

Abstract
Objective
Lung cancer is the leading cause of cancer death in both men and women. Non-small cell lung cancer (NSCLC) has an overall 5-year survival rate of 15%. While aberrant STAT3 activation has previously been observed in NSCLC, the scope of its contribution is uncertain and agents that target STAT3 for treatment are not available clinically.
Methods
We determined levels of activated STAT3 (STAT3 phosphorylated on Y705, pSTAT3) and the two major isoforms of STAT3 (α and β) in protein extracts of 8 NSCLC cell lines, as well as the effects of targeting STAT3 in vitro and in vivo in NSCLC cells using short hairpin (sh) RNA and two novel small-molecule STAT3 inhibitors, C188-9 and piperlongumine (PL).
Results
Levels of pSTAT3, STAT3α, and STATβ were increased in 7 of 8 NSCLC cell lines. Of note, levels of pSTAT3 were tightly correlated with levels of STAT3β, but not STAT3α. Targeting of STAT3 in A549 cells using shRNA decreased tSTAT3 by 75%; this was accompanied by a 47–78% reduction in anchorage-dependent and anchorage-independent growth and a 28–45% reduction in mRNA levels for anti-apoptotic STAT3 gene targets. C188-9 and PL (@30μM) each reduced pSTAT3 levels in all NSCLC cell lines tested by ≥50%, reduced anti-apoptotic protein mRNA levels by 25–60%, and reduced both anchorage-dependent and anchorage-independent growth of NSCLC cell lines with IC50 values ranging from 3.06–52.44μM and 0.86–11.66μM, respectively. Treatment of nude mice bearing A549 tumor xenografts with C188-9 or PL blocked tumor growth and reduced levels of pSTAT3 and mRNA encoding anti-apoptotic proteins.
Conclusion
STAT3 is essential for growth of NSCLC cell lines and tumors and its targeting using C188-9 or PL may be a useful strategy for treatment.
Keywords: STAT3, NSCLC, small molecule inhibitors
1. Introduction
Lung cancer is the leading cause of cancer-related death in men and women in Western countries (1). Non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancer cases. Approximately 70% of patients present with locally advanced or metastatic disease (1, 2). Despite novel molecular therapies, the prognosis remains poor with an overall 5 year survival rate of ~15%.
Elevated STAT3 was found in the majority (91–94%) of resected human NSCLC tumors (3, 4) and increased pSTAT3 and nuclear STAT3 were found in ~55–62% of NSCLC tumors as measured by immunohistochemical analyses (5, 6). Downstream transcriptional targets of STAT3 have been found to be upregulated in resected NSCLC (7, 8). However, our understanding of the contribution of STAT3 to NSCLC oncogenesis is incomplete and it remains uncertain whether targeting STAT3 in NSCLC is technically feasible or therapeutically beneficial.
In a drug development program involving virtual ligand screening, 2-D similarity screening, 3-D pharmacophore analysis, and SAR-based medicinal chemistry, we identified C188-9 (9–11) as a potent small-molecule probe that targets the Src-homology (SH) 2 domain of STAT3, thus blocking two steps in its activation—recruitment to activated receptors and homodimerization. We also identified piperlongumine (PL), a natural product isolated from the fruit of the pepper Piper longum, as a direct STAT3 inhibitor in a drug repurposing screen (12). Our objectives were to determine the frequency of activation of STAT3, and of increased levels of STAT3 isoforms (α and β), in a panel of human NSCLC cell lines. In addition, we wanted to determine the effect of targeting STAT3 with shRNA, C188-9, or PL on growth of NSCLC cells in vitro and in vivo.
2. Materials and Methods
2.1 Cell culture
Eight NSCLC human cell lines (A549, H1299, H1563, H1437, H661, H2126, H1573, and H1975) were obtained from the American Type Culture Collection (ATCC, Rockville, MD). A normal human bronchial cell line, HBEC3-KT, was a gift from John Minna (UT-Southwestern). Cells were maintained in complete media with 10% FBS, antibiotics/antimycotics and not passed continuously more than 4 weeks. All cell lines were originally authenticated by ATCC.
2.2 Compounds
C188-9 was obtained from StemMed, Ltd., who developed it from an initial hit (C188) that was identified using computer-based docking of ~ 1 million compounds into the phosphotyrosyl-peptide binding pocket of the STAT3 SH2 domain (9–11). PL, previously identified in a drug repurposing screen (12), was obtained from Indofine Chemical Company. Treatments with C188-9 and PL were performed as described in the text.
2.3 Luminex Assay
All samples were lysed using buffer containing protease (Roche, catalog # 05892791001) and phosphatase inhibitors (Roche, catalog #04906837001). The protein extracts were collected after centrifugation at 12,000g at 4°C for 10 minutes. Protein was plated in a 96 well filter plate pre-loaded with beads (Millipore, Danvers, MA) coupled to antibody against the indicated analytes and incubated overnight at 4°C. Bead-bound analytes were measured using biotinylated detection antibody specific for a different epitope and streptavidin-phycoerythrin (streptavidin-PE). Data were collected and analyzed using the Bio-Plex suspension array system (Luminex 100 system, Bio-Rad Laboratories, Hercules, CA). Where indicated, GAPDH-normalized pSTAT3 values from each treatment condition were corrected for untreated cells, expressed as percentage untreated, and used to determine the IC50 using GraphPad.
2.4 Immunoblotting
All specimens were lysed as described in section 2.3. Fifty μg of protein was separated on a 4–15% SDS-PAGE gel, transferred onto PVDF membrane (Bio-Rad, catalog #162-0174) in Tris-glycine buffer (BioRad, catalog# 161-0771) containing 20% v/v methanol. The PVDF membrane was blocked with 5% non-fat dry milk in TBS containing 0.05% Tween 20 (TBST). The blots incubated with primary antibody overnight at 4°C. The primary STAT3β mouse monoclonal antibody was generated in our laboratory against the C-terminal 7 amino acid residues unique to STAT3β (13) and used at a dilution of 1:1000. The membranes were incubated with horseradish-peroxidase (HRP)-conjugated secondary goat anti-mouse antibody (Santa Cruz, catalog# sc2005) at 1:5,000. The bands were visualized with enhanced chemiluminescence (Pierce ECL Western Blotting Substrate, Thermo Scientific, catalog# 32106). Densitometry analysis was performed using Image-J software (NIH).
2.5 Construction of A549 cells stably expressing STAT3-specific or control shRNA
A puromycin titration identified the optimal inhibitory concentration of 3.0 μg/mL. Stable STAT3 knockdown cell lines were created by infecting A549 cells with pLK0.1-based lentiviral particles containing shRNAs targeted to STAT3 (#TRCN0000329888; Sigma-Aldrich, St. Louis, MO) or Mission TRC2 Transduction Particle containing control shRNA (#SHC216V). Forty-eight hours post-infection, cells were passaged and selected with 3.0 μg/mL puromycin for 10 days to eliminate uninfected cells.
2.6 Quantitative RT-PCR
Total RNA was isolated from samples using RNeasy (Qiagen), reverse transcribed, and amplified by real time PCR. Levels of STAT3 mRNA (Hs00374280_m1) and 18S RNA (Hs03928985_g1) were determined using TaqMan Universal Master Mix II (Life Technologies) and TaqMan primers (Life Technologies). Levels of Bcl-2, Bcl-xL, Cyclin D1, Survivin and GAPDH were determined using Sybr Green Master Mix (Life Technologies) and primers from Invitrogen. The results were expressed as relative mRNA levels normalized to the 18S or GAPDH mRNA level using the formula [2^[Ct(18S or GAPDH)–Ct(Gene)].
2.7 Anchorage-dependent and anchorage-independent cell growth assays
Cell growth was determined by measuring viability using an MTT assay, as described (12). Cells were grown in triplicates in 96 well plates (or in ultra low attachment plates for the anchorage independent assay) in the recommended media with 10% FBS ± C188-9 or PL for 24–72 hours and cell number measured by determining the optical density (OD) at 590 nm using a 96-well multi-scanner (EL-800 universal microplate reader, BioTek Inc, VT, USA). Relative % proliferation (OD590 after any treatment ÷ OD590 of untreated cells x 100) was plotted along Y-axis. At least 2 replicate experiments were performed and used for IC50 calculation using GraphPad software.
2.8 Flow cytometry
A549 cells were treated with C188-9 or PL (@30μM) for up to 48 hrs. Cells were dissociated into singlets followed by staining with FITC-conjugated antibodies for Annexin V analysis (Becton Dickinson, catalog #556547) or propidium iodide stain solution for cell cycle analysis (Becton Dickinson, catalog #340242). Cells were analyzed on an Attune flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
2.9 Nude mouse tumor xenograft assay
A549 cells were suspended in serum free media at a concentration of 3x106 and were subcutaneously inoculated into the right flank of 5–6 week old athymic nude mice (11 mice/group). When the average tumor size was ~100mm3, mice received DMSO (50μL), C188-9 (50mg/kg twice daily in 25μL DMSO) or PL (30 mg/kg once daily in 50μl DMSO) intraperitoneally 5 days/week for 3 weeks. Subcutaneous tumor volume (mm3) was calculated as 0.5 x (long dimension) x (short dimension)2. At the end of 3 weeks, mice were euthanized. Tumors were excised, weighed and frozen in liquid nitrogen. Whole blood was obtained by cardiac puncture and anticoagulated. Peripheral blood mononuclear cells (PBMC) were isolated from whole blood by sedimentation over Ficoll-Paque PREMIUM 1.084 according to the manufacturer’s instruction (GE Healthcare Life Sciences, cat #17-5442-02). Total protein was extracted from tumor pieces following sonication as described in section 2.3.
2.10 Statistical Analysis
Student’s t-test or a paired t-test was used to compare differences between groups as indicated.
3. Results
3.1 STAT3 is constitutively activated in most human NSCLC cell lines; levels of pSTAT3 correlate with levels of STAT3β
Constitutively activated STAT3 has been demonstrated to be increased compared to corresponding normal tissues in multiple cancers, including NSCLC tumors cell lines (14). To more thoroughly explore the frequency of increased levels of constitutively activated STAT3 in NSCLC cell lines, we first determined the level of activated STAT3 i.e. STAT3 phosphorylated on Y705 (pSTAT3), within protein extracts of a panel of eight NSCLC cell lines using a Luminex bead-based assay (Figure 1A) and compared each to the level of pSTAT3 in the non-tumorigenic bronchial epithelial cell line (HBEC-3KT). One NSCLC cell line (H1563) had levels of pSTAT3 similar to HBEC-3KT, while each of the other seven cell lines (H1437, H661, H2126, A549, H1573, H1299, H1975) had pSTAT3 levels 2–6 fold greater than HBEC-3KT.
Figure 1. STAT3 is constitutively activated in most NSCLC cell lines, which correlates with levels of STAT3β.

A) Level of pSTAT3 were determined by Luminex beads in human NSCLC cell lines and in a normal human bronchial epithelial cell line (HBEC3-KT); values were corrected for GAPDH levels, expressed as a fraction of that obtained in HBEC3-KT cells, and the mean ± SD of 3 determinations shown; an asterisk (*) indicates those cell lines increased compared to HBEC3-KT cells (p<0.04). B) Levels of STAT3α, STAT3β and GAPDH were determined by immunoblot in lysates of murine embryonic fibroblast cells (MEF) lines in which the STAT3 gene was deleted followed by transient expression of STAT3α (MEF-STAT3α) or STAT3β (MEF-STAT3β)(13), HBEC3-KT cells, and human NSCLC cell lines (representative gel shown). Levels of STAT3α (C) and STAT3β (D) were quantified by densitometry using Image software. Each value was corrected using its corresponding GAPDH level and the mean ± SD of 2 determinations shown for each cell line (left panels) or as a function of its corresponding pSTAT3 level (right panels). An asterisk (*) indicates those cell lines increased compared to HBEC3-KT cells (p<0.02). pSTAT3 levels did not significantly correlate with levels of STAT3α (R2=0.1856, p=0.247), but did with levels STAT3β levels (R2=0.7348, p=0.0031).
Two isoforms of STAT3 (α/p92 and β/p83), resulting from alternative splicing, are present within most cell types at a 4:1 ratio (15). STAT3α has been more tightly linked to oncogenesis, while STAT3β has been shown to antagonize the pro-oncogenic functions of STAT3α (15). The contribution of each isoform to levels of STAT3 activation in NSCLC has not been reported. Using commercially available antibodies, we found the level of STAT3α was increased in 6 of 8 NSCLC cell lines each by 2–5 fold compared to HBEC-3KT (Figure 1B and 1C). However, to our surprise, levels of STAT3α did not correlate with levels of pSTAT3 (Figure 1C).
Compared to pSTAT3α homodimers, studies demonstrated that STAT3β homodimers have been found to bind DNA more avidly (16), to be resistant to dephosphorylation (16, 17), and to have markedly prolonged nuclear retention (18). More recent studies demonstrated that increased co-expression of STAT3β relative to STAT3α was shown to increase levels of pSTAT3 and to prolong its phosphorylation (17), presumably through resistance to dephosphorylation of STAT3β homodimers and STAT3α/β heterodimers (17). We developed monoclonal antibodies that recognize the 7 C-terminal amino acids unique to STAT3β (CT7) and were specific for STAT3β (13). Using our STAT3β-specific antibodies, we found that STAT3β protein levels, similar to STAT3α proteins levels, were elevated in 7 of 8 NSCLC cell lines each by 2–6 fold compared to HBEC-3KT (Figure 1B and 1D). However, in contrast to levels of STAT3α, levels of STAT3β positively correlated with pSTAT3 levels (Figures 1D). These findings suggest that increased expression of STAT3β protein in lung cancer cell lines may directly contribute to the increased constitutive levels of pSTAT3 through increasing resistance of pSTAT3 to dephosphorylation.
3.2 Effect of shRNA targeting of STAT3 on NSCLC proliferation
To determine the contribution of STAT3 to tumor cell growth, we targeted it using shRNA. A549 cells were transfected with lentivirus expressing STAT3 specific shRNA or control shRNA. STAT3 mRNA was reduced by 78–84% in A549 cells stably transfected with STAT3 shRNA (A549/shSTAT3) vs. control shRNA (A549/shControl; Figure 2A). Similarly, levels of pSTAT3 and total STAT3 protein were reduced by 58% and 75%, respectively, in A549/shSTAT3 vs. A549/shControl cells (Figure 2B). Anchorage-dependent and anchorage-independent cell proliferation each was reduced by 78% and 47%, respectively in A549/shSTAT3 cells vs. A549/shControl cells (Figure 2C). Silencing of STAT3 reduced mRNA levels of STAT3 gene targets, including genes encoding anti-apoptotic molecules Bcl-2, Bcl-xL, and Survivin, as well as the cell cycle protein, Cyclin D1, each by 28–45% (Figure 2D). These findings indicate STAT3 is necessary for proliferation of the A549 cell line likely through increasing levels of anti-apoptotic and cell cycle proteins.
Figure 2. Effect of shRNA targeting of STAT3 on STAT3 gene targets and A549 cell proliferation.

Total RNA (A) or protein (B) was extracted from A549 cells stably transfected with either control shRNA or 2 distinct STAT3 shRNA constructs and used to determine levels of STAT3 mRNA by quantitative RT-PCR (A) or levels of pSTAT3, total STAT3 and GAPDH protein using Luminex beads (B); mean ± SD of 3 separate determinations are shown; *, p<0.003 for each. C) Cell proliferation in A549 cells stably transfected with either control shRNA or 2 distinct STAT3 shRNA constructs was determined by MTT assay using either anchorage-dependent (left panel) and anchorage- independent (right panel) conditions. Data shown are mean ± SD of 3 determinations; *, p<0.0001 for each. D) Levels of four STAT3 downstream gene targets were determined in total RNA of A549 cells stably transfected with either control shRNA or 2 distinct STAT3 shRNA constructs using quantitative RT-PCR. Data shown are mean ± SD of 3 determinations; *, p<0.02, p<0.04, p<0.03 and p<0.02, as shown.
3.3 STAT3 targeting with C188-9 or PL decreased pSTAT3 levels, reduced proliferation, and induced apoptosis in a panel of NSCLC cell lines
We previously identified C188-9 and PL as small molecule STAT3 inhibitors. To begin to determine their potential for treating NSCLC, we incubated a panel of human NSCLC cell lines with C188-9 or PL @30μM for various times (0, 0.25, 0.5, 1, 2, 2.5, 3, and 4 hours). C188-9 and PL each reduced pSTAT3 levels in all cell lines tested, as early as 1 hour and maximally by 2.5 hours (Figure 3A; data not shown). In addition, C188-9 and PL exposure (2.5 hours) each decreased pSTAT3 levels in a concentration-dependent manner starting at concentrations as low as 3 nM (Figure 3B). However, inhibition only exceeded 50% at much higher concentrations of C188-9 and PL (100 μM and 1 μM, respectively). To determine the effect of C188-9 and PL on proliferation of NSCLC cell lines, we incubated each cell line with C188-9 or PL at various concentrations (0, 0.1, 0.3, 1, 3, 10, and 100 μM) for 72 hours under both anchorage-dependent and anchorage- independent conditions. C188-9 and PL each reduced both anchorage-dependent and anchorage-independent cell viability of 8 human NSCLC cell lines after 72hr incubation with IC50 values ranging from 3.06–52.44μM and 0.86–11.66μM, respectively (Figure 3C). In addition, similar to targeting of STAT3 with shRNA, targeting of STAT3 in A549 cells with C188-9 or PL (@30μM) for 4hrs reduced levels of STAT3 downstream transcriptional targets, including Bcl-2, Bcl-xL, Survivin and Cyclin D1, by 25–60% compared to untreated cells (Figure 3D).
Figure 3. C188-9 or PL treatment decreased levels of pSTAT3, reduced proliferation, and induced apoptosis of A549 cells.

A549 cells were incubated with C188-9 (left panel) or PL (right panel; @30μM) for the indicated time periods (A) or for 2.5 hrs at the indicated concentrations (B). pSTAT3 levels were determined in duplicate using Luminex beads, corrected for GAPDH. Data presented are mean ± SD of duplicate determinations; * indicates treatment time intervals (A) and concentrations (B) significantly reduced from time 0 or concentration=0 (p<0.04). C) A549 cells were incubated in C188-9 (left panel) or PL (right panel) at the concentrations indicated for 72 hrs under anchorage-dependent (top panel) and anchorage-independent (bottom panel) conditions. Cell proliferation was assayed by MTT assay and the results used to generated the curves and IC50 values shown. D) Levels of four STAT3 downstream gene targets were determined in total RNA of A549 cells treated for 4 hours with C188-9 or PL (@30μM) using quantitative RT-PCR. Data shown are mean ± SD of 3 determinations; an asterisk (*) indicates a significant difference from untreated cells with p values indicated.
To determine whether the reduction in anti-apoptotic and cell cycle mRNA resulted in apoptosis vs. cessation of progression of the cell cycle, flow cytometry was performed on cells treated with each inhibitor (@ 30μM for 2, 4, 8, 12, 24 and 48hrs). Both C188-9 and PL increased the rate of apoptosis in A549 cells from 3.25–3.5% in the untreated cells (N=7) to 11.75% in C188-9-treated cells (N=4; p<0.0012) and 33.76% in PL-treated cells (N=3; p<0.022) at 24hrs (Figure 4). At 48hrs incubation, treatment with C188-9 increased the percentage of apoptotic cells to 82.7% (n=2; p<0.0012). Neither inhibitor significantly affected cell cycle progression (data not shown).
Figure 4. Annexin V staining of A549 cells following incubation with C188-9 or PL.
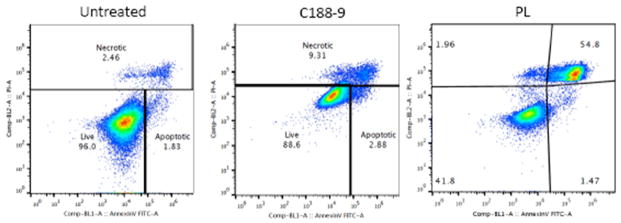
A549 cells were incubated with C188-9 and PL (@30μM) for 24 hours then stained with Annexin V and analyzed by flow cytometry. The percentage of cells in the necrotic, live, and apoptotic-gated areas are indicated.
3.4 Effect of C188-9 or PL on A549 tumor xenograft volume, weight, pSTAT3 levels and expression of STAT3 downstream targets
To examine the ability of C188-9 and PL to inhibit growth of human NSCLC tumors, C188-9, PL or DMSO (vehicle) was administered for three weeks by IP injection into athymic nude mice bearing human A549 tumor xenografts starting when the average tumor size in each group was ~100 mm3. C188-9 and PL each inhibited tumor growth with the difference in tumor volume of drug-treated vs. vehicle-treated mice becoming significant within two days and continuing until the end of treatment (Figure 5A). Similarly, C188-9 and PL treatment reduced tumor weights at the end of treatment by 50% compared to vehicle controls (p<0.05; Figure 5B). Importantly, levels of pSTAT3 protein were reduced by 65% in tumors from drug-treated mice compared to tumors from vehicle treated mice (p<0.05; Figure 5C), which was accompanied by a 35% reduction in mRNA levels of STAT3 gene targets (P<0.05 for each; Figure 5D). Thus both C188-9 and PL hit target in tumors resulting in stasis of tumor growth through downregulation of STAT3 anti-apoptotic and cell cycle gene targets.
Figure 5. Effect of C188-9 and PL on A549 xenograft tumor volume, tumor weight, pSTAT3 levels, and expression of downstream STAT3 gene targets.

Tumor volumes (A) or weights (B) were measured on the days indicated or following treatment for 3 weeks, respectively, with C188-9 (50mg/kg IP twice daily; n=12), PL (30mg/kg IP daily; n=11), or vehicle (n=17). The mean ± SD for each group is shown. In panel A, asterisk (*) or caret (^) indicates a reduction compared to vehicle in animals treated with C188-9 (p<0.04) or with PL (p<0.04), respectively. In panel B, the asterisk (*) indicates p<0.05. C) Levels of pSTAT3 and GAPDH levels were determined using Luminex beads in protein extracts of tumors; pSTAT3 levels were normalized for GAPDH and the mean ± SD shown; the asterisk (*) indicates a reduction in pSTAT3 levels in C188-9- or PL-treated tumors compared to vehicle (p<0.039). D) Levels of STAT3 downstream genes targets were assessed by RT-PCR in total tumor RNA. Mean ± SD is shown; an asterisk (*) indicates a significant reduction in Bcl-2 (p<0.05), Bcl-xL (p<0.045), Cyclin D1 (p<0.022) and Survivin (p<0.02) RNA levels in C188-9- and PL-treated tumors compared to vehicle-treated tumors.
3.5 Effect of C188-9 or PL on levels of pSTAT3 in peripheral blood mononuclear cells
Increased pSTAT3 has previously been observed in PBMC of humans and mice with tumors(19), consequently, reduction of PBMC pSTAT3 could serve as a useful biomarker in patients receiving a STAT3 inhibitor. To determine if pSTAT3 was reduced in the PBMC of tumor-bearing mice receiving C188-9 or PL, we isolated PBMC from whole blood of mice bearing A549 xenografts at the end of treatment with C188-9, PL, or vehicle. Levels of pSTAT3 were reduced by 25% (p=0.039) in PBMC of mice treated with C188-9 vs. vehicle (Figure 6).
Figure 6. Effect of C188-9 or PL treatment on levels of pSTAT3 levels in PBMC.
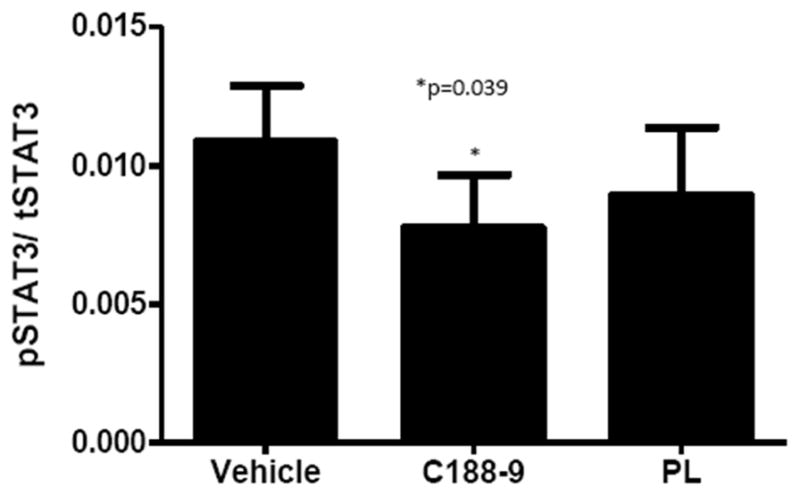
Levels of pSTAT3 and total STAT3 were determined using Luminex beads in protein extracts of PBMC isolated from whole blood of tumor-bearing mice after 3 weeks of treatment with vehicle, C188-9, or PL. Level of pSTAT3 were normalized to total STAT3. The mean ± SD is shown; an asterisk (*) indicates a reduction in pSTAT3 level compared to vehicle (p=0.039).
4. Discussion
We demonstrated that the level of pSTAT3, STAT3α, and STAT3β were increased in 88% of NSCLC cells lines and that levels of activated STAT3 (pSTAT3) were tightly correlated with levels of STAT3β, but not STAT3α. Targeted decrease of total STAT3 in a representative NSCLC cell line, A549, using shRNA reduced anchorage-dependent and anchorage-independent growth and was accompanied by reductions in mRNA levels for each of four anti-apoptotic and cell cycle proteins. Two small-molecule STAT3 inhibitors, C188-9 and PL, reduced pSTAT3 levels in all NSCLC cell lines tested and potently inhibited their anchorage-dependent and anchorage-independent growth. Treatment of A549 cells with each inhibitor increased cell apoptosis by decreasing mRNA levels of known STAT3 gene targets encoding anti-apoptotic proteins. Treatment of nude mice bearing A549 tumor xenografts with either C188-9 or PL reduced tumor volumes, tumor weights, and levels of tumor pSTAT3 and mRNA encoding anti-apoptotic proteins. Thus, STAT3 is essential for survival and growth of NSCLC cell lines and tumors and its targeting using C188-9 or PL may be a useful strategy for treatment of NSCLC patients.
Activation of STAT3 through phosphorylation on Y705 (pSTAT3) leads to its dimerization, nuclear translocation, DNA binding and gene transcription (20). STAT3 is known to regulate the expression in many tumor types of genes involved in survival, cell cycle progression, angiogenesis, immune evasion, epithelial mesenchymal transformation and metastasis (8, 20–25). In resected NSCLC samples and human NSCLC cell lines, elevated STAT3 has been inversely correlated with tumor apoptosis, associated with resistance to chemotherapy and radiation, and associated with tumor initiating colonies or stem cell-like properties (23, 26, 27). Furthermore, several studies have found that elevated pSTAT3 in NSCLC is associated with decreased survival (8, 28). STAT3 activation in solid tumors, including NSCLC, has been proposed to be induced by hyperactive growth factor receptors or autocrine cytokine signaling by members of the IL-6 family (3, 29, 30).
Several strategies have been employed to identify agents that target STAT3 to treat NSCLC, including small molecule inhibitors and oligonucleotides that target the IL-6/JAK/STAT pathway. Targeting the STAT pathway with these inhibitors resulted in decreased cancer cell viability, decreased stem cell-like properties and increased sensitivity to chemotherapy, radiation, and targeted therapies such as erlotinib (26, 31–34). Specifically, two JAK inhibitors (momelotinib and ruxolitinib) previously used to treat myelofibrosis are now in early clinical trials in combination with other chemotherapeutic agents for late stage and recurrent NSCLC. One potential drawback of these inhibitors is that they work by reducing levels of pSTAT3 indirectly and leave other pathways leading to increased pSTAT3 unaffected. Clinical results with these agents may help determine the importance of other STAT3-activating pathways to NSCLC tumor growth, in addition to determining whether JAK inhibition will be an effective clinical strategy for treatment of at least a subset of patients with NSCLC.
Of the two STAT3 inhibitors examined in our studies, C188-9, in particular, has several features that make it an attractive candidate for transition to clinical use. It binds to STAT3 with high affinity (KD=4.7 ± 0.4 nM)(11), has a favorable safety profile, and excellent oral pharmacokinetic properties in mice, rats and dogs (35). In addition, we identified a safe, scalable, 3-step process for producing C188-9 for use in Good Laboratory Practice (GLP) and current Good Manufacturing Practice (cGMP) testing using commercially available starting materials (35).
In addition to reducing STAT3 in A549 xenograft tumors, C188-9 treatment also reduced levels of pSTAT3 within their PBMC. These findings suggest the possibility that PBMC pSTAT3 levels may serve as a readily accessible biomarker useful for determining the biological effect of STAT3 inhibitors such as C188-9 in clinical trials.
Highlights.
pSTAT3, STAT3α and STAT3β are elevated in multiple NSCLC cell lines.
Silencing of STAT3 results in reduced cell viability in A549 cells.
C188-9 and PL reduce cell viability in multiple NSCLC cell lines.
C188-9 and PL increase apoptosis and reduce levels of anti-apoptotic proteins.
C188-9 and PL reduce tumor size, weight and pSTAT3 levels in a tumor xenograft model
Acknowledgments
This project was supported by a grant from the Dan L. Duncan Cancer Center to DJT, a stipend to KML from NIH T32 HL007747, and funding from the NIH grants P30 AI036211, P30 CA125123, and S10 RR024574) to the Cytometry and Cell Sorting Core at Baylor College of Medicine. We would like to thank John Minna, MD of the University of Texas Southwestern Medical Center at Dallas for his generously providing the HBEC3-KT cell line.
Footnotes
Grant support: This project was supported by a grant from the Dan L. Duncan Cancer Center to DJT, a stipend to KML from NIH T32 HL007747, and funding from the NIH grants P30 AI036211, P30 CA125123, and S10 RR024574) to the Cytometry and Cell Sorting Core at Baylor College of Medicine.
Conflicts of Interest
Baylor College of Medicine (BCM), with David J. Tweardy as inventor, were issued patents from the U.S., Canada, and Australia covering composition and use of C188-9 for cancer treatment. BCM has licensed these patents to StemMed, Ltd. Tweardy is President and CEO of StemMed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.American Cancer Society. Global Cancer Facts & Figures. 2. Atlanta: American Cancer Society; 2011. [Google Scholar]
- 2.Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, et al., editors. SEER Cancer Statistics Review, 1975–2011. National Cancer Institute; Bethesda, MD: Apr, 2014. http://seer.cancer.gov/csr/1975_2011/, based on November 2013 SEER data submission, posted to the SEER web site. [Google Scholar]
- 3.Looyenga BD, Hutchings D, Cherni I, Kingsley C, Weiss GJ, MacKeigan JP. STAT3 Is Activated by JAK2 Independent of Key Oncogenic Driver Mutations in Non-Small Cell Lung Carcinoma. PLoS ONE. 2012;7(2):e30820. doi: 10.1371/journal.pone.0030820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Achcar ROD, Cagle PT, Jagirdar J. Expression of Activated and Latent Signal Transducer and Activator of Transcription 3 in 303 Non–Small Cell Lung Carcinomas and 44 Malignant Mesotheliomas. Archives of Pthology and Laboratory Medicine. 2007;131(9):1350–60. doi: 10.5858/2007-131-1350-EOAALS. [DOI] [PubMed] [Google Scholar]
- 5.Lai SY, Johnson FM. Defining the role of the JAK-STAT pathway in head and neck and thoracic malignancies: Implications for future therapeutic approaches. Drug Resistance Updates. 2010;13(3):67–78. doi: 10.1016/j.drup.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Yang Q, Shen SS, Zhou S, Ni J, Chen D, Wang G, et al. STAT3 activation and aberrant ligand-dependent sonic hedgehog signaling in human pulmonary adenocarcinoma. Experimental and Molecular Pathology. 2012;93(2):227–36. doi: 10.1016/j.yexmp.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Ai T, Wang Z, Zhang M, Zhang L, Wang N, Li W, et al. Expression and prognostic relevance of STAT3 and cyclin D1 in non-small cell lung cancer. International Journal of Biological Markers. 2012;27(2):e132–8. doi: 10.5301/JBM.2012.9146. [DOI] [PubMed] [Google Scholar]
- 8.Zhao M, Gao F-H, Wang J-Y, Liu F, Yuan H-H, Zhang W-Y, et al. JAK2/STAT3 signaling pathway activation mediates tumor angiogenesis by upregulation of VEGF and bFGF in non-small-cell lung cancer. Lung Cancer. 2011;73:366–74. doi: 10.1016/j.lungcan.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Xu X, Kasembeli M, Jiang X, Tweardy BJ, Tweardy DJ. Chemical Probes that Competitively and Selectively Inhibit Stat3 Activation. PLoS ONE. 2009;4(3):e4783. doi: 10.1371/journal.pone.0004783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB, Tweardy DJ. Stat3 signaling in acute myeloid leukemia: ligand-dependent and -independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood. 2011;117(21):5701–9. doi: 10.1182/blood-2010-04-280123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Pan J, Dong Y, Tweardy DJ, Dong Y, Garibotto G, et al. Stat3 Activation Links a C/EBPd to Myostatin Pathway to Stimulate Loss of Muscle Mass. Cell Metabolism. 2013;18:368–79. doi: 10.1016/j.cmet.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bharadwaj U, Eckols TK, Kolosov M, Kasembeli MM, Adam A, Torres D, et al. Drug-repositioning screening identified piperlongumine as a direct STAT3 inhibitor with potent activity against breast cancer. Oncogene. 2014 Mar 31; doi: 10.1038/onc.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bharadwaj U, Kasemebeli MM, Eckols TK, Kolosov M, Lang P, Christensen K, et al. Monoclonal Antibodies Specific for STAT3β Reveal Its Contribution to Constitutive STAT3 Phosphorylation in Breast Cancer. Cancer. 2014;6(4):2012–34. doi: 10.3390/cancers6042012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Letters. 2007;251:199–210. doi: 10.1016/j.canlet.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 15.Shao H, Quintero AJ, Tweardy DJ. Identification and characterization of cis elements in the STAT3 gene regulating STAT3alpha and STAT3beta messenger RNA splicing. Blood. 2001;98:3853–6. doi: 10.1182/blood.v98.13.3853. [DOI] [PubMed] [Google Scholar]
- 16.Schaefer TS, Sanders LK, Park OK, Nathans D. Functional Differences between Stat3a and Stat3b. Molecular and Cellular Biology. 1997;17(9):5307–16. doi: 10.1128/mcb.17.9.5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng IHW, Ng DCH, Jans DA, Bogoyevitch MA. Selective STAT3-α or -β expression reveals spliceform-specific phosphorylation kinetics, nuclear retention and distinct gene expression outcomes. Biochemical Journal. 2012;447:125–36. doi: 10.1042/BJ20120941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang Y, Qiu J, Dong S, Redell MS, Poli V, Mancini MA, et al. Stat3 Isoforms, a and b, Demonstrate Distinct Intracellular Dynamics with Prolonged Nuclear Retention of Stat3b Mapping to Its Unique C-terminal End. J Biol Chem. 2007;282(48):34958–67. doi: 10.1074/jbc.M704548200. [DOI] [PubMed] [Google Scholar]
- 19.Humphries W, Wang Y, Qiao W, Reina-Ortiz C, Abou-Ghazal MK, Crutcher LM, et al. yuDetecting the percent of peripheral blood mononuclear cells displaying p-STAT-3 in malignant glioma patients. Journal of Translational Medicine. 2009;7(92) doi: 10.1186/1479-5876-7-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jing N, Tweardy DJ. Targeting Stat3 in cancer therapy. Anticancer Drugs. 2005;16:601–7. doi: 10.1097/00001813-200507000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, Pitt AS, et al. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth In vitro. The Journal of Clinical Investigation. 1998;102(7):1385–92. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu H, Jove R. The STATs of cancer - new molecular targets come of age. Nature Reviews Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 23.Haura EB, Zheng Z, Song L, Cantor A, Bepler G. Activated Epidermal Growth Factor Receptor - Stat-3 Signaling Promotes Tumor Survival In vivo in Non - Small Cell Lung Cancer. Clinical Cancer Research. 2005;11:8288–94. doi: 10.1158/1078-0432.CCR-05-0827. [DOI] [PubMed] [Google Scholar]
- 24.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature Reviews Immunology. 2007;7(1):41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 25.Xiong H, Hong J, Du W, Lin Y-W, Ren L-L, Wang Y-C, et al. Roles of STAT3 and ZEB1 proteins in E--cadherin down--regulation and human colorectal cancer epithelial--mesenchymal transition. Journal of Biological Chemistry. 2012;287:5819–32. doi: 10.1074/jbc.M111.295964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu H-S, Huang P-I, Chang Y-L, Tzao C, Chen Y-W, Shih H-C, et al. Cucurbitacin I Inhibits Tumorigenic Ability and Enhances Radiochemosensitivity in Nonsmall Cell Lung Cancer-Derived CD133-Positive Cells. Cancer. 2011;117(13):2970–85. doi: 10.1002/cncr.25869. [DOI] [PubMed] [Google Scholar]
- 27.Gottschling S, Schnabel PA, Herth FJF, Herpel E. Are we Missing the Target? – Cancer Stem Cells and Drug Resistance in Non-small Cell Lung Cancer. Cancer Genomics & Proteomics. 2012;9:275–86. [PubMed] [Google Scholar]
- 28.Mostertz W, Stevenson M, Acharya C, Chan I, Walters K, Lamlertthon W, et al. Age- and Sex-Specific Genomic Profiles in Non–Small Cell Lung Cancer. JAMA. 2010;303(6):535–43. doi: 10.1001/jama.2010.80. [DOI] [PubMed] [Google Scholar]
- 29.Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. Journal of Clinical Investigation. 2007;117(12):3846–56. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeh H-H, Lai W-W, Chen HHW, Liu H-S, Su W-C. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene. 2006;25:4300–9. doi: 10.1038/sj.onc.1209464. [DOI] [PubMed] [Google Scholar]
- 31.Lin C-C, Yeh H-H, Huang W-L, Yan J-J, Lai W-W, Su W-P, et al. Metformin Enhances Cisplatin Cytotoxicity by Suppressing Signal Transducer and Activator of Transcription–3 Activity Independently of the Liver Kinase B1–AMP-Activated Protein Kinase Pathway. American Journal of Respiratory Cell and Molecular Biology. 2013;49(2):241–50. doi: 10.1165/rcmb.2012-0244OC. [DOI] [PubMed] [Google Scholar]
- 32.Harada D, Takigawa N, Ochi N, Ninomiya T, Yasugi M, Kubo T, et al. JAK2-related pathway induces acquired erlotinib resistance in lung cancer cells harboring an epidermal growth factor receptor-activating mutation. Cancer Science. 2012;103(10):1795–802. doi: 10.1111/j.1349-7006.2012.02363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu X, Guo W, Wu S, Wang L, Wang J, Dai B, et al. Antitumor activity of a novel STAT3 inhibitor and redox modulator in non-small cell lung cancer cells. Biochemical Pharmacology. 2012;83:1456–64. doi: 10.1016/j.bcp.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun Y, Moretti L, Giacalone NJ, Schleicher S, Speirs CK, Carbone DP, et al. Inhibition of JAK2 Signaling by TG101209 Enhances Radiotherapy in Lung Cancer Models. Journal of Thoracic Oncology. 2011;6(4):699–706. doi: 10.1097/JTO.0b013e31820d9d11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bharadwaj U, Eckols TK, Xu X, Kasembeli MK, Chen Y, Adachi M, et al. Hit-to-lead identification of C188-9, a potent, small-molecule inhibitor of STAT3 and STAT1, highly active against radioresistant head and neck squamous cell carcinoma. Cancer Research. 2015 Submitted. [Google Scholar]