

Abstract
Expression of adenosine deaminase acting on RNA1 (ADAR1) is driven by alternative promoters. Promoter PA, activated by interferon (IFN), produces transcripts that encode the inducible p150 ADAR1 protein, whereas PB specifies the constitutively expressed p110 protein. We show using Stat1−/−, Stat2−/− and IRF9−/− MEFs that induction of ADAR1 p150 occurs by STAT2- and IRF9-dependent signaling that is enhanced by, but not obligatorily dependent upon, STAT1. Chromatin immunoprecipitation analysis demonstrated STAT2 at the PA promoter in IFN-treated Stat1−/− cells, whereas IFN-treated wild-type cells showed both STAT1 and STAT2 bound at PA. By contrast, with human 2fTGH cells and mutants U3A or U6A, ADAR1 induction by IFN was dependent upon both STAT1 and STAT2. These results suggest that transcriptional activation of Adar1 by IFN occurs in the absence of STAT1 by a non-canonical STAT2-dependent pathway in mouse but not human cells.
Keywords: adenosine deaminase acting on RNA (ADAR), innate immunity, interferon (IFN), signal transducers and activators of transcription (STAT)
Introduction
Adenosine deaminase acting on double-stranded RNA1 (ADAR1) is an interferon (IFN) inducible A-to-I RNA editing enzyme (Patterson and Samuel, 1995; Samuel, 2011). Expression of the Adar1 gene is regulated by multiple promoters (George and Samuel, 1999b; Samuel, 2011; Toth et al., 2006). The synthesis of transcripts encoding the p150 isoform of ADAR1 inducible by IFN is driven by the PA promoter possessing an interferon-stimulated response element (ISRE) characteristic of type I IFNα/β-inducible genes (George et al., 2008; George and Samuel, 1999b).
Binding of type I IFNs to their cognate cell surface receptor, which is present on most types of cells, triggers the activation of Jak-STAT signaling (Stark and Darnell, 2012). In the canonical IFNα/β signaling response, phosphorylation of STAT1 and STAT2 transcription factors leads to their dimerization and association with transcription factor IRF9 to form the heterotrimeric ISGF3 complex. ISGF3 then translocates to the nucleus and binds DNA at the ISRE element of type I interferon-stimulated genes (Schindler et al., 2007; Stark and Darnell, 2012). In the case of type II IFNγ signaling, STAT2 is dispensable. IFNγ inducible genes possessing the gamma IFN-activated sequence (GAS) require STAT1 for transcriptional activation. Upon phosphorylation, STAT1 homodimerizes to form GAF that translocates to the nucleus, binds GAS elements and drives inducible transcription (Borden et al., 2007; Stark and Darnell, 2012). The importance of STAT1 in IFN signaling leading to innate and adaptive immune defense responses is illustrated by the effects of genetic disruption of the Stat1 gene and by viruses and pathogens that antagonize STAT1 function (Durbin et al., 1996; Meraz et al., 1996; Randall and Goodbourn, 2008; Samuel, 2001). Viral gene products impair STAT1 function by causing its degradation, by preventing its activation or by inhibiting its nuclear translocation (Randall and Goodbourn, 2008; Stark and Darnell, 2012).
ADAR proteins are dsRNA adenosine deaminases and they fulfill multiple functions (Gelinas et al., 2011; Samuel, 2011; Toth et al., 2006). Among these, ADAR1 is anti-apoptotic and protects cells from adverse cytotoxic effects following viral infection (Toth et al., 2009; Ward et al., 2011). The inducible ADAR1 p150 protein is found in both the cytoplasm and nucleus, whereas the constitutively expressed ADAR1 p110 and ADAR2 are predominantly if not exclusively nuclear proteins (Bass, 2002; Samuel, 2011). ADAR1, even though inducible by IFN (George and Samuel, 1999a; Patterson and Samuel, 1995; Patterson et al., 1995; Schoggins et al., 2014), suppresses both IFN induction and IFN action in cell culture and intact animals (Hartner et al., 2009; John and Samuel, 2014; Li et al., 2012; Okonski and Samuel, 2013; Rice et al., 2012; Toth et al., 2009). Genetic disruption of the mouse Adar1 gene by a strategy in which both p150 and p110 are knocked out leads to uncontrolled apoptosis and impaired maintenance of stem cells necessary for proper development of the hematopoietic system, thereby leading to embryonic lethality (Hartner et al., 2004; Hartner et al., 2009; Wang et al., 2004). The selective knockout of the p150 isoform of ADAR1, while retaining expression of the constitutive p110 protein, also results in embryonic lethality (Ward et al., 2011).
Mutations in the human Adar1 gene are associated with two human diseases, dyschromatosis symmetrica hereditaria and Aicardi-Goutieres (AG) syndrome (Rice et al., 2012; Zhang et al., 2013). Mutation of Adar1 in AG syndrome patients (Rice et al., 2012), genetic knockout of Adar1 in mice (Hartner et al., 2009) and stable knockdown of ADAR1 protein in cultured human cells (Li et al., 2012) all are characterized by elevated production of type I IFN and a type I IFN-stimulated gene expression signature that together likely contributes to the pathogenesis and diseases seen in mice and humans with ADAR1 deficiency. Therefore, proper control of ADAR1 expression appears essential for normal physiology, both in mice and humans, in order to prevent interferonopathies.
We earlier observed that the IFN-induced expression of ADAR1 p150 protein in mouse embryo fibroblast cells is independent of STAT1 but dependent upon STAT2, the JAK1 kinase and IFNAR receptor (George et al., 2008). Because the STAT1-independent phenotype for p150 expression in MEFs was unexpected, and because of ongoing studies with measles virus which displays tropism for human cells (Li et al., 2012; Okonski and Samuel, 2013; Toth et al., 2009), we further examined the requirement for STAT1 in the induction of ADAR1 by type I IFN in human cells as well as mouse cells. We found that in human 2fTGH cells and derived U3A and U6A mutant cell lines, the induction of p150 ADAR1 followed the canonical pathway and both STAT1 and STAT2 were essential, while in mouse cells STAT2 and IRF9 were sufficient and STAT1 dispensable for ADAR1 induction. However, ADAR1 induction by IFN in STAT1-sufficient MEFs was greater than in Stat1−/− null mutant MEFs.
Materials and Methods
Cells and Maintenance
Wild-type (WT) mouse embryo fibroblast (MEF) cells and Stat1−/− (Durbin et al., 1996; Meraz et al., 1996) and Stat2−/− (Park et al., 2000) mutant MEFs were generously provided by Robert Schreiber (Washington University, St. Louis) and Joan Durbin (New York Univeristy, NYC), and Christopher Schindler (Columbia University, NY), respectively. These MEFs were maintained in DMEM (GIBCO) supplemented with 10% FBS (HyClone), 100 U of penicillin/mL and 100 μg of streptomycin/mL, and 1% sodium pyruvate. IRF9−/− mutant primary MEFs and the corresponding WT cells (Kimura et al., 1996) were generously provided by Karen Mossman (McMaster University, Canada). These cells were cultured using alpha MEMGlutaMax (GIBCO) supplemented with 15% FBS (HyClone), 100 U of penicillin/mL, 100 μg of streptomycin/mL and 2 mM L-glutamine. Human 2fTGH parental cells, STAT1 (U3A) and STAT2 (U6A) mutant cells, and U3A cells reconstituted to express STAT1 (U3A-RH) (John et al., 1991; McKendry et al., 1991) were generously provided by George Stark (Cleveland Clinic, Ohio). These cells were maintained in DMEM supplemented with 10% FBS as well as penicillin and streptomycin as above and 1 mM sodium pyruvate and 250 μg/ml hygromycin B.
Interferon treatment
Cells seeded in 6-well plates were treated with IFN 24 h after seeding, or left untreated. Treatment was with IFNαA/D (PBL) at 1000 U/mL for 24 h unless otherwise stated. For ChIP analyses, human cells were primed with 100 U/ml of human IFNγ (PBL) for 18 h followed by treatment with 1000 U/mL IFNαA/D (PBL) for 30 min (Levy et al., 1990; Ward and Samuel, 2003); mouse cells were primed with 100 U/mL of mouse IFNγ (PBL) for 18h followed by 1000 U/ml of IFNαA/D for 30 min.
Western immnoblot analysis
Whole cell extracts were prepared using high salt lysis buffer containing a protease inhibitor cocktail (Sigma) and western analysis carried out as previously described (George et al., 2011; Taghavi and Samuel, 2012), except that the membranes following transfer were blocked using 5% w/v BSA in Tris-buffered saline. Protein concentrations were determined by the Bradford assay using the BioRad reagent. The primary antibodies used for the immunoblotting were as follows: human ADAR1, Kin88 #2 (Patterson and Samuel, 1995); mouse ADAR1 (sc 73408 15.8.6), human STAT1 (sc 592 M-22), mouse STAT1 (sc 346 E-23), human STAT2 (sc 1668 A-7 and sc 476 C-20) and mouse STAT2 (sc 950 L-20) were all from Santa Cruz Biotechnology; β-actin, mouse monoclonal antibody (Sigma); and, for tubulin, mouse monoclonal antibody (Sigma-Aldrich). Protein bands were detected by scanning with Li-Cor Odessey infrared imager system (Li-Cor Biosciences) and quantitated using the Odyssey image processing software (Version 3.0, LiCor).
RNA isolation and PCR analysis
Isolation and analysis of RNA by PCR was performed essentially as previously described (McAllister et al., 2012; Toth et al., 2006). Total RNA was isolated by the TriZol method following the manufacturer's recommendations from untreated and IFN-treated cells grown in 6-well plates. RNAs, dissolved in DEPC-water, were quantified by a UV spectrophotometer (Nanodrop) and stored at −80° C until used. cDNA was prepared from total RNA (1.0 μg) by reverse transcription using Superscript II reverse transcriptase (Invitrogen) and random hexamer primers. cDNA products were amplified using Taq polymerase (Roche) according to the manufacturer's protocol using gene-specific forward and reverse primers; amplification products were analyzed by agarose gel electrophoresis and visualized by ethidium bromide staining. For qPCR, quantification of ADAR1 transcripts relative to GAPDH was carried out using the iQ SYBR green supermix (Bio-Rad) and a MyIQ single-color real-time qPCR instrument and software (version 1.0, Bio-Rad). The following primer-pairs were used: human ADAR1 Exon 1A-containing transcripts, forward- Exon 1A plus 170. 5’-AATGCCTCGCGGGCGCAATGAATC-3’and reverse- Exon 2 minus 273. 5’-TGACTTCCGAGATGCACG-3’; human ADAR1 exon 1B transcripts, forward- Exon 1B plus 12. 5’-GAGAAGGCTACGTGGTGG-3’ and reverse - Exon 2 minus 273 primer as above; mouse Adar1 Exon 1A-containing transcripts, forward –Exon 1A plus 19. 5’-GTCTCAAGGGTTCAGGGGACCC-3’ and reverse- Exon 2 minus 646. 5’-CCTCTAGGGAATTCCTGGAT-3’; mouse Exon 1B-containing transcripts, forward -Exon 1B Plus 72. 5’-TCACGAGTGGGCAGCGTCCGAGG-3’and reverse- Exon2 Minus 646 primer as above; human Adar2 transcripts, forward- 5’-GTGTAAGCACGCGTTGTACTG-3’and reverse- 5’-CGTAGTAAGTGGGAGGGAACC-3’; human GAPDH, forward- 5’-GCCTTCCGTGTCCCCACTG-3’ and reverse 5’-CGCCTGCTTCACCACCTTC-3’ ; mouse GAPDH, forward - 5’-GCCTTCCGTGTTCCTACCC-3’ and reverse- 5’-TGCCTGCTTCACCACCTTC-3’.
Chromatin immune precipitation analysis
Chromatin immunoprecipitation (ChIP) analysis of STAT1 and STAT2 binding was carried out essentially following the protocol of Cell Signaling using their ChIP-kit. Briefly, confluent monolayers, either untreated or IFN-treated, were fixed with formaldehyde (1%) for 10 min at room temperature (RT). Glycine was then added to a final concentration of 125 mM and incubation continued for 5 min at room temperature (Ward and Samuel, 2003). Fixed cells were harvested after washing the monolayers 3X with cold PBS, scrapped into cold PBS, and collected by centrifugation and suspended in ice-cold Buffer A containing DTT, PMSF and protease inhibitor cocktail (Cell Signaling). Nuclei were pelleted by centrifugation and the pellet suspended in nuclear lysis buffer B containing DTT and then treated with micrococcal nuclease for 15 min at 37° C followed by sonication. The sonicated sample was clarified by centrifugation. The fragmented chromosomal DNA cross-linked to protein was incubated overnight at 4°C with antibody against STAT1 (for mouse, E-23X, sc-346X, Santa Cruz Biotechnology or 9172S of Cell Signaling; for human, E-23X, sc-346X, Santa Cruz Biotechnology) or antibody against STAT2 (for mouse, LS-C50070, Lifespan Biosciences; for human L-20X, sc-950X, Santa Cruz Biotechnology). Antibody-protein-DNA complexes were then recovered using Protein G-conjugated Dyna beads (Magnetic beads- Life Technologies). After extensive washing, the cross-linking was reversed and the DNA purified using a QIAquick PCR purification kit (Qiagen). Analysis of the immunoprecipitated purified DNA was by PCR using Taq Polymerase (Roche) or Real Time qPCR using SYBR green supermix, and promoter specific forward and reverse primer pairs. The primers were as follows: for the mouse ADAR1 PA inducible promoter, Forward – 5’-GCGGCCCAGCCCTTATGG-3’, and Reverse – 5’-ACCTGTGGGTCCCCTGAAC-3’; and, for the human ADAR1 PA promoter, Forward – 5’-GACTTGTAACCGGCCTGAAACC-3’, and Reverse – 5’-GCCTCCGCTACTCCGCACTG-3’. PCR products were characterized by agarose gel electrophoresis when Taq polymerase was used.
Results
Induction of ADAR1 RNA and p150 protein are dependent upon both STAT1 and STAT2 in human 2fTGH cells
We earlier established by western immunoblot analysis, using wild-type and null mutant MEF cells, that induction by type I IFN of the ADAR1 p150 protein isoform is dependent upon STAT2, JAK1 and IFNAR, but independent of STAT1 in mouse cells (George et al., 2008). Because ADAR1 plays a role in suppression of dsRNA-dependent innate immune responses in cultured cells following infection with measles virus that displays tropism for human cells (Li et al., 2012; Okonski and Samuel, 2013; Pfaller et al., 2015; Toth et al., 2009), and because mutation of the human Adar1 gene is implicated as a cause of Aicardi-Goutieres Syndrome that is associated with a type I interferon signature (Rice et al., 2012), we sought to determine the requirements for the STAT1 and STAT2 factors in the induction of p150 ADAR1 by IFN in human cells. The IFN inducible p150 protein isoform of ADAR1 is encoded by exon 1A-containing transcripts expressed from the inducible PA promoter, both in mouse and in human cells (George and Samuel, 1999b; George et al., 2005; Samuel, 2011).
As shown in figure 1, induction of exon 1A-containing Adar1 RNA transcripts in parental human fibrosarcoma 2fTGH cells by IFNα was robust and substantially elevated compared to the levels found in untreated cells as measured by PCR (Fig. 1A). By contrast, in mutant U3A cells lacking STAT1 protein and mutant U6A cells lacking STAT2 protein (Fig. 1B), exon 1A RNA levels were low and comparable to that of untreated cells (Fig. 1A). When mutant U3A cells complemented to express STAT1 (Fig. 1B) were examined, these cells (U3A-RH) displayed restored induction by IFN of exon 1A-containing RNA (Fig. 1A). As further controls, the transcript levels of Adar1 (exon 1B), Adar2 and Gapdh also were measured. They were comparable between untreated and IFN-treated cells in parental 2fTGH cells, the mutant U3A and U6A cells, and the U3A-RH cells (Fig. 1A). Exon 1B-containing Adar1 transcripts are produced by alternative promoter (PB) usage and alternative exon 1 splicing in human cells and they encode the constitutively expressed p110 ADAR1 protein (George and Samuel, 1999b; Samuel, 2011).
Figure 1. Interferon induced expression of ADAR1 RNA and p150 protein in human cells requires both STAT1 and STAT2.


Parental wild-type (2fTGH) fibrosarcoma cells, derived STAT1 (U3A) and STAT2 (U6A) mutant lines, and U3A mutant cells reconstituted for STAT1 expression (U3A-RH) were left untreated or treated with IFN-αA/D for 24h. (A) Total RNA was isolated and analyzed by PCR using specific primer pairs for human Adar1 exon 1A-containing RNA (Ex1A) or exon 1B-containing RNA (Ex1B), Adar2 or Gapdh. PCR products were analyzed on 1% agarose gels and visualized by ethidium bromide staining. (B) Total cell extracts were prepared and analyzed by western immunoblot assay with antibodies against ADAR1 (P150 and p110), STAT1, STAT2, actin and tubulin as described under Materials and Methods.
When whole cell extracts prepared from IFN-treated and parallel untreated cells were examined by immunoblot analysis, IFN treatment did not increase the level of p150 ADAR1 in either U3A (STAT1 mutant) or U6A (STAT2 mutant) cells compared to the p150 induction seen in parental 2fTGH cells (Fig. 1B). As a further control, the level of the constitutively expressed p110 ADAR1 isoform was comparable in all cells, untreated or IFN-treated (Fig. 1B). Likewise, IFN treatment did not increase the level of either PKR or ISG15 in either U3A or U6A cells, but both proteins were induced in parental 2fTGH and U3A-RH cells reconstituted with STAT1 (data not shown). Thus, in human 2fTGH cells, induction by IFN of ADAR1 p150 appears to follow the canonical JAKSTAT signaling pathway and is dependent upon both STAT1 and STAT2 (Fig. 1), unlike the p150 induction observed in mouse MEF cells that was STAT1 independent (George et al., 2008).
Induction of ADAR1 by IFN does not display an obligate requirement for STAT1 in Stat1(Δ1-124)−/− MEFs but is less robust than in STAT1-sufficient MEFs
The STAT1 independence of mouse ADAR1 p150 protein expression described earlier (George et al., 2008) was observed using Stat1−/− MEF cells in which the first three translated exons corresponding to amino acids 1 to 124 along with 0.7 kb of upstream sequence was deleted in the knockout (Meraz et al., 1996), hereafter designated Stat1(Δ1-124)−/−. We now additionally have examined the requirement for STAT1 in the induction by IFN of exon 1A-containing Adar1 RNA in these MEFs. As shown in figure 2, exon 1A-containing Adar1 transcripts were inducible by IFN in Stat1(Δ1-124)−/− cells as measured by qPCR (Fig. 2A). However, in wild-type MEFs that express STAT1, the induction was about 2 to 3-fold greater than in the Stat1(Δ1-124)−/− mutant MEFs when normalized to Gapdh transcript levels (Fig. 2A). These results suggest that the induction by type I IFN of Adar1 is not dependent upon STAT1, but when STAT1 is present as in wild-type MEFs, the induction achieved by IFN is elevated over the STAT1-independent induction seen in Stat1(Δ1-124)−/− MEFs. Consistent with earlier observations (George et al., 2008), the p150 protein likewise was inducible by IFN treatment of Stat1(Δ1-124)−/− MEFs as measured by western blotting (Fig. 2B).
Figure 2. ADAR1 RNA and p150 protein are induced by IFN in Stat1(Δ1-124)−/− mouse cells.


Wild-type (WT) and Stat1(Δ1-124)−/− mutant MEFs were left untreated or treated with IFN-αA/D for 24h. (A) Total RNA was isolated and Adar1 exon 1A-containing RNA transcript levels determined by qPCR with a Bio-Rad MyIQ instrument as described under Materials and Methods. Adar1 Ex1A-transcript levels were normalized to Gapdh transcript levels. (B) Total cell extracts were prepared and analyzed by western immunoblot assay with antibodies against ADAR1 and β-actin. Quantitation of blots for p150 ADAR1 protein amount was carried out using a Li-Cor Odessey imaging system and software as described under Materials and Methods. The results shown are means and standard errors of three independent experiments. Student's t-test for comparison of ADAR1 expression in IFN-treated cells and untreated cells: *, p < 0.05; **, p < 0.005; ***; p < 0.0005.
Induction of ADAR1 by interferon occurs in occurs in Stat1(Δ221-365)−/− MEFs but not in Stat2−/− or IRF9−/− MEFs
The differing requirement for the STAT1 transcription factor for induction of ADAR1 observed between mouse cells (Stat1(Δ1-124)−/− mutant, Fig. 2) and human cells (U3A mutant, Fig. 1) was unexpected. We therefore examined an independent Stat1−/− knockout generated by deletion of amino acids 221-365 that results in loss of a portion of the DNA binding domain of STAT1 (Durbin et al., 1996), hereafter designated Stat1(Δ221-365)−/−. As shown in figure 3, the IFN inducible expression of exon 1A-containing RNA transcripts was observed both with Stat1(Δ221-365)−/− MEFs and wild-type MEFs, but not with Stat2−/− MEFs, as measured by PCR (Fig. 3A,B). Likewise, the p150 isoform of ADAR1 protein was inducible by IFN treatment of Stat1(Δ221-365)−/− mutant MEFs but not detectably by treatment of Stat2−/− MEFs as revealed by quantitation of western immunoblots (Fig. 3C). The levels of Adar1 exon 1B-containing transcripts and Gapdh RNA were comparable in the three MEFs, both untreated and IFN-treated (Fig. 3A). While exon 1A-containing transcripts that encode p150 were inducible in both Stat1 mutants, the Stat1(Δ221-365)−/− cells (Fig. 3B) and the Stat1(Δ1-124)−/− cells (Fig. 2A), a further enhanced induction was observed with wild-type MEFs that express STAT1 (Fig. 2A, 3B).
Figure 3. ADAR1 RNA transcripts and p150 protein are induced by IFN in Stat1(Δ221-365)−/− mouse cells but not as efficiently as in wild-type mouse cells whereas ADAR1 induction is STAT2 dependent.
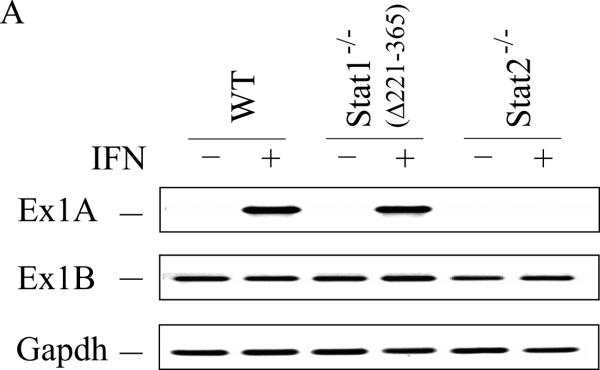


Wild-type (WT) MEFs and mutant MEFs derived from Stat1(Δ221-365)−/− or Stat2−/− knockouts were left untreated or treated with IFN-αA/D for 24h. (A) Total RNA was isolated and analyzed by PCR using specific primer pairs for mouse Adar1 exon 1A-containing RNA (Ex1A) or exon 1B-containing RNA (Ex1B), or Gapdh. PCR products were analyzed on 1% agarose gels and visualized by ethidium bromide staining and were the predicted sizes. (B) Quantification of Adar1 exon 1A-containing RNA (Ex1A) was carried out by qPCR using total RNA isolated from WT, Stat1(Δ221-365)−/− and Stat2−/− mutant MEFs, either untreated or IFN-treated, as indicated under Materials and Methods. (C) Total cell extracts were prepared and analyzed by western immunoblot assay with antibodies against ADAR1 and actin. Upper: representative blot. Lower: the results shown are means and standard errors of three independent experiments. Quantitation of blots for p150 ADAR1 protein was carried out as in Figure 2B. Student's t-test for comparison of ADAR1 expression in IFN-treated cells and untreated cells; *, P < 0.05; **, P < 0.005.
The induction of ADAR1 by type I IFN was also dependent upon IRF9 in MEFs when measured by either exon 1A-containing RNA (Fig. 4A,B) or p150 protein (Fig. 4C). By contrast, the level of p110 protein was comparable in wild-type and IRF9−/− MEFs, both untreated and treated (Fig. 4C). These results suggest that both STAT2 (Fig. 3) and IRF9 (Fig. 4) are required for induction of ADAR1, but that in Stat1(Δ221-365)−/− MEFs (Fig. 3) like in Stat1(Δ1-124)−/− MEFs (Fig. 2) the induction of ADAR1 is not dependent in an obligatory manner on STAT1. Furthermore, the results suggest that if all three components of the canonical ISGF3 signaling complex (STAT1, STAT2 and IRF9) are present as in wild-type MEFs, then the induction of Adar1 achieved by type I IFN is further elevated over the STAT1-independent induction level seen for Stat1−/− mutant MEF cells.
Figure 4. Induction by IFN of ADAR1 RNA transcripts and encoded p150 is IRF9 dependent.
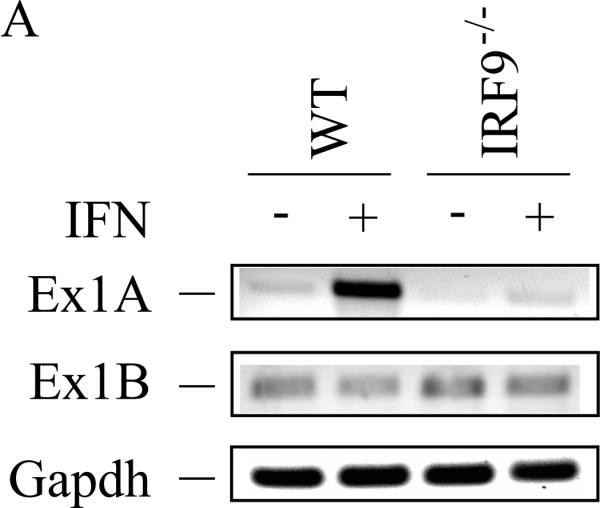


Primary MEFs genetically null for IRF9 (IRF9−/−) or the corresponding wild-type (WT) parental MEFs were left untreated or treated with IFN-αA/D for 24h. (A) Total RNA was isolated and analyzed by PCR using specific primer pairs for mouse Adar1 exon 1A-containing RNA (Ex1A) or exon 1B-containing RNA (Ex1B), or Gapdh. PCR products were analyzed as in Figure 3. (B) Quantification of Adar1 exon 1A-containing RNA (Ex1A) was carried out by qPCR using total RNA isolated from WT and IRF9−/− mutant MEFs, either untreated or IFN-treated, as indicated. *, p < 0.05 by student's t-test for comparison of ADAR1 expression in IFN-treated cells and untreated cells; n.s., no statistical significance. (C) Total cell extracts were prepared and analyzed by western immunoblot assay with antibodies against ADAR1 and β–actin.
STAT2 protein binds to the IFN-inducible Adar1 promoter region in vivo in the absence of STAT1
Our results obtained using mutant MEFs revealed a STAT1 independence, but STAT2 dependence, for the induction by IFN of Adar1 exon 1A RNA and p150 protein in mouse cells. Furthermore, although Adar1 induction occurred in the absence of STAT1 both in Stat1(Δ1-124)−/− MEFs and Stat1(Δ221-365)−/− MEFs, the induction observed in IFN-treated wild-type MEFs was typically greater than that seen in the Stat1−/− MEFs. This finding prompted us to examine, in wild-type and Stat1−/− mutant MEFs, the binding of STAT2 protein to the inducible Adar1 promoter region which possesses an ISRE element (George et al., 2008; George and Samuel, 1999a). For this purpose, chromatin immune precipitation (ChIP) assays were performed. As shown in figure 5, both STAT1 (Fig. 5A) and STAT2 (Fig. 5B) were bound to the Adar1 PA promoter region in wild-type MEFs in an IFN-dependent manner as anticipated. STAT2 also bound at the Adar1 inducible promoter in Stat1(Δ221-365)−/− mutant MEFs in an IFN-dependent manner, although the IFN-induced STAT2 binding in the mutant MEFs lacking STAT1 was less than that observed in wild-type MEFs possessing STAT1 (Fig. 5B). No specific binding of either STAT1 or STAT2 was observed to the PA promoter region with untreated MEFs above the control level without specific antibody seen for untreated wild-type or Stat1(Δ221-365)−/− MEFs (Fig. 5A, 5B). Finally, when ChIP analysis was carried out with the human 2fTGH cells, binding of STAT2 to the Adar1 PA promoter region was IFN-dependent and observed only in the parental 2fTGH cells and not in U3A mutant cells (Fig. 6), consistent with the results showing STAT1 and STAT2 dependence for induction of the exon 1A RNA and p150 protein analyses (Fig.1).
Figure 5. STAT2 binds to the ISRE-containing IFN-inducible Adar1 promoter region even in the absence of STAT1 in Stat1(Δ221-365)−/− MEFs whereas both STAT1 and STAT2 bind in wild-type MEFs.


ChIP analysis was performed using chromatin prepared from wild-type (WT) and Stat1(Δ221-365)−/− MEFs, using either no antibody (−) or with antibody (+) against STAT1 (A) or STAT2 (B). Both chromatin from untreated (−) and IFN-treated (+) MEF cells were analyzed. DNA isolated from input chromatin was amplified as a control. *, p < 0.05 by student's t-test.
Figure 6. STAT1 and STAT2 bind to the ISRE-containing IFN-inducible Adar1 PA promoter region in parental 2fTGH but not mutant U3A cells.

ChIP analysis was performed using chromatin prepared from parental 2fTGH and STAT1 (U3A) mutant cells, using either no antibody (−) or with antibody (+) against STAT1 or STAT2 as indicated. Chromatin from untreated (−) and IFN-treated (+) cells were analyzed. DNA isolated from imput chromatin was analyzed as a control.
Discussion
We report herein that the dependency on STAT1 for induction of RNA adenosine deaminase ADAR1 by type I IFN differs between mouse and human cell lines commonly utilized in interferon signal transduction studies. While the presence of STAT1 enhanced induction of ADAR1 in mouse cells, STAT1 was not required for IFN-induced Adar1 transcriptional activation based on analyses of MEFs derived from two independent Stat1−/− knockouts. By contrast, ADAR1 induction in human 2fTGH cells was strictly dependent on the presence of STAT1. And, in both mouse Stat2−/− MEFs and human U6A mutant cells, ADAR1 induction by type I IFNα was not observed.
We earlier reported that the IFNα-induced expression of the ADAR1 p150 in Stat1(Δ1-124)−/− MEFs was independent of the STAT1 transcription factor (George et al., 2008). However, induction by IFNα of PKR in the Stat1(Δ1-124)−/− MEF cells was STAT1-dependent (Das et al., 2006). Furthermore, induction of both ADAR1 p150 and PKR in MEFs displayed requirements for STAT2, JAK1 and IFNAR pathway components (George et al., 2008). Subsequently, we carried out similar studies with human 2fTGH parental and U3A and U6A mutant cells respectively lacking STAT1 and STAT2. This was done because the innate immune response is suppressed by ADAR1 (Hartner et al., 2009) including in measles virus-infected cells (Li et al., 2012; Toth et al., 2009), because some viruses antagonize STAT1 function (Randall and Goodbourn, 2008), and because some viruses including measles virus display tropism for human cells (Mateo et al., 2014). In contrast to the induction of Adar1 exon 1A-containing RNA and p150 protein seen in Stat1(Δ1-124)−/− MEFs as well as in Stat1(Δ221-365)−/− MEFs by IFNα, the IFN-induced expression of Adar1 exon 1A RNA and p150 protein were not observed in mutant human cells lacking either STAT1 (U3A) or STAT2 (U6A). ADAR1 induction was observed both in the parental 2fTGH cells and U3A-RH cells complemented to express STAT1. These findings suggest a species difference between mouse and human in the STAT1 requirement for induction of Adar1 by type I IFN.
The transcription factor STAT1 was identified originally as an essential factor for the canonical signaling by type I IFNs, functioning as a component of the heterotrimeric ISGF3 complex (Durbin et al., 1996; Meraz et al., 1996; Stark and Darnell, 2012). Following receptor binding of type I IFN, STAT1 and STAT2 are phosphorylated and form heterodimers that associate with IRF9 to form ISGF3 that translocates to the nucleus and binds the ISRE element to drive transcription. STAT1-dependent IFNα induced genes in MEFs include ISG54, GBP1, C3 and IRF1 (Durbin et al., 1996; Meraz et al., 1996; Randall and Goodbourn, 2008; Samuel, 2001) and PKR (Das et al., 2006). However, more recent studies have revealed STAT1-independent type I IFN responses, both in cultured cells and intact animals. Among them are our earlier finding, based on the analyses using Stat1(Δ1-124)−/− and Stat2−/− MEFs, that IFNα induction of p150 is STAT1- independent and STAT2-dependent (George et al., 2008). This conclusion is also true for Stat1(Δ221-365)−/− MEFs as shown herein. Induction of mouse Oas1b and IRF7 by type I IFNs likewise has been described to be STAT1-independent. IFNβ induction of the Oas1b gene in Stat1(Δ1-124)−/− MEFs is STAT1-independent but STAT2-dependent (Pulit-Penaloza et al., 2012), and induction of IRF7 following lymphocytic choriomeningitis virus (LCMV) infection or IFNα treatment is STAT2- and IRF9-dependent but independent of STAT1 based on Stat1(Δ1-124)−/− knockout (Ousman et al., 2005). LCMV also has been seen to evade the mouse immune system through a type I IFN-mediated response that is STAT1-independent but STAT2-dependent (Hahm et al., 2005).
The inducible Adar1 PA promoter region, both mouse and human, includes an ISRE element (George et al., 2008). ChIP results established STAT2 binding to PA; the binding of STAT2 was observed in Stat1−/− MEFs following IFNα treatment (Fig. 5). Likewise, binding of STAT2 to ISRE elements of Oas1b and Irf7 genes was observed in STAT1-deficient mice following Dengue virus infection (Perry et al., 2011). STAT2 also bound the promoter region of Oas1b in Stat1−/− MEFs (Pulit-Penaloza et al., 2012). These results, together with our finding that Adar1 transcriptional activation was STAT2 and IRF9 dependent but STAT1-independent in mouse cells, suggest a role for the STAT2 factor independent of its role as a subunit of ISGF3. While IRF9 lacks an intrinsic ability to mediate transcriptional activation, a hybrid IRF9-STAT2 fusion protein with the trans activation domain (TAD) of STAT2 is a robust stimulator of ISRE-dependent transcription (Kraus et al., 2003). Possibly, in the case of Adar1, Oas1b and Irf7 induction by IFN, STAT2 functions with IRF9 as a STAT2 homodimer in type I IFN-treated Stat1−/− MEFs to activate transcription. Overexpression of STAT2, both in Stat1−/− MEFs and human U3C (STAT1 mutant) cells, was recently described to produce a STAT1-independent IFNα inducible expression of Oas2 and Ifit1 (Blaszczyk et al., 2015).
While the central importance of the canonical JAK-STAT signaling pathway is well established, additional signaling mechanisms have been described for IFN signaling including the p38 mitogen-activated protein kinase pathway and the phosphatidyl inositol 3-kinase signaling pathway (Platanias, 2005). These pathways are independent of STATs, and since ADAR1 induction by IFN is tightly dependent on STAT2, they seem unlikely to be operative for ADAR1.
Our finding that, with human 2fTGH derived mutant cells, Adar1 induction by IFNα to be strictly dependent upon both STAT1 (U3A) and STAT2 (U6A), whereas two different mouse Stat1−/− knockouts displayed STAT1-independent induction of Adar1 that was STAT2-dependent, suggests a species difference between mouse and human cells in the type I IFN signaling response leading to transcriptional activation of Adar1 expression. That is, in human cells STAT1 is required in addition to STAT2 to activate Adar1 inducible transcription, whereas in mouse cells STAT2 is able to function in the absence of STAT1. However, a potential contributing factor to the STAT1-dependence for ADAR1 induction seen in U3A cells may be the low level of STAT2 in U3A observed by us (Fig. 1B) and others (Lou et al., 2011). Other studies likewise have suggested a species-specific activity of STAT2. For example, the NS5 protein of Dengue virus targets STAT2 for degradation in species-specific manner: NS5 binds and mediates the degradation of STAT2 protein from human cells but not mouse cells (Ashour et al., 2010). STAT2 also displays a species-specific effect in simian virus 5 (SV5) infections. Differences between human and mouse STAT2 are the determinant of SV5-induced STAT1 degradation seen in human cells but not in mouse cells (Parisien et al., 2002). Conceivably, the STAT2 factor is also the determinant of the species difference in the STAT1 requirement between mouse and human cells for ADAR1 induction by type I IFNs. Interestingly, the STAT2 in the mouse (925 aa) is about 10% larger than STAT2 in the human (850 aa) (Schindler et al., 2007).
In summary, transcriptional activation of the Adar1 gene by type I IFN displays a different requirement for the STAT1 factor between human and mouse cells in culture. Why this difference exists is unclear. The mouse may possess a more stringent requirement for the ADAR1 p150 protein compared to the human, and hence the existence of an alternate pathway for Adar1 induction in the event that STAT1 protein function is disrupted in the mouse due to pathogen infection or other physiologic stresses. ADAR1 p150 is critically important for normal physiology. We have shown that embryos of mice genetically disrupted for ADAR1 p150 expression but retaining constitutive p110 expression die (Ward et al., 2011), similar to the E11.5-12.0 embryonic lethality seen following disruption of both p150 and p110 expression (Hartner et al., 2004; Wang et al., 2004). The embryonic lethality during mouse development recently has been found to be rescued to live birth in Adar1:Mavs double mutants, in which IFN induction in response to double-stranded RNA (dsRNA) is prevented (Mannion et al., 2014). ADAR1 is a feedback suppressor of IFN production (Hartner et al., 2009; Li et al., 2012; Rice et al., 2012). ADAR1 deficiency leads to enhanced type I IFN production, both in cultured cells (Li et al., 2012) and mice (Hartner et al., 2009). Interestingly, in humans the Aicardi-Goutieres Syndrome (AGS), a rare neurological disorder linked to mutations in the Adar1 gene leading to the dysfunction of ADAR1 proteins, is characterized by a type I IFN signature (Livingston et al., 2014; Rice et al., 2012), providing another example of the critical importance to normal physiology of proper expression of ADAR1.
Highlights.
We examined the induction of RNA adenosine deaminase ADAR1 by interferon. 74
ADAR1 induction was STAT1-independent but STAT2- and IRF9-dependent in mouse cells. 84
ADAR1 induction was dependent on both STAT1 and STAT2 in human cells. 70
STAT2 bound at the inducible promoter in the absence of STAT1 in Stat1−/− MEFs. 80
ADAR1 induction in STAT1-sufficient MEFs was greater than in STAT1 null MEFs. 78
Acknowledgments
This work was supported in part by research grants AI-12520 and AI-20611 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, U.S. Public Health Service.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashour J, Morrison J, Laurent-Rolle M, Belicha-Villanueva A, Plumlee CR, Bernal-Rubio D, Williams KL, Harris E, Fernandez-Sesma A, Schindler C, Garcia-Sastre A. Mouse STAT2 restricts early dengue virus replication. Cell Host Microbe. 2010;8:410–421. doi: 10.1016/j.chom.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk K, Olejnik A, Nowicka H, Ozgyin L, Chen YL, Chmielewski S, Kostyrko K, Wesoly J, Balint BL, Lee CK, Bluyssen HA. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem J. 2015;466:511–524. doi: 10.1042/BJ20140644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Ward SV, Tacke RS, Suske G, Samuel CE. Activation of the RNA-dependent protein kinase PKR promoter in the absence of interferon is dependent upon Sp proteins. J Biol Chem. 2006;281:3244–3253. doi: 10.1074/jbc.M510612200. [DOI] [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Gelinas JF, Clerzius G, Shaw E, Gatignol A. Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase. J Virol. 2011;85:8460–8466. doi: 10.1128/JVI.00240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Das S, Samuel CE. Organization of the mouse RNA-specific adenosine deaminase Adar1 gene 5′-region and demonstration of STAT1-independent, STAT2-dependent transcriptional activation by interferon. Virology. 2008;380:338–343. doi: 10.1016/j.virol.2008.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Gan Z, Liu Y, Samuel CE. Adenosine deaminases acting on RNA, RNA editing, and interferon action. J Interferon Cytokine Res. 2011;31:99–117. doi: 10.1089/jir.2010.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Samuel CE. Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene. 1999a;229:203–213. doi: 10.1016/s0378-1119(99)00017-7. [DOI] [PubMed] [Google Scholar]
- George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci U S A. 1999b;96:4621–4626. doi: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Wagner MV, Samuel CE. Expression of interferon-inducible RNA adenosine deaminase ADAR1 during pathogen infection and mouse embryo development involves tissue-selective promoter utilization and alternative splicing. J Biol Chem. 2005;280:15020–15028. doi: 10.1074/jbc.M500476200. [DOI] [PubMed] [Google Scholar]
- Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22:247–257. doi: 10.1016/j.immuni.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem. 2004;279:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John J, McKendry R, Pellegrini S, Flavell D, Kerr IM, Stark GR. Isolation and characterization of a new mutant human cell line unresponsive to alpha and beta interferons. Mol Cell Biol. 1991;11:4189–4195. doi: 10.1128/mcb.11.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John L, Samuel CE. Induction of stress granules by interferon and down-regulation by the cellular RNA adenosine deaminase ADAR1. Virology. 2014;454-455:299–310. doi: 10.1016/j.virol.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Kadokawa Y, Harada H, Matsumoto M, Sato M, Kashiwazaki Y, Tarutani M, Tan RS, Takasugi T, Matsuyama T, Mak TW, Noguchi S, Taniguchi T. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells. 1996;1:115–124. doi: 10.1046/j.1365-2443.1996.08008.x. [DOI] [PubMed] [Google Scholar]
- Kraus TA, Lau JF, Parisien JP, Horvath CM. A hybrid IRF9-STAT2 protein recapitulates interferon-stimulated gene expression and antiviral response. J Biol Chem. 2003;278:13033–13038. doi: 10.1074/jbc.M212972200. [DOI] [PubMed] [Google Scholar]
- Levy DE, Lew DJ, Decker T, Kessler DS, Darnell JE., Jr. Synergistic interaction between interferon-alpha and interferon-gamma through induced synthesis of one subunit of the transcription factor ISGF3. EMBO J. 1990;9:1105–1111. doi: 10.1002/j.1460-2075.1990.tb08216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Okonski KM, Samuel CE. Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. J Virol. 2012;86:3787–3794. doi: 10.1128/JVI.06307-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston JH, Lin JP, Dale RC, Gill D, Brogan P, Munnich A, Kurian MA, Gonzalez-Martinez V, De Goede CG, Falconer A, Forte G, Jenkinson EM, Kasher PR, Szynkiewicz M, Rice GI, Crow YJ. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J Med Genet. 2014;51:76–82. doi: 10.1136/jmedgenet-2013-102038. [DOI] [PubMed] [Google Scholar]
- Lou YJ, Zhang ZL, Pan XR, Xu GP, Jia PM, Li D, Tong JH. Intact JAK-STAT signaling pathway is a prerequisite for STAT1 to reinforce the expression of RIG-G gene. Exp Cell Res. 2011;317:513–520. doi: 10.1016/j.yexcr.2010.10.025. [DOI] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, Jantsch MF, Dorin J, Adams IR, Scadden AD, Ohman M, Keegan LP, O'Connell MA. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014;9:1482–1494. doi: 10.1016/j.celrep.2014.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo M, Navaratnarajah CK, Willenbring RC, Maroun JW, Iankov I, Lopez M, Sinn PL, Cattaneo R. Different roles of the three loops forming the adhesive interface of nectin-4 in measles virus binding and cell entry, nectin-4 homodimerization, and heterodimerization with nectin-1. J Virol. 2014;88:14161–14171. doi: 10.1128/JVI.02379-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister CS, Taghavi N, Samuel CE. Protein kinase PKR amplification of interferon beta induction occurs through initiation factor eIF-2alpha-mediated translational control. J Biol Chem. 2012;287:36384–36392. doi: 10.1074/jbc.M112.390039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKendry R, John J, Flavell D, Muller M, Kerr IM, Stark GR. High-frequency mutagenesis of human cells and characterization of a mutant unresponsive to both alpha and gamma interferons. Proc Natl Acad Sci U S A. 1991;88:11455–11459. doi: 10.1073/pnas.88.24.11455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Okonski KM, Samuel CE. Stress granule formation induced by measles virus is protein kinase PKR dependent and impaired by RNA adenosine deaminase ADAR1. J Virol. 2013;87:756–766. doi: 10.1128/JVI.02270-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ousman SS, Wang J, Campbell IL. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J Virol. 2005;79:7514–7527. doi: 10.1128/JVI.79.12.7514-7527.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien JP, Lau JF, Horvath CM. STAT2 acts as a host range determinant for species-specific paramyxovirus interferon antagonism and simian virus 5 replication. J Virol. 2002;76:6435–6441. doi: 10.1128/JVI.76.13.6435-6441.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity. 2000;13:795–804. doi: 10.1016/s1074-7613(00)00077-7. [DOI] [PubMed] [Google Scholar]
- Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol Cell Biol. 1995;15:5376–5388. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson JB, Thomis DC, Hans SL, Samuel CE. Mechanism of interferon action: double-stranded RNA-specific adenosine deaminase from human cells is inducible by alpha and gamma interferons. Virology. 1995;210:508–511. doi: 10.1006/viro.1995.1370. [DOI] [PubMed] [Google Scholar]
- Perry ST, Buck MD, Lada SM, Schindler C, Shresta S. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog. 2011;7:e1001297. doi: 10.1371/journal.ppat.1001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller CK, Mastorakos GM, Matchett WE, Ma X, Samuel CE, Cattaneo R. Measles Virus Defective-interfering RNAs are Generated Frequently and Early in the Absence of C Protein and can be Destabilized by Adenosine Deaminase Acting on RNA 1-like Hypermutations. J Virol. 2015 doi: 10.1128/JVI.01017-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Pulit-Penaloza JA, Scherbik SV, Brinton MA. Activation of Oas1a gene expression by type I IFN requires both STAT1 and STAT2 while only STAT2 is required for Oas1b activation. Virology. 2012;425:71–81. doi: 10.1016/j.virol.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol. 2008;89:1–47. doi: 10.1099/vir.0.83391-0. [DOI] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin JP, Lourenco CM, Male AM, Marques W, Jr., Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O'Connell MA, Lovell SC, Crow YJ. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology. 2011;411:180–193. doi: 10.1016/j.virol.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505:691–695. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, Darnell JE., Jr. The JAK-STAT pathway at twenty. Immunity. 2012;36:503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghavi N, Samuel CE. Protein kinase PKR catalytic activity is required for the PKR-dependent activation of mitogen-activated protein kinases and amplification of interferon beta induction following virus infection. Virology. 2012;427:208–216. doi: 10.1016/j.virol.2012.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth AM, Li Z, Cattaneo R, Samuel CE. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J Biol Chem. 2009;284:29350–29356. doi: 10.1074/jbc.M109.045146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth AM, Zhang P, Das S, George CX, Samuel CE. Interferon action and the double-stranded RNA-dependent enzymes ADAR1 adenosine deaminase and PKR protein kinase. Prog Nucleic Acid Res Mol Biol. 2006;81:369–434. doi: 10.1016/S0079-6603(06)81010-X. [DOI] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem. 2004;279:4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- Ward SV, George CX, Welch MJ, Liou LY, Hahm B, Lewicki H, de la Torre JC, Samuel CE, Oldstone MB. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci U S A. 2011;108:331–336. doi: 10.1073/pnas.1017241108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SV, Samuel CE. The PKR kinase promoter binds both Sp1 and Sp3, but only Sp3 functions as part of the interferon-inducible complex with ISGF-3 proteins. Virology. 2003;313:553–566. doi: 10.1016/s0042-6822(03)00347-7. [DOI] [PubMed] [Google Scholar]
- Zhang JY, Chen XD, Zhang Z, Wang HL, Guo L, Liu Y, Zhao XZ, Cao W, Xing QH, Shao FM. The adenosine deaminase acting on RNA 1 p150 isoform is involved in the pathogenesis of dyschromatosis symmetrica hereditaria. Br J Dermatol. 2013;169:637–644. doi: 10.1111/bjd.12401. [DOI] [PubMed] [Google Scholar]