease animal models may be associated with glutamate overactivation [72], examining early stages of nigrostriatal pathway loss, wherein intervention may occur remains a clinically relevant approach. As such, ceftriaxone treatment was initiated on the day of 6-OHDA infusion, due to the fact that its ability to increase GLT-1 expression requires a minimum of 5 days of consecutive treatments [43]. For clinical translation, intervention with ceftriaxone could be feasible if a reliable biomarker for Parkinson’s disease was detected in a patient, prior to locomotor symptom presentation. The ability for ceftriaxone to attenuate TH loss, after the lesion is well underway, also indicates a potential for the antibiotic to be effective in later stages of Parkinson’s disease progression as well. It is not known if our present findings showing TH loss protection may, however, translate into an increase in cell number or protect against their further loss. Thus, subsequent immunohistochemistry studies examining TH-positive cells may reveal an increase in cells or increase in TH per remaining cells and indicate how TH protein loss is mitigated. Current strides in identifying viable Parkinson’s disease biomarkers [79] provides the potential for earlier detection, giving further credence to the neuroprotective capability of ceftriaxone in the treatment of Parkinson’s disease, especially from the standpoint of extending the period of time a susceptible individual may realize prior to the manifestation of locomotor symptoms.
Ca2+-Dependent Mechanism: Calpain and Serine 19 Phosphorylation
Ceftriaxone has been thought to increase GLT-1 transcription through the nuclear factor-κβ signaling pathway, implicating increased transcriptional activity to enhance synaptic glutamate clearing capacity. Here, however, we demonstrate a potentially novel mechanism through Ca2+-dependent means by which ceftriaxone is mitigating TH loss. Glutamate-mediated calcium influx through glutamate receptors such as NMDA has long been considered an essential component of glutamate excitotoxicity [22]. The Ca2+ overload resulting from the overactivation of glutamate receptors can induce the activation of calpain, a family of cysteine proteases activated by calcium, triggering the cleavage of several downstream substrates leading ultimately, to neuronal death [80]. Although we did not see significant differences in calpain breakdown between treatments of ceftriaxone and vehicle, it does not exclude the possibility of calpain activation given that the striatal tissue analyzed also contains GABAergic, glutamatergic, and glial components. Furthermore, our preparation technique to analyze glutamate uptake in conjunction with all protein measures mandated a homogenization step to produce the crude synaptosomes. This physical stress in the neuronal entities may have affected overall calpain activity to a degree that may have affected the sensitivity of the measures between the striatal samples and treatment groups.
Another calcium-dependent marker, and notably only in catecholaminergic neurons, TH phosphorylation at serine19, was decreased in ceftriaxone-treated compared to vehicle-treated rats by ~30 % in lesioned striatum. NMDA agonists may increase Ser19 TH phosphorylation [81], which has been shown to depend upon Ca2+ influx [54]. Furthermore, we have recently reported that increased glutamate uptake capacity in striatum may be associated with decreased ser19 TH phosphorylation [42]. In addition, changes in TH phosphorylation may be induced by NMDA receptor activation [81]. In particular, phosphorylation at Ser19 has been seen to alter the conformation of TH and may increase TH degradation susceptibility through the ubiquitin–proteasome pathway [63]. Ceftriaxone-mediated reduction of Ser19 TH phosphorylation may consequently minimize TH degradation vulnerability and serve as a mechanism through which TH protection is occurring and, importantly, signify less Ca2+ influx in the nigrostriatal terminals. Taken together, our data demonstrate the utility of ceftriaxone in protecting from further TH loss when given at the time of 6-OHDA lesion, potentially through a calcium-mediated mechanism.
Transporter Plasticity
Although there is evidence to support that increasing GLT-1 expression can protect nigrostriatal neurons, other glutamate transporters may also provide possible protection of nigrostriatal neurons. For example, EAAC1 function may also be important for nigrostriatal function, due to it being indirectly involved in glutathione biosynthesis [82]. Another glial glutamate transporter, GLAST, may increase function with GLT-1 compromise [41, 42], although ceftriaxone does not seem to affect GLAST expression levels in our hands as well as others [43].
Clinical Translation and Future Work
The establishment of predictive Parkinson’s disease biomarkers has been at the forefront of recent research [83, 84]. As such, with the eventual establishment of biomarkers, we show evidence suggesting that ceftriaxone could mitigate or diminish the rate of TH protein loss after a lesion has begun or prior to the emergence of locomotor impairment. If this approach is successfully translated into the clinic, ceftriaxone could be used to treat patients identified to be at high risk for Parkinson’s disease due to the presence of such biomarkers, and reduce the rate of dopaminergic neuropil loss in these patients. Although the duration and pharmacological approach for treatment required to protect against Parkinson’s disease progression remains to be determined, the preexisting approval of ceftriaxone by the Food and Drug Administration would make clinical translation even more feasible and thus accelerate its availability to Parkinson’s disease patients.
The potential of ceftriaxone in Parkinson’s disease progression may not be limited to extending the function of nigrostriatal neurons. Ceftriaxone could also conceivably be useful to counteract complications that come with the current gold standard of treatment, L-DOPA. L-DOPA-induced dyskinesia (LID), a debilitating setback for Parkinson’s disease patients who rely on L-DOPA for its therapeutic effects occurs in 90 % of patients with 10 or more years of therapy. There is evidence for glutamatergic involvement in LID pathology [85–87], so treatment with ceftriaxone early in Parkinson’s disease progression may not only minimize TH loss but also, indirectly, postpone the initial administration of L-DOPA, thereby minimizing or delaying L-DOPA-induced dyskinesia expression. Future experiments will be needed to examine the potential for ceftriaxone to be of benefit in this scenario, as well as examining its potential to prolong the time for attaining protection against or restore TH loss in different Parkinson’s disease stages with additional therapies, such as glial cell line-derived neurotrophic factor.
Conclusions
Glutamate excitotoxicity has been long proposed to have some level of involvement in Parkinson’s disease progression. Here, we provide evidence to further support a role for glutamate excitotoxicity in the loss of tyrosine hydroxylase, the rate-limiting enzyme for dopamine biosynthesis, in the 6-OHDA model of Parkinson’s disease. In association with loss of TH, there is a decrease in glutamate uptake. Pharmacologically induced increases in GLT-1 expression from ceftriaxone (seven consecutive daily administrations) resulted in increased glutamate uptake in the striatum, which was concurrent with attenuating the loss of TH caused by 6-OHDA. This protection was associated with a decrease in ser19 TH phosphorylation, serving as evidence that the attenuation of TH loss by 6-OHDA was related to a decrease in Ca2+-influx produced in association with the lesion. The enhanced GLT-1 expression properties of ceftriaxone has been established to take a minimum of five consecutive injections, and we therefore initiated delivery at the time of 6-OHDA, given the rapid rate of TH loss it produces. The translatability of our work may therefore be applicable to a clinical scenario wherein Parkinson’s disease progression is suspected prior to presentation of locomotor impairment. Thus, increasing glutamate uptake could serve to prolong the period of time that a susceptible individual could be free from eventual locomotor impairment, thus increasing the therapeutic window for additional treatments that could further halt or reverse loss of the nigrostriatal neurons.
Acknowledgments
The authors wish to thank Victoria Fields for her outstanding technical assistance at various points throughout the study. This study was funded by the Edward P. Stiles Trust Fund-LSUHSC-Shreveport and Biomedical Research Foundation of NW Louisiana awards to MFS and the Ike Muslow Predoctoral Fellowship to TC.
Footnotes
Conflict of interest The authors declare they have no conflict of interest.
Contributor Information
Tanya Chotibut, Department of Pharmacology, Toxicology, & Neuroscience, Louisiana State University Health Sciences Center-Shreveport, Shreveport, LA 71130, USA.
Richard W. Davis, Department of Pharmacology, Toxicology, & Neuroscience, Louisiana State University Health Sciences Center-Shreveport, Shreveport, LA 71130, USA
Jennifer C. Arnold, Department of Pharmacology, Toxicology, & Neuroscience, Louisiana State University Health Sciences Center-Shreveport, Shreveport, LA 71130, USA
Zachary Frenchek, Department of Pharmacology, Toxicology, & Neuroscience, Louisiana State University Health Sciences Center-Shreveport, Shreveport, LA 71130, USA.
Shawn Gurwara, Department of Pharmacology, Toxicology, & Neuroscience, Louisiana State University Health Sciences Center-Shreveport, Shreveport, LA 71130, USA.
Vimala Bondada, Spinal Cord & Brain Injury Research Center, University of Kentucky Medical Center, Lexington, KY 40536, USA.
James W. Geddes, Spinal Cord & Brain Injury Research Center, University of Kentucky Medical Center, Lexington, KY 40536, USA
Michael F. Salvatore, Email: msalva@lsuhsc.edu, Department of Pharmacology, Toxicology, & Neuroscience, Louisiana State University Health Sciences Center, 1501 Kings Highway, Shreveport, LA 71106, USA
References
- 1.Fonnum F. Glutamate: a neurotransmitter in the mammalian brain. J Neurochem. 1984;42:1–11. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- 2.Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain. 2002;125:1908–1922. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- 3.Lievens JC, Salin P, Nieoullon A, Kerkerian-Le Goff L. Nigrostriatal denervation does not affect glutamate transporter mRNA expression but subsequent levodopa treatment selectively increases GLT1 mRNA and protein expression in the rat striatum. J Neurochem. 2001;79:893–902. doi: 10.1046/j.1471-4159.2001.00644.x. [DOI] [PubMed] [Google Scholar]
- 4.Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, et al. Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience. 2008;153:329–337. doi: 10.1016/j.neuroscience.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 6.Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol. 2011;226:2484–2493. doi: 10.1002/jcp.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 8.Albin RL, Young AB, Penney JB. The functional anatomy of disorders of the basal ganglia. Trends Neurosci. 1995;18:63–64. [PubMed] [Google Scholar]
- 9.Chung EK, Chen LW, Chan YS, Yung KK. Downregulation of glial glutamate transporters after dopamine denervation in the striatum of 6-hydroxydopamine-lesioned rats. J Comp Neurol. 2008;511:421–437. doi: 10.1002/cne.21852. [DOI] [PubMed] [Google Scholar]
- 10.Hazell AS, Itzhak Y, Liu H, Norenberg MD. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) decreases glutamate uptake in cultured astrocytes. J Neurochem. 1997;68:2216–2219. doi: 10.1046/j.1471-4159.1997.68052216.x. [DOI] [PubMed] [Google Scholar]
- 11.Plaitakis A, Shashidharan Glutamate transport and metabolism in dopaminergic neurons of substantia nigra: implications for the pathogenesis of Parkinson’s disease. J Neurol. 2000;247(Suppl 2):S1125–S1135. doi: 10.1007/pl00007757. [DOI] [PubMed] [Google Scholar]
- 12.Lindefors N, Ungerstedt U. Bilateral regulation of glutamate tissue and extra-cellular levels in caudate-putamen by midbrain dopamine neurons. Neurosci Lett. 1990;115:248–252. doi: 10.1016/0304-3940(90)90463-j. [DOI] [PubMed] [Google Scholar]
- 13.Meshul CK, Emre N, Nakamura CM, Allen C, Donohue MK, Buckman JF. Time-dependent changes in striatal glutamate synapses following a 6-hydroxydopamine lesion. Neuroscience. 1999;88:1–16. doi: 10.1016/s0306-4522(98)00189-4. [DOI] [PubMed] [Google Scholar]
- 14.Robinson S, Freeman P, Moore C, Touchon JC, Krentz L, Meshul CK. Acute and subchronic MPTP administration differentially affects striatal glutamate synaptic function. Exp Neurol. 2003;180:74–87. doi: 10.1016/s0014-4886(02)00050-x. [DOI] [PubMed] [Google Scholar]
- 15.Touchon JC, Holmer HK, Moore C, McKee BL, Frederickson J, Meshul CK. Apomorphine induced alterations in striatal and substantia nigra pars reticulata glutamate following unilateral loss of striatal dopamine. Exp Neurol. 2005;193:131–140. doi: 10.1016/j.expneurol.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 16.Meredith GE, Totterdell S, Beales M, Meshul CK. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp Neurol. 2009;219:334–340. doi: 10.1016/j.expneurol.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weihmuller FB, Ulas J, Nguyen L, Cotman CW, Marshall JF. Elevated NMDA receptors in parkinsonian striatum. Neuroreport. 1992;3:977–980. doi: 10.1097/00001756-199211000-00007. [DOI] [PubMed] [Google Scholar]
- 18.Ulas J, Weihmuller FB, Brunner LC, Joyce JN, Marshall JF, Cotman CW. Selective increase of NMDA-sensitive glutamate binding in the striatum of Parkinson’s disease, Alzheimer’s disease, and mixed Parkinson’s disease/Alzheimer’s disease patients. J Neurosci. 1994;14:6317–6324. doi: 10.1523/JNEUROSCI.14-11-06317.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez-Pernaute R, Wang JQ, Kuruppu D, Cao L, Tueckmantel W, Kozikowski A, et al. Enhanced binding of metabotropic glutamate receptor type 5 (mGluR5) PET tracers in the brain of parkinsonian primates. Neuroimage. 2008;42:248–251. doi: 10.1016/j.neuroimage.2008.04.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:789–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- 21.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 22.Choi D. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293–297. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- 23.Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci. 1990;11:379–387. doi: 10.1016/0165-6147(90)90184-a. [DOI] [PubMed] [Google Scholar]
- 24.Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–542. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- 25.Geddes JW, Saatman KE. Targeting individual calpain isoforms for neuroprotection. Exp Neurol. 2010;226:6–7. doi: 10.1016/j.expneurol.2010.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grant RJ, Sellings LH, Crocker SJ, Melloni E, Park DS, Clarke PB. Effects of calpain inhibition on dopaminergic markers and motor function following intrastriatal 6-hydroxydopamine administration in rats. Neuroscience. 2009;158:558–569. doi: 10.1016/j.neuroscience.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 27.Levesque S, Wilson B, Gregoria V, Thorpe LB, Dallas S, Polikov VS, et al. Reactive microgliosis: extracellular micro-calpain and microglia-mediated dopaminergic neurotoxicity. Brain. 2010;133:808–821. doi: 10.1093/brain/awp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ambrosi G, Armentero MT, Levandis G, Bramanti P, Nappi G, Blandini F. Effects of early and delayed treatment with an mGluR5 antagonist on motor impairment, nigrostriatal damage and neuroinflammation in a rodent model of Parkinson’s disease. Brain Res Bull. 2010;82:29–38. doi: 10.1016/j.brainresbull.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 29.Armentero MT, Fancellu R, Nappi G, Bramanti P, Blandini F. Prolonged blockade of NMDA or mGluR5 glutamate receptors reduces nigrostriatal degeneration while inducing selective metabolic changes in the basal ganglia circuitry in a rodent model of Parkinson’s disease. Neurobiol Dis. 2006;22:1–9. doi: 10.1016/j.nbd.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 30.Vernon AC, Zbarsky V, Datla KP, Croucher MJ, Dexter DT. Subtype selective antagonism of substantia nigra pars compacta Group I metabotropic glutamate receptors protects the nigrostriatal system against 6-hydroxydopamine toxicity in vivo. J Neurochem. 2007;103:1075–1091. doi: 10.1111/j.1471-4159.2007.04860.x. [DOI] [PubMed] [Google Scholar]
- 31.Hallert PJ, Standaert DG. Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharmacol Ther. 2004;102:155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 32.Nutt JG, Gunzler SA, Kirchhoff T, Hogarth P, Weaver JL, Krams M, Jamerson B, Menniti FS, Landen JW. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and Parkinsonism. Mov Disord. 2008;15:1860–1866. doi: 10.1002/mds.22169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hubsher G, Haider M, Okun MS. Amantadine: the journey from fighting flu to treating Parkinson disease. Neurology. 2012;78:1096–1099. doi: 10.1212/WNL.0b013e31824e8f0d. [DOI] [PubMed] [Google Scholar]
- 34.Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- 35.Koller M, Urwyler S. Novel N-methyl-D-aspartate receptor antagonists: a review of compounds patented since 2006. Expert Opin Ther Pat. 2006;20:1683–1702. doi: 10.1517/13543776.2010.533656. [DOI] [PubMed] [Google Scholar]
- 36.Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- 37.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 39.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 40.Selkirk JV, Nottebaum LM, Vana AM, Verge GM, Mackay KB, Stiefel TH, et al. Role of the GLT-1 subtype of glutamate transporter in glutamate homeostasis: the GLT-1-preferring inhibitor WAY-855 produces marginal neurotoxicity in the rat hippocampus. Eur J Neurosci. 2005;21:3217–3228. doi: 10.1111/j.1460-9568.2005.04162.x. [DOI] [PubMed] [Google Scholar]
- 41.Duan S, Anderson CM, Stein BA, Swanson RA. Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci. 1999;19:10193–10200. doi: 10.1523/JNEUROSCI.19-23-10193.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salvatore MF, Davis RW, Arnold JC, Chotibut T. Transient striatal GLT-1 blockade increases EAAC1 expression, glutamate reuptake, and decreases tyrosine hydroxylase phosphorylation at ser19. Exp Neur. 2012;234:428–436. doi: 10.1016/j.expneurol.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 43.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 44.van Landeghem FK, Weiss T, Oehmichen M, von Deimling A. Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J Neurotrauma. 2006;23:1518–1528. doi: 10.1089/neu.2006.23.1518. [DOI] [PubMed] [Google Scholar]
- 45.Rao VL, Dogan A, Bowen KK, et al. Antisense knockdown of the glial glutamate transporter GLT-1 exacerbates hippocampal neuronal damage following traumatic injury to rat brain. Eur J Neurosci. 2001;13:119–28. [PubMed] [Google Scholar]
- 46.Hirsch EC. Nigrostriatal system plasticity in Parkinson’s disease: effect of dopaminergic denervation and treatment. Ann Neurol. 2000;47:S115–20. [PubMed] [Google Scholar]
- 47.Smeyne RJ, Jackson-Lewis V. The MPTP model of Parkinson’s disease. Brain Res Mol Brain Res. 2005;134:57–66. doi: 10.1016/j.molbrainres.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 48.Leung TC, Lui CN, Chen LW, et al. Ceftriaxone ameliorates motor deficits and protects dopaminergic neurons in 6-hydroxydopamine-lesioned rats. ACS Chem Neurosci. 2012;3:22–30. doi: 10.1021/cn200072h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sari Y, Prieto A, Barton SJ, et al. Ceftriaxone-induced up-regulation of cortical and striatal GLT1 in the R6/2 model of Huntington’s disease. J Biomed Sci. 2010;17:62. doi: 10.1186/1423-0127-17-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown RH., Jr Amyotrophic lateral sclerosis—a new role for old drugs. N Engl J Med. 2005;352:1376–1378. doi: 10.1056/NEJMcibr050274. [DOI] [PubMed] [Google Scholar]
- 51.Chotibut T, Apple DM, Jefferis R, et al. Dopamine transporter loss in 6-OHDA Parkinson’s model is unmet by parallel reduction in dopamine uptake. PLos One. 2012 doi: 10.1371/journal.pone.0052322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 2. Academic; Sydney: 1986. [DOI] [PubMed] [Google Scholar]
- 53.Hudson JL, van Horne CG, Stromberg I, Brock S, Clayton J, Masserano J, et al. Correlation of apomorphine- and amphetamine-induced turning with nigrostriatal dopamine content in unilateral 6-hydroxydopamine lesioned rats. Brain Res. 1993;626:167–174. doi: 10.1016/0006-8993(93)90576-9. [DOI] [PubMed] [Google Scholar]
- 54.Salvatore MF, Waymire JC, Haycock JW. Depolarization-stimulated catecholamine biosynthesis: involvement of protein kinases and tyrosine hydroxylase phosphorylation sites in situ. J Neurochem. 2001;79:349–360. doi: 10.1046/j.1471-4159.2001.00593.x. [DOI] [PubMed] [Google Scholar]
- 55.Salvatore MF, Apparsundaram S, Gerhardt GA. Decreased plasma membrane expression of striatal dopamine transporter in aging. Neurobiol Aging. 2003;24:1147–1154. doi: 10.1016/s0197-4580(03)00129-5. [DOI] [PubMed] [Google Scholar]
- 56.Henn FA, Anderson DJ, Rustad DG. Glial contamination of synaptosomal fractions. Brain Res. 1976;101:341–344. doi: 10.1016/0006-8993(76)90274-2. [DOI] [PubMed] [Google Scholar]
- 57.Suchak SK, Baloyianni NV, Perkinton MS, Williams RJ, Meldrum BS, Rattray M. The ‘glial’ glutamate transporter, EAAT2 (Glt-1) accounts for high affinity glutamate uptake into adult rodent nerve endings. J Neurochem. 2003;84:522–532. doi: 10.1046/j.1471-4159.2003.01553.x. [DOI] [PubMed] [Google Scholar]
- 58.Minger SL, Geddes JW, Holtz ML, Craddock SD, Whiteheart SW, Siman RG, Pettigrew LC. Glutamate receptor antagonists inhibit calpain-mediated cytoskeletal proteolysis in focal cerebral ischemia. 1998;810:181–199. doi: 10.1016/s0006-8993(98)00921-4. [DOI] [PubMed] [Google Scholar]
- 59.Nickell J, Pomerleau F, Allen J, Gerhardt GA. Age-related changes in the dynamics of potassium-evoked L-glutamate release in the striatum of Fisher 344 rats. J Neural Transm. 2005;112:87–96. doi: 10.1007/s00702-004-0151-x. [DOI] [PubMed] [Google Scholar]
- 60.Wassum KM, Tolosa VM, Wang J, Walker E, Monbouquette HG, Maidment NT. Silicon wafer-based platinum microelectrode array biosensor for near real-time measurement of glutamate in vivo. Sens Basel Sens. 2008;8:5023–5036. doi: 10.3390/s8085023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salvatore MF, Pruett BS, Dempsey C, Fields V. Comprehensive profiling of dopamine regulation in substantia nigra and ventral tegmental area. J Vis Exp. 2012 doi: 10.3791/4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci. 1973;20:415–455. doi: 10.1016/0022-510x(73)90175-5. [DOI] [PubMed] [Google Scholar]
- 63.Nakashima A, Mori K, Kaneko YS, Nobuhiro H, Toshiharu N, Ota A. Phosphorylation of the N-terminal portion of tyrosine hydroxylase triggers proteasomal digestion of the enzyme. Bio Biophys Res Com. 2011;407:343–347. doi: 10.1016/j.bbrc.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 64.DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci. 1990;13:286–289. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- 65.Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 66.Andre VM, Cepeda C, Levine MS. Dopamine and glutamate in Huntington’s disease: a balancing act. CNS Neurosci Ther. 2010;16:163–178. doi: 10.1111/j.1755-5949.2010.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, Meshul CK. Dopamine modulates release from corticostriatal terminals. J Neurosci. 2004;24:9541–9552. doi: 10.1523/JNEUROSCI.2891-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.David HN, Ansseau M, Abraini JH. Dopamine-glutamate reciprocal modulation of release and motor responses in the rat caudate-putamen and nucleus accumbens of “intact” animals. Brain Res Rev. 2005;50:336–360. doi: 10.1016/j.brainresrev.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 69.Greenamyre JT. Glutamate-dopamine interactions in the basal ganglia: relationship to Parkinson’s disease. J Neural Transm Gen Sect. 1993;91:255–269. doi: 10.1007/BF01245235. [DOI] [PubMed] [Google Scholar]
- 70.Starr MS. Glutamate/dopamine D1/D2 balance in the basal ganglia and its relevance to Parkinson’s disease. Synapse. 1995;19:264–293. doi: 10.1002/syn.890190405. [DOI] [PubMed] [Google Scholar]
- 71.Yamamoto BK, Davy S. Dopaminergic modulation of glutamate release in striatum as measured by microdialysis. J Neurochem. 1992;58:1736–1742. doi: 10.1111/j.1471-4159.1992.tb10048.x. [DOI] [PubMed] [Google Scholar]
- 72.Greenamyre JT, Hastings TG. Biomedicine. Parkinson’s—divergent causes, convergent mechanisms. Science. 2004;304:1120–1122. doi: 10.1126/science.1098966. [DOI] [PubMed] [Google Scholar]
- 73.Blandini F, Greenamyre JT, Nappi G. The role of glutamate in the pathophysiology of Parkinson’s disease. Funct Neurol. 1996;11:3–15. [PubMed] [Google Scholar]
- 74.Izumi Y, Yamamoto N, Matsuo T, Wakita S, Takeuchi H, Kume T, et al. Vulnerability to glutamate toxicity of dopaminergic neurons is dependent on endogenous dopamine and MAPK activation. J Neurochem. 2009;110:745–755. doi: 10.1111/j.1471-4159.2009.06178.x. [DOI] [PubMed] [Google Scholar]
- 75.Black YD, Xiao D, Pellegrino D, Kachroo A, Brownell AL, Schwarzschild MA. Protective effect of metabotropic glutamate mGluR5 receptor elimination in 6-hydroxydopamine model of Parkinson’s disease. Neurosci Lett. 2010;486:161–165. doi: 10.1016/j.neulet.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schlag BD, Vondrasek JR, Munir M, Kalandadze A, Zelenaia OA, Rothstein JD, et al. Regulation of the glial Na+-dependent glutamate transporters by cyclic AMP analogs and neurons. Mol Pharmacol. 1998;53:355–369. doi: 10.1124/mol.53.3.355. [DOI] [PubMed] [Google Scholar]
- 77.Tian G, Lai L, Guo H, Lin Y, Butchbach ME, Chang Y, et al. Translational control of glial glutamate transporter EAAT2 expression. J Biol Chem. 2007;282:1727–1737. doi: 10.1074/jbc.M609822200. [DOI] [PubMed] [Google Scholar]
- 78.Dervan AG, Meshul CK, Beales M, McBean GJ, Moore C, Totterdell S, et al. Astroglial plasticity and glutamate function in a chronic mouse model of Parkinson’s disease. Exp Neurol. 2004;190:145–156. doi: 10.1016/j.expneurol.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 79.Wang J, Hoekstra JG, Zuo C, Cook TJ, Zhang J. Biomarkers of Parkinson’s disease: current status and future perspectives. Drug Discov Today. 2013;18:155–162. doi: 10.1016/j.drudis.2012.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bevers MB, Neumar RW. Mechanistic role of calpains in postischemic neurodegeneration. J Cereb Blood Flow Metab. 2008;28:655–673. doi: 10.1038/sj.jcbfm.9600595. [DOI] [PubMed] [Google Scholar]
- 81.Lindgren N, Xu ZQ, Lindskog M, Herrera-Marschitz M, Goiny M, Haycock JW, et al. Regulation of tyrosine hydroxylase activity and phosphorylation at ser19 and ser40 via activation of glutamate NMDA receptors in rat striatum. J Neurochem. 2000;74:2470–2477. doi: 10.1046/j.1471-4159.2000.0742470.x. [DOI] [PubMed] [Google Scholar]
- 82.Nafia I, Re DB, Masmejean F, Melon C, Kachidian P, Kerkerian-Le Goff L, Nieoullon A, Had-Aissouni L. Preferential vulnerability of mesencephalic dopamine neurons to glutamate transporter dysfunction. J Neurochem. 2008;105:484–496. doi: 10.1111/j.1471-4159.2007.05146.x. [DOI] [PubMed] [Google Scholar]
- 83.Maetzler W, Liepelt I, Berg D. Progression of Parkinson’s disease in the clinical phase: potential markers. The Lancent Neurology. 2009;8:1158–1171. doi: 10.1016/S1474-4422(09)70291-1. [DOI] [PubMed] [Google Scholar]
- 84.Wu Y, Le W, Jankovic J. Preclinical biomarkers of Parkinson disease. Arch Neurol. 2011;68:22–30. doi: 10.1001/archneurol.2010.321. [DOI] [PubMed] [Google Scholar]
- 85.Cenci MA, Lindgren HS. Advances in under standing L-DOPA-induced dyskinesia. Current Opinion in Neurobiology. 2007;17(6):665–671. doi: 10.1016/j.conb.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 86.Cenci MA, Lundblad M. Post-versus presynap tic plasticity in L-DOPA-induced dyskinesia. Journal of Neurochemistry. 2006;99(2):381–392. doi: 10.1111/j.1471-4159.2006.04124.x. [DOI] [PubMed] [Google Scholar]
- 87.Chase TN, Oh JD. Striatal dopamine- and glutamate-mediated dysregulation in experimental parkinsonism. Trends in Neurosciences. 2000;23(10 Suppl):S86–S91. doi: 10.1016/s1471-1931(00)00018-5. [DOI] [PubMed] [Google Scholar]