

Abstract
The highly active antiretroviral therapy reduces HIV-1 RNA in plasma to undetectable levels. However, the virus continues to persist in the long-lived resting CD4+ T cells, macrophages and astrocytes which form a viral reservoir in infected individuals. Reactivation of viral transcription is critical since the host immune response in combination with antiretroviral therapy may eradicate the virus. Using the chronically HIV-1 infected T lymphoblastoid and monocytic cell lines, primary quiescent CD4+ T cells and humanized mice infected with dual-tropic HIV-1 89.6, we examined the effect of various X-ray irradiation (IR) doses (used for HIV-related lymphoma treatment and lower doses) on HIV-1 transcription and viability of infected cells. Treatment of both T cells and monocytes with IR, a well-defined stress signal, led to increase of HIV-1 transcription, as evidenced by the presence of RNA polymerase II and reduction of HDAC1 and methyl transferase SUV39H1 on the HIV-1 promoter. This correlated with the increased GFP signal and elevated level of intracellular HIV-1 RNA in the IR-treated quiescent CD4+ T cells infected with GFP-encoding HIV-1. Exposition of latently HIV-1infected monocytes treated with PKC agonist bryostatin 1 to IR enhanced transcription activation effect of this latency-reversing agent. Increased HIV-1 replication after IR correlated with higher cell death: the level of phosphorylated Ser46 in p53, responsible for apoptosis induction, was markedly higher in the HIV-1 infected cells following IR treatment. Exposure of HIV-1 infected humanized mice with undetectable viral RNA level to IR resulted in a significant increase of HIV-1 RNA in plasma, lung and brain tissues. Collectively, these data point to the use of low to moderate dose of IR alone or in combination with HIV-1 transcription activators as a potential application for the “Shock and Kill” strategy for latently HIV-1 infected cells.
Keywords: HIV-1 latency, CD4+ T cells, Monocytes, X-ray, Irradiation, Transcription, Apoptosis, HIV-1 reactivation, Humanized mice
Background
Besides productive infection in active CD4+ T lymphocytes and macrophages, HIV-1 is capable of maintaining latent infection in stable reservoirs such as resting CD4+ T cells, naïve T cells and CD34+ multipotent hematopoietic stem cells (Carter et al., 2010; Eisele and Siliciano, 2012; Wightman et al., 2010). HIV-1 can also persist in such long-lived reservoirs as perivascular macrophages, microglia and astrocytes, which together form the central nervous system (CNS) tissue viral reservoir in infected individuals (Alexaki et al., 2008; Churchill et al., 2009; Clements et al., 2011). The latent infection reservoirs may persist for many years even though the patients are usually treated with highly active antiretroviral therapy (HAART). Thus, the virus can be activated rapidly once HAART is stopped (Chun et al., 1999; Finzi et al., 1999; Richman et al., 2009). Therefore, precise detection of the latently HIV-1 infected cells and elicitation of the treatment that can directly eliminate latently HIV-1 infected cells within the pool of resting T cells and macrophages are important for effective depletion of the viral reservoirs and eventual eradication of infection.
One of the major factors that determine HIV-1 latency, i.e. silencing of integrated viral genome, is down regulation or repression of HIV-1 transcription. Suppressed expression of Tat, multiple transformations of a positive transcription elongation factor b (P-TEFb) (Bisgrove et al., 2007; Chen et al., 2004; Hargreaves et al., 2009; Yang et al., 2005), as well as such epigenetic mechanisms as deacetylation of lysine residues in LTR-bound histones by histone deacetylases (HDAC) (Tyagi et al., 2010), methylation of histone H3 (Pearson et al., 2008) and direct methylation of cytosine residues in two CpG islands flanking HIV-1 transcription start site (Kauder et al., 2009) are critical factors of the repression of LTR transcription (Furia et al., 2002; Williams et al., 2007). Thus, the activation of latent HIV-1 proviral genomes in the infected cells, that results in viral replication is critical, since the virus itself as well as the host immune response in combination with antiretroviral drugs (“shock and kill” strategy) may eliminate the virus following reactivation. Current approaches to use pharmacologic induction of HIV-1 transcription for virus reactivation are promising. However, recently performed testing of such latency-reversing agents (LRAs) as HDAC inhibitors (vorinostat, romidepsin and panobinostat), NF-κB activator, disulfiram; and the bromodomain-containing protein 4 inhibitor JQ1 using ex vivo viral outgrowth assay (VOA) showed that none of the tested LRAs induced outgrowth of HIV-1 from the latent reservoir of patients on ART. Only the protein kinase C (PKC) modulator bryostatin-1 caused significantly increased transcription of HIV-1 genome in reactivated CD4+ Tcells (Bullen et al., 2014). These data suggest that for successful “shock and kill” virus eradication, additional non-pharmacological approaches are required.
Ionizing irradiation (IR) is the well-known and effective stress signal that induces DNA damage and activates cellular stress response. The typical X-ray- and γ-IR-induced DNA lesions cause double strand brakes (DSB) that can result in induction of up to four independent repair pathways: homologous recombination, non-homologous end joining, (NHEJ), alternative-NHEJ, and single-strand annealing (reviewed in Curtin (2012)). X-ray IR stress usually activates NHEJ machinery (Hartlerode and Scully, 2009), which is associated with enhanced activity of many cellular factors involved in transcription activation, such as NFκB (Aggarwal, 2004), Sp1 (Iwahori et al., 2008), HAT1 (Lebel et al., 2010) and CBP/p300 whose activation has been shown to lead to SWI/SNF-mediated chromatin remodeling (Agbottah et al., 2006; Ogiwara et al., 2011; Van Duyne et al., 2011). For HIV-infected cells, both X-ray and γ-IR have been shown to activate LTR-driven transcription via the activation of NF-κB DNA binding (Faure et al., 1995; Smith et al., 2001), and to induce cell death via chromatin DNA-damage (Ogawa et al., 2003). Our earlier studies indicated that single dose of γ-IR differently activated infected T cells and their parental uninfected cell lines, with respect to the cell cycle and gene expression (Clark et al., 2000). Other studies reported that the X-ray-treated human colonic carcinoma cells could activate NF-κB-mediated HIV-1 transcription in non-irradiated cells through the secretion of cell activating cytokines (Faure, 1998) indicating indirect effect of IR on HIV-1 infected cells.
In the present study, we examined effect of various X-ray doses (starting from the doses which are equivalent to the typically used for therapy of HIV-related lymphoma (Altschuler et al., 1989; Haas, 2009; Yukl et al., 2013) and lower) on HIV-1 replication and viability of chronically and latently infected CD4+ T cells and monocytes. We observed that treatment of both peripheral blood mononuclear cells (PBMCs) and monocytes with different IR doses led to significant increase of HIV-1 transcription. Combination of IR with PKC agonist bryostatin 1 resulted in enhancement of HIV-1 transcription in monocytes as compared to individual treatments. Both chronically HIV-1 infected T cell lines and latently infected resting CD4+ T lymphocytes displayed elevated level of apoptosis in response to IR that correlated with the increased level of Ser46 phosphorylation on the p53 protein. Finally, exposure of HIV-1 infected NSG humanized mice with undetected basal levels of viral replication showed dramatic increase of HIV-1 RNA in plasma, lung and brain tissues. Taken together, these data suggest that IR-induced cellular stress activates HIV-1 expression and facilitates the apoptotic death of infected T cells possibly via phosphorylation of p53 protein.
Results
X-ray irradiation activates HIV-1 transcription in chronically-infected cell lines and peripheral blood mononuclear cells
In our earlier published study (Clark et al., 2000), we showed that treatment of chronically HIV-1 infected T cell lines with γ-IR resulted in activation of virus replication and apoptosis, whereas the uninfected parental cells demonstrated repair with minimal to no apoptosis. In the present study we tested the effect of either the doses of X-ray IR typically used for HIV-related lymphoma treatment (Altschuler et al., 1989; Haas, 2009; Yukl et al., 2013) or the lower doses, non-pathogenic for uninfected cells, on the transcription of HIV-1 provirus in chronically-infected T cell line and promonocytes. We found that exposure of the T cell line to 5 Gy dose of IR led to 2.5-3-fold increase of RT activity in the culture media of the treated cells 24 h post-IR, suggesting that IR induced the expression of the integrated HIV-1 genome and virion production (Fig. 1A). The promonocytes displayed slower response with IR consisting of two-fold increase of the RT activity 48 h post-IR.
Fig. 1. IR activates viral transcription in chronically HIV-1 infected cell lines and primary CD4+ T cells through the enhancement of LTR-driven transcription.
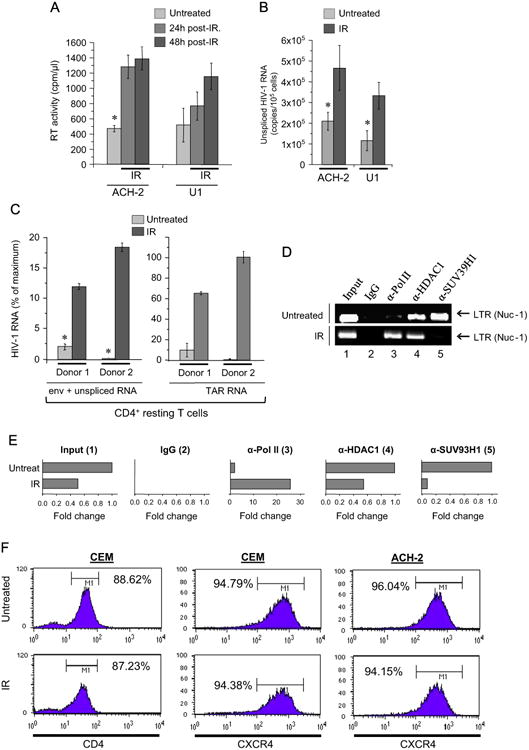
(A) Chronically HIV-1 infected T cell line ACH-2 and promonocytes U1 were exposed to 5 Gy X-ray irradiation (IR). Reverse transcriptase (RT) activity was measured in culture medium at indicated time points. Results are shown as a mean of three independent measurements ± SD. Asterisk indicates p ≤0.05. (B) Same cell lines were pretreated with ART cocktail (indinavir, tenofovir, lamivudine/emitricitabine, 10 μM of each) for 10 days, washed and then exposed to 5 Gy X-ray IR in a fresh culture medium. HIV-1 RNA was measured in the cytoplasmic extracts at 24 h post-IR by quantitative RT-PCR using gag-specific primers. Results are shown as a mean of three independent measurements ± SD. Asterisk indicates p ≤ 0.01. (C) PBMCs isolated from two healthy donors were infected with HIV-1 89.6 dual-tropic strain (10 ng p24/105 primary activated PBMCs), and then cultured for 45 days with IL-7 (added every 5 days) to place cells into the resting phase. Then the cells were exposed to 5 Gy dose of IR. HIV-1 RNA was measured in the cytoplasmic extracts at 72 h post-IR by quantitative RT-PCR using env-specific and TAR-specific primers. The RNA count is shown as a percentage of maximal level of TAR RNA in the cell lysate. The results are shown as a mean of triplicate measurements ± SD. Asterisk indicates p ≤ 0.01. (D) Chromatin immunoprecipitation (ChIP) assay of the nuclear extracts from ACH-2 cells was performed with indicated antibodies at 24 h post-IR as described in Materials and Methods. Specific DNA sequences in the immunoprecipitates were detected by PCR using primers specific for the HIV-1 LTR. E) The data of ChIP assay (panel D) were quantified using ImageJ software. Results are presented as a fold change of grey values for the LTR DNA bands in each immunocomplex; the value of untreated control is shown as 1. (F) IR does not affect surface presentation of CD4 and CXCR4 receptors in HIV-1 infected and uninfected cells. The uninfected (CEM) and chronically HIV-1 infected (ACH-2) T cells were exposed to 5 Gy IR, and then analyzed by flow cytometry with FITC-anti-human CD4 and PE-anti-human CXCR4 labeling performed 24 h post IR treatment.
The chronically infected U1 and especially ACH-2 cell lines support basal level of HIV-1 transcription (Poli et al., 1989). ACH-2 produces the virions that potentially may reinfect the cells and, therefore, induce super-infection. To bring our model to the latent HIV-1 infected cells, we pre-incubated cells with the cocktail of antiretrovirals (ART) containing both protease inhibitor (indinavir) and mixture of RT inhibitors including tenofovir and lamivudine/emtricitabine for 10 days. The HIV-1 RNA was not detected in the culture media (data not shown), but the basic level of HIV-1 persisted on the level around 1–2 copies per cell (Fig. 1B). Exposure of these cells to 5 Gy IR dose led to twofold increase of HIV-1 transcription in ACH-2 and threefold in U1 cells within first 24 h.
Unlike the immortalized cell lines, the PBMCs isolated from healthy donor, infected with HIV-1, treated with IL-7 and then cultured for 45 days (to obtain quiescent cells) displayed 5–50 fold increase of unspliced viral RNA 48 h after IR in the samples from different donors (Fig. 1C, left panel). Interestingly, quantification of HIV-1 TAR RNA in the lysates of irradiated T cells revealed a similar trend. However, the absolute count of TAR was approximately fivefold higher than the count of unspliced HIV-1 RNA (Fig. 1C, right panel). Taken together, these data indicate that IR reactivates transcription of the inactive HIV-1 genome in primary blood mononuclear cells. The abundance of TAR RNA in HIV-1 infected cells, as well as detectable level of this small viral non-coding RNA in the lysates of latently HIV-1 infected cells, was reported earlier by us and other colleagues (Klase et al., 2009; Narayanan et al., 2013; Ouellet et al., 2008, 2013; Yeung et al., 2009). Since the basic transcription of short viral RNA fragments which include TAR sequence does not require Tat activity (Kao et al., 1987), the enhanced level of both TAR and coding HIV-1 RNA in the cells exposed to IR suggests that IR itself activated both Tat-dependent and independent transcription from the LTR promoter.
To elucidate activation effect of IR on the HIV-1 LTR-driven transcription, we performed ChIP assays to test the occupancy of factors including Pol II as well HDAC1 and SUV39H1 (both markers of suppression) on the HIV-1 LTR promoter (du Chene et al., 2007; He and Margolis, 2002). The data in Fig. 1D and E indicate that the untreated ACH-2 cells contain both suppressive markers of transcription, HDAC1 (deacetylase) and SUV39H1 (methyl transferase) and do not have much Pol II associated with the LTR promoter. Treatment of the cells with IR showed almost complete removal of SUV39H1 from the HIV-1 promoter and decrease of HDAC1 binding when cells were treated with ALLN. Importantly, Pol II binding displayed from 50 to 100-fold increase to the LTR promoter (panel E).
Finally, to test whether IR can affect presentation of CD4 receptor as well as CXCR4 co-receptor on the surface of treated cells and, therefore, potentially enhance or weaken reinfection of the cells with the virus released after reactivation, we tested CD4 and CXCR4 in irradiated uninfected CEM and HIV-1 infected ACH-2 cells via flow cytometry. Histogram plots in Fig. 1F indicate that IR did not have detectable effect on the presentation of the receptors important for HIV-1 entry on the surface of exposed cells. Collectively, these data imply that activation effect of X-ray treatment on HIV-1 transcription involves epigenetic mechanisms.
Exposure of HIV-1 infected T cells and monocytes treated with PKC agonist bryostatin 1 to IR enhances the LRA activation effect on viral transcription in latently infected monocytes
Bryostatin 1 is a potent PKC modulator. Recent studies have shown that at low nanomolar concentrations bryostatin 1 dramatically reactivated latent HIV-1 in monocytic and lymphocytic reservoirs via activation of PKCα and δ (Mehla et al., 2010). Since multiple studies demonstrated effectiveness of bryostatin-1 for the reactivation of HIV-1 latency in different types of infected cells (DeChristopher et al., 2012; Perez et al., 2010), we then tested whether the exposure to IR enhanced bryostatin 1-induced activation of HIV-1 transcription. Bryostatin 1 has been shown to be cytotoxic in the doses exceeding 10 nM (Bullen et al., 2014). Here we tested effect of different IR doses on the ACH-2 and U1 cells, pre-incubated with ART cocktail for 10 days, as described above, and followed by treatment with three concentrations of bryostatin (Fig. 2A). Quantitative analysis of unspliced HIV-1 RNA at 48 h post-treatment with bryostatin 1 and 44 h after IR showed not dramatic but significant impact of bryostatin 1 on the enhancement of HIV-1 transcription in ACH-2 cells. However, exposure of these cells to IR did not contribute to the bryostatin 1 effect. In U1 cells, all doses of bryostatin 1 drastically induced transcription of HIV-1 RNA. Moreover, exposure to 1 and 3 Gy X-ray doses enhanced the bryostatin 1 effect by 2.5–3 fold. The cell viability analysis demonstrated that all bryostatin 1 doses tested indicated decreased viability (Fig. 2B). Interestingly, while 3 Gy dose of IR slightly reduced viability of bryostatin 1 untreated T cells, the viability of the monocytes did not alter following irradiation. Analysis of viability of parental uninfected (U-937) and chronically HIV-1 infected monocytes (U1) stimulated to differentiate into macrophages (Biswas et al., 1992; Poli et al., 1991), showed that after treatment with various doses of IR the cell viability was not altered among the various groups of cells (Fig. 2C). Both uninfected and HIV-1 infected macrophage model cells showed no difference in viability even when they were exposed to high single X-ray doses up to 20 Gy. In fact, macrophages may not be sensitive to IR, and when radiotherapy is applied, the macrophages have been shown to be radioresistant, survive and activated (Narang and Krishna, 2008).
Fig. 2. IR enhances bryostatin 1-induced activation of HIV-1 transcription in latently infected monocytes.

(A) Effect of IR on ACH-2 (left panel) and U1 (right panel) cells treated with bryostatin 1. The cells were cultured with ART cocktail for 10 days as described in Fig. 1B and then treated with 3 nM, 10 nM or 30 nM bryostatin-1 in PBS. At 4 h post-treatment the cells were exposed to indicated doses of IR. Quantitative RT-PCR analysis of unspliced HIV-1 RNA with gag-specific primers was performed 44 h after IR. Results are shown as a mean of three independent measurements ± SD. Asterisks indicate p ≤ 0.01 between indicated samples and untreated (No bryostatin) control or between the samples connected by bracket. (B) Viability of ACH-2 (left panel) and U1 (right panel) cells treated with bryostatin 1 and exposed to IR. The cells were treated as described in (A), harvested 44 h after IR and subjected to CellTiter Glo assay (Promega). Results are shown as a mean of three independent experiments ± SD. Asterisks indicate p ≤ 0.01 between indicated samples and untreated (No bryostatin) control. (C) The parental uninfected U937 and chronically HIV-1 infected U1 promonocytic cells were stimulated with 10 nM PMA to differentiate into the macrophage phenotype. After 72 h incubation, the cells were exposed to X-ray IR. Cell viability was assessed 72 h post-treatment using CellTiter Glo assay (Promega). Results are shown as a mean of triplicate experiment ± SD.
Since HIV-1 genome in non-activated U1 cells displays very low level of transcription and bryostatin 1 reactivates the virus from “latent” stage, our data indicate that additional exposure to a moderate (1 Gy) or higher doses of IR (≥ 1 Gy) may significantly increase reactivation effect of this PKC agonist on myeloid cells. In ACH-2 the bryostatin 1 treatment enhanced transcription, but additional effect of IR was not detected probably because of the relatively high basic level of HIV-1 transcription in these cells.
X-ray IR activates HIV-1 replication and enhances apoptosis in latently HIV-1 infected quiescent T cells
To test the effect of IR on the latently HIV-1 infected CD4+ T cells, the cells were initially isolated from PBMCs, activated with monocyte derived dendritic cells (MDDC) for four days and then infected with full length, replication competent, CCR5-tropic HIV-1 expressing GFP. Infection was expanded for two weeks and then cells were placed in the medium containing IL-7 to achieve quiescence and latency for an additional two weeks as described earlier (Marini et al., 2008). After IR, the cells were cultured with or without ALLN, harvested 96 h post-IR and then stained to determine percentage of GFP positive (HIV-1 expressing) and Annexin V positive (dying) cells. The flow cytometry analysis of remaining viable cells indicated that IR dose 5 Gy was cytotoxic, since it decreased the population of quiescent CD4+ T cells (Fig. 3A). IR led to 1.5-fold increase of the level of GFP expression. Additional qPCR of total DNA from these cells did not reveal 2-LTR circles within the samples (data not shown) that indicates the lack of reinfection and expression of the viral genes only from provirus.
Fig. 3. IR activates HIV-1 replication and enhances apoptosis in latently infected primary quiescent CD4+ T cells.

(A) Flow cytometry analysis of HIV-1 replication and apoptosis in activated (memory-phenotype) primary CD4+ T cells enriched by negative selection from CD4+ T cells and monocyte-derived dendritic cell cultures and infected with replication competent HIV-1 strain expressing GFP. The HIV-1 infected quiescent CD4+ T cells were treated with single 5 Gy X-ray IR dose, cultured for 96 h and then subjected to flow cytometry. Left panels show gating of live cells (R1) with numbers representing percent of total counts in plot (based on FSC-H vs. SSC-H). Other plots represent analysis of R1 sub-population. The HIV-1 replication was measured as a percent of GFP-positive cells within the R1 population (second column of panels); the percent of apoptotic cells was measured using Annexin V staining separately in subpopulations of GFP-negative and GFP-positive cells (right two columns of panels). (B) Flow cytometry analysis of apoptosis in IR-treated uninfected primary CD4+ T cells (memory-phenotype) prepared as described in panel A. The cells were exposed to indicated doses of IR and then analyzed at 48 h post-IR. The upper row indicates gating of living cells (R1) with numbers representing percent of total counts in plot (based on FSC-H vs SSC-H). The bottom row shows the percent of apoptotic cells measured using Annexin V staining in the gated R1 sub-population. (C) The count of unspliced HIV-1 RNA in the lysates of infected resting CD4+ T cells treated with indicated X-ray IR doses, measured by RT-qPCR with gag-specific primers 72 h after IR. Results are shown as a mean of three independent measurements ± SD. Asterisk indicates p ≤ 0.05 between indicated sample and unexposed (No IR) or 0.25 Gy IR exposed cells.
Indeed, IR had a profound effect on apoptosis induction and cell survival. The percentage of Annexin V positive cells after IR was twofold greater in the GFP-positive than in GFP-negative subsets (Fig. 3A). It should be noted that basic level of apoptosis in GFP positive cells was six fold higher, than in the negative control cells, and that the absolute proportion of dying cells after IR is about 40% in the infected cell population and only 18.2% in uninfected cells. Therefore cytopathogenic effect of IR on reactivated HIV-1 infected T cells is higher than on the uninfected cells.
Despite the higher cytopathogenicity of IR for HIV-1 infected CD4+ T lymphocytes, the 20–30% cell death in the population of uninfected CD4+T lymphocytes appeared high. To better define the maximal dose of IR which is capable of reactivating HIV-1 transcription and does not activate apoptotic response in uninfected resting CD4+ T cells, we tested lower IR doses on the apoptosis and HIV-1 RNA transcription in the cells. We found, that doses ≤ 0.5 Gy did not dramatically increase apoptosis of the uninfected cells (Fig. 3B), but increased level of HIV-1 transcription (1.6 fold for 0.25 Gy dose of IR), in the infected cells (Fig. 3C), suggesting that the doses between 0.25 and 0.5 Gy may be considered as a potential reactivator of HIV-1 transcription in resting CD4+ T cells.
IR activates p53 in both uninfected and chronically HIV-1 infected T cells but enhances apoptotic pathways mostly in the infected cells
Our data above indicate that IR induces higher level of apoptosis in HIV-1 infected primary cells, than in uninfected cells. Indeed, analysis of viability of PBMCs, infected or not with 89.6 strain of HIV-1, cultured for 45 days with IL-7 and then irradiated with X-ray IR displayed four- to five-fold reduction in viability of uninfected cells and 10-fold decrease viability of the infected cells (Fig. 4A).
Fig. 4. IR activates p53 in both uninfected and chronically HIV-1 infected T cells, but enhances apoptosis in the infected cells.

(A) IR differentially induces apoptosis in HIV-1 infected and uninfected PBMCs: the cells from two donors were infected or not with 89.6 dual-tropic strain of HIV-1, cultured for 45 days with IL-7, irradiated with 5 Gy X-ray dose and then cultured for 48 h. Cell viability was measured using CellTiter Glo assay (Promega). Results are shown as a mean of three independent experiments ± SD. Asterisk indicates p ≤ 0.05 between indicated IR-exposed samples and non-irradiated cells. (B). Western blot analysis of p53 phosphorylation in the lysates of parental uninfected (CEM) and chronically HIV-1 infected T cells (ACH-2) in response to X-ray IR (5 Gy). The cells were harvested 48 h after exposure, lysed in cell lysis buffer containing proteasomal inhibitor cocktail (Roche). Lysates were then normalized according to the total protein concentration and subjected to SDS-PAGE and Western blot analysis with rabbit polyclonal antibodies against total p53 and indicated phosphorylated forms of p53 protein. (C) Quantitation of Western blot bands. Western blot bands were quantified using ImageJ software and results are presented as percentage of the peak value for each form of p53 protein (total or phosphorylated at Ser9, Ser15 or Ser46) in analyzed cell lysates. (D) Western blot of lysates of HIV-1 chronically-infected (ACH-2) and parental uninfected (CEM) T cell lines treated with X-ray IR (5 Gy). The cells were harvested at 48 h time point, lysed, normalized as described in panel B, and then subjected to SDS-PAGE and western blot analysis with rabbit polyclonal antibodies against PARP-1 and mouse monoclonal antibodies against HIV-1 p24 and actin.
The cells respond to IR-induced genotoxic stress by activation of DNA repair (reviewed in Curtin (2012)) and/or induction of apoptosis in the case of severe DNA damage which cannot be repaired by the Ataxia telangiectasia mutated (ATM)-induced DNA damage repair pathways (Collis et al., 2004). Earlier studies indicated that low linear energy transfer IR, such as X-rays and γ-rays, induce p53-dependent apoptosis in both normal and cancer cells, whereas high linear energy transfer IR (α-particles) mostly induce p53-independent apoptotic pathway (Haro et al., 2012; Mori et al., 2009). For HIV-infected cells, both X-ray and γ-IR have been shown to induce cell death via chromatin DNA-damage (Ogawa et al., 2003). To test whether the enhanced apoptosis in HIV-1 infected T cells is dependent on p53 activation, we analyzed p53 phosphorylation at different sites critical for its activity to induce the DNA repair pathways or apoptosis. Data in Fig. 4B indicate phosphorylation of the Ser9 and Ser15 residues (both trigger p53 activity to induce DNA repair pathways in response to DNA damage (Haas, 2009; Oda et al., 2000; Shahbazi et al., 2013; Xu et al., 2012; Yamaguchi et al., 2001)), as well as Ser46, whose phosphorylation regulates the ability of p53 to induce apoptosis (Oda et al., 2000). The western blot analysis showed that X-ray IR induced phosphorylation of all analyzed serine residues in both HIV-1 chronically infected and parental uninfected cells. However, while the level of phosphorylation of Ser9 and Ser15 was the same in both infected and uninfected cells, phosphorylation of Ser46 was approximately 40% higher in the infected T cells (Fig. 4C, top panel). These data indicate that IR equally induced p53-mediated DNA repair pathways in HIV-1 infected and uninfected cells, but activated pro-apoptotic p53 activity predominantly in the infected cells.
To further test this hypothesis, we performed western blot analysis of the cellular factor PARP-1 involved in the DNA damage induced apoptotic pathway. Both infected and uninfected cells were X-ray irradiated. We found that the level of cleaved PARP-1 (indicator of apoptosis (Lazebnik et al., 1994)) was dramatically higher in HIV-1 infected cells 48 h after IR, suggesting higher ratio of DNA damage response in infected cells (Fig. 4D). Collectively, these results imply that the HIV-1 chronically-infected cells are more susceptible to DNA damage stress-induced apoptosis when exposed to IR than uninfected cells.
Exposure to X-ray reactivates HIV-1 replication in various tissues of infected humanized mice.
To test the effect of IR on viral replication in different tissues of the infected animal, we used the model of NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG) humanized mice infected with dual-tropic HIV-1 89.6 strain. Different colleagues (Choudhary et al., 2012; Kassu et al., 2009; Marsden et al., 2012; Palmer et al., 2013), as well as our group (Duyne et al., 2011) earlier showed that HIV-1-infected BLT, hu-Rag2−/− γ(c)−/− and NSG mice can develop latent infection – the virus is integrated, activation inducible, and replication competent.
For analysis of IR on virus replication, we used mice that were engrafted with human fetal liver derived CD34+ cells and infected at 16 weeks after birth. We then waited for 6 months and then irradiated with 4 Gy non-lethal X-ray dose (Guo et al., 2009; Wang et al., 2013). The quantitative RT-PCR (qRT-PCR) analysis did not reveal any HIV-1 RNA in the plasma samples before IR, whereas qPCR detected various amounts of proviral DNA in the blood cells from all infected animals (mice 1–6, Fig. 5A). The blood samples obtained on day 10 after IR displayed from 2000 to 27,000 copies of unspliced viral RNA per μg of total RNA (Fig. 5B). Analysis of the ratio of unspliced viral RNA and viral DNA in the total pellet of blood cells displayed from 7 to 100 fold increase of HIV-1 RNA proportion within 10 days after IR (Fig. 5C). The qPCR of the RNA isolated from the blood cells, liver, lung, brain and spleen of the mice sacrificed on day 10 after IR revealed various detectable levels of viral RNA. The RNA levels in lung and brain specimens were similar to the levels in blood cells, whereas the liver and spleen tissues showed lower levels of viral RNA (Fig. 5D). Finally, to test toxicity of IR for the mice, we performed analysis of the viability of other group of mice within 90 days after exposure to doses 1.0, 2.5, 5.0, 7.5 and 10.0 Gy of X-ray IR. We found that 7.5 and 10 Gy doses of IR resulted in death of the mice in 6 and 9 days post IR, whereas the mice subjected to 5 Gy and lower IR doses did not die during the whole period of observation (data not shown).
Fig. 5. IR activates HIV-1 transcription in various tissues of infected humanized mice.

The NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG) humanized mice were infected with dual-tropic HIV-1 89.6 strain as described in Materials and Methods. The latent animals (6 months after initial infection) were irradiated with 4 Gy non-lethal X-ray dose and sacrificed 10 days after IR. The blood, brain, liver, spleen and lung were harvested, homogenized and subjected to total RNA isolation. Pure RNA samples were analyzed by quantitative RT-real-time PCR with HIV-1 specific primers. (A) HIV-1 provirus counts in blood cells of infected animals, 10 days post-IR. The cellular fraction was isolated from 0.5 ml blood samples; total DNA was purified and subjected to real-time PCR with R-U5 LTR-specific primers to detect HIV-1 DNA. Results are shown as a mean of three independent measurements ± SD. (B) Viral RNA in the blood samples of animals – 28 days before and 10 days after IR. The total purified RNA was subjected to quantitative RT-PCR with gag-specific primers. Results are shown as a mean of three independent measurements ± SD. Asterisks indicate p ≤ 0.01 between RNA samples from the same animal before and after IR. (C) The HIV-1 RNA/DNA ratio in the blood of infected animals, 10 days post-IR. Total RNA and DNA were purified as in panels A and B, and then subjected to RT-qPCR with (RNA) or qPCR (DNA) with gag-specific primers. Results are shown as a mean of three independent measurements ± SD. Asterisks indicate p ≤ 0.01 between samples from the same animal before and after IR. (D) The total RNA extracted from indicated organs at day 10 after IR was subjected to HIV-1 RNA quantitation using qRT-PCR as described in (B). Results are shown as a mean of three independent measurements ± SD. The dash line indicates the cutoff level of HIV-1 RNA.
To model latent HIV-1 infection in the humanized mice, new set of mice infected with dual-tropic 89.6 virus were then injected intraperitoneally three times with the cocktail of four antiretrovirals in weeks 8, 9 and 10 after infection. Quantitative RT-PCR analysis of whole blood samples indicated rather high amount of viral RNA (from 1500 to 16,000 copies per 0.2 ml sample as shown in Fig. 6A, right panel) within two months post-infection. Dramatic reduction of HIV-1 RNA to very low level (from 45 to 600 copies per 0.2 ml sample) was detected in the blood of ART-treated mice as compared with the same animals before treatment (from 4000 to 17,000 copies/sample), whereas the count of viral DNA in the lysates of total blood cells was reduced by an average of 4.6 times (Fig. 6A, middle panel). Exposure of the ART-treated mice to two 1 Gy doses of IR on the 7th and 9th days after last ART injection resulted in 20–1000-fold increase of viral RNA count in the blood on day 2 after second X-ray IR exposure (Fig. 6A, right panel). The count of viral DNA has also been approximately fivefold increased probably due to the reverse transcription and accumulation of cDNA in newly infected cells. It is important to note that the count of human β globin DNA (marker of human cells) was not significantly changed within the whole time of experiment (Fig. 6A, left panel) Analysis of the ratio of unspliced viral RNA to viral DNA in the total blood samples showed that HIV-1 infected females (Fig. 6B, # 603–605) demonstrated higher level of viral RNA transcription prior to ART treatment, but relatively low increase after post-ART irradiation (5–30 ×) which is lower than the RNA level prior to ART treatment. Whereas the males displayed relatively low initial level of HIV-1 transcription, but in 50% cases showed dramatic increase of the viral transcription after IR: the RNA level after IR was the same (#606, 609, and 610) or 10-fold higher (#607, 608, and 611) compared to pre-ART RNA count. These data suggest that the HIV-1 infected females had initially higher level of virus transcription, probably due to better engraftment of human CD34+ cells than in NSG males as reported earlier (Denton and Garcia, 2011; Martin-Padura et al., 2010; Notta et al., 2010). Taken together, our results indicate that IR significantly activates HIV-1 expression in the blood and some tissue reservoirs including brain and lung of the infected humanized mice.
Fig. 6. IR doses induce viral transcription in ART-treated latently HIV-1 infected humanized mice.

Nine NSG humanized mice were infected with dual-tropic HIV-1 89.6 strain and two mice were left as an uninfected control. The infected and uninfected mice were injected i.p. three times with ART cocktail in week 8, 9 and 10 after infection time point. The mice were then irradiated with double 1 Gy doses of X-ray on days 7th and 9th after last ART injection. The 0.2 ml specimens of blood were collected before (Untreated), after ART treatment (ART) and 48 h after second IR (IR, post-ART). (A) Statistical analysis of HIV-1 response to ART treatment and IR in the blood samples from 9 infected animals. Total RNA and DNA was purified from the blood specimens and then subjected to RT-qPCR with oligo-dT and gag primers (RNA) to measure count of unspliced viral RNA, and to qPCR with human β globin and HIV-1 gag primers (DNA) to measure human and viral DNA count, respectively. Asterisk shows p value ≤ 0.05 between the samples connected by bracket. (B) The HIV-1 RNA/DNA ratio in the blood specimens of infected and control uninfected mice, 2 days post-second IR. Total RNA and DNA were purified and subjected to RT-qPCR and qPCR with HIV-1 gag-specific primers as described in panel A. Results are shown as a mean of three independent measurements ± SD. Asterisks indicate p ≤ 0.01 between RNA/DNA count in indicated samples (IR post-ART) and RNA/DNA count in the samples collected after ART from the same animal.
Discussion
Despite the wide use of IR for therapy for different types of cancers and application of the whole body IR used for HIV-1 infected individual known as “Berlin patient“ for destruction of his lymphoma prior to CCR5Δ32/Δ32 donor hematopoietic stem cell transplantation, the effect of therapeutic doses of IR on HIV-1 has not been fully recognized. Meanwhile, the total body IR treatment with the doses of 2 Gy and 4 Gy (Allers et al., 2011) that were applied to Berlin patient prior to the transplantation did not have documented negative effect on other tissues (Yukl et al., 2013). Importantly, the virus was likely eradicated from latent reservoirs, since further prolonged tests could not detect HIV-1 in PBMCs, spinal fluid, lymph node, or terminal ileum, and no replication-competent virus could be cultured from PBMCs (Yukl et al., 2013). Therefore, these data point to the importance of studying the impact of IR treatment on HIV-1 replication, specifically within the context of reactivation and depletion of the latent viral reservoirs.
Our data presented above indicated that a single dose of X-ray IR activated HIV-1 replication in both chronically infected lymphoblastoid and monocytic cell lines as well as in CD4+ resting T cells. We found that IR treatment of cells correlated with dissociation of the histone deacetylase HDAC1, and methyl transferase SUV39H1 from the HIV-1 LTR promoter. Direct involvement of these factors in HIV-1 latency was shown previously by a number of colleagues (Pearson et al., 2008; Tyagi et al., 2010). Our data further suggest a triggering effect of IR on HIV-1 transcription via epigenetic mechanisms. In fact, the studies of the last decade revealed involvement of various epigenetic factors in IR stress-induced transcription activation associated with DNA damage response. In IR-sensitive tumor cells, the γ-IR has been shown to induce the global demethylation of various promoter sequences (Kumar et al., 2011). Inhibitory effect of IR-induced DNA damage on HDAC1 followed by transcriptional activation was shown for both cellular and viral promoters such as HBV (Chung and Tsai, 2009; Peng et al., 2007). At the same time, typical for IR response activation of NHEJ pathway to repair DNA double-strand breaks leads to activation of the homologous HATs, including CBP and p300 factors (Ogiwara et al., 2011). The CBP/p300 proteins are recruited to the sites of doublestrand brakes and contribute to acetylation of lysine 18 within histone H3, and lysines 5, 8, 12, and 16 within histone H4. This also correlates with the recruitment of BRM, a catalytic subunit of the SWI/SNF complex involved in chromatin remodeling (Agbottah et al., 2006; Ogiwara et al., 2011; Van Duyne et al., 2011). Together, these published data indicate important role of epigenetic events such as inhibition of promoter methylation and enhancement of histone acetylation in the cellular response to IR-induced DNA damage. Since acetylation of LTR-bound nucleosomes by HATs is involved in activation of HIV-1 transcription (Deng et al., 2000, 2001), inhibition of HDACs is critical for LTR transcription and important for reactivation of latent HIV-1 provirus (Archin et al., 2009, 2012; Blazkova et al., 2012; Quivy et al., 2002; Van Lint et al., 1996).
Our data that the irradiation of HIV-1 infected cells enhanced level of apoptosis in the irradiated cells suggest that activation of HIV-1 replication in response to IR can trigger apoptotic pathways likely via the potential effect of HIV-1 Tat on p53 phosphorylation (Kim et al., 2010). In fact, we have observed presence of Tat after IR when transfecting an epi-Tat construct into lymphocytes (CEM) following IP/WB for presence of Tat after 48 h (data not shown). IR-induced p53 phosphorylation at Ser15 and Ser20 are utilized for DNA repair and release, whereas Ser46 phosphorylation is critical for apoptosis (Haas, 2009; Shahbazi et al., 2013; Yamaguchi et al., 2001). Western blot analysis of chronically-HIV-1 infected T cell line and parental uninfected cells revealed involvement of the mechanism of p53-dependent apoptosis in both infected and uninfected irradiated cells, with the level of phosphorylation of Ser46 residue markedly higher on the p53 protein from infected cells. This may potentially explain lower viability of HIV-1 infected cells after IR. Since Tat has been shown to activate PKCδ in HIV-1 infected cells (Bennasser and Bahraoui, 2002; Leghmari et al., 2008a, 2008b), this interaction might facilitate p53-dependent apoptosis in response to IR induced HIV-1 activation. Moreover, it has also been found that Tat protein binds directly to p53 and hence down-regulates p53-dependent transcription (Harrod et al., 2003; Li et al., 1995; Longo et al., 1995) and therefore IR-induced Tat expression may potentially deregulate p53-mediated DNA repair (Fig. 7A). Other recently published data indicate that the apoptosis induced in ACH-2 and U1 cells by such cytotoxic agents as doxorubicin, etoposide, fludarabine phosphate, or vincristine significantly activated latent HIV-1 infection and resulted in increased level of coding viral RNA and p24 production (Khan et al., 2015). This suggests that along with induction of p53-dependent apoptosis in response to IR-activated HIV-1 expression, the IR stress-induced apoptosis in turn may enhance HIV-1 transcription and replication.
Fig. 7. Summarized data of the effect of X-ray irradiation on the cell viability and virus transcription in HIV-1 infected cells.

(A) IR-induced DNA damage in HIV-1 infected and uninfected cells mediates phosphorylation of various Ser residues on p53 protein resulting in DNA repair or apoptosis (adapted from Shahbazi et al. (2013) with modifications). Low IR doses cause DNA damage that activates p53 via phosphorylation of Ser9, Ser15 and Ser46 on p53 in uninfected cells as shown in Fig. 4B. Phosphorylated p53 activates p21WAF1 and p53-dependent ribonucleotide reductase p53R2 which induce G1 arrest and DNA repair pathway, respectively. IR of HIV-1 infected cells activates production of Tat and viral accessory proteins which cause up-regulation of DNA damage machinery (Kim et al., 2012; Koyama et al., 2013). Tat can also activate PKCδ (Bennasser and Bahraoui, 2002; Leghmari et al., 2008a, 2008b) that induces enhanced phosphorylation of Ser46 on p53 which results in p53-dependent apoptosis. (B) Consolidated data on the effect of IR and IR with latency-reversing agent bryostatin 1 on HIV-1 transcription in latently infected T cells and monocytes.
A number of colleagues have previously shown that HIV-1 replication induces apoptosis via p53-independent pathways (Bartz and Emerman, 1999; Ferri et al., 2000; Jacotot et al., 2000; Rasola et al., 2001; Sastry et al., 1996; Stewart et al., 2000; Terai et al., 1991). Since IR is able to activate expression of HIV-1 genome, viral proteins such as Env, Tat, Vpr, Vpu, Nef, and Vif whose pro-apoptotic activity is well described (reviewed in Cossarizza (2008)), may activate apoptosis in the IR treated cells. Moreover, earlier studies have also shown that IR itself might induce the same mechanism of apoptosis as viral Vpr protein, which involves the Wee-1 kinase depletion (Yuan et al., 2003).
Modulators of PKC activity, such as PMA and prostratin were earlier investigated for their capacity to reactivate latent HIV-1 in T-lymphocytes and monocyte/macrophages (Biancotto et al., 2004; Brooks et al., 2003; Trushin et al., 2005). PKC signaling may activate transcription of the latent HIV-1 provirus via recruitment of cellular transcription factors such as NF-κB, NF-AT and AP1 (Beans et al., 2013; Williams et al., 2004). Bryostatin 1, like prostratin also targets PKC but it is significantly more potent (DeChristopher et al., 2012). In our experiments we observed enhancement of bryostain 1-induced HIV-1 reactivation in latently-infected monocytes by single IR doses of 1 and 3 Gy. Since low doses of bryostatin 1 have been shown to be capable of activating PKCδ (Mehla et al., 2010), we hypothesize that IR-induced expression of Tat could strengthen impact of bryostatin 1 via additional activation of PKCδ by Tat protein (as previously described (Bennasser and Bahraoui, 2002; Leghmari et al., 2008a, 2008b)). Summarized data on the effect of IR and bryostatin 1 treatment on the latently HIV-1 infected T cells and monocytes are shown in Fig. 7B. Taken together, our results indicate that low doses of IR had a moderate effect on HIV-1 reactivation in latently-infected CD4+ T cells, but affected the infection dramatically in monocytes/macrophages, especially when IR was in combination with bryostatin 1 treatment.
Our experiments with the model of humanized mice, where the basal level of virus replication was virtually undetectable either after six months of infection or after triplicate injection of antiretrovirals, indicated that single dose of IR (4 Gy) or double 1 Gy dose could activate HIV-1 transcription in peripheral blood cells as well as in the lung and brain tissues. This observation is especially interesting since both brain and lung tissues are rich in macrophages, including alveolar and microglia (Eilbott et al., 1989; Pollard, 2009; Wiley et al., 1986). It is also important to note that blood was not removed from these organs prior to RNA isolation, which may contribute to presence of viral RNA in these samples. Finally, potent effect of IR on latently HIV-1 infected monocytes after PKC activation with bryostatin 1, transferring these cells to macrophage phenotype, may explain observed dramatic reactivation of HIV-1 transcription in macrophage-rich tissues in the mice after IR.
Conclusions
Collectively, our results demonstrate activating effect of therapeutic doses of X-ray IR on HIV-1 LTR-driven transcription and viral replication in major HIV-1 host cells, and increased apoptosis in HIV-1 infected T cells. Use of low doses of IR (less than 0.5 Gy) in combination with chemical agents leading to reactivation of HIV-1 transcription, such as HDAC inhibitors (SAHA, vorinostat) or PKC modulators like prostratin or bryostatin 1, holds great promise for reactivation of latent HIV-1 in reservoirs in vivo. Unlike the chemical compounds, IR affects various tissues independently of their location in the body and does not have the complication of delivery into various body compartments. Thus, our data suggest an alternative strategy to activate and eradicate HIV-1 from persistently infected reservoirs to subsequently eliminate the activated virus using traditional therapeutic approaches. Future experiments will better define the possible combination use of IR and other agents for the “shock and kill” strategy.
Materials and methods
Cells and viruses
The human acute T leukemia lymphoblastoid cell line CCRF-CEM was purchased from ATCC (Manassas, VA). Chronically HIV-1 infected U-937 subclone U1 and CEM subclone ACH-2 (Folks et al., 1987; Perez et al., 1991) were provided by the NIH AIDS Research & Reference Reagent Program. All cells were maintained at 37 °C and 5% CO2 in 25 cm2 and 75 cm2 tissue culture flasks with RPMI-1640 culture media supplemented with 10% Fetal Bovine Serum, penicillin/streptomycin (100 μg/ml), and L-Glutamine (lymphoid and myeloid cells). To reduce basic level of HIV-1 replication and exclude reinfection, the chronically HIV-1 infected cells were incubated for 7–10 days with the cocktail of three antiretrovirals (lamivudine/emtricitabine, tenofovir and indinavir, each in concentration 10 μM) provided by NIH AIDS Research & Reference Reagent Program. Then the cells were washed with PBS and cultured in regular medium for 16–18 h until IR exposure. Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood from healthy, anonymous donors using Ficoll gradient centrifugation and then expanded in medium containing 1 μg/ml PHA-L and 30 or 50 IU/mL rhIL-2. After two days of cultivation the cells were washed and then cultured in the medium containing 30 IU/mL rhIL-2 without PHA-L.
The stocks of NL4-3BalEnv R5 virus or HIV-1 89.6 dual-tropic strain (Collman et al., 1992) (provided by Dr. Ronald Collman via NIH AIDS Research & Reference Reagent Program) were used for infection of PHA-activated PBMCs (800 ng of p24 for 40 × 106 cells/ml). Cells were incubated with virus for 4 h and after washes, the cells were cultured in complete RPMI 1640 medium supplemented with 50 U/ml rhIL-2 (30 U/ml) for 10 days, treated with 1 ng/ml IL-7 and then cultured for 45 days to place the cells in quiescent phase.
CD 4+ T lymphocytes and latent infection with HIV-1
Primary cells latently infected with HIV-1 were generated essentially as described in Marini et al. (2008), with some modifications. Total CD4+ T cells were enriched by negative selection with the CD4+ T Cell Isolation Kit (Miltenyi Biotec) from PBMC of healthy seronegative donors. CD4+ T cells were activated with monocyte-derived dendritic cells (MDDC; 1 MDDC10 CD4+ T cells), 500 ng/ml SEB and 50 U/ml IL-2. After 4 days half of the cells were infected with a full length, replication competent HIV-1 strain expressing GFP (Brown et al., 2006) by spinoculation at 1200g for 2 h at room temperature. The other half of the cells was left uninfected. Infected and uninfected cultures were expanded for 10–15 days in medium containing 50 U/ml IL-2. When the infected culture contained 10–15% infected cells as determined by GFP positivity in flow cytometry, both infected and uninfected cultures were placed in quiescent phase for 7 days in medium containing 1 ng/ml IL-7. At the end of the 7 day incubation, the cultures were irradiated and then cultured for 96 h.
Flow cytometry
Analysis of live/dead cells after irradiation was performed with the Annexin V Apoptosis Detection Kit I from BD Biosciences following the manufacturer's instructions. Briefly, cells were harvested, washed with PBS and resuspended at 1 × 106/ml in 1 × Annexin V Binding Buffer. Annexin V (5 μl/100 μl of cell suspension) was added to each sample, followed by incubation for 15 min at room temperature. Finally, an equal volume of 1 × Annexin V Binding Buffer was added, and the samples were analyzed by flow cytometry within 1 h using a FACScalibur and the software CellQuest (BD Biosciences). Surface staining of CD4 and CXCR4 receptors exposed on the plasma membrane of CEM and ACH-2 cells was performed with mouse monoclonal FITC-anti-human CD4 (BD Pharmingen) and PE-anti-human CXCR4 (Biolegend). Flow cytometry files were analyzed using FlowJo (Tree Star, Ashland, OR).
Reverse transcriptase (RT) activity analysis
RT analyses were performed as published previously (Easley et al., 2010). Briefly, 10 μl of cell culture supernatants were incubated in a 96-well plate with RT reaction mixture containing 1 × RT buffer (50 mM Tris–HCl, 1 mM DTT, 5 mM MgCl2, 20 mM KCl, 0.1%Triton, poly-r(A) 1U/ml, oligo-d(T) 1 U/ml, and [3H] dTTP). The mixture was incubated for 2 h at 37 ° C, and 5 μl of the reaction mix was spotted on a DEAE Filtermat paper, washed four times with 5% Na2HPO4 and four times with water, and dried. RT activity was measured in a Betaplate counter (Wallac, Gaithersburg, MD).
DNA isolation and quantitative real-time PCR
The cells from blood samples of the uninfected control and HIV-1 infected humanized mice were normalized to the total protein count using DC Protein Assay (BioRad, Hercules, CA), lysed in the Nuclei Lysis Solution (Promega, Madison, WI), and the total DNA was then isolated from lysates using Wizard Genomic DNA Purification Kit (Promega) according to manufacturer's protocol. After isolation, the cellular DNA samples were analyzed by quantitative TaqMan realtime PCR. The primers For-Late: 5′-TGTGTGCCCGTCTGTTGTGT-3′, Rev-Late: 5′-GAGTCCTGCGTCGAGAGATC-3′, and probe 1312-Lt-LTR: 5′-FAM-CAGTGGCGCCCGAACAGGGA-TAMRA-3′, were used to recognize the U5-Ψ HIV-1 LTR region. Set of primers specific for human p-globin gene has been used to assess presence of human cells in the mice blood samples: forward primer BGF1 (5′-CAACCTCAAACAGA-CACCATGG-3′), reverse primer BGR1 (5′-TCCACGTTCACCTTGCCC-3′), and probe BGX1 (5′-FAM-CTCCTGAGGAGAAGTCTGCCGTTACTGCC-TAMRA-3′). PCR reactions were performed with PerfeCTa qPCR FastMix, UNG (Quanta Biosciences, Gaithersburg, MD) using 300 nM of each primer and 200 nM of probe according to the manufacturer's protocol. Serial dilutions of DNA from 8E5 cells (CEM cell line containing a single copy of HIV-1 LAV provirus per cell) were used as the quantitative standards. Real-time PCR reactions were carried out at least in triplicate using the CFX96 Real-Time PCR System (BioRad) and SFX Manager Software ver. 2.0.
RNA isolation and quantitative RT-PCR
For quantitative analysis of HIV-1 RNA, total RNA was isolated from various samples including: PBMC lysates, lysates of primary CD4+ T cells and chronically HIV-1 infected cell lines (ACH-2, U1), blood specimens of HIV-1-infected humanized mice and homogenized mice tissues (liver, lung, brain, and spleen). RNA was isolated using Trizol Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. A total of 0.5 μg of RNA from the RNA fraction was treated with 0.25 mg/ml DNase I RNase-free (Roche, Mannheim, Germany) for 60 min in the presence of 5 mM MgCl2, followed by heat inactivation at 65 ° C for 15 min. A 200–250 ng aliquot of total RNA was used to generate cDNA with the GoScript Reverse Transcription System (Promega, Madison, WI) using TAR-specific reverse primer TARfll-R: 5′-GTGGGTTCCCTAGTTAGC-3′ or oligo-dT reverse primers. Subsequent quantitative real-time PCR analysis was performed with 2 μl of undiluted and 10−1 and 10−2 diluted aliquots of RT reaction mixes. The iQ SYBR Green Supermix (Bio-Rad, Hercules, CA) was used with the primers specific for (1) HIV-1 TAR: TARfll-F: 5′-GGTCTCTCTGGTTAGACC-3′ and TARfll-R as above amplified 60 nt. TAR sequence; (2) HIV-1 env gene: Env2019F: 5′-GGCAAGTCTGTGGAATTGG-3′ and Env2187R: 5′-TGGGA-TAAGGGTCTGAAACG-3′ amplified 168 nt. fragment of HIV-1 env and (3) HIV-1 gag gene: Gag1483-F (5′-AAGGGGAAGTGACATAGCAG-3′) and Gag1625-R (5′-GCTGGTAGGGCTATACATTCTTAC-3′) amplifying 143 nt. fragment of HIV-1 gag sequence. The quantitation of U5-Ψ HIV-1 LTR sequences was performed by quantitative TaqMan realtime PCR using PerfeCTa qPCR FastMix, UNG (Quanta Biosciences) using the primers described in previous section. Real-time PCR reactions were carried out at least in triplicate as described above.
X-ray irradiation and HIV-1 reactivation in cell cultures
The HIV-1 latently infected, treated or not with ART cocktail, and control uninfected cells were plated onto 24-well or 12-well plates in 1 or 1.5 ml of complete culture medium respectively. After overnight incubation the cells were treated or not with various concentrations of bryostatin 1 (Sigma-Aldrich, St. Louis, MO) diluted in 1 × PBS and then exposed to various doses of X-ray irradiation using RS 2000 X-ray Irradiator (Rad Source, Suwanee, GA). The cell cultures were then incubated for 24, 48 or 72 h for virus activation.
Chromatin immunoprecipitation (ChIP) assay
Cell cultures were collected 48 h post-irradiation and then processed for ChIP analysis. Approximately 5 × 106 cells were used for immunoprecipitation. ChIP assays were performed as previously described (Klase et al., 2007). Briefly, cells were harvested, washed with PBS, and resuspended in 1% formaldehyde for 10 min at 37 °C. Then the cells were washed twice with PBS and resuspended in 500 μl of SDS Lysis Buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl, pH 8.1) per sample. Cells were sonicated for six 10 s pulses and clarified by centrifugation at 14,000 rpm for 10 min at 4 °C. Supernatants were collected and diluted 10 fold in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100,1.2 mM EDTA, 16.7 mM Tris–HCl, pH 8.1,167 mM NaCl). Extracts were precleared with ChIP prepared A/G beads (Protein A/G beads with 10 mg/mL salmon sperm DNA and 10 mg/mL BSA) for 1 h at 4 °C. After preincubation, the extracts were spun at 3000 rpm for 10 min at 4 °C and the supernatants were transferred to a new tube. Specific antibodies (5 μg) were incubated overnight at 4 °C on a rotator. ChIP prepared A/G beads were added the next day and allowed to rotate for 2 h at 4 °C. Samples were spun for 5 min at 3000 rpm at 4 °C and washed successively with 1 × low salt buffer (0.1%SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1,150 mM NaCl), 2 × high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 500 mM NaCl), 1 × LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris–HCl, pH 8.1), and 1 × TE. Complexes were eluted off of beads twice with elution buffer (1% SDS, 0.1 M NaHCO3). Eluates were then reverse crosslinked using 5 M NaCl and 50 μg/mL proteinase K for 5 h at 55 °C. DNA was extracted with phenol:chloroform, precipitated with isopropanol, washed and resuspended in 50 μl of TE buffer. Specific DNA sequences in the immunoprecipitates were detected by PCR using primers specific for the HIV-1 LTR between −92 to +180 nt (forward primer, 5′-ACTTTTCCGGGGAGGCGC-GATC-3′; reverse primer, 5′-GCCACTGCTAGAGATTTCCACACTG-3′) (Zhou et al., 2003).
SDS-PAGE and Western blot analysis
Proteins from total cell extracts were separated by SDS-PAGE on 4–20% Tris–glycine gels (Invitrogen). For Western blot analyses, proteins were transferred to PVDF membranes (BioRad) at 80 mA for 4 h. Membranes were blocked with Dulbecco's phosphate-buffered saline (PBS)+0.1% tween-20 + 5% dry milk for 1 h at room temperature. Primary antibodies against specified proteins were incubated with the membranes overnight at 4 °C. Anti-HIV-1 p24 Gag monoclonal antibody #24-3 (from Michael H. Malim) were provided by the NIH AIDS Research & Reference Reagent Program. Anti-PARP-1/2 (H-250) rabbit polyclonal antibody was from Santa Cruz Biotechnology (Dallas, TX). For analysis of p53 phosphorylation, the rabbit polyclonal antibodies against S9-P, S15-P and S46-P from P-p53 Ab Sampler Kit (Cell Signaling, Danvers, MA) were used. Membranes were then washed twice with PBS+0.1% tween-20 and incubated with HRP-conjugated secondary antibody for 2 h at 4°C. Membranes were washed two times with PBS+0.1% Tween-20, and once with PBS prior to imaging. HRP luminescence was elicited with Super Signal West Dura Extended Duration Substrate (Pierce) and visualized by a Bio-Rad Molecular Imager ChemiDoc XRS system (BioRad).
Cell viability assay
Cell viability was assessed using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) following the manufacturer's recommendations. Briefly, the reagent was added to the wells of the 96-well plate with cells (1:1 reagent: media) and incubated at room temperature for 10 min protected from light. The luminescence signal was recorded using the GloMax-Multi Detection System (Promega). The viable cell number was normalized with control group and the results shown as relative cell viability.
Preparation and infection of humanized mice
The NSG mice were humanized as described earlier (Shultz et al., 2005; Van Duyne et al., 2008, 2009). Briefly, neonates were injected intraperitoneally (i.p.) with 1 × 106 fetal liver derived CD34+ hematopoietic stem cells and were maintained for no less than 16 weeks to allow cell differentiation. Animals exhibiting significant levels of engraftment, as measured by CD45+ and CD4+ cells in the peripheral blood, were then infected i.p. with 100 μl of dual-tropic HIV-1 89.6 (3000 TCID). The levels of HIV-1 RNA in serum and HIV-1 provirus in the white blood cells were tested at 8 weeks after inoculation using qRT-PCR with the primers specific for HIV-1 gag and by qPCR with the R-U5 LTR-specific primer set, as described above. The mice were kept for 6 months to ensure absence of any viral RNA in the serum (latent animals) or i.p. injected with ART cocktail (emtricitabine 210 mg/kg, tenofovir 210 mg/kg, raltegravir 60 mg/kg, and maraviroc 60 mg/kg (Denton et al., 2012; Neff et al., 2010)) three times with intervals 5–7 days. Then the mice were irradiated with non-lethal single dose of 4 Gy (Guo et al., 2009; Wang et al., 2013) or double 1 Gy doses of X-ray (on days 7th and 9th after last ART injection) using RS 2000 X-ray Irradiator (Rad Source). The blood was collected 48 h after second IR treatment. The animals were sacrificed in 10 days after IR; the blood, liver, lung, brain and spleen (0.25–0.75 cm3 grinded tissue) were collected for further analyses. Reactivation of HIV-1 was assessed by qRT-PCR of unspliced viral RNA with gag-specific primers in the blood and lysates of homogenized tissue specimens from HIV-1 infected and uninfected control mice. All mouse experiments were approved by the George Mason University Institutional Animal Care and Use Committee (IACUC).
Statistical analysis
Quantitative data were analyzed by two-way ANOVA (OriginPro v. 8.0) and Student's t test (Microsoft Excel; OriginPro). Standard deviation was calculated in all quantitative experiments for at least three independent preparations. The difference was considered to be statistically significant when p ≤ 0.05.
Acknowledgments
The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: U1 and ACH-2 cells from Dr. Thomas Folks; HIV-1 89.6 dual-tropic viral strain from Dr. Ronald Collman; anti-HIV-1 p24 mouse monoclonal antibody from Dr. Michael Malim. The antiretrovirals lamivudine/emtricitabine, tenofovir, indinavir, raltegravir, and maraviroc as well as LRA bryostatin 1 were also from AIDS Research and Reference Reagent Program. The HIV-1 proviral clone NL was kindly provided by Dr. Lee Ratner. Authors are grateful to Dr. Shabana Shabbeer Meyering for reading of the manuscript. This work was supported by NIH grants AI043894, AI114490, and AI113140 to FK. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- ALLN
N-Acetyl-Leu-Leu-Nle-al, Calpain 1 inhibitor
- ATM
ataxia telangiectasia mutated
- BAF
BRG1 or hbrm-associated factor
- BBB
blood–brain barrier
- ChIP
chromatin immune precipitation
- CNS
central nervous system
- DTT
dithiothreitol
- GFP
green fluorescence protein
- HAART
highly active antiretroviral therapy
- HAT
histone acetyltransferase
- HBV
hepatitis B virus
- HDAC
histone deacetylase
- IR
ionizing radiation
- LRA
latency-reversing agent
- MDDC
monocyte derived dendritic cells
- MDM
monocyte derived macrophages
- NHEJ
non-homologous end joining pathway
- PARP
poly (ADP-ribose) polymerase
- PBAF
polybromo-associated BAF
- PBMC
peripheral blood mononuclear cells
- PKC
proteinkinase C
- PMA
phorbol 12-myristate 13-acetate
- PML
promyelocytic leukemia
- SAHA
suberoylanilide hydroxamic acid
References
- Agbottah E, Deng L, Dannenberg LO, Pumfery A, Kashanchi F. Effect of SWI/SNF chromatin remodeling complex on HIV-1 Tat activated transcription. Retrovirology. 2006;3:48. doi: 10.1186/1742-4690-3-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–208. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Alexaki A, Liu Y, Wigdahl B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr HIV Res. 2008;6:388–400. doi: 10.2174/157016208785861195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allers K, Hutter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, Schneider T. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood. 2011;117:2791–2799. doi: 10.1182/blood-2010-09-309591. [DOI] [PubMed] [Google Scholar]
- Altschuler C, Resbeut M, Maraninchi D, Guillet JP, Blaise D, Stoppa AM, Carcassonne Y. Fractionated total body irradiation and allogeneic bone marrow transplantation for standard risk leukemia. Radiother Oncol. 1989;16:289–295. doi: 10.1016/0167-8140(89)90041-8. [DOI] [PubMed] [Google Scholar]
- Archin NM, Espeseth A, Parker D, Cheema M, Hazuda D, Margolis DM. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res Hum Retrovir. 2009;25:207–212. doi: 10.1089/aid.2008.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartz SR, Emerman M. Human immunodeficiency virus type 1 Tat induces apoptosis and increases sensitivity to apoptotic signals by up-regulating FLICE/caspase-8. J Virol. 1999;73:1956–1963. doi: 10.1128/jvi.73.3.1956-1963.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beans EJ, Fournogerakis D, Gauntlett C, Heumann LV, Kramer R, Marsden MD, Murray D, Chun TW, Zack JA, Wender PA. Highly potent, synthetically accessible prostratin analogs induce latent HIV expression in vitro and ex vivo. Proc Natl Acad Sci USA. 2013;110:11698–11703. doi: 10.1073/pnas.1302634110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennasser Y, Bahraoui E. HIV-1 Tat protein induces interleukin-10 in human peripheral blood monocytes: involvement of protein kinase C-betaII and -delta. FASEB J. 2002;16:546–554. doi: 10.1096/fj.01-0775com. [DOI] [PubMed] [Google Scholar]
- Biancotto A, Grivel JC, Gondois-Rey F, Bettendroffer L, Vigne R, Brown S, Margolis LB, Hirsch I. Dual role of prostratin in inhibition of infection and reactivation of human immunodeficiency virus from latency in primary blood lymphocytes and lymphoid tissue. J Virol. 2004;78:10507–10515. doi: 10.1128/JVI.78.19.10507-10515.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc Natl Acad Sci USA. 2007;104:13690–13695. doi: 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas P, Poli G, Kinter AL, Justement JS, Stanley SK, Maury WJ, Bressler P, Orenstein JM, Fauci AS. Interferon gamma induces the expression of human immunodeficiency virus in persistently infected promonocytic cells (U1) and redirects the production of virions to intracytoplasmic vacuoles in phorbol myristate acetate-differentiated U1 cells. J Exp Med. 1992;176:739–750. doi: 10.1084/jem.176.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazkova J, Chun TW, Belay BW, Murray D, Justement JS, Funk EK, Nelson A, Hallahan CW, Moir S, Wender PA, Fauci AS. Effect of histone deacetylase inhibitors on HIV production in latently infected, resting CD4(+) T cells from infected individuals receiving effective antiretroviral therapy. J Infect Dis. 2012;206:765–769. doi: 10.1093/infdis/jis412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Hamer DH, Arlen PA, Gao L, Bristol G, Kitchen CM, Berger EA, Zack JA. Molecular characterization, reactivation, and depletion of latent HIV. Immunity. 2003;19:413–423. doi: 10.1016/s1074-7613(03)00236-x. [DOI] [PubMed] [Google Scholar]
- Brown A, Zhang H, Lopez P, Pardo CA, Gartner S. In vitro modeling of the HIV-macrophage reservoir. J Leukoc Biol. 2006;80:1127–1135. doi: 10.1189/jlb.0206126. [DOI] [PubMed] [Google Scholar]
- Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat Med. 2014;20:425–429. doi: 10.1038/nm.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CC, Onafuwa-Nuga A, McNamara LA, Riddell Jt, Bixby D, Savona MR, Collins KL. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med. 2010;16:446–451. doi: 10.1038/nm.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Yang Z, Zhou Q. Phosphorylated positive transcription elongation factor b (P-TEFb) is tagged for inhibition through association with 7SK snRNA. J Biol Chem. 2004;279:4153–4160. doi: 10.1074/jbc.M310044200. [DOI] [PubMed] [Google Scholar]
- Choudhary SK, Archin NM, Cheema M, Dahl NP, Garcia JV, Margolis DM. Latent HIV-1 infection of resting CD4(+) T cells in the humanized Rag2 (−)/(−) gammac(−)/(−) mouse. J Virol. 2012;86:114–120. doi: 10.1128/JVI.05590-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Davey RT, Jr, Engel D, Lane HC, Fauci AS. Re-emergence of HIV after stopping therapy. Nature. 1999;401:874–875. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- Chung YL, Tsai TY. Promyelocytic leukemia nuclear bodies link the DNA damage repair pathway with hepatitis B virus replication: implications for hepatitis B virus exacerbation during chemotherapy and radiotherapy. Mol Cancer Res. 2009;7:1672–1685. doi: 10.1158/1541-7786.MCR-09-0112. [DOI] [PubMed] [Google Scholar]
- Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, Gorry PR. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol. 2009;66:253–258. doi: 10.1002/ana.21697. [DOI] [PubMed] [Google Scholar]
- Clark E, Santiago F, Deng L, Chong S, de La Fuente C, Wang L, Fu P, Stein D, Denny T, Lanka V, Mozafari F, Okamoto T, Kashanchi F. Loss of G (1)/S checkpoint in human immunodeficiency virus type 1-infected cells is associated with a lack of cyclin-dependent kinase inhibitor p21/Waf1. J Virol. 2000;74:5040–5052. doi: 10.1128/jvi.74.11.5040-5052.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JE, Gama L, Graham DR, Mankowski JL, Zink MC. A simian immunodeficiency virus macaque model of highly active antiretroviral treatment: viral latency in the periphery and the central nervous system. Curr Opin HIV AIDS. 2011;6:37–42. doi: 10.1097/COH.0b013e3283412413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collis SJ, Schwaninger JM, Ntambi AJ, Keller TW, Nelson WG, Dillehay LE, Deweese TL. Evasion of early cellular response mechanisms following low level radiation-induced DNA damage. J Biol Chem. 2004;279:49624–49632. doi: 10.1074/jbc.M409600200. [DOI] [PubMed] [Google Scholar]
- Collman R, Balliet JW, Gregory SA, Friedman H, Kolson DL, Nathanson N, Srinivasan A. An infectious molecular clone of an unusual macrophagetropic and highly cytopathic strain of human immunodeficiency virus type 1. J Virol. 1992;66:7517–7521. doi: 10.1128/jvi.66.12.7517-7521.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossarizza A. Apoptosis and HIV infection: about molecules and genes. Curr Pharm Des. 2008;14:237–244. doi: 10.2174/138161208783413293. [DOI] [PubMed] [Google Scholar]
- Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–817. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- DeChristopher BA, Loy BA, Marsden MD, Schrier AJ, Zack JA, Wender PA. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat Chem. 2012;4:705–710. doi: 10.1038/nchem.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, de la Fuente C, Fu P, Wang L, Donnelly R, Wade JD, Lambert P, Li H, Lee CG, Kashanchi F. Acetylation of HIV-1 Tat by CBP/P300 increases transcription of integrated HIV-1 genome and enhances binding to core histones. Virology. 2000;277:278–295. doi: 10.1006/viro.2000.0593. [DOI] [PubMed] [Google Scholar]
- Deng L, Wang D, de la Fuente C, Wang L, Li H, Lee CG, Donnelly R, Wade JD, Lambert P, Kashanchi F. Enhancement of the p300 HAT activity by HIV-1 Tat on chromatin DNA. Virology. 2001;289:312–326. doi: 10.1006/viro.2001.1129. [DOI] [PubMed] [Google Scholar]
- Denton PW, Garcia JV. Humanized mouse models of HIV infection. AIDS Rev. 2011;13:135–148. [PMC free article] [PubMed] [Google Scholar]
- Denton PW, Olesen R, Choudhary SK, Archin NM, Wahl A, Swanson MD, Chateau M, Nochi T, Krisko JF, Spagnuolo RA, Margolis DM, Garcia JV. Generation of HIV latency in humanized BLT mice. J Virol. 2012;86:630–634. doi: 10.1128/JVI.06120-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007;26:424–435. doi: 10.1038/sj.emboj.7601517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duyne RV, Narayanan A, Kehn-Hall K, Saifuddin M, Shultz L, Kashanchi F. Humanized mouse models of HIV-1 latency. Curr HIV Res. 2011;9:595–605. doi: 10.2174/157016211798998781. [DOI] [PubMed] [Google Scholar]
- Easley R, Carpio L, Dannenberg L, Choi S, Alani D, Van Duyne R, Guendel I, Klase Z, Agbottah E, Kehn-Hall K, Kashanchi F. Transcription through the HIV-1 nucleosomes: effects of the PBAF complex in Tat activated transcription. Virology. 2010;405:322–333. doi: 10.1016/j.virol.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilbott DJ, Peress N, Burger H, LaNeve D, Orenstein J, Gendelman HE, Seidman R, Weiser B. Human immunodeficiency virus type 1 in spinal cords of acquired immunodeficiency syndrome patients with myelopathy: expression and replication in macrophages. Proc Natl Acad Sci USA. 1989;86:3337–3341. doi: 10.1073/pnas.86.9.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012;37:377–388. doi: 10.1016/j.immuni.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure E. X-rays-induced secretion of cellular factor(s) that enhance(s) HIV-1 promoter transcription in various non-irradiated transfected cell lines. Cell Mol Biol (Noisy-le-grand) 1998;44:1275–1292. [PubMed] [Google Scholar]
- Faure E, Cavard C, Zider A, Guillet JP, Resbeut M, Champion S. X irradiation-induced transcription from the HIV type 1 long terminal repeat. AIDS Res Hum Retrovir. 1995;11:41–43. doi: 10.1089/aid.1995.11.41. [DOI] [PubMed] [Google Scholar]
- Ferri KF, Jacotot E, Blanco J, Este JA, Zamzami N, Susin SA, Xie Z, Brothers G, Reed JC, Penninger JM, Kroemer G. Apoptosis control in syncytia induced by the HIV type 1-envelope glycoprotein complex: role of mitochondria and caspases. J Exp Med. 2000;192:1081–1092. doi: 10.1084/jem.192.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. Latent infection of CD4 + T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- Folks TM, Justement J, Kinter A, Dinarello CA, Fauci AS. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science. 1987;238:800–802. doi: 10.1126/science.3313729. [DOI] [PubMed] [Google Scholar]
- Furia B, Deng L, Wu K, Baylor S, Kehn K, Li H, Donnelly R, Coleman T, Kashanchi F. Enhancement of nuclear factor-kappa B acetylation by coactivator p300 and HIV-1 Tat proteins. J Biol Chem. 2002;277:4973–4980. doi: 10.1074/jbc.M107848200. [DOI] [PubMed] [Google Scholar]
- Guo MF, Zhao Y, Tian R, Li L, Guo L, Xu F, Liu YM, He YB, Bai S, Wang J. In vivo99mTc-HYNIC-annexin V imaging of early tumor apoptosis in mice after single dose irradiation. J Exp Clin Cancer Res. 2009;28:136. doi: 10.1186/1756-9966-28-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas RL. Low dose radiotherapy in indolent lymphomas, enough is enough. Hematol Oncol. 2009;27:71–81. doi: 10.1002/hon.882. [DOI] [PubMed] [Google Scholar]
- Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haro KJ, Scott AC, Scheinberg DA. Mechanisms of resistance to high and low linear energy transfer radiation in myeloid leukemia cells. Blood. 2012;120:2087–2097. doi: 10.1182/blood-2012-01-404509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrod R, Nacsa J, Van Lint C, Hansen J, Karpova T, McNally J, Franchini G. Human immunodeficiency virus type-1 Tat/co-activator acetyltransferase interactions inhibit p53Lys-320 acetylation and p53-responsive transcription. J Biol Chem. 2003;278:12310–12318. doi: 10.1074/jbc.M211167200. [DOI] [PubMed] [Google Scholar]
- Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J. 2009;423:157–168. doi: 10.1042/BJ20090942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, Margolis DM. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol Cell Biol. 2002;22:2965–2973. doi: 10.1128/MCB.22.9.2965-2973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwahori S, Yasui Y, Kudoh A, Sato Y, Nakayama S, Murata T, Isomura H, Tsurumi T. Identification of phosphorylation sites on transcription factor Sp1 in response to DNA damage and its accumulation at damaged sites. Cell Signal. 2008;20:1795–1803. doi: 10.1016/j.cellsig.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HL, Zamzami N, Costantini P, Druillennec S, Hoebeke J, Briand JP, Irinopoulou T, Daugas E, Susin SA, Cointe D, Xie ZH, Reed JC, Roques BP, Kroemer G. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med. 2000;191:33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SY, Calman AF, Luciw PA, Peterlin BM. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature. 1987;330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- Kassu A, D'Souza M, O'Connor BP, Kelly-McKnight E, Akkina R, Fontenot AP, Palmer BE. Decreased 4-1BB expression on HIV-specific CD4+ T cells is associated with sustained viral replication and reduced IL-2 production. Clin Immunol. 2009;132:234–245. doi: 10.1016/j.clim.2009.03.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauder SE, Bosque A, Lindqvist A, Planelles V, Verdin E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009;5:e1000495. doi: 10.1371/journal.ppat.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SZ, Hand N, Zeichner SL. Apoptosis-induced activation of HIV-1 in latently infected cell lines. Retrovirology. 2015;12:42. doi: 10.1186/s12977-015-0169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Heo K, Choi J, Jackson S, Kim H, Xiong Y, An W. Vpr-binding protein antagonizes p53-mediated transcription via direct interaction with H3 tail. Mol Cell Biol. 2012;32:783–796. doi: 10.1128/MCB.06037-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Kukkonen S, Gupta S, Aldovini A. Association of Tat with promoters of PTEN and PP2A subunits is key to transcriptional activation of apoptotic pathways in HIV-infected CD4+ T cells. PLoS Pathog. 2010;6:e1001103. doi: 10.1371/journal.ppat.1001103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klase Z, Kale P, Winograd R, Gupta MV, Heydarian M, Berro R, McCaffrey T, Kashanchi F. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol Biol. 2007;8:63. doi: 10.1186/1471-2199-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klase Z, Winograd R, Davis J, Carpio L, Hildreth R, Heydarian M, Fu S, McCaffrey T, Meiri E, Ayash-Rashkovsky M, Gilad S, Bentwich Z, Kashanchi F. HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology. 2009;6:18. doi: 10.1186/1742-4690-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama T, Sun B, Tokunaga K, Tatsumi M, Ishizaka Y. DNA damage enhances integration of HIV-1 into macrophages by overcoming integrase inhibition. Retrovirology. 2013;10:21. doi: 10.1186/1742-4690-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Rai PS, Upadhya R, Vishwanatha, Prasada KS, Rao BS, Satyamoorthy K. gamma-radiation induces cellular sensitivity and aberrant methylation in human tumor cell lines. Int J Radiat Biol. 2011;87:1086–1096. doi: 10.3109/09553002.2011.605417. [DOI] [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- Lebel EA, Boukamp P, Tafrov ST. Irradiation with heavy-ion particles changes the cellular distribution of human histone acetyltransferase HAT1. Mol Cell Biochem. 2010;339:271–284. doi: 10.1007/s11010-010-0390-0. [DOI] [PubMed] [Google Scholar]
- Leghmari K, Bennasser Y, Tkaczuk J, Bahraoui E. HIV-1 Tat protein induces IL-10 production by an alternative TNF-alpha-independent pathway in monocytes: role of PKC-delta and p38 MAP kinase. Cell Immunol. 2008a;253:45–53. doi: 10.1016/j.cellimm.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Leghmari K, Contreras X, Moureau C, Bahraoui E. HIV-1 Tat protein induces TNF-alpha and IL-10 production by human macrophages: differential implication of PKC-betaII and -delta isozymes and MAP kinases ERK1/2 and p38. Cell Immunol. 2008b;254:46–55. doi: 10.1016/j.cellimm.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Li CJ, Wang C, Friedman DJ, Pardee AB. Reciprocal modulations between p53 and Tat of human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1995;92:5461–5464. doi: 10.1073/pnas.92.12.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo F, Marchetti MA, Castagnoli L, Battaglia PA, Gigliani F. A novel approach to protein-protein interaction: complex formation between the p53 tumor suppressor and the HIV Tat proteins. Biochem Biophys Res Commun. 1995;206:326–334. doi: 10.1006/bbrc.1995.1045. [DOI] [PubMed] [Google Scholar]
- Marini A, Harper JM, Romerio F. An in vitro system to model the establishment and reactivation of HIV-1 latency. J Immunol. 2008;181:7713–7720. doi: 10.4049/jimmunol.181.11.7713. [DOI] [PubMed] [Google Scholar]
- Marsden MD, Kovochich M, Suree N, Shimizu S, Mehta R, Cortado R, Bristol G, An DS, Zack JA. HIV latency in the humanized BLT mouse. J Virol. 2012;86:339–347. doi: 10.1128/JVI.06366-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Padura I, Agliano A, Marighetti P, Porretti L, Bertolini F. Sex-related efficiency in NSG mouse engraftment. Blood. 2010;116:2616–2617. doi: 10.1182/blood-2010-07-295584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehla R, Bivalkar-Mehla S, Zhang R, Handy I, Albrecht H, Giri S, Nagarkatti P, Nagarkatti M, Chauhan A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS One. 2010;5:e11160. doi: 10.1371/journal.pone.0011160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori E, Takahashi A, Yamakawa N, Kirita T, Ohnishi T. High LET heavy ion radiation induces p53-independent apoptosis. J Radiat Res. 2009;50:37–42. doi: 10.1269/jrr.08075. [DOI] [PubMed] [Google Scholar]
- Narang H, Krishna M. Effect of nitric oxide donor and gamma irradiation on MAPK signaling in murine peritoneal macrophages. J Cell Biochem. 2008;103:576–587. doi: 10.1002/jcb.21429. [DOI] [PubMed] [Google Scholar]
- Narayanan A, Iordanskiy S, Das R, Van Duyne R, Santos S, Jaworski E, Guendel I, Sampey G, Dalby E, Iglesias-Ussel M, Popratiloff A, Hakami R, Kehn-Hall K, Young M, Subra C, Gilbert C, Bailey C, Romerio F, Kashanchi F. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J Biol Chem. 2013;288:20014–20033. doi: 10.1074/jbc.M112.438895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff CP, Ndolo T, Tandon A, Habu Y, Akkina R. Oral pre-exposure prophylaxis by anti-retrovirals raltegravir and maraviroc protects against HIV-1 vaginal transmission in a humanized mouse model. PLoS One. 2010;5:e15257. doi: 10.1371/journal.pone.0015257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Doulatov S, Dick JE. Engraftment of human hematopoietic stem cells is more efficient in female NOD/SCID/IL-2Rgc-null recipients. Blood. 2010;115:3704–3707. doi: 10.1182/blood-2009-10-249326. [DOI] [PubMed] [Google Scholar]
- Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Kobayashi T, Nishioka A, Kariya S, Hamasato S, Seguchi H, Yoshida S. Radiation-induced oxidative DNA damage, 8-oxoguanine, in human peripheral T cells. Int J Mol Med. 2003;11:27–32. [PubMed] [Google Scholar]
- Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, Yasui A, Yokota J, Kohno T. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of nonhomologous end joining factors. Oncogene. 2011;30:2135–2146. doi: 10.1038/onc.2010.592. [DOI] [PubMed] [Google Scholar]
- Ouellet DL, Plante I, Landry P, Barat C, Janelle ME, Flamand L, Tremblay MJ, Provost P. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucl Acids Res. 2008;36:2353–2365. doi: 10.1093/nar/gkn076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet DL, Vigneault-Edwards J, Letourneau K, Gobeil LA, Plante I, Burnett JC, Rossi JJ, Provost P. Regulation of host gene expression by HIV-1 TAR microRNAs. Retrovirology. 2013;10:86. doi: 10.1186/1742-4690-10-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer BE, Neff CP, Lecureux J, Ehler A, Dsouza M, Remling-Mulder L, Korman AJ, Fontenot AP, Akkina R. In vivo blockade of the PD-1 receptor suppresses HIV-1 viral loads and improves CD4+ T cell levels in humanized mice. J Immunol. 2013;190:211–219. doi: 10.4049/jimmunol.1201108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol. 2008;82:12291–12303. doi: 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Stewart D, Li W, Hawkins M, Kulak S, Ballermann B, Jahroudi N. Irradiation modulates association of NF-Y with histone-modifying cofactors PCAF and HDAC. Oncogene. 2007;26:7576–7583. doi: 10.1038/sj.onc.1210565. [DOI] [PubMed] [Google Scholar]
- Perez M, de Vinuesa AG, Sanchez-Duffhues G, Marquez N, Bellido ML, Munoz-Fernandez MA, Moreno S, Castor TP, Calzado MA, Munoz E. Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency. Curr HIV Res. 2010;8:418–429. doi: 10.2174/157016210793499312. [DOI] [PubMed] [Google Scholar]
- Perez VL, Rowe T, Justement JS, Butera ST, June CH, Folks TM. An HIV-1-infected T cell clone defective in IL-2 production and Ca2+ mobilization after CD3 stimulation. J Immunol. 1991;147:3145–3148. [PubMed] [Google Scholar]
- Poli G, Kinter AL, Justement JS, Bressler P, Kehrl JH, Fauci AS. Transforming growth factor beta suppresses human immunodeficiency virus expression and replication in infected cells of the monocyte/macrophage lineage. J Exp Med. 1991;173:589–597. doi: 10.1084/jem.173.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli G, Orenstein JM, Kinter A, Folks TM, Fauci AS. Interferon-alpha but not AZT suppresses HIV expression in chronically infected cell lines. Science. 1989;244:575–577. doi: 10.1126/science.2470148. [DOI] [PubMed] [Google Scholar]
- Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259–270. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quivy V, Adam E, Collette Y, Demonte D, Chariot A, Vanhulle C, Berkhout B, Castellano R, de Launoit Y, Burny A, Piette J, Bours V, Van Lint C. Synergistic activation of human immunodeficiency virus type 1 promoter activity by NF-kappaB and inhibitors of deacetylases: potential perspectives for the development of therapeutic strategies. J Virol. 2002;76:11091–11103. doi: 10.1128/JVI.76.21.11091-11103.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Gramaglia D, Boccaccio C, Comoglio PM. Apoptosis enhancement by the HIV-1 Nef protein. J Immunol. 2001;166:81–88. doi: 10.4049/jimmunol.166.1.81. [DOI] [PubMed] [Google Scholar]
- Richman DD, Margolis DM, Delaney M, Greene WC, Hazuda D, Pomerantz RJ. The challenge of finding a cure for HIV infection. Science. 2009;323:1304–1307. doi: 10.1126/science.1165706. [DOI] [PubMed] [Google Scholar]
- Sastry KJ, Marin MC, Nehete PN, McConnell K, el-Naggar AK, McDonnell TJ. Expression of human immunodeficiency virus type I tat results in down-regulation of bcl-2 and induction of apoptosis in hematopoietic cells. Oncogene. 1996;13:487–493. [PubMed] [Google Scholar]
- Shahbazi J, Lock R, Liu T. Tumor protein 53-induced nuclear protein 1 enhances p53 function and represses tumorigenesis. Front Genet. 2013;4:80. doi: 10.3389/fgene.2013.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, Greiner DL, Handgretinger R. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- Smith RA, Ingels J, Lochemes JJ, Dutkowsky JP, Pifer LL. Gamma irradiation of HIV-1. J Orthop Res. 2001;19:815–819. doi: 10.1016/S0736-0266(01)00018-3. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Poon B, Song JY, Chen IS. Human immunodeficiency virus type 1 vpr induces apoptosis through caspase activation. J Virol. 2000;74:3105–3111. doi: 10.1128/jvi.74.7.3105-3111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terai C, Kornbluth RS, Pauza CD, Richman DD, Carson DA. Apoptosis as a mechanism of cell death in cultured T lymphoblasts acutely infected with HIV-1. J Clin Invest. 1991;87:1710–1715. doi: 10.1172/JCI115188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushin SA, Bren GD, Asin S, Pennington KN, Paya CV, Badley AD. Human immunodeficiency virus reactivation by phorbol esters or T-cell receptor ligation requires both PKCalpha and PKCtheta. J Virol. 2005;79:9821–9830. doi: 10.1128/JVI.79.15.9821-9830.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi M, Pearson RJ, Karn J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol. 2010;84:6425–6437. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne R, Cardenas J, Easley R, Wu W, Kehn-Hall K, Klase Z, Mendez S, Zeng C, Chen H, Saifuddin M, Kashanchi F. Effect of transcription peptide inhibitors on HIV-1 replication. Virology. 2008;376:308–322. doi: 10.1016/j.virol.2008.02.036. [DOI] [PubMed] [Google Scholar]
- Van Duyne R, Guendel I, Narayanan A, Gregg E, Shafagati N, Tyagi M, Easley R, Klase Z, Nekhai S, Kehn-Hall K, Kashanchi F. Varying modulation of HIV-1 LTR activity by Baf complexes. J Mol Biol. 2011;411:581–596. doi: 10.1016/j.jmb.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne R, Pedati C, Guendel I, Carpio L, Kehn-Hall K, Saifuddin M, Kashanchi F. The utilization of humanized mouse models for the study of human retroviral infections. Retrovirology. 2009;6:76. doi: 10.1186/1742-4690-6-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBOJ. 1996;15:1112–1120. [PMC free article] [PubMed] [Google Scholar]
- Wang C, Nakamura S, Oshima M, Mochizuki-Kashio M, Nakajima-Takagi Y, Osawa M, Kusunoki Y, Kyoizumi S, Imai K, Nakachi K, Iwama A. Compromised hematopoiesis and increased DNA damage following non-lethal ionizing radiation of a human hematopoietic system reconstituted in immunodeficient mice. Int J Radiat Biol. 2013;89:132–137. doi: 10.3109/09553002.2013.734947. [DOI] [PubMed] [Google Scholar]
- Wightman F, Solomon A, Khoury G, Green JA, Gray L, Gorry PR, Ho YS, Saksena NK, Hoy J, Crowe SM, Cameron PU, Lewin SR. Both CD31 (+) and CD31(−) naive CD4(+) T cells are persistent HIV type 1-infected reservoirs in individuals receiving antiretroviral therapy. J Infect Dis. 2010;202:1738–1748. doi: 10.1086/656721. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB. Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci USA. 1986;83:7089–7093. doi: 10.1073/pnas.83.18.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Chen LF, Kwon H, Fenard D, Bisgrove D, Verdin E, Greene WC. Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem. 2004;279:42008–42017. doi: 10.1074/jbc.M402124200. [DOI] [PubMed] [Google Scholar]
- Williams SA, Kwon H, Chen LF, Greene WC. Sustained induction of NF-kappa B is required for efficient expression of latent human immunodeficiency virus type 1. J Virol. 2007;81:6043–6056. doi: 10.1128/JVI.02074-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Hu J, Zhao Y, Xiao J, Wang Y, Ma D, Chen Y. PDCD5 interacts with p53 and functions as a positive regulator in the p53 pathway. Apoptosis. 2012;17:1235–1245. doi: 10.1007/s10495-012-0754-x. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Matsuda K, Sagiya Y, Iwadate M, Fujino MA, Nakamura Y, Arakawa H. p53R2-dependent pathway for DNA synthesis in a p53-regulated cell cycle checkpoint. Cancer Res. 2001;61:8256–8262. [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell. 2005;19:535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Yeung ML, Bennasser Y, Watashi K, Le SY, Houzet L, Jeang KT. Pyrosequencing of small non-coding RNAs in HIV-1 infected cells: evidence for the processing of a viral-cellular double-stranded RNA hybrid. Nucl Acids Res. 2009;37:6575–6586. doi: 10.1093/nar/gkp707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Xie YM, Chen IS. Depletion of Wee-1 kinase is necessary for both human immunodeficiency virus type 1 Vpr- and gamma irradiation-induced apoptosis. J Virol. 2003;77:2063–2070. doi: 10.1128/JVI.77.3.2063-2070.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukl SA, Boritz E, Busch M, Bentsen C, Chun TW, Douek D, Eisele E, Haase A, Ho YC, Hutter G, Justement JS, Keating S, Lee TH, Li P, Murray D, Palmer S, Pilcher C, Pillai S, Price RW, Rothenberger M, Schacker T, Siliciano J, Siliciano R, Sinclair E, Strain M, Wong J, Richman D, Deeks SG. Challenges in detecting HIV persistence during potentially curative interventions: a study of the Berlin patient. PLoS Pathog. 2013;9:e1003347. doi: 10.1371/journal.ppat.1003347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Deng L, Kashanchi F, Brady JN, Shatkin AJ, Kumar A. The Tat/TAR-dependent phosphorylation of RNA polymerase II C-terminal domain stimulates cotranscriptional capping of HIV-1 mRNA. Proc Natl Acad Sci USA. 2003;100:12666–12671. doi: 10.1073/pnas.1835726100. [DOI] [PMC free article] [PubMed] [Google Scholar]