

Abstract
Palatal shelf elevation is an essential morphogenetic process during secondary palate closure and failure or delay of palatal shelf elevation is a common cause of cleft palate, one of the most common birth defects in humans. Here, we studied the role of mesenchymal fibroblast growth factor receptor (FGFR) signaling during palate development by conditional inactivation of Fgfrs using a mesenchyme-specific Dermo1-Cre driver. We showed that Fgfr1 is expressed throughout the palatal mesenchyme and Fgfr2 is expressed in the medial aspect of the posterior palatal mesenchyme overlapping with Fgfr1. Mesenchyme-specific disruption of Fgfr1 and Fgfr2 affected palatal shelf elevation and resulted in cleft palate. We further showed that both Fgfr1 and Fgfr2 are expressed in mesenchymal tissues of the mandibular process but display distinct expression patterns. Loss of mesenchymal FGFR signaling reduced mandibular ossification and lower jaw growth resulting in abnormal tongue insertion in the oral-nasal cavity. We propose a model to explain how redundant Fgfr1 and Fgfr2 expression in the palatal and mandibular mesenchyme regulates shelf medial wall protrusion and growth of the mandible to coordinate the craniofacial tissue movements that are required for palatal shelf elevation.
Keywords: FGFR gene expression, Palatal shelf elevation, Cleft palate, Secondary palate development, Mandible development, Conditional gene knockout
Introduction
Cleft palate, as an isolated anomaly, is among the most common craniofacial malformations, affecting one in every 1500–2000 live births worldwide. Cleft palate is also commonly seen in combination with cleft lip and is associated with over 400 known human syndromes (Murray and Schutte, 2004). Non-syndromic cleft palate is a complex trait with genetic and environmental etiologies (Dixon et al., 2011). Cleft palate occurs when the paired palatal shelves fail to join and fuse with each other during embryonic development. After outgrowth from the maxillary processes, the palatal shelves grow vertically on either side of the tongue and undergo an elevation process to become horizontally oriented above the tongue. After elevation, the medial edge epithelium (MEE) of the opposing shelves approximate for contact and adhesion to form a medial epithelial seam (MES). During palatal shelf fusion, degeneration of the MES results in a confluent palatal mesenchyme and formation of an intact secondary palate (Greene and Pratt, 1976; Ferguson, 1988). Phenotypic analysis of cleft palate in mouse models suggests that cleft palate could result from defects in any stage of palate development, i.e. palatal shelf outgrowth, elevation, adhesion, and fusion (Gritli-Linde, 2007; Gritli-Linde, 2008; Bush and Jiang, 2012).
The Fibroblast Growth Factor Receptors (FGFRs) are receptor tyrosine kinases that interact with eighteen signaling Fibroblast Growth Factor (FGF) ligands and heparan sulfate proteoglycan (HSPG) cofactors. The activated FGFR binds to and phosphorylates adaptor proteins for four major intracellular signaling pathways, RAS-MAPK, PI3K-AKT, PLCγ, and signal transducer and activator of transcription (STAT) to initiate intracellular signaling cascades (Ornitz and Itoh, 2015). The structure of the FGFR includes two to three immunoglobulin-like domains and for FGFR1-3, the second half of the third immunoglobulin-like domain undergoes alternative mRNA splicing resulting in IIIb and IIIc splice variants. In general, the IIIc receptor isoforms, such as FGFR2c, are expressed in the mesenchyme and bind epithelially-expressed FGF ligands such as members of the FGF8 subfamily (FGF8, FGF17, and FGF18), while the IIIb isoforms such as FGFR2b are expressed in the epithelium and exclusively bind members of the FGF7 subfamily of ligands (FGF7, FGF10, and FGF22), which are commonly expressed in mesenchymal tissues adjacent to the epithelium.
Abnormal craniofacial development is associated with dominantly expressed FGFR1 and FGFR2 mutations in human craniosynostosis syndromes, such as Apert, Crouzon, Pfeiffer, Muenke, and Jackson-Weiss syndromes. Apert syndrome FGFR2 mutations also cause variable palatal anomalies such as high arched palate, cleft soft palate, or bifid uvula in humans (Wilkie et al., 1995; Slaney et al., 1996; Lajeunie et al., 1999) and lead to severe palate dysmorphologies in mice (Wang et al., 2005; Martinez-Abadias et al., 2013a). Although mice heterozygous for a Crouzon syndrome FGFR2 mutation (Fgfr2C342Y/+ mice) do not show significant palatal anomalies (Martinez-Abadias et al., 2013b), homozygous (Fgfr2C342Y/C342Y) mice have a high incidence of cleft palate (Snyder-Warwick et al., 2010; Snyder-Warwick and Perlyn, 2012). Both syndromic and non-syndromic cleft palate have been linked to FGFR mutations. Cleft palate is associated with Kallmann syndrome type 2 (KAL2), an autosomal dominant developmental disorder characterized by hypogonadotropic hypogonadism due to mutations in FGFR1 (Dode et al., 2003). Mutations in FGFR1, FGFR2, FGFR3, and a ligand, FGF8 have also been identified as the etiology of up to 5% of non-syndromic cleft lip and palate (NS-CLP) cases in humans (Riley et al., 2007; Riley and Murray, 2007).
We previously characterized cleft palate phenotypes in Fgfr2C342Y/C342Y mice (Snyder-Warwick et al., 2010). The C342Y substitution leads to gain-of-function receptor activity that affects mesenchymal FGFR2c without affecting epithelial FGFR2b (Eswarakumar et al., 2004). During palate development, Fgfr2 is expressed in both the palatal epithelium and mesenchyme. While disruption of epithelial-expressed Fgfr2b or conditional disruption of Fgfr2 in the palatal epithelium leads to cleft palate in mice (Rice et al., 2004; Hosokawa et al., 2009), disruption of mesenchymal-expressed Fgfr2c or conditional disruption of Fgfr2 in mesenchymal tissues does not affect palate development (Eswarakumar et al., 2002; Yu et al., 2003). On the other hand, Kallmann syndrome FGFR1 mutations (KAL2) reduce receptor activity (Dode et al., 2003; Pitteloud et al., 2005; Pitteloud et al., 2006; Thurman et al., 2012). As only 25–30% of Kallmann syndrome patients with FGFR1 mutations have cleft palate (Dode and Hardelin, 2009), it suggests that loss of FGFR1 activity could be compensated for by FGFR2, both of which are commonly co-expressed in mesenchymal tissues during development (Orr-Urtreger et al., 1991; Peters et al., 1992). In this study, we examined expression patterns of Fgfr1 and Fgfr2 during murine palate development and used the conditional gene targeting approach to disrupt Fgfr1 and Fgfr2 in mesenchymal tissues. The results indicated that Fgfr1 and Fgfr2 expression partially overlap in the palatal mesenchyme and together maintained mesenchymal FGFR signaling to regulate palatal shelf elevation during palate development. Moreover, we found that both Fgfr1 and Fgfr2 are required for mandibular ossification and growth, which we propose is essential for removal of tongue obstruction before palatal shelf elevation occurs.
Results
Fgfr1 and Fgfr2 are redundantly expressed in the palatal mesenchyme
To determine if FGFR1 and FGFR2 function redundantly during palate development, we examined Fgfr1 and Fgfr2 expression at different stages of palate development along the anterior-posterior axis. During palate shelf outgrowth at E13.5, Fgfr1 was expressed in the entire palatal mesenchyme but not in the palatal epithelium (Figure 1A–D and M). In the posterior regions, Fgfr1 expression was more intense on the medial aspect of the palatal mesenchyme compared to the lateral aspect (Figure 1A and B). In the middle region, Fgfr1 expression was intense on both the medial and lateral aspects of the palatal mesenchyme but was weak in the central region (Figure 1C). In the anterior region, Fgfr1 was strongly expressed in the palatal mesenchyme at the tip but weakly expressed in the other areas of the palatal shelves (Figure 1D). At E13.5, Fgfr2 was expressed in the entire palate epithelium without showing differences between the medial and lateral aspects (Figure 1E–H and O). Fgfr2 was only weakly expressed in the medial aspect of the palatal mesenchyme in the posterior region of the developing palate (Figure 1F and O).
Figure 1.

Fgfr1 and Fgfr2 expression in different regions along the anterior-posterior axis of the secondary palate. (A–D) Fgfr1 expression and (E–H) Fgfr2 expression detected by in situ hybridization (red) on coronal sections of the posterior end, posterior, middle and anterior regions of an E13.5 palate. (I–L) Fgfr1 expression in an E14.5 palate before elevation (V, vertical shelves). Noted that Fgfr2 expression at E14.5 before elevation was shown in a previous study (Snyder-Warwick et al., 2010). (M and N) Magnified images comparing Fgfr1 and Fgfr2 expression in the palatal epithelium and mesenchyme of the posterior region at E13.5. (O and P) Magnified images comparing Fgfr1 and Fgfr2 expression in the palatal epithelium and mesenchyme of the posterior region at E14.5 before elevation. (Q) Fgfr1 expression in the posterior region of an E14.5 palate after elevation (H, horizontal shelves). (R) Fgfr1 expression in the posterior region of an E15 palate after fusion of the palatal shelves. (S) Fgfr2 expression in the posterior region of an E14.5 palate after elevation. (T) Fgfr2 expression in the posterior region of an E15 palate after fusion of the palatal shelves. Arrows in R and T indicate mesenchymal condensation of the palatine bones. n, nasal cavity; o, oral cavity; p, palatal shelf; t, tongue. Scale bars, 200μm.
In mice, palatal shelf elevation occurs in a three-hour period at E14.5 (Walker and Fraser, 1956) and therefore the palatal shelves at E14.5 show a spectrum of morphologies that range from vertically oriented shelves to horizontally oriented and fused shelves (Yu and Ornitz, 2011). In vertically oriented E14.5 (E14.5 V) palatal shelves, Fgfr1 expression showed reduced intensity in the posterior regions (Figure 1I, J and N) and in the anterior region (Figure 1L). In the middle region, Fgfr1 expression became more intense in the lateral aspect of the palatal mesenchyme than that of the medial aspect (Figure 1K). Fgfr2 expression remained in the palatal epithelium and in the medial aspect of the posterior palatal mesenchyme (Figure 1P) (Snyder-Warwick et al., 2010). In horizontally oriented E14.5 (E14.5 H) palatal shelves, Fgfr1 was weakly expressed throughout the entire palatal mesenchyme (Figure 1Q) and Fgfr2 was expressed in the nasal aspect of the palatal mesenchyme (Figure 1S). At E15, when palate shelf fusion occurs, Fgfr1 expression was intensified throughout the confluent palatal mesenchyme (Figure 1R) and Fgfr2 expression remained in the nasal aspect of the confluent palatal mesenchyme (Figure 1T). Interestingly, Fgfr2 expression was found in mesenchymal tissues surrounding the ossifying palatine bones but not in bony tissues at the center of the mesenchymal condensation where Fgfr1 was weakly expressed (arrows in Figure 1R and T).
Conditional disruption of Fgfrs in the palatal mesenchyme leads to cleft secondary palate
We previously reported generation of the Dermo1(Twist2)-Cre driver, which specifically targets mesenchymal tissues during mouse development (Yu et al., 2003). During craniofacial development, Dermo1-Cre activity was present throughout the entire mesenchyme of the first pharyngeal arch at E10.5 (Huh and Ornitz, 2010). By mating Dermo1-Cre mice with double-fluorescent Cre reporter mT/mG mice (Muzumdar et al., 2007), we found that Dermo1-Cre activity was present throughout the maxilla, mandible, and tongue at E13.5 (green fluorescence in Figure 2A), but absent in neural tissues of the brain and the nasal region (red fluorescence in Figure 2A). By mating Dermo1-Cre mice with ROSA26 reporter (ROSA26R) mice (Soriano, 1999), we further examined Dermo1-Cre activity in the developing palate by detecting β-galactosidase activity. At E13.5, β-galactosidase activity was found in the entire palatal mesenchyme but was excluded from the palatal epithelium (Figure 2B). β-galactosidase activity was also found in the Meckel’s cartilages (arrowhead in Figure 2C) and mesenchymal tissues of the developing mandible (arrow in Figure 2C). We previously used the Dermo1-Cre driver to generate Fgfr2 conditional knockout (Fgfr2cko) mice (Table 1), which showed defects in skeletal development but had normal palate development (Yu et al., 2003). Similar results were found in mice that lost mesenchymal expressed Fgfr2c (Eswarakumar et al., 2002).
Figure 2.

Dermo1-Cre activity in the developing craniofacial regions and conditional disruption of Fgfr1 and Fgfr2 using the Dermo1-Cre driver. (A) Fluorescent images showing expression of GFP and dtTomato fluorescent protein in the craniofacial regions of an E13.5 Dermo1-Cre, mT/mG embryo. (B) LacZ staining showing β-galatosidase activity in the palatal mesenchyme of an E13.5 Dermo1-Cre, Rosa26r embryo. Noted the absence of β-galatosidase activity in the palatal epithelium and tooth germ. (C) LacZ staining showing β-galatosidase activity in the Meckel’s cartilages (arrowhead) and the mandibular mesenchyme (arrow) of an E13.5 Dermo1-Cre, Rosa26r embryo. (D and E) Palate and tongue of a P0 wild type pup. (F and G) Cleft palate and normal tongue of a P0 Fgfr1ccko pup. (H and I) Cleft palate and abnormal tongue of a P0 Fgfr1/2dcko pup. (J) H&E stained coronal section of the anterior part of a P0 wild type palate. (K) H&E stained coronal section of the anterior part of a P0 Fgfr1/2dcko palate. Asterisks indicate unfused palatal shelves. mc, Meckel’s cartilages; n, nasal cavity; o, oral cavity; p, palatal shelf; t, tongue; tg, tooth germ. Scale bars in A and J, 50μm and in B, 200μm.
Table 1.
Genotypes and phenotypes of Fgfr1 and/or Fgfr2 conditional knockout mice using the Dermo1-Cre driver.
| Fgfrs conditional knockout (cko) | Fgfr1cko | Fgfr2cko | Compound cko Fgfr1ccko | Compound cko Fgfr2ccko | Double cko Fgfr1/2dcko |
|---|---|---|---|---|---|
| Genotypes | Dermo1Cre/+, Fgfr1Δ/fl | Dermo1Cre/+, Fgfr2Δ/fl | Dermo1Cre/+, Fgfr1Δ/fl, Fgfr2+/fl | Dermo1Cre/+, Fgfr1+/fl, Fgfr2Δ/fl | Dermo1Cre/+, Fgfr1Δ/fl, Fgfr2Δ/fl |
| Cleft palate incidence | 16% (3 of 19) | 0 | 84% (16 of 19) | 0 | 100% (20 of 20) |
| Small mandible | no | no | no | no | yes |
| Tongue anomaly | no | no | no | no | yes |
To further determine the function of FGFR signaling during palate development, we used the Dermo1-Cre driver to generate Fgfr1 conditional knockout (Fgfr1cko) mice and compound Fgfr1 conditional knockout (Fgfr1ccko) mice that also lack one allele of Fgfr2 (Table 1). Fgfr1cko and Fgfr1ccko mice developed complete clefting of the secondary palate when compared with wild type control mice (Figure 2D and F). Of mice that only lost Fgfr1 (Fgfr1cko mice), 16% (3 of 19) developed cleft palate. However, removal of one allele of Fgfr2 greatly increased the incidence of cleft palate to 84% (16 of 19) in Fgfr1ccko mice. In contrast, compound Fgfr2 conditional knockout (Fgfr2ccko) mice that lack one allele of Fgfr1 did not show defects in palate development (Table 1). We further used the Dermo1-Cre driver to generate Fgfr1 and Fgfr2 double conditional knockout (Fgfr1/2dcko) mice (Table 1), all of which developed cleft palate (Figure 2H) and died within several hours after birth (20 of 20). Additionally, the cleft in Fgfr1/2dcko mice was considerably wider that that of compound Fgfr1ccko mice (Figure 2F and H). Histological examination showed that at P0 unfused palatal shelves in Fgfr1/2dcko mice displayed an abnormal shape (asterisks in Figure 2K) and maintained a large gap between the nasal and oral cavity (Figure 2K), which are normally separated by a fused secondary palate in wild type mice (Figure 2J). Because the palate develops in contact with the tongue, examination of tongue morphology also can give insight into palate development. While the tongue showed a flattened shape in P0 wild type and Fgfr1ccko mice (Figure 2E and G), the tongue in P0 Fgfr1/2dcko mice remained inserted between the opposing palatal shelves and displayed an abnormally narrow shape (Figure 2H and I).
FGFR signaling in the palatal mesenchyme is required for palatal shelf elevation
After outgrowth from the maxillary processes at E12, the palatal shelves increase their size through substantial growth in a vertical direction to form a wedge-shaped structure at E13.5. Histological examination indicated that at E13.5, the palatal shelves of Fgfr1/2dcko mice showed a similar size and shape to that of wild type mice in both the anterior (Figure 3A and A′) and posterior (Figure 3B and B′) regions, suggesting that loss of FGFR signaling in the palatal mesenchyme did not significantly affect vertical shelf growth. Since the palatal shelves of all E14.5 Fgfr1/2dcko mice examined were vertically oriented, we selected wild type E14.5 mice with the vertically oriented shelves to compare shelf morphology immediately before elevation. The palatal shelves of Fgfr1/2dcko mice had a narrower and more pointed tip in the anterior region and were smaller in the posterior region when compared with that of wild type mice (arrowheads in Figure 3C, C′, D and D′). Such shelf morphological changes were also observed in E14.5 Fgfr1ccko mice (arrowheads in Figure 3G and I). It should also be noted that at E13.5 and E14.5, prior to elevation, the tongue of Fgfr1/2dcko mice showed similar morphology to that of wild type mice. At E15.5, the palatal shelves were elevated and fused in wild type mice (Figure 3E and F). However, the palatal shelves of Fgfr1/2dcko and Fgfr1ccko mice remained in the vertical position at E15.5 (Figure 3E′, F′, H and J). The tongue of E15.5 Fgfr1/2dcko mice remained inserted between the opposing shelves in the anterior region potentially physically preventing the shelves from moving into the horizontal position (Figure 3E′). In the posterior region, the palatal shelves and tongue of E15.5 Fgfr1/2dcko mice did not show significant morphological changes when compared with those of E14.5 Fgfr1/2dcko mice (Figure 3D′ and F′). Unlike that of Fgfr1/2dcko mice, the tongue of Fgfr1ccko mice has moved out of the nasal cavity at E15.5 (Figure 3H and J). However, palatal shelf elevation in Fgfr1ccko mice either did not occur in the anterior region or proceeded partially in the posterior region, which was not enough to allow the opposing shelves to contact each other (Figure 3H and J). While tongue anomalies in Fgfr1/2dcko mice could result in failure of palatal shelf elevation, shelf morphology in Fgfr1ccko mice suggested that failure of palatal shelf elevation could also result from intrinsic shelf defects due to reduced FGFR signaling in the palatal mesenchyme. To test this possibility, we examined shelf behaviors in an ex vivo condition in which the tongue and lower jaw were removed immediately after specimen dissection. We found that tongue excision did not affect shelf orientation in post-fixed E14.5 wild type mice (Figure 4A), but induced rapid shelf movement in freshly dissected E14.5 wild type mice, which caused the vertical shelves of the anterior palate to approximate each other separated by a narrow gap (Figure 4B). In freshly dissected E14.5 Fgfr1ccko and Fgfr1/2dcko mice, tongue excision did not lead to significant changes in shelf orientation (Figure 4C and D), and the gap between the opposing shelves of the anterior palate was significantly larger than that of wild type mice (Figure 4E). Palatal shelf movement during elevation is thought to be driven by an elevating force that result from accumulation of glycosaminoglycans (GAGs) especially hyaluronic acid (HA) in the palatal mesenchyme (Ferguson, 1988). We therefore used alcian blue staining to examine the level of total GAGs in the anterior palate prior to elevation. However, we did not detect significant changes of mesenchymal GAGs in the palatal shelves of both Fgfr1ccko and Fgfr1/2dcko mice (Figure 4F–H), suggesting that mesenchymal FGFR signaling may regulate factors other than mesenchymal GAGs to promote shelf movement in the anterior palate during elevation.
Figure 3.

Palate development in wild type, Fgfr1ccko and Fgfr1/2dcko mice. (A and B) E13.5, (C and D) E14.5, and (E and F) E15.5 wild type palates shown on H&E stained coronal sections. (A′ and B′) E13.5, (C′ and D′) E14.5, and (E′ and F′) E15.5 Fgfr1/2dcko palates shown on H&E stained coronal sections. (G and I) E14.5 and (H and J) E15.5 Fgfr1ccko palates shown on H&E stained coronal sections. Noted that the palatal shelves start to show abnormal shapes in Fgfr1/2dcko and Fgfr1ccko mice at E14.5 before elevation (arrowheads in C, C′, D, D′, G and I). p, palatal shelf; t, tongue. At least three specimens of each genotype were examined at each stage. Scale bar, 200μm.
Figure 4.

Ex vivo palatal shelf elevation and examination of total GAGs in the palatal shelves. (A) H&E stained coronal section of the anterior part of an E14.5 wild type palate with the tongue and lower jaw removed after fixation. (B) H&E stained coronal section of the anterior part of an E14.5 wild type palate with the tongue and lower jaw immediately removed after embryo dissection. (C) H&E stained coronal section of the anterior part of an E14.5 Fgfr1ccko palate with the tongue and lower jaw immediately removed after embryo dissection. (D) H&E stained coronal section of the anterior part of an E14.5 Fgfr1/2dcko palate with the tongue and lower jaw removed before fixation. Blue dashed lines in A and B show measurement of the gap distance between the opposing shelves without and with immediate ex vivo tongue excision, respectively. (E) Graph showing average gap distance between the opposing shelves of wild type palates with and without immediate ex vivo tongue excision and of Fgfr1ccko and Fgfr1/2dcko palates with immediate ex vivo tongue excision. (F) Alcian blue stained coronal section of the anterior part of an E14.5 wild type palate. (G) Alcian blue stained coronal section of the anterior part of an E14.5 Fgfr1ccko palate. (H) Alcian blue stained coronal section of the anterior part of an E14.5 Fgfr1/2dcko palate. At least three specimens were used in each experimental group.
Mesenchymal FGFR signaling regulates mandibular ossification
The association of cleft palate with tongue anomalies in Fgfr1/2dcko mice resembled the phenotypes of Pierre-Robin sequence (PRS), a congenital condition characterized by an unusually small mandible (micrognathia), posterior displacement or retraction of the tongue (glossoptosis), and cleft palate (Cohen, 1999). We therefore examined lower jaw development in Fgfr1/2dcko mice. Skeletal preparations showed that at E14.5, formation of Meckel’s cartilages in the mandibular process of Fgfr1/2dcko mice was comparable to that of wild type mice (Figure 5A and A′). At E15, Fgfr1/2dcko mice showed a curved Meckel’s cartilages and significant reduction of ossification in the mandibular process when compared with that of wild type mice (Figure 5B and B′). At P0, the mandible of Fgfr1/2dcko mice was shorter and smaller than that of wild type mice (Figure 5D and D′). In contrast, formation of Meckel’s cartilage and ossification in the mandibular process appeared to be normal in Fgfr1ccko mice when compared with that of wild type mice (Figure 5B, C, D and E).
Figure 5.

Mandible development in wild type, Fgfr1ccko and Fgfr1/2dcko mice. (A) E14.5, (B) E15, and (D) P0 wild type mouse heads stained with alizarin red & alcian blue. (A′) E14.5, (B′) E15 and (D′) P0 Fgfr1/2dcko heads stained with alizarin red & alcian blue. (C) E15 and (E) P0 Fgfr1ccko heads stained with alizarin red & alcian blue. Arrows indicate the developing mandible with Mechel’s cartilages stained blue and ossified bones stained red. Noted the same magnification of specimens shown in A–E. At least three specimens of each genotype were examined at each stage.
During mandibular development, bone formation does not use Meckel’s cartilages as a cartilaginous template like that of other endochondral bones but rather mesenchymal cells lateral to Meckel’s cartilages directly differentiate into osteoblasts to form a sheet-like bony structure (arrows in Figure 6A, D and G), which becomes mineralized at E15.5 (arrow in Figure 6I). Both Fgfr1 and Fgfr2 were expressed in the developing mandibular mesenchyme (Figure 6B, C, E and F). At E13.5, Fgfr1 was strongly expressed in the entire mandibular mesenchyme but only weakly in Meckel’s cartilages (asterisk in Figure 6B). Fgfr2 was expressed in the periosteum of the developing mandible and Meckel’s cartilages (asterisk in Figure 6C). At E14.5, Fgfr1 expression was maintained in the bony tissues of the developing mandible (arrow in Figure 6E) but was reduced in other mandibular mesenchymal tissues and no longer expressed in Meckel’s cartilages (asterisk in Figure 6E). Fgfr2 expression was maintained in Meckel’s cartilages (asterisk in Figure 6F) and was intensified in the periosteum of the developing mandible and in the perichondrium of Meckel’s cartilages (arrowheads in Figure 6F). It should be noted that Fgfr2 was not expressed in the bony tissues of the developing mandible (between arrowheads in Figure 6F). At E14.5, Fgfr1 and Fgfr2 expression showed a clear complementary pattern in the developing mandible, of which Fgfr1 was expressed in the more differentiated central area and Fgfr2 was expressed in the less differentiated surrounding periosteum. Histological examination and von Kossa staining indicated that mandibular ossification in Fgfr1/2dcko mice was greatly reduced and did not occur in the middle region (arrowheads in Figure 6G′ and I′). By contrast, mandibular ossification in Fgfr1ccko mice was relatively normal when compared with that of wild type mice (Figure 6G, H, I and J). These results suggested that both Fgfr1 and Fgfr2 are required for maintaining mesenchymal FGFR signaling to regulate osteoblast differentiation and patterning during mandibular development.
Figure 6.

Fgfr1 and Fgfr2 expression in the mandibular mesenchyme and regulation of mandibular ossification by mesenchymal FGFR signaling. (A) H&E stained coronal section of an E13.5 wild type mandible. (B) Fgfr1 expression and (C) Fgfr2 expression detected by in situ hybridization (dark field) on coronal sections of an E13.5 wild type mandible. (D) H&E stained coronal section of an E14.5 wild type mandible. (E) Fgfr1 expression and (F) Fgfr2 expression detected by in situ hybridization (dark field) on coronal sections of an E14.5 wild type mandible. Arrow in E indicates Fgfr1 expression in bony tissues and arrowheads in F indicate Fgfr2 expression in the periosteal tissues. Asterisks in B, C, E and F indicate the position of Meckel’s cartilages. (G and I) E15.5 wild type mandible, (G′ and I′) E15.5 Fgfr1/2dcko mandible, and (H and J) E15.5 Fgfr1ccko mandible shown on H&E stained (G, G′ and H) and von Kossa stained (I, I′ and J) coronal sections. Arrowheads in G′ and I′ indicate the absence of mandibular ossification in Fgfr1/2dcko mice. Arrows in A, C, G and I indicate the developing mandible. mc, Meckel’s cartilages. At least three specimens of each genotype were examined at each stage. Scale bar, 200μm.
Discussion
The importance of Fgfr1 in palate development was first revealed in the study of mutant mice carrying a hypomorphic Fgfr1 allele, which showed multiple craniofacial defects including cleft palate (Trokovic et al., 2003). Conditional disruption of Fgfr1 using a neural crest cell-specific Wnt1-Cre driver resulted in severe midfacial clefting that included cleft lip and palate (Wang et al., 2013). These results suggested that Fgfr1 function is required for development of neural crest cells, from which most craniofacial structures are derived (Chai et al., 2000; Jiang et al., 2002; Yoshida et al., 2008). As Fgfr1/2dcko mice did not show obvious craniofacial anomalies at E13.5, it suggested that the Dermo1-Cre driver would not disrupt the function of FGFR signaling during early craniofacial development, which makes it a suitable tool to study the function of mesenchymal FGFR signaling during palatogenesis. While molecular controls of palatal shelf outgrowth and fusion have been elucidated through genetic studies in mice, molecular events during palatal shelf elevation remain largely undefined (Bush and Jiang, 2012). Our finding that mesenchymal FGFR signaling is indispensable for palatal shelf elevation is a first step towards delineating the molecular mechanism of this critical morphological event during palatogenesis.
How does mesenchymal FGFR signaling regulate palatal shelf elevation?
Past studies suggested that palatal shelf elevation is regulated by an elevating force, which is commonly thought to result from accumulation of GAGs, especially HA, in the palatal mesenchyme (Ferguson, 1988). Hydration of HA-rich extracellular matrix (ECM) increases turgidity within the palatal shelves to result in rapid shelf movement in the anterior palate, which has been referred to as “flipping-up” (Ferguson, 1978; Ferguson, 1988). As we found that shelf movement in the anterior palate was abolished in both Fgfr1ccko and Fgfr1/2dcko mice, it suggested that mesenchymal FGFR signaling may be important for regulating the elevating force. Interestingly, mesenchymal GAGs were not significantly reduced in the palatal shelves of both Fgfr1ccko and Fgfr1/2dcko mice, suggesting that the mesenchymal ECM may not be essential for generating the elevating force in the anterior palate. In a previous study, Brinkley and Vickerman used a pharmacological approach to enhance HA degradation and reduce the level of mesenchymal HA in the palatal shelves (Brinkley and Vickerman, 1982). They found that palatal shelf elevation in the posterior region was delayed during palatal organ culture but was not affected in the anterior region, further suggesting that hydration of HA-rich ECM may not play a predominant role in promoting shelf movement in the anterior palate.
Previous studies suggested that palatal shelf elevation in the anterior region could depend on epithelial cells located on the lateral aspect of the palatal shelves (Bulleit and Zimmerman, 1985). When lateral epithelial cells were surgically removed, palatal shelf elevation in the anterior region was abolished during palatal organ culture, but removal of medial epithelial cells did not affect palatal shelf elevation. In a previous study of limb bud development, we found that epithelial cells can be ablated by disrupting FGFR2b signaling using genetic approaches to result in developmental defects that are comparable with those of using surgical approaches (Yu and Ornitz, 2008). We therefore compared cleft palate phenotypes that resulted from loss of mesenchymal FGFR signaling with those from loss of epithelial FGFR2 signaling during palate development. It appeared that complete loss of epithelial FGFR2 signaling affected palate shelf outgrowth resulting in much smaller palatal shelves in Fgfr2b knockout mice that failed to contact with each other at E15.5 (Rice et al., 2004). Conditional disruption of Fgfr2 using an epithelial-specific keratin 14 (K14)-Cre driver overcame early outgrowth defects in Fgfr2b knockout mice. At E15.5, the palatal shelves of K14-Cre, Fgfr2f/f mice were elevated but failed to contact with each other owing to their small size (Hosokawa et al., 2009). By contrast, the palatal shelves of Fgfr1ccko mice remained in a vertical position at E15.5 in the absence of tongue obstruction (Figure 3H). These results argued against in vitro findings that palatal epithelial cells could influence palatal shelf elevation and suggested that palatal shelf elevation in the anterior region could also rely on mesenchymal factors that remain to be identified but that would be regulated by mesenchymal FGFR signaling, especially that of FGFR1, which was the predominant receptor present in the anterior palatal mesenchyme.
How does mandible growth regulate palatal shelf elevation?
Palatal shelf elevation not only requires the elevating force to direct shelf movement but also requires the tongue to be displaced from the center of the oral-nasal cavity to accommodate shelf movement. It is not known if the elevating force is required for tongue displacement. It has been suggested that the tongue descends or moves downward as the result of lower jaw downward growth or movement (Greene and Pratt, 1976). However, the evidence for coordinated downward movement of the tongue and lower jaw during palatal shelf elevation is inconclusive (Diewert, 1978; Ferguson, 1978). Tongue and lower jaw defects of Fgfr1/2dcko mice suggested that lower jaw growth is essential for tongue displacement during palatal shelf elevation. Meckel’s cartilages form about one day before palatal shelf elevation (Bhaskar, 1953) and retardation in growth of Meckel’s cartilages has been found to cause reduction in mandibular length and result in cleft palate (Seegmiller and Fraser, 1977; Murray et al., 2007). The results of our study suggest that growth of Meckel’s cartilages depends on ossification at the lateral aspect of the mandibular process, which may provide rigidity to the lower jaw and allow Meckel’s cartilages to continually extend longitudinally.
During in vivo elevation, the palatal shelves in the posterior region display an intermediate morphology of medial wall protrusion, of which the palatal mesenchyme in the medial aspect increases in volume to cause medial expansion of the palatal shelves (Yu and Ornitz, 2011). Using morphological analysis, we suggest that shelf medial wall protrusion could coordinate lower jaw growth and tongue morphological changes during palatal shelf elevation (Figure 7). As the posterior half of the tongue shows a descending curvature, anterior growth of the lower jaw could create a space superior to the dorsum of the tongue (arrow and asterisk in Figure 7A). This space would allow shelf medial wall protrusion to occur (asterisk and arrowhead in Figure 7B), which allows the tongue to expand laterally (arrow in Figure 7B) and eventually move out of the nasal cavity (Yu and Ornitz, 2011). Therefore, medial wall protrusion not only leads to shelf movement in the posterior palate during elevation but also is important for removing tongue obstruction by coordinating growth or movement of craniofacial tissues that occurs on different anatomic axes. As we did not observe medial wall protrusion in Fgfr1ccko and Fgfr1/2dcko mice (Figure 3), it is possible that Fgfr1 and Fgfr2 together maintain mesenchymal FGFR signaling to regulate this unique shelf morphological change during elevation. Our model thus explains why disruption of one allele of Fgfr2 in the palatal mesenchyme dramatically increases cleft palate incidence in Fgfr1ccko mice, as reduced mesenchymal FGFR signaling in the medial aspect of the posterior palate (blue-colored area in Figure 7B) could abolish medial wall protrusion and disrupt coordinated craniofacial tissue movements that are required for palatal shelf elevation.
Figure 7.
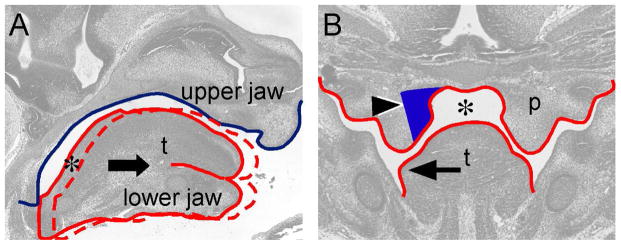
Coordinated craniofacial tissue interactions during palatal shelf elevation. (A) Diagram showing space formation (asterisk) in the posterior palatal region through lower jaw-mediated tongue movement on a sagittal plane. The blue line contours the cranial base and upper jaw. The solid red line contours the tongue and lower jaw in the oral-nasal cavity before palatal shelf elevation. The arrow indicates anterior movement of the lower jaw and tongue during developmental progression. The dashed red line contours the tongue and lower jaw after anterior movement. (B) Diagram showing that space (asterisk) allows coordinated medial wall protrusion (arrowhead) and tongue flattening (arrow) occurred in the posterior palatal region. Noted that medial wall protrusion occurs in the region where both Fgfr1 and Fgfr2 are expressed (blue-colored area). p, palatal shelf; t, tongue.
Experimental procedures
Mice
Mice carrying genetically engineered alleles were maintained on a C57BL/6J x 129X1 mixed genetic background. The following mouse lines were used: Dermo1-Cre (Dermo1Cre/+) (Yu et al., 2003), mT/mG (Muzumdar et al., 2007), ROSA26R (Soriano, 1999), floxed Fgfr1 (Fgfr1f/+) and null Fgfr1 (Fgfr1Δ/+) (Jacob et al., 2006), floxed Fgfr2 (Fgfr2f/+) and null Fgfr2 (Fgfr2Δ/+) (Yu et al., 2003). Fgfr1 conditional knockout (Dermo1Cre/+; Fgfr1Δ/f) mice were generated by mating Dermo1Cre/+; Fgfr1Δ/+ mice with Fgfr1f/f mice. Breeding of Dermo1Cre/+; Fgfr1Δ/+; Fgfr2Δ/+ compound heterozygous mice with Fgfr1f/f; Fgfr2f/f double homozygous mice generated Fgfr1/2 double conditional knockout (Dermo1Cre/+; Fgfr1Δ/f; Fgfr2Δ/f), compound Fgfr1 conditional knockout (Dermo1Cre/+; Fgfr1Δ/f; Fgfr2+/f) and compound Fgfr2 conditional knockout (Dermo1Cre/+; Fgfr1+/f; Fgfr2Δ/f) mice. Mouse skeletons were prepared as described previously (Yu et al., 2003).
Histology, fluorescent imaging, LacZ staining, and in situ hybridization
Embryos were dissected from the uterus in PBS at a designated post-coitum day (or embryonic day). Embryos were decapitated and the heads were fixed in 4% paraformaldehyde overnight at 4°C and embedded in paraffin. Specimens were sectioned coronally (4 μm) and stained with hematoxylin and eosin (H&E). Von Kossa staining was used to detect minerized bone tissues (Yu et al., 2003). Alcian blue staining was used to detect total GAGs in the palatal mesenchyme (Snyder-Warwick et al., 2010). To visualize Cre-mediated recombination in the craniofacial regions of Dermo1-Cre, mT/mG mice, post-fixed heads were embedded in OCT and 100μm-thick coronal tissue slices were used for fluorescent imaging to detect expression of GFP (after Cre-mediated recombination) or dtTomato fluorescent protein (without Cre-mediated recombination). For β-galactosidase histochemistry, embryos were fixed in 0.2% glutaraldehyde in PBS for 60 minutes, washed in PBS and stained in β-gal staining buffer (5 mM K3Fe (CN)6,5 mM K4FE (CN)6*3H2O, 1 mM MgCl2, 0.01% sodium desoxycholate, 0.009% NP40, 0.002% X-Gal) for 8 hours at 4°C. After post-fixing in 4% paraformaldehyde, embryos were embedded in paraffin for sectioning and counterstained with nuclear fast red. In situ hybridization using 33P-UTP-labeled Fgfr1 or Fgfr2 riboprobes was carried out as described previously (Yu et al., 2003).
Ex vivo palatal shelf elevation
The tongue and lower jaw were removed from the heads of freshly dissected E14.5 embryos by making two transverse incisions along the junction of the maxilla and mandible. The dissected heads were placed in PBS for 10–20 minutes before being transferred to 4% PFA for fixation. During the procedure and following incubation, the vertical shelves of wild type mice changed their orientation to become elevated. As a control, the tongue and lower jaw were also removed from the heads of post-fixed E14.5 embryos, which did not lead to shelf orientation changes. The degree of ex vivo elevation was quantified by measuring the gap distance between the opposing palatal shelves on H&E stained histological sections.
Acknowledgments
Grant support information:
NIH grant HD049808 (DMO)
Internal grants from Craniofacial Center of Seattle Children’s Hospital and from Center for Developmental Biology and Regenerative Medicine of Seattle Children’s Research Institute (KY).
We thank Yongjun Yin and Sung-ho Huh for collecting mouse embryos used in this study.
References
- Bhaskar SN. Growth pattern of the rat mandible from 13 days insemination age to 30 days after birth. Am J Anat. 1953;92:1–53. doi: 10.1002/aja.1000920102. [DOI] [PubMed] [Google Scholar]
- Brinkley LL, Vickerman MM. The effects of chlorcyclizine-induced alterations of glycosaminoglycans on mouse palatal shelf elevation in vivo and in vitro. Journal of Embryology and Experimental Morphology. 1982;69:193–213. [PubMed] [Google Scholar]
- Bulleit RF, Zimmerman EF. The influence of the epithelium on palate shelf reorientation. Journal of Embryology and Experimental Morphology. 1985;88:265–279. [PubMed] [Google Scholar]
- Bush JO, Jiang R. Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development. 2012;139:231–243. doi: 10.1242/dev.067082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas P, Jr, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127:1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- Cohen MM., Jr Robin sequences and complexes: causal heterogeneity and pathogenetic/phenotypic variability. Am J Med Genet. 1999;84:311–315. [PubMed] [Google Scholar]
- Diewert VM. A quantitative coronal plane evaluation of craniofacial growth and spatial relations during secondary palate development in the rat. Arch Oral Biol. 1978;23:607–629. doi: 10.1016/0003-9969(78)90186-3. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12:167–178. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dode C, Hardelin JP. Kallmann syndrome. Eur J Hum Genet. 2009;17:139–146. doi: 10.1038/ejhg.2008.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-Van De Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P. A gain-of-function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci U S A. 2004;101:12555–12560. doi: 10.1073/pnas.0405031101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarakumar VP, Monsonego-Ornan E, Pines M, Antonopoulou I, Morriss-Kay GM, Lonai P. The IIIc alternative of Fgfr2 is a positive regulator of bone formation. Development. 2002;129:3783–3793. doi: 10.1242/dev.129.16.3783. [DOI] [PubMed] [Google Scholar]
- Ferguson MW. Palatal shelf elevation in the Wistar rat fetus. Journal of Anatomy. 1978;125:555–577. [PMC free article] [PubMed] [Google Scholar]
- Ferguson MW. Palate development. Development. 1988;103(Suppl):41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- Greene RM, Pratt RM. Developmental aspects of secondary palate formation. Journal of Embryology and Experimental Morphology. 1976;36:225–245. [PubMed] [Google Scholar]
- Gritli-Linde A. Molecular control of secondary palate development. Dev Biol. 2007;301:309–326. doi: 10.1016/j.ydbio.2006.07.042. [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A. The etiopathogenesis of cleft lip and cleft palate: usefulness and caveats of mouse models. Current Topics in Developmental Biology. 2008;84:37–138. doi: 10.1016/S0070-2153(08)00602-9. [DOI] [PubMed] [Google Scholar]
- Hosokawa R, Deng X, Takamori K, Xu X, Urata M, Bringas P, Jr, Chai Y. Epithelial-specific requirement of FGFR2 signaling during tooth and palate development. J Exp Zool B Mol Dev Evol. 2009;312B:343–350. doi: 10.1002/jez.b.21274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh SH, Ornitz DM. Beta-catenin deficiency causes DiGeorge syndrome-like phenotypes through regulation of Tbx1. Development. 2010;137:1137–1147. doi: 10.1242/dev.045534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob AL, Smith C, Partanen J, Ornitz DM. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev Biol. 2006;296:315–328. doi: 10.1016/j.ydbio.2006.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–116. doi: 10.1006/dbio.2001.0487. [DOI] [PubMed] [Google Scholar]
- Lajeunie E, Cameron R, El Ghouzzi V, de Parseval N, Journeau P, Gonzales M, Delezoide AL, Bonaventure J, Le Merrer M, Renier D. Clinical variability in patients with Apert’s syndrome. J Neurosurg. 1999;90:443–447. doi: 10.3171/jns.1999.90.3.0443. [DOI] [PubMed] [Google Scholar]
- Martinez-Abadias N, Holmes G, Pankratz T, Wang Y, Zhou X, Jabs EW, Richtsmeier JT. From shape to cells: mouse models reveal mechanisms altering palate development in Apert syndrome. Dis Model Mech. 2013a;6:768–779. doi: 10.1242/dmm.010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Abadias N, Motch SM, Pankratz TL, Wang Y, Aldridge K, Jabs EW, Richtsmeier JT. Tissue-specific responses to aberrant FGF signaling in complex head phenotypes. Dev Dyn. 2013b;242:80–94. doi: 10.1002/dvdy.23903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JC, Schutte BC. Cleft palate: players, pathways, and pursuits. Journal of Clinical Investigation. 2004;113:1676–1678. doi: 10.1172/JCI22154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray SA, Oram KF, Gridley T. Multiple functions of Snail family genes during palate development in mice. Development. 2007;134:1789–1797. doi: 10.1242/dev.02837. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015;4:215–266. doi: 10.1002/wdev.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P. Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development. 1991;113:1419–1434. doi: 10.1242/dev.113.4.1419. [DOI] [PubMed] [Google Scholar]
- Peters KG, Werner S, Chen G, Williams LT. Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development. 1992;114:233–243. doi: 10.1242/dev.114.1.233. [DOI] [PubMed] [Google Scholar]
- Pitteloud N, Acierno JS, Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, Yialamas M, Hall JE, Grant E, Mohammadi M, Crowley WF., Jr Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A. 2006;103:6281–6286. doi: 10.1073/pnas.0600962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitteloud N, Acierno JS, Jr, Meysing AU, Dwyer AA, Hayes FJ, Crowley WF., Jr Reversible kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J Clin Endocrinol Metab. 2005;90:1317–1322. doi: 10.1210/jc.2004-1361. [DOI] [PubMed] [Google Scholar]
- Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113:1692–1700. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BM, Mansilla MA, Ma J, Daack-Hirsch S, Maher BS, Raffensperger LM, Russo ET, Vieira AR, Dode C, Mohammadi M, Marazita ML, Murray JC. Impaired FGF signaling contributes to cleft lip and palate. Proc Natl Acad Sci U S A. 2007;104:4512–4517. doi: 10.1073/pnas.0607956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BM, Murray JC. Sequence evaluation of FGF and FGFR gene conserved non-coding elements in non-syndromic cleft lip and palate cases. Am J Med Genet A. 2007;143A:3228–3234. doi: 10.1002/ajmg.a.31965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seegmiller RE, Fraser FC. Mandibular growth retardation as a cause of cleft palate in mice homozygous for the chondrodysplasia gene. J Embryol Exp Morphol. 1977;38:227–238. [PubMed] [Google Scholar]
- Slaney SF, Oldridge M, Hurst JA, Moriss-Kay GM, Hall CM, Poole MD, Wilkie AO. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. 1996;58:923–932. [PMC free article] [PubMed] [Google Scholar]
- Snyder-Warwick AK, Perlyn CA. Coordinated events: FGF signaling and other related pathways in palatogenesis. J Craniofac Surg. 2012;23:397–400. doi: 10.1097/SCS.0b013e318240fed7. [DOI] [PubMed] [Google Scholar]
- Snyder-Warwick AK, Perlyn CA, Pan J, Yu K, Zhang L, Ornitz DM. Analysis of a gain-of-function FGFR2 Crouzon mutation provides evidence of loss of function activity in the etiology of cleft palate. Proc Natl Acad Sci U S A. 2010;107:2515–2520. doi: 10.1073/pnas.0913985107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Thurman RD, Kathir KM, Rajalingam D, Kumar TK. Molecular basis for the Kallmann syndrome-linked fibroblast growth factor receptor mutation. Biochem Biophys Res Commun. 2012;425:673–678. doi: 10.1016/j.bbrc.2012.07.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trokovic N, Trokovic R, Mai P, Partanen J. Fgfr1 regulates patterning of the pharyngeal region. Genes Dev. 2003;17:141–153. doi: 10.1101/gad.250703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BE, Fraser FC. Closure of the Secondary Palate in Three Strains of Mice. J Embryol Exp Morphol. 1956;4:176–189. [Google Scholar]
- Wang C, Chang JY, Yang C, Huang Y, Liu J, You P, McKeehan WL, Wang F, Li X. Type 1 fibroblast growth factor receptor in cranial neural crest cell-derived mesenchyme is required for palatogenesis. J Biol Chem. 2013;288:22174–22183. doi: 10.1074/jbc.M113.463620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xiao R, Yang F, Karim BO, Iacovelli AJ, Cai J, Lerner CP, Richtsmeier JT, Leszl JM, Hill CA, Yu K, Ornitz DM, Elisseeff J, Huso DL, Jabs EW. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2+/S252W mouse. Development. 2005;132:3537–3548. doi: 10.1242/dev.01914. [DOI] [PubMed] [Google Scholar]
- Wilkie AOM, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD, Hayward RD, David DJ, Pulleyn LJ, Rutland P, Malcolm S, Winter RM, Reardon W. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Vivatbutsiri P, Morriss-Kay G, Saga Y, Iseki S. Cell lineage in mammalian craniofacial mesenchyme. Mech Dev. 2008 doi: 10.1016/j.mod.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Yu K, Ornitz DM. FGF signaling regulates mesenchymal differentiation and skeletal patterning along the limb bud proximodistal axis. Development. 2008;135:483–491. doi: 10.1242/dev.013268. [DOI] [PubMed] [Google Scholar]
- Yu K, Ornitz DM. Histomorphological study of palatal shelf elevation during murine secondary palate formation. Dev Dyn. 2011;240:1737–1744. doi: 10.1002/dvdy.22670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]