

Abstract
Upon apoptotic stimuli, epithelial cells compensate the gaps left by dead cells by activating proliferation. This has led to the proposal that dying cells signal to surrounding living cells to maintain homeostasis. Although the nature of these signals is not clear, reactive oxygen species (ROS) could act as a signaling mechanism as they can trigger pro-inflammatory responses to protect epithelia from environmental insults. Whether ROS emerge from dead cells and what is the genetic response triggered by ROS is pivotal to understand regeneration of Drosophila imaginal discs. We genetically induced cell death in wing imaginal discs, monitored the production of ROS and analyzed the signals required for repair. We found that cell death generates a burst of ROS that propagate to the nearby surviving cells. Propagated ROS activate p38 and induce tolerable levels of JNK. The activation of JNK and p38 results in the expression of the cytokines Unpaired (Upd), which triggers the JAK/STAT signaling pathway required for regeneration. Our findings demonstrate that this ROS/JNK/p38/Upd stress responsive module restores tissue homeostasis. This module is not only activated after cell death induction but also after physical damage and reveals one of the earliest responses for imaginal disc regeneration.
Author Summary
Regenerative biology pursues to unveil the genetic networks triggered by tissue damage. Regeneration can occur after damage by cell death or by injury. We used the imaginal disc of Drosophila in which we genetically activated apoptosis or physically removed some parts and monitored the capacity to repair the damage. We found that dying cells generate a burst of reactive oxygen species (ROS) necessary to activate JNK and p38 signaling pathways in the surrounding living cells. The action of these pathways is necessary for the activation of the cytokines Unpaired (Upd). Eventually, Upd will turn on the JAK/STAT signaling pathway to induce regenerative growth. Thus, we present here a module of signals that depends on oxidative stress and that, through the p38-JNK interplay, will activate cytokine-dependent regeneration.
Introduction
Tissues and organs need to function reliably regardless of adverse environmental conditions. Injuries, disease, infection and environmental insults are stressors causing cell damage that can be repaired via homeostatic machinery. Thus, optimal health is largely dependent upon tissue homeostasis, which involves cell replacement and tissue repair. Although many signaling pathways have been proposed to respond to environmental insults, the early activation of those signals is poorly understood.
Response to damage can involve oxidative stress and, subsequently, the stimulation of stress-activated protein kinases. The production of reactive oxygen species (ROS) by various redox metabolic reactions, which has generally been considered to be deleterious, is now emerging as an active participant in cell signaling events [1,2]. ROS are byproducts of aerobic metabolism that include superoxide O2 -, peroxide H2O2 and hydroxyl radicals OH·. ROS, and in particular H2O2 are required for inflammatory cell recruitment [3,4]. Amphibian and zebrafish injuries produce the ROS necessary to promote proliferation and regeneration [5–8]. In mammalian cells, ROS are known to act as second messengers to activate diverse redox-sensitive signaling transduction cascades, including the stress-activated MAP kinases p38 and the Jun-N Terminal kinase (JNK) [9–11]. ROS-mediated p38 activation occurs during the inflammatory response in rats [12] and during the loss of self-renewal and differentiation in glioma-initiating cells [13]. It has also been found that p38 and JNK are differentially required during repair. In endothelial cells, TNF-α stimulates repair through the positive action of JNK and negative regulation of p38 [14], whereas in corneal repair, p38, and not JNK, is required for epithelial migration [15]. In Drosophila both MAPK have been associated with stress responses [16]. Drosophila p38 pathway responds to different environmental stimuli and stressors [17,18]. Moreover, increasing ROS beyond basal level triggers precocious differentiation of Drosophila hematopoietic progenitors through JNK signaling [19].
The JNK signaling pathway has emerged as an early response to cell death and physical damage and appears to play a critical role in compensatory proliferation, regeneration and wound healing [20–28]. Moreover, upon apoptotic stimulus p53 and JNK are activated by the caspase Dronc and function upstream of pro-apoptotic genes, creating an amplifying loop that ensures cell death [29–33]. One of the early known responses to cell death is the transcriptional activation of the phosphatase puckered (puc), a downstream effector of the JNK pathway and a powerful negative regulator of the same pathway. Interestingly puc has been found in surviving cells of nearby tissue after cell death [23,27] and after physical injury [22,34]. JNK activation of the cytokines unpaired (upd), a family of cytokines linked to the human interleukin-6, is necessary for hyperproliferation in Drosophila tumors and for wound healing [34–36]. Thus, we hypothesize here that the activation of JNK, which is amplified in dying cells, is in some way propagated to nearby surviving tissue where beneficial low levels of JNK promote upd expression.
Apoptotic cells have been observed in the early regeneration of different animals and are thought to provide signals that regulate wound healing and regeneration [37–39]. As apoptosis has been associated with oxidative stress and cytokines act as a functional link between oxidative stress and compensatory proliferation in mammals [40], we decided to investigate whether ROS occur upstream from the stress-activated protein kinases p38 and JNK and cytokines during tissue repair. We took advantage of the regeneration capacity of Drosophila imaginal disc epithelium (reviewed in [41,42]) to address these questions. Imaginal discs are larval epithelial sacs that possess a robust ability for homeostatic cell renewal to overcome the effect of stressors. We report here that, either by inflicting a physical lesion or after inducing cell death, imaginal disc cells produce ROS that are linked to the activation of p38 and JNK stress MAP kinases. In addition, JNK and p38 activity in the living tissue triggers transcription of the cytokine unpaired (upd), which acts as a ligand of the JAK/STAT signaling pathway and promotes regeneration of the missing part.
Results
ROS are produced after tissue damage
To monitor ROS after tissue damage we used CellROX Green, a cell-permeant fluorogenic probe that is non-fluorescent in the reduced state and exhibits bright fluorescence upon oxidation. We found high levels of CellROX Green near the wound edges of physically cut wing imaginal discs. Only a few of the CellROX Green positive cells were TO-PRO-3 positive cells (dying cells), indicating that most ROS-producing cells were alive (Fig 1A). We examined the production and propagation of ROS over time. Few minutes after cut (0–5’) some cells at the wound edges were CellROX Green positive, indicating that the oxidative burst is rapidly occurring after damage (Figs 1B and 1C and S1). Ex vivo imaging showed that fluorescence propagates to the neighboring cells during the first 30’ after damage (Figs 1B and S1).
Fig 1. ROS produced after physical injury and after cell death.

(A) Cut disc cultured ex vivo (white wedge indicates cut edges) and thermal LUT of CellROX Green. (B) Cut disc cultured ex vivo, imaged at just after cut (0–5’) and 30’ later. Thermal scale indicates pixel intensity. (C) Sketch of wing imaginal discs with the area (black square) shown in A, B and E. (D) Fixed disc stained for nuclei to show disc contour (TP-3: TO-PRO-3) and caspase-3 after ptc>rpr activation for 11h at 29°C. (E) ptc>rpr disc cultured ex vivo; basal images at the bottom, apical at the top. Left, cell death (TO-PRO-3). Right, thermal LUT taken from the ROS channel (CellROX Green) of the same preparation. Note that most dead cells (TO-PRO-3 positive) show high ROS (red in thermal image) whereas living cells (TO-PRO-3 negative) had low ROS (green-cyan in thermal image). (F) Mean pixel intensity (grey value) of the indicated zones in control discs without cell death (ptc>rpr OFF) and discs with cell death (ptc>rpr ON). The pixel intensity in the ptc domain in the absence of cell death (ptc>rpr OFF) was 1.76 ± 0.55 (SD; from 48 regions of interest [ROI] in n = 5 discs). The mean pixel intensity for the apoptotic region (basal; ptc>rpr ON) was 33.14±8.18 (SD), measured in 27 ROI on confocal images taken from n = 6 discs. Living cells adjacent to the apoptotic zone showed a mean grey value of 8.51±2.12 (SD; 15 ROI from 6 discs taken from cells near the ptc domain). White rectangles in E: example ROI for Basal Apoptotic Zone (1) and Apical Living Zone (2). The ROI’s for the ptc Zone, in discs in which ptc>rpr is OFF, were placed as (1). ***P<0.001. Thermal scale indicates sample values from raw images.
We next monitored ROS production after controlled induction of cell death (also known as genetic ablation), which can be used as a type of insult to study cellular responses. Apoptosis was induced using patched (ptc)-Gal4 to drive expression of the pro-apoptotic gene reaper (rpr) under the control of a UAS (henceforth ptc>rpr); the Gal4/UAS system was controlled by the temperature-sensitive form of Gal80 (Gal80 TS), which inhibits Gal4 and enables examination of regeneration after cell death [23,24]. As previously described, ptc>rpr discs show a stripe of apoptotic cells that eventually extrudes basally and is replaced apically by living cells (Fig 1D) [23]. CellROX Green was strongly incorporated into the ptc>rpr apoptotic cells (TO-PRO-3 positive) (Fig 1E). Strikingly, living cells adjacent to the apoptotic zone also showed ROS, albeit at much lower levels than in dead cells (Fig 1E and 1F). Similar observations were obtained using 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA) which upon oxidation is converted to the highly fluorescent DCF. Indeed, cut or rpr-ablated discs, showed high levels of fluorescence on the wound edges, in the apoptotic cells and also in the living cells near the apoptotic (S1B, S1C and S1D Fig).
Thus, these results showed that both physical injury and genetically induced apoptosis are insults that result in the production of ROS.
ROS are required for tissue repair
Oxidative burst following death or damage could propagate from dying to living cells in sub-toxic doses and initiate repair. To explore this issue, we decided to deplete ROS production and examine adult wings after cell death. We first checked whether antioxidants (vitamin C, Trolox or N-acetyl cysteine [NAC]) are capable of blocking ROS production. We incubated cut discs in Schneider’s medium containing antioxidants, and found strong reduction of CellROX Green fluorescence (S2 Fig).
Next, we studied the effects of ROS scavenging on regeneration. We used a Gal4 construct under the control of a wing-specific enhancer (sal E/Pv -Gal4), which allows analysis in adult wings while not affecting the rest of the organism, to activate UAS-rpr (henceforth sal E/Pv >rpr). To deplete intracellular ROS, sal E/Pv >rpr larvae were fed with food supplemented with antioxidants (Fig 2A). ROS scavengers in sal E/Pv >rpr controls kept at 17°C to prevent cell death did not show any alteration of wing morphology (S2B Fig). Conversely, a sal E/Pv >rpr control group without scavengers moved to 29°C for 11 h showed complete wing regeneration (Fig 2B). However, the sal E/Pv >rpr experimental group with ROS scavengers and induced cell death showed incomplete regeneration in about 50% of the cases (Fig 2B and 2C). We considered incomplete regeneration when some veins or intervein sectors were missing. To discard that these effects could be caused by differences in survival or developmental delay, we checked whether proliferation is impaired after ROS depletion. We counted the number of mitoses after cell death induction in discs from NAC-fed larvae and found a significant decrease compared to discs from larvae fed in the absence of antioxidants (Fig 2D). The number of mitoses in controls fed with or without antioxidants and kept at 17°C to block cell death did not vary (Fig 2D).
Fig 2. ROS are required for tissue repair.

(A) Design for chemical antioxidant intake and cell death induction. At 17°C and 24 h before cell death induction, larvae were transferred to a vial with standard food supplemented with antioxidant. Cell death (sal E/Pv >rpr ON) was induced by shifting temperature to 29°C for 11 h (blue stripes in the disc). Larvae were transferred to 17°C where they regenerated and emerged into adults, in which wings were scored. Blue color in the wing: area emerged from sal E/Pv. Controls sal E/Pv >rpr OFF were kept at 17°C to avoid cell death. (B) Percentage of regenerated wings after cell death (sal E/Pv >rpr ON) in the absence of antioxidant (Std Food), or in the presence of antioxidants (NAC, Trolox or Vitamin C). (C) Examples of sal E/Pv >rpr ON wings with the indicated food supplement. In controls without antioxidants (Std Food), the complete wing recovered. For each antioxidant an example of incomplete regeneration after cell death induction is shown. (D) Mitosis number in ptc>rpr discs from larvae fed with and without NAC and with or without rpr-ablation (ON versus OFF). Ptc>rpr OFF: 41.86 ± 9.84 (S.D.); NAC ptc>rpr OFF: 39.9 ± 4.68 (S.D.); ptc>rpr ON: 49.73 ± 8.18 (S.D.); NAC ptc>rpr ON: 29.52 ± 9.41 (S.D.) (E) Design for ectopic expression of enzymatic antioxidant transgenes and simultaneous cell death induction when shifted to 29°C for 11 h. The Gal4/UAS (red) activate Cat, Sod or Cat+Sod transgenes. Blue striped area: sal E/Pv -LHG lexO-rpr. Adult wings were scored for complete regeneration of the missing zone. Red coloration indicates zone influenced by the enzymatic antioxidant; purple: zone influenced by enzymatic antioxidant and cell death. sal E/Pv -LHG and nub-Gal4 are under the control of tubGal80 TS. (F) Percentage of regenerated wings in Cat, Sod or Cat and Sod ectopically expressed transgenes. (G) Wings from individuals after cell death and transgene activation (ON). For Cat, Sod or Sod:Cat and example of incomplete regeneration is shown. TP-3: TO-PRO-3. ***P<0.001
In addition, we used enzymatic manipulation of ROS. Superoxide dismutase (Sod) catalyzes the dismutation of superoxide anion into oxygen and hydrogen peroxide. In the presence of hydrogen peroxide, Catalase (Cat) catalyzes its breakdown into water and oxygen. Thus, overexpression of Sod or Cat will remove their respective ROS substrates, whereas simultaneous activation of Sod and Cat will enhance the depletion of both O2 - and H2O2. UAS-Sod, UAS-Cat or UAS-Sod:UAS-Cat were ectopically expressed under the nub-Gal4 driver, which operates throughout the wing pouch (Fig 2E). To induce cell death, we used an independent transactivator based on the LexA/lexO binary system. We generated a sal E/Pv -LHG transgene, which includes a Gal80 suppressible form of LexA [43], to conditionally express lexO-rpr in the sal E/Pv domain. This combination permits control of the temporary expression of two binary systems (sal E/Pv -LHG lexO-rpr and nub-Gal4 UAS-transgene) by tubGal80 TS (Fig 2E). This design has the advantage of simultaneously activating two wing-specific transgenes (nub-Gal4 and sal E/Pv -LHG) in overlapping domains, therefore hindering early ROS. After sal E/Pv -LHG lexO-rpr genetic ablation and nub-Gal4 UAS-transgene expression we allowed the larvae to develop to adulthood and found a drop in the number of regenerated wings (Figs 2F and 2G and S2C and S2D). Together, these results indicate that chemical and enzymatic ROS scavengers interfere with regeneration.
ROS control JNK activity
To determine whether ROS act on JNK during wing disc repair, we first monitored the activity of this pathway in wing discs after cell death. We used two different reporters to monitor JNK activity: puc-lacZ, which marks puc-expressing cells [44], and the TRE-DsRed.T4 reporter, which monitors the JNK substrate AP1 transcription factor (hereafter TRE-red reporter) (Fig 3A and S3A Fig) [45]. In ptc>rpr discs, we found high levels of TRE-red reporter in the basal apoptotic zone and, to a lesser extent, in the apical living cells (Fig 3A and 3B). In contrast, puc-lacZ positive cells were found in the apical zone, as described previously [23], and rarely in the apoptotic zone. Some puc positive cells incorporated EdU, supporting that JNK is also induced in living cells (Fig 3C).
Fig 3. ROS control JNK activity.

(A) Test of JNK reporters. All images in A correspond to the same disc after ptc>rpr induction. Top row: apical sections. Bottom row: basal sections. Note that puc is more abundant in apical than basal sections, particularly in the notum (n; arrowhead) and wing pouch (wp; arrow). Cell death (TUNEL) and high TRE-red are more abundant in basal sections. (B) Zoom of a digital cross section of the zone marked with a white line in A. Endogenous puc-lacZ is found in the outer layer of peripodial membrane cells (pm). Puc-lacZ cells in the disc columnar epithelium are apical (white), most apoptotic cells are basal (red), and TRE-red positive cells are apical and basal (blue). (C) Three digital cross section in an apical puc-lacZ zone of the wing pouch (wp) and notum (n1, n2). Each example contains three to four cells with co-localization of ß-galactosidase and EdU. (D) TRE-red reporter in ptc>rpr discs of larvae fed with standard food or NAC-supplemented food (NAC). TP-3: TO-PRO-3. (E) Mean pixel intensities of TRE-red reporter in ptc>rpr discs with standard or NAC food. The pixel intensity for standard food was 26.06 ± 7.22 (S.D.; n = 15) and for NAC 18.12 ± 8.32 (S.D.; n = 25). (F) TRE-red reporter expression in physically injured discs, cultured for 7 h ex vivo in Schneider’s culture medium with or without NAC. Outline: disc contour. Wedges: cut. (G) Mean pixel intensities of TRE-red reporter in ex vivo cultured discs with or without NAC. The pixel intensity for standard culture was 88.98 ± 22.25 (S.D.; n = 6) and for NAC 23.98 ± 10.26 (S.D.; n = 16). **P<0.01, ***P<0.001.
As NAC is an excellent source of sulfhydryl SH- groups and efficiently promotes scavenging of free radicals [46], it was the most suitable antioxidant to determine the relationship between ROS and JNK in stressed imaginal discs. To test NAC effects on JNK, we used the TRE-red reporter because is more rapidly and extensively expressed than puc-lacZ (Figs 3A and S3B) and because its activity is blocked in JNK mutants [45] or after chemical JNK inhibitors (S3C Fig). We found that the mean pixel intensity of the TRE-red reporter in ptc>rpr wing discs from animals grown in NAC-supplemented food was lower than in the same zone of individuals fed with standard food (Fig 3D and 3E). Moreover, discs cultured ex vivo in which NAC was added into the medium resulted in a drop of TRE-red activity after physical injury (Fig 3F and 3G). These observations indicate that activation of JNK is ROS dependent.
ROS stimulates p38 phosphorylation
Another potential response to ROS increase is the activation of the p38 signaling cascade [10,17,18]. Active p-38 signaling can be monitored using anti-phosphorylated p38 (P-p38). We found that discs fixed and incubated with anti-P-p38 a few minutes after physical injury showed P-p38 staining around the wound. P-p38 localization was variable and depended on the severity of the injury. In contrast, intact discs immediately stained after fixing did not show P-p38 (Fig 4A). However, discs cultured for 3 to 8 hours with or without injury showed P-p38 staining throughout the disc. This general staining is likely due to the stress generated by culturing, and contrasts with the fast local P-p38 response around the damaged zone. We next wondered whether the boost in ROS that propagates to the surviving tissue triggers p38 activation. We observed that the early P-p38 staining was blocked in discs cut and cultured ex vivo in medium containing NAC (Fig 4B and 4C).
Fig 4. ROS stimulate p38 phosphorylation.

(A) P-p38 staining of intact (uncut, controls) and cut discs cultured for the indicated times after injury. White lines: wound edges; white arrowhead: small incision. (B) Discs cultured with or without NAC, cut and stained for P-p38. (C) Mean pixel intensities of P-p38 fluorescence from cut discs cultured with standard medium (95.29 ± 17.52; S.D.) or NAC-supplemented (22.45±2.56; S.D.). (D) Apical and basal images of P-p38 after ptc>rpr induction. (E) Apical and basal images of ptc>rpr after NAC supplementation showing reduction of P-p38 localization. (F) Mean pixel intensities of P-p38 fluorescent labeling from ptc>rpr discs fed with standard (52.17±19.96; S.D.) or NAC-supplemented food (7,85 ± 2,42; S.D.). (G) Genetic ROS scavenging using ci>Sod:Cat, activated in the anterior compartment (ci, black in the sketch). Sal EPv >rpr cell death (blue in the sketch) in the same disc results in inhibition of P-p38 only in anterior compartment. TP-3: TO-PRO-3 nuclei staining. Outlined white in D, E and G: apoptotic zone. ***P<0.001.
We also analyzed p38 activation after inducing cell death and found P-p38 only in living cells but never in the basal apoptotic zone (Fig 4D). In the absence of cell death, no P-p38 was detected. Blocking of ROS production with NAC resulted in a significant drop in P-p38-labeled cells (Fig 4D and 4E). In addition, we used the double transcriptional trans-activator system consisting of the sal E/Pv -LHG lexO-rpr to induce apoptosis and simultaneously interfere with ROS production by inducing UAS-Sod:UAS-Cat in the anterior (ci-Gal4) compartment (Fig 4G). The results showed a strong reduction of P-p38 in the anterior (ci-Gal4 UAS-Sod:UAS-Cat) compartment in comparison to the posterior.
To test whether an independent source of ROS could activate P-p38 in discs, we fed larvae for 2h with food supplemented with 1% H2O2 and checked for P-p38. Intact discs (no cut, no cell death) from these larvae resulted in high levels of P-p38 as well as high CellROX Green fluorescence (S4 Fig).
Together, these observations show that chemical (NAC) or genetic (UAS-Sod:UAS-Cat) ROS scavengers inhibit P-p38 and therefore indicate that oxidative stress is required for p38 activation.
p38 signaling is required for tissue repair
We next scored wing regeneration after sal E/Pv >rpr induction of cell death in different mutant backgrounds of the p38 pathway. As most of the alleles are lethal or semilethal in homozygosis [47], we tested them in heterozygosis. Alleles of two Drosophila p38 genes, p38a and p38b, were used in this work. We found that heterozygous p38b d27 animals regenerated entire wings (Fig 5A). However, a severe effect was observed with p38a 1 as the resulting wings lacked some sectors and presented notches in the margin. Drosophila p38 signaling is activated by MKK3/licorne (lic)-mediated phosphorylation [48]. Heterozygous lic d13 showed all wing sectors albeit wings were smaller than controls. However, double heterozygotes for lic d13 and p38b d27 were unable to regenerate some wing sectors. We also tested Atf2 PB, a hypomorphic allele of the ATF2 transcription factor downstream of p38 [49], either in homozygosis or in double heterozygous combinations (lic d13 Atf2 PB or p38b d27 Atf2 PB). We found defects in size and pattern after sal E/Pv >rpr induction. Regeneration was severely impaired in double heterozygotes for p38a 1 (MAPK) and Atf2 PB (Fig 5A).
Fig 5. p38 inhibition impairs tissue repair.

(A) Adult wing parameters in p38 signaling mutant backgrounds after genetic ablation. Left: ratios of the wing areas between experimental groups (rpr induction sal E/Pv >rpr ON) and control (no rpr induction sal E/Pv >rpr OFF). Right: percentage of fully regenerated wings. Far right: examples of wings with full regeneration (control) and incomplete regeneration (indicated genotypes) after sal E/Pv >rpr. (B) Percentage of fully regenerated wings after SB202190 intake in sal E/Pv >rpr flies. Right: wing fully regenerated (top) and examples of incomplete regeneration for each SB202190 concentration.
We also blocked the pathway with UAS-RNAi constructs for lic, p38b, p38a and Atf2 and analyzed the adult wings. These transgenes were activated in the anterior compartment (ci>RNAi) and cell death was induced in the sal E/Pv domain (sal E/Pv -LHG lexO-rpr). We found a reduction of individuals capable to fully regenerate wings for those RNAi’s (S5A Fig).
To gain further insight into the requirement for p38, we chemically blocked the pathway using the imidazole drug SB202190, a specific cell permeable p38 MAP kinase inhibitor that has been reported to do not interfere JNK or ERK kinases and is known to prevent phosphorylation of Atf2 in Drosophila S2 cells [50]. We first tested the specificity of the SB202190 on P-p38 in rpr-ablated discs and found significant differences between individuals fed with the drug and controls. In contrast, the differences on TRE-red reporter were not significant (S5C Fig). This indicates that SB202190 strongly blocked P-p38 and weakly the TRE-red. Sal E/Pv >rpr larvae grown at 17°C to prevent cell death and fed with food containing SB202190 (0.12, 1.0 or 5.0 μM) emerged into normal adults (S5B Fig). However, sal E/Pv >rpr-induced larvae fed with SB202190 developed wings lacking some sectors. The highest percentage of aberrant wings was found using 5 μM SB202190 (Fig 5B). This observation confirms that activation of p38 is required for wing repair.
p38 and JNK act independently
To assess the relationship between JNK and p38, we tested p38 activation in wounded null hemizygous JNKK hemipterous (hep r75) discs. P-p38 was localized near the wound after physical injury (Fig 6A). This contrasts with the decrease in P-p38 when the MAPK kinase lic, which is the p38 activating kinase, was interfered with RNAi in injured discs (S6A Fig).
Fig 6. p38 and JNK are activated independently.

(A) Hep r75 hemizygous disc cut (wedge) and stained with P-p38. Sketch of wing discs with square indicate location of images. (B) Hep r75 hemizygous disc after ptc>rpr induction and stained for P-p38. Dead domain is outlined white. TP-3; TO-PRO-3. (C) Wild type and p38a 1-/- discs, cultured for 7h showing TRE-red activation close to the cut edges. (D) Mean pixel intensity for TRE-red measured in discs with physical injury in wild type (88.24 ± 22.58; S.D.) and p38a 1-/- (70.80 ± 19.14; S.D.). P = 0.33 n.s.
Moreover, P-p38 staining was localized in hep r75 discs after ptc>rpr induction, as in the wild type (Fig 6B, compare with Fig 4), indicating that JNK and p38 act independently. In addition, we fed animals with the JNK Inhibitor IX, which abolishes TRE-red reporter expression and inhibits regeneration (S3C and S3D Fig), and found that P-p38 after rpr-ablation was not affected (S6B Fig).
To confirm that JNK and p38 act independently, we blocked the p38 pathway and checked for TRE-red reporter activity. As the p38a 1 allele in heterozygosis strongly affects regeneration (Fig 5), we used this null allele in homozygosis and tested TRE-red activity after physical injury. Our results showed that TRE-red is induced at the wound edges of p38a 1-/- mutant discs (Fig 6C and 6D). Together, these results demonstrate that p38 and JNK stress responses act independently in damaged imaginal discs.
Upd expression is triggered by ROS
The evidence that JNK is active in the living tissue located near damaged zones arises from the expression of puc and TRE-red reporters (Fig 3A and 3B), and also because inhibition of JNK results in defects in repair [21,23,25,26,51]. Moreover, JNK activation promotes upd expression in different contexts [28,52]. We wondered whether those low non-deleterious JNK levels are capable of triggering tissue repair through upd expression. Upd cytokines are ligands that associate with the receptor domeless (dome) to stimulate the kinase activity of the receptor associated protein kinase hopscotch (hop), which in turn phosphorylates dimers of the transcription factor STAT92E [53]. We found upd and upd3 expression near the wound after both physical injury and cell death (Fig 7A, 7B, 7F and 7G). This injury-induced upd expression was blocked in JNKK hep r75 mutants (Fig 7C and 7D) and by JNK Inhibitor IX (S3C Fig), which is consistent with previous observations [28,35].
Fig 7. Cytokine signaling is controlled by ROS and JNK.

(A, B) In situ hybridization of upd (A) and upd3 (B) in wild type (wt) discs and JNKK hep r75 hemizygotes (C) after injury. (D) hep r75 hemizygote stained with anti-Upd after injury. (E) Mean pixel intensities of upd reporter (upd>myrtom Std food: 16.68 ± 3.22 and NAC: 9.01 ± 3.41, S.D.) and STAT92E reporter (10xSTAT92E-GFP Std food: 18.24 ± 2.73 and NAC: 8.63 ± 4.07, S.D.) after standard or NAC-supplemented food. White wedges indicate zone of injury. (F, G) Upd (anti-Upd) is mainly expressed in living cells and not in dead cells after ptc>rpr or sal>rpr. (H) Upd expression declines after NAC intake. TP-3: TP-PRO-3 nuclei staining. (I) Mean pixel intensities of Upd stained ptc>rpr discs with or without NAC feeding (150.29 ± 7.11 and 96.69 ± 18.97, S.D.). (J) Inhibition of the JAK/STAT signaling within dome DN impairs wing regeneration. Genetic design (J) using double transactivator system (as in Fig 2) to induce death (blue) and activate dome DN (red). (K) Percentage of regenerated wings for controls (rpr or dome DN expression only) and experimental (rpr and dome DN dual expression). Note that dome DN wings were not able to regenerate (rpr and dome DN dual expression), whereas dome DN wings in the absence of cell death are normal. Examples of wings (left) of controls and experimental. (L) Experimental design for testing the rescue of NAC effects by ectopic activation of upd. (M) NAC effect on repair ability was rescued by upd overexpression. Quantification of the percentage of wings that regenerate after NAC feeding for the indicated genotypes. (N) Examples of wings from NAC-feeding with rpr-ablation defects (upper) and with rescue after rpr-ablation and upd activation (lower). *P<0.05 **P<0.01.
To study the requirement for JAK/STAT for regeneration, we used the sal E/Pv -LHG lexO-rpr to induce apoptosis and simultaneously interfered with the receptor dome using the dominant negative form UAS-dome DN driven by ci-Gal4. These wings lacked most of the tissue where cell death was induced and dome was blocked (Fig 7J and 7K), indicating that JAK/STAT signaling is needed for tissue recovery. Moreover, heterozygous alleles for the JAK/STAT pathway resulted in partial disruption of adult wing recovery after cell death (S7 Fig).
We next analyzed if JAK/STAT signaling requires ROS in this context. Two different reporters (10XSTAT92E-GFP and upd-Gal4 UAS-myrtomato) were used in physically injured discs and showed reduced expression after NAC feeding (Fig 7E). In addition, ptc>rpr induced discs from NAC fed larvae showed a reduction of upd expression (Fig 7H and 7I). Thus, this expression is ROS dependent after both physical injury and cell death. We speculated that if ROS operate upstream upd, the impairment of regeneration resulting from NAC feeding should be rescued by activating Upd. To this aim, we used the double transactivation system to induce cell death in NAC-fed individuals and concomitantly activate upd expression (Fig 7L). Analysis of the resulting wings showed that upd ectopic expression rescued the NAC inhibition phenotype (Fig 7M and 7N). These observations demonstrate that ROS function upstream of JAK/STAT during repair.
We wondered whether p38 is also required for upd expression in damaged discs. Expression of upd or upd3 was severely reduced in p38a 1-/- wound edges (Figs 8A–8C and S8). This suggests that in addition to JNK, p38 is essential for upd expression upon stress. Finally, we argued that if p38 is required for repair through upd, its ectopic expression should rescue the impaired regeneration after inhibition of p38. We, again, used the double transactivation system to induce cell death in SB202190-fed individuals, to block p38 phosphorylation and alongside activate upd expression (Fig 8D). Indeed, we found that the number of wings that regenerated after p38 inhibition increased (Fig 8E and 8F). Altogether these results position Upd cytokines downstream from the ROS/p38/JNK module.
Fig 8. p38 controls upd expression.

(A) In situ hybridization of upd in wild type (wt) and p38a 1-/- cut discs. (B) In situ hybridization of upd3 in wild type (wt) and p38a 1-/- cut discs. (C) Immunostaining with anti-Upd in wild type (wt) and p38a 1-/- cut discs. White lines and wedges indicate the position of the cut (D) Experimental design for testing the rescue of SB202190 effects by ectopic activation of upd. (E) SB202190 effect on repair ability was rescued by upd overexpression. Quantification of the percentage of wings that regenerate after SB202190 feeding for the indicated genotypes. (F) Examples of wings from SB202190-feeding with rpr-ablation defects (upper) and with rescue after rpr-ablation and upd activation (lower).
Discussion
In this work, we demonstrate a stress-responsive module activated upon cell death or physical damage. This module consists of ROS dependent stimulation of non-deleterious levels of JNK and p38 MAP kinases necessary for the expression of Upd and JAK/STAT signaling which drives regeneration. Non-lethal levels of JNK may have multiple functions, among them cytoskeleton organization [44,54], healing and initiation of regenerative growth [21,23–26,51,55,56]. Thus, this early responsive module is crucial to maintain tissue in a healthy condition, trigger tissue repair and restore homeostasis.
In an apoptotic context, Rpr dimerizes and, through direct binding, brings the Drosophila inhibitor of apoptosis protein-1 (DIAP1) to mitochondria, concomitantly promoting DIAP1 auto-ubiquitination and destruction [57,58]. Rpr action on the mitochondria results in alteration of cytochrome C driven by caspases [59] and in mitochondrial disruption [60]. The ROS dyes used here detect a wide range of ROS, and therefore we cannot discriminate between membrane oxidases or mitochondrial origin. However, since Rpr acts on mitochondria, mitochondrial alterations could cause the burst of ROS in apoptotic cells. Of note, we observed that high ROS levels are associated with high levels of JNK in apoptotic cells. It has been proposed that ROS can mediate the activation of JNK [61] by quenching the MAP kinase phosphatases [62]. Conversely, low levels of ROS detected in nearby surviving tissue correlate with low non-deleterious levels of JNK and activation of MAP kinase phosphatases. Thus, puc MAP kinase phosphatase could protect the living cells close to the damage from the noxious effects of high JNK. Indeed, living cells near the wound retain low levels of JNK, not sufficient to kill but necessary for tissue recovery.
Additionally, the caspase Dronc, which acts downstream from Rpr, has functions beyond apoptosis [63]. Dronc is involved in the activation of JNK and p53, which activate the pro-apoptotic genes, creating an amplification loop that ensures apoptosis [27,29–31,64]. The JNK/p53 driven apoptosis stimulates proliferation of the nearby tissues [29–31,65]. Although still unclear, it has been proposed that apoptotic cells can release the products of mitogenic genes such as wingless (wg) and decapentaplegic (dpp) [33,66,67][31,68]. Alternatively, we show here that ROS operate as signals responding to insults (apoptosis, mechanical stress) that turn on the homeostatic machinery to compensate the epithelial damage. This fits with a scenario in which ROS are able to either diffuse from cell to cell or perhaps to propagate their production to several rows of cells. Indeed, ROS have been proven to cross cell membranes, to spread through gap junctions [69–71] and to enter into the cell through specific membrane aquaporin channels [71,72]. Therefore, ROS behave as an efficient paracrine signal that ultimately will result in Upd activation.
In addition to JNK, ROS are stressors involved in p38 activation [73]. ROS may activate the p38 pathway through the oxidative modification of intracellular kinases such as redox-sensitive activating protein-1 ASK1 [74]. We showed here that not only JNK but also p38 is required for regeneration. Moreover, the p38a 1 allele seems to particularly affect upd expression and regeneration. This concurs with the finding that Drosophila p38a is more susceptible to environmental stressors, such as oxidative stress [18]. However, other p38 kinases could contribute to tissue regeneration. Indeed, heterozygous alleles of the p38 activating kinase lic, which normally do not show patterning defects after rpr-mediated ablation, can result in incomplete regeneration when a dose of p38b is missing (Fig 5A). Moreover, RNAi of p38b also can show defective regeneration individuals (S2 Fig). In addition, we cannot discard that p38c, which has been recently found involved in intestinal immune homeostasis [75], may also function in imaginal disc regeneration.
We have found that both hep r75 and p38a 1 inhibit upd expression. But hep r75 mutants, which block JNK signaling, do not affect p38 phosphorylation and viceversa, p38a 1 mutants, which block at least the p38a branch of the p38 kinase, do not interfere with the TRE-red reporter expression. This suggests that ROS activate p38 and JNK independently and that both MAP kinases act on upd expression to drive tissue repair. Thus, ROS signaling operates through these two MAP kinase pathways that in turn will converge to stimulate the transcriptional expression of the cytokines (Fig 9).
Fig 9. Cell protection module activated by injury or cell death.
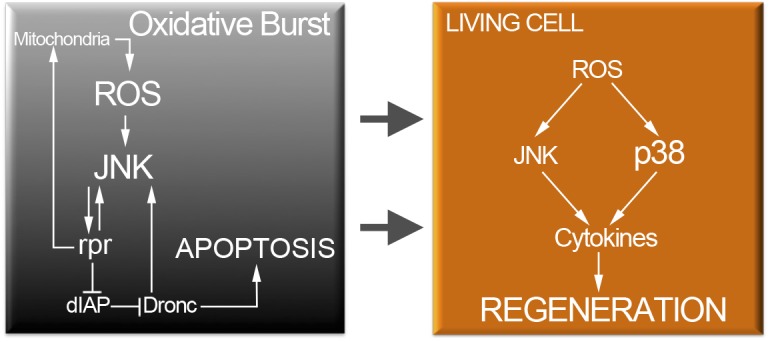
Oxidative stress in dying cells is likely of mitochondrial origin and results in highly toxic JNK. However, low levels of ROS propagate to adjacent surviving cells (arrows). Non-deleterious ROS will activate moderate levels of JNK and p38 only in surviving cells. P38 and JNK are required for cytokine activation and tissue repair.
JNK and p38 are not only activated after cell death but also after physical injury. Beneficial ROS production is an ubiquitous reaction associated with inflammatory responses to wounding [4,6,76]. Recent findings show that ROS produced in dynamic epithelia operate as a tuning mechanism for reorganization of epithelia [77]. Therefore, it could be that changes in mechanical stress generated during wounding and epithelial disruption (mechanical stretching) results in ROS production. Some dead cells were also found after physical injury. Thus, a partial contribution of dead cells in addition to the stress due to epithelial disruption can account for the oxidative burst generated after physical injury.
In summary, an early boost of oxidative stress is required to activate p38 and JNK in apoptotic cells or near the wound. Moreover, upd is turned on downstream JNK and p38. Thus, downstream of the stress response module, cytokines operate to control tissue growth during regeneration.
Materials and Methods
Drosophila strains
The Drosophila melanogaster strains used were ptc-Gal4 [78], tubGal80 TS [79], UAS-rpr [80], ci-Gal4 [81], nub-Gal4 [82] sal-Gal4 and sal E/Pv -Gal4 [83], p38b d27, lic d13 [47], dATF2 PB [49], p38a 1, [17], LexO-rCD2::GFP [43], TRE-DsRed.T4 [45] as AP1 reporter, puc-lacZ [44], puc E69-A -Gal4 [84], UAS-upd [85], upd-Gal4 (from D. Harrison), 10XSTAT92E-GFP [86], en-Gal4, UAS-GFP, UAS-myrtomato, UAS-Sod.A (sod1), UAS-Cat.A, UAS-dome DN, hop2, hop 27, stat92e 06346 (Bloomington Stock center), stat92e 397 [87], and hep r75 [88]. Transgenic Drosophila shRNAi lines were obtained from the Vienna Drosophila RNAi Center (VDRC). Canton S was used as the wild type control.
Imaginal disc culture and physical injury
Wing discs were dissected from third instar larvae in Schneider’s insect medium (Sigma-Aldrich) and a small fragment was removed with tungsten needles. Discs were cultured in Schneider’s insect medium supplemented with 2% heat activated fetal calf serum, 2.5% fly extract and 5 μg/ml insulin, for different periods of time (from 1 to 10 hours) at 25°C. Ex vivo images were taken using a Leica SPE confocal microscope and processed with Fiji software.
Generation of LexA/lexO strains for genetic ablation
The sal E/Pv -LHG construct was created cutting the wing specific enhancer of spalt, sal E/Pv [83] from pC4LacZ-Spalt PE EcoRI/BamHI and cloning this fragment into the plasmid attB-LHG containing a Gal80-suppressible form of LexA transcriptional activator (LHG) [43]. LHG contains both the binding domain of LexA and the activator domain of Gal4, which is recognized by the inhibitor Gal80 TS.The LexO-rpr strain was obtained subcloning the pro-apoptotic gene reaper (rpr) from pOT2-rpr (IP02529) EcoRI/XhoI in the pLOTattB plasmid [89] carrying the lexA operator LexO. Transgenic flies were performed with standard protocols.
Genetic ablation and dual Gal4/LexA transactivator system
Cell death was genetically induced as previously described [23,90]. We used two different drivers to induce cell death. The first, ptc-Gal4 which is expressed in a narrow stripe in the center of the disc. This strain was used to induce cell death in imaginal discs (UAS-rpr), because the dead domain can be easily discerned from the neighboring living domain. The second, sal E/Pv -Gal4 strain, which consists of sal wing enhancer with expression confined to the wing [83] has been used in this work to score adult wing parameters.
The UAS line used to promote cell death was UAS-rpr, and the system was controlled by the thermo sensitive repressor tubGal80 TS. We also used the sal E/Pv -LHG and LexO-rpr strains for genetic ablation using the same design as for Gal4/UAS.
Embryos were kept at 17°C until the 8th day/192 h after egg laying (equivalent to 96 hours at 25°C) to prevent rpr expression. They were subsequently moved to 29°C for 11 hours and then back to 17°C until adulthood. Controls without rpr expression were always treated in parallel.
In dual transactivation experiments, we used the sal E/Pv -LHG LexO-rpr to ablate the sal E/Pv domain, whereas Gal4 was used to express different transgenes under the control of nub-Gal4 or ci-Gal4.
In the experiments on antioxidants (Fig 2) and upd (Figs 7 and 8) overexpression, larvae were transferred to NAC- (100 μg/ml) or SB202190- (5 μM) supplemented food 24 h before cell death induction.
ROS detection ex vivo
All experiments for ROS detection were done in living conditions. To detect the presence of ROS we used CellROX Green Reagent (Life Technologies), which is an indicator of oxidative stress in living cells. For both genetic ablation and physical injury experiments, third instar discs were dissected in Schneider´s medium immediately after cell death or injury and incubated for 15 minutes in medium containing 5 μM CellROX Green Reagent, followed by three washes. Samples were protected from light throughout. Then they were mounted using culture medium supplemented with 1 μM TO-PRO-3 (Life Technologies) nucleic acid stain. As TO-PRO-3 only enters dead cells, we used it to distinguish dead cells from living cells in the ex vivo experiments. Images were taken using a Leica SPE and SPII confocal microscope. Grey values of regions of interest (ROI) were measured using Fiji software. ROIs were established at the wound edges of injured discs (examples in S2 Fig), or in rectangles as indicated in Fig 1E. Pixel intensities were collected and analyzed from raw images taken under the same laser confocal conditions. Thermal LUT images were rendered from slices taken from the confocal using the Interactive 3D Surface Plot tool of the Fiji software (ImageJ). We also used the cell-permeant 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA 5μM, Life Technologies) which upon oxidation is converted to the highly fluorescent 2',7'-dichlorofluorescein (DCF).
To visualize the ROS images in Fig 1 after genetic ablation, the whole stacks were subject to the Enhance Contrast tool at 0.4 pixel saturation in whole stack normalization. For physical injury images, thermal LUT images were obtained from raw stacks.
ROS scavenging
To prevent ROS production, we used two protocols. The first was mainly used for rpr-ablation discs. It consisted in that antioxidants were supplemented into standard fly food. As antioxidants we used vitamin C (250 μg/ml), Trolox (an analog of vitamin E; 20 μg/ml) and N-acetyl cysteine (NAC) (100 μg/ml), all from Sigma-Aldrich. To score adult wings, larvae were transferred from vials containing standard food to vials containing food with the desired antioxidant concentration. Antioxidant treatment was administered at 168 h of development at 17°C (equivalent to 84 h AEL at 25°C). After 24 hours, experimental larvae were moved to 29°C for 11 hours to promote rpr apoptosis. Meanwhile one control consisted of larvae maintained at 17°C and another control consisted of larvae transferred to a vial with standard food and moved to 29°C for the same period as in the experimental group. After rpr induction temperature was returned to 17°C to allow tissue recovery. This protocol was applied for Figs 2A–2D, 3D, 3E, 4E, 4F and 7E.
The second was used for ex vivo cultured discs. Wing imaginal discs were incubated for 30 minutes in Schneider’s insect medium supplemented with NAC 100 μg/ml. Then, they were transferred to Schneider’s containing CellROX Green (S2 Fig). NAC incubated discs were used for monitoring TRE-red (Fig 3F and 3G) or for P-p38 antibody staining (Fig 4B and 4C). In S2 Fig medium was supplemented with NAC, Trolox or Vit C.
Chemical inhibition of p38 and JNK pathway
The imidazole drug SB202190 (Sigma-Aldrich) was added to standard fly food to prevent p38 activation. We used three different concentrations (0.12 μM, 1 μM and 5 μM), and DMSO as the control. To inhibit chemically JNK we used the JNK Inhibitor IX (5 μM, Selleckchem) which is a thienylnaphthamide compound that is a selective and potent inhibitor of the ATP binding site of JNK. The timing and protocol followed to inhibit both pathways was the same as that to scavenge ROS.
Oxidative stress induction
Third instar larvae were transferred to vials containing 1% H2O2, 1,3% low melting agarose and 5% sucrose. To avoid loss of oxidative capacity, H2O2 was added at a temperature under 45°C. Larvae were fed for 2h in this medium prior dissection and fixation of the discs. Controls without H2O2 were done in parallel.
Test for regenerated adult wings and statistics
For testing the capacity to regenerate we used adult wings emerged from sal E/Pv >rpr individuals, in which patterning defects can be easily scored. Flies were fixed in glycerol:ethanol (1:2) for 24 h. Wings were dissected on water and then washed with ethanol. Then they were mounted on 6:5 lactic acid:ethanol and analyzed and imaged under a microscope.
Definition of regenerated/non-regenerated wings: when veins or interveins were missing, we considered them as defective in their capacity to restore the normal pattern. Therefore, the % of regenerated wings (Figs 2, 5, 7, 8, S2, S3, S5 and S7) was calculated after the number of wings with the complete set of veins and interveins. For each sample of “regenerated wings” we scored the percentage of individuals that belong to the “regenerated wings” class and calculated the standard error of sample proportion based on binomial distribution (regenerate complete wing or not) SE = √p (1-p)/n, where p is the proportion of successes in the population.
Ratios between wing areas (Fig 5) were used as an indication of the size achieved after cell death for each genetic background, and consisted of a comparison between wing size with and without rpr induction.
Immunochemistry and fluorescence in situ hybridization (FISH)
Immunostaining and FISH were performed using standard protocols. Primary antibodies used in this work were P-p38 (rabbit 1:50, Cell Signaling Technology), phospho-Histone-H3 (rabbit 1:1000, Millipore), ß-galactosidase (rabbit 1:1000, ICN Biomedicals), Upd (rabbit 1:800, gift from D. Harrison) and cleaved caspase-3 (rabbit 1:100, Cappel).
Fluorescently labeled secondary antibodies were from Life Technologies and Jackson Immunochemicals. Discs were mounted in SlowFade (Life Technologies) supplemented with 1 μM TO-PRO-3 (Life Technologies) to label nuclei. Note that in fixed tissues all nuclei are TO-PRO-3 labeled, whereas in ex-vivo culture only nuclei of dead or dying cells are TO-PRO-3 labeled.
The number of mitosis after analyzing the stacks of confocal images was calculated using Fiji software (Cell counter plug-in). Mitosis were counted for the entire anterior compartment of the wing pouch for each disc.
For apoptotic cell detection, we used both anti cleaved caspase 3 or TUNEL assay. For TUNEL we used the fluorescently labeled dUTP ChromaTide BODIPY FL-14-dUTP (Life Technologies) and incorporated using terminal deoxynucleotidyl transferase (Roche).
EdU was incorporated using the Click-iT EdU Imaging Kit (Life Technologies). Wing discs were dissected after cell death induction and incubated in Schneider’s insect medium supplemented with 1 mg/ml EdU for 5 minutes. Following EdU incorporation, discs were fixed and immunostained.
Riboprobes for upd and upd3 were synthesized using cDNA clones from DGRC AT1366 and FI03911.
Genotypes
A, B. Wild type
D, E, F. ptc>rpr: UAS-rpr/+; ptc-Gal4/+; tubGal80TS/+
A, B, C. salE/Pv>rpr: UAS-rpr/+; salE/Pv-Gal4/+; tubGal80TS/+
D. ptc>rpr: UAS-rpr/+; ptc-Gal4:tubGal80TS/+
E, F, G. salE/Pv>rpr nub>GFP: w; nub-Gal4/UAS-GFP; salE/Pv-LHG:tubGal80TS/lexO-rpr
sal E/Pv >rpr nub>Cat: w; nub-Gal4/UAS-Cat; salE/Pv-LHG:tubGal80TS/lexO-rpr
sal E/Pv >rpr nub>Sod: w; nub-Gal4/UAS-Sod; salE/Pv-LHG:tubGal80TS/lexO-rpr
sal E/Pv >rpr nub>Sod:Cat: w; nub-Gal4/UAS-Sod:Cat; salE/Pv-LHG:tubGal80TS/lexO-rpr
A, B. ptc>rpr: UAS-rpr/+; ptc-Gal4:tubGal80TS/TRE-DsRed.T4; puc-LacZ/+
C. ptc>rpr: UAS-rpr/+; ptc-Gal4:tubGal80TS/+; puc-LacZ/+
D, E. ptc>rpr: UAS-rpr/+; ptc-Gal4:tubGal80TS/TRE-DsRed.T4
F, G. w; TRE-DsRed.T4
A, B, C. Wild type.
D, E, F. ptc>rpr: UAS-rpr/+; ptc-Gal4/+; tubGal80TS/+
G. w; ci-Gal4/UAS-Sod:Cat; salE/Pv-LHG:tubGal80TS/lexO-rpr
A. Control: w; +; salE/Pv-LHG:tubGal80TS/lexO-rpr (control for lexO-rpr on the third chromosome) and w; lexO-rpr/+; salE/Pv-LHG:tubGal80TS/+ (control for lexO-rpr on the second chromosome)
lic d13/+: licd13/+; +; salE/Pv-LHG:tubGal80TS/lexO-rpr
p38b d27/+: w; p38bd27/+; salE/Pv-LHG:tubGal80TS/lexO-rpr
p38a 1/+: w; lexO-rpr/+; p38a1/salEPv-LHG:tubGal80TS
licd13/+p38bd27/+: licd13/+; p38bd27/+; salEPv-LHG:tubGal80TS/lexO-rpr
dATF2PB/+: w; Atf2PB/+; salE/Pv-LHG:tubGal80TS/lexO-rpr
dATF2 PB-/-: w; Atf2PB/Atf2PB; salE/Pv-LHG:tubGal80TS/lexO-rpr
lic d13/+ dATF2 PB/+: licd13/+; Atf2PB/+; salE/Pv-LHG:tubGal80TS/lexO-rpr
p38b d27/+ dATF2 PB/+: w; p38bd27/Atf2PB; salE/Pv-LHG:tubGal80TS/lexO-rpr
dATF2 PB/+ p38a 1/+: w; Atf2PB/lexO-rpr; p38a1/salEPv-LHG:tubGal80TS
B. salE/Pv>rpr: UAS-rpr/+; salE/Pv-Gal4/+; tubGal80TS/+
A. hepr75: hepr75/Y
B. ptc>rpr hepr75: hepr75/Y; ptc-Gal4:tubGal80TS/+; UAS-rpr/+
C. wt: w; TRE-DsRed.T4
p38a -/-: w; TRE-DsRed.T4; p38a1/p38a1
A, B. Wild type.
C, D. hepr75: hepr75/Y
E. upd-Gal4/+; UAS-myrtomato/10XSTAT92E-GFP
F, G, H, I. ptc>rpr: UAS-rpr/+; ptc-Gal4/+; tubGal80TS/+
sal E/Pv >rpr: UAS-rpr/+; sal-Gal4/+; tubGal80TS/+
J, K. salE/Pv>rpr ci>GFP: w; ci-Gal4/lexO-rpr; salE/Pv-LHG:tubGal80TS/UAS-GFP
ci>dome DN sal E/Pv >GFP: w; ci-Gal4/lexO-rCD2::GFP; salE/Pv-LHG:tubGal80TS/UAS-domeDN
sal E/Pv >rpr ci> dome DN: w; ci-Gal4/lexO-rpr; salE/Pv-LHG:tubGal80TS/UAS-domeDN
L, M, N. ci-Gal4 UAS-upd: w; ci-Gal4/UAS-upd; salE/Pv-LHG:tubGal80TS/lexO-rCD2::GFP
sal E/Pv -LHG lexO-rpr: w; ci-Gal4/UAS-GFP; salE/Pv-LHG:tubGal80TS/lexO-rpr
ci-Gal4 UAS-upd sal E/Pv -LHG lexO-rpr: w; ci-Gal4/UAS-upd; salE/Pv-LHG:tubGal80TS/lexO-rpr
A, B, C. Wild type.
p38a -/-: w; +; p38a1/p38a1
D, E, F. ci-Gal4 UAS-upd: w; ci-Gal4/UAS-upd; salE/Pv-LHG:tubGal80TS/lexO-rCD2::GFP
sal E/Pv -LHG lexO-rpr: w; ci-Gal4/UAS-GFP; salE/Pv-LHG:tubGal80TS/lexO-rpr
ci-Gal4 UAS-upd sal E/Pv -LHG lexO-rpr: w; ci-Gal4/UAS-upd; salE/Pv-LHG:tubGal80TS/lexO-rpr
A, C, D. Wild type.
B. ptc>rpr: UAS-rpr/+; ptc-Gal4:tubGal80TS/+
A. wt
B. salE/Pv>rpr: UAS-rpr/+; salE/Pv-Gal4/+; tubGal80TS/+
C. salE/Pv>rpr nub>GFP: w; nub-Gal4/UAS-GFP; salE/Pv-LHG:tubGal80TS/lexO-rpr
sal E/Pv >rpr nub>Cat: w; nub-Gal4/UAS-Cat; salE/Pv-LHG:tubGal80TS/lexO-rpr
sal E/Pv >rpr nub>Sod: w; nub-Gal4/UAS-Sod; salE/Pv-LHG:tubGal80TS/lexO-rpr
sal E/Pv >rpr nub>Sod:Cat: w; nub-Gal4/UAS-Sod:Cat; salE/Pv-LHG:tubGal80TS/lexO-rpr
D. salE/Pv>rpr nub>GFP: w; nub-Gal4/UAS-GFP; salE/Pv-LHG:tubGal80TS/lexO-rpr
sal E/Pv >rpr nub>Cat: w; nub-Gal4/UAS-Cat; salE/Pv-LHG:tubGal80TS/lexO-rpr
nub>Cat: w; nub-Gal4/UAS-Cat; salE/Pv-LHG:tubGal80TS/lexO-GFP
sal E/Pv >rpr nub>Sod: w; nub-Gal4/UAS-Sod; salE/Pv-LHG:tubGal80TS/lexO-rpr
nub>Sod: w; nub-Gal4/UAS-Sod; salE/Pv-LHG:tubGal80TS/lexO-GFP
sal E/Pv >rpr nub>Sod:Cat: w; nub-Gal4/UAS-Sod:Cat; salE/Pv-LHG:tubGal80TS/lexO-rpr
nub>Sod:Cat: w; nub-Gal4/UAS-Sod:Cat; salE/Pv-LHG:tubGal80TS/lexO-GFP
A. Wild type.
B. w; TRE-DsRed.T4/+; puc-Gal4:UAS-GFP/+
C. ptc>rpr: UAS-rpr/+; ptc-Gal4:tubGal80TS/TRE-DsRed.T4
D. salE/Pv>rpr: UAS-rpr/+; salE/Pv-Gal4/+; tubGal80TS/+
Wild type
A. salE/Pv>rpr: w; ci-Gal4/UAS-GFP; salE/Pv-LHG:tubGal80TS/lexO-rpr
ci>RNAi p38a: w; ci-Gal4/UAS-RNAi p38a; salE/Pv-LHG:tubGal80TS/lexO-GFP
sal E/Pv >rpr ci>RNAi p38a: w; ci-Gal4/UAS-RNAi p38a; salE/Pv-LHG:tubGal80TS/lexO-rpr
ci>RNAi p38b: w; ci-Gal4/UAS-RNAi p38b; salE/Pv-LHG:tubGal80TS/lexO-GFP
sal E/Pv >rpr ci>RNAi p38b: w; ci-Gal4/UAS-RNAi p38b; salE/Pv-LHG:tubGal80TS/lexO-rpr
ci>RNAi Atf2: w; ci-Gal4/UAS-RNAi Atf2; salE/Pv-LHG:tubGal80TS/lexO-GFP
sal E/Pv >rpr ci>RNAi Atf2: w; ci-Gal4/UAS-RNAi Atf2; salE/Pv-LHG:tubGal80TS/lexO-rpr
ci>RNAi lic: w; ci-Gal4/UAS-RNAi lic; salE/Pv-LHG:tubGal80TS/lexO-GFP
sal E/Pv >rpr ci>RNAi lic: w; ci-Gal4/UAS-RNAi lic; salE/Pv-LHG:tubGal80TS/lexO-rpr
B. salE/Pv>rpr: UAS-rpr/+; salE/Pv-Gal4/+; tubGal80TS/+
C. salE/Pv>rpr: UAS-rpr/+; salE/Pv-Gal4/+; tubGal80TS/+
ptc>rpr: UAS-rpr/+; ptc-Gal4:tubGal80TS/TRE-DsRed.T4
en>RNAi lic: w; en-Gal4:UAS-GFP/UAS-RNAi lic
Control: UAS-rpr/+; salE/Pv-Gal4/+; tubGal80TS/+
hop 2/+: UAS-rpr/hop2; salE/Pv-Gal4/+; tubGal80TS/+
hop 27/+: UAS-rpr/hop27; salE/Pv-Gal4/+; tubGal80TS/+
stat92e 397/+: UAS-rpr/+; salE/Pv-Gal4/+; tubGal80TS/stat92e397
stat92e 06346/+: UAS-rpr/+; salE/Pv-Gal4/+; tubGal80TS/stat92e06346
Wild type.
p38a -/-: w; +; p38a1/p38a1
Supporting Information
(A) Propagation of ROS labeled with CellROX Green towards the adjacent tissue during the first 15’ after injury. Thermal scale corresponds to the same as in Fig 1B. White line indicates cut edge. (B) ROS detected with H2DCFDA after ptc>rpr. ROS are found in dead cells and in adjacent living cells. TP-3: TO-PRO-3. (C, D) ROS detected with H2DCFDA after physical injury (white wedge).
(TIF)
(A) Ex vivo analysis of cut imaginal discs cultured in Schneider’s medium, incubated with NAC, Trolox or VitC. Top row shows images of control discs (no antioxidant). Lower row shows images of discs incubated with the indicated antioxidant. Dotted lines indicate zones used as ROI for pixel intensity measurements (below). White wedges indicate the position of the cut. *P<0.05 **P<0.01. (B) Examples of control wings kept at 17°C (sal EPv >rpr OFF) that grew in food supplemented with antioxidant. All cases, showed normal set of interveins and veins. (C) Examples of control wings kept at 17°C (sal EPv >rpr OFF) that grew from the indicated genotypes. (D) Controls for transgenes of Fig 2F. Activation of transgenes (nub>Cat; nub>Sod; nub>Sod:Cat) in the absence of cell death results in normal wings.
(TIF)
(A) Endogenous expression of the TRE-red and puc-lacZ reporters. Note that both reporters are expressed only at the tip of the notum (n: notum; wp: wing pouch). (B) TRE-red and puc>GFP expression after physical injury. Note that TRE-red expression is activated earlier and more extensive than puc>GFP. White wedges indicate the position of the cut. Dotted line indicates the edges of the disc. (C) The JNK Inhibitor IX eliminates TRE-red activity and upd expression. Top row: rpr–ablated disc from larvae fed with standard food stained for nuclei (TP3: TO-PRO-3), TRE-red reporter, and anti-Upd. Bottom row: rpr–ablated disc from larvae supplemented with JNK Inhibitor IX. (D) JNK Inhibitor IX inhibits regeneration. Quantification of regenerated sal EPv >rpr wings after feeding with standard food or JNK Inhibitor IX supplemented.
(TIF)
To test whether P-p38 is activated after an independent mechanism of oxidative stress in the absence of damage, larvae were fed with 1% H2O2 for 2 h before processed for imaging. (A) Live imaging showing high ROS in the entire disc. (B) Fixed disc stained with P-p38.
(TIF)
(A) Inhibition of p38 with RNAi constructs prevents tissue repair. Ectopic expression of p38 RNAis under the control of ci-Gal4 and simultaneous cell death induction with sal E/Pv -LHG LexO-rpr when shifted to 29°C for 11 h. Adult wing size was measured after ectopic expression of the indicated RNAi transgenes (red). The experiments with rpr-ablation are indicated in blue. (B) Examples of control wings of Fig 5B, in which no rpr-ablation was induced (kept at 17°C) for the indicated concentrations of the p38 inhibitor SB202190. (C) Test for the reliability of the SB202190. Discs were dissected from rpr-ablated larvae that were fed with 5 μM SB202190, fixed and imaged. SB202190 intake reduces P-p38 activation after cell death as measured from the Mean Pixel Intensity in comparison to DMSO fed larvae. TRE-red in individuals fed with 5 μM SB202190 is active. Right: Mean Pixel Intensities for both experiments. For p38: control 48.77 ± 30.82 (S.D.); SB202190 10.52 ± 8.09 (S.D.). For TRE-red: control 136.46 ± 44.5 (S.D.); SB202190 93.5 ± 23.19 (S.D.). ***P<0.001 for the P-p38 and P = 0,15 n.s. for TRE-red.
(TIF)
(A) RNA interference of MKK lic inhibits p38 phosphorilation after injury. The UAS-RNAi lic was activated in the posterior compartment together with UAS-GFP (white). Two injuries were inflected with tungsten needles, one in the anterior and one in the posterior compartment. The cuts were performed in Schneider’s medium, and fixation for immunostaining 20’ after injury. P-p38 activation was localized in the anterior compartment and almost absent around the posterior cut. (B) Blocking JNK with JNK Inhibitor IX does not affect P-p38 after rpr-ablation. The domain of dead cells is outlined in white.
(TIF)
Percentages of regenerated wings for the indicated genetic background after sal E/Pv >rpr ablation. Right wings: sal E/Pv >rpr OFF column: wings of those genetic backgrounds without cell death (kept at 17°C). All wings raised in those conditions contain the normal set of veins and interveins. sal E/Pv >rpr ON column: top (wt) is an example of fully regenerated wing. The rest of wings are examples of non-regenerated or incomplete regeneration in the heterozygous condition indicated.
(TIF)
Quantification of in situ hybridizations of upd mRNA (A) and upd3 mRNA (B) and antibody localization for Upd (C). Regions of interest were determined around the wound edges (as in S2 Fig) of wild type discs (wt) and p38a 1-/- mutants. Images in Fig 8A–8C are examples of the quantification shown here. ***P<0.001 **P<0.01. Bars indicate standard deviation.
(TIF)
Acknowledgments
We thank R. Yagi and K. Basler for providing us with the vectors for the LexA/lexO system. We thank H. Stocker for stocks and discussion. We also thank H. Bellen, A. Nebreda, E. Vizcaya, M. Salicrú, M. Giralt and F. Villaroya, for discussion and suggestions. We also thank the Confocal Unit of the CCiT-UB and in particular M. Bosch. We are also grateful to N. Ferreira and E. Alcañiz for their help in the early stages of this work.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This project was funded by grants BFU2009-09781, CSD2007-00008, and BFU2012-36888, Ministerio de Economía y Competitividad, Spain (http://www.mineco.gob.es/portal/site/mineco/) to the FS and MC labs and by grant BFU2010-21123, PCIN-2013-048 and CSD2007-00008 to the MM lab. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194: 7–15. 10.1083/jcb.201102095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bigarella CL, Liang R, Ghaffari S. Stem cells and the impact of ROS signaling. Development. 2014;141: 4206–4218. 10.1242/dev.107086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moreira S, Stramer B, Evans I, Wood W, Martin P. Prioritization of competing damage and developmental signals by migrating macrophages in the Drosophila embryo. Curr Biol. 2010;20: 464–70. 10.1016/j.cub.2010.01.047 [DOI] [PubMed] [Google Scholar]
- 4. Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459: 996–9. 10.1038/nature08119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gauron C, Rampon C, Bouzaffour M, Ipendey E, Teillon J, Volovitch M, et al. Sustained production of ROS triggers compensatory proliferation and is required for regeneration to proceed. Sci Rep. 2013;3: 2084 10.1038/srep02084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Love NR, Chen Y, Ishibashi S, Kritsiligkou P, Lea R, Koh Y, et al. Amputation-induced reactive oxygen species are required for successful Xenopus tadpole tail regeneration. Nat Cell Biol. 2013;15: 222–8. 10.1038/ncb2659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seyfried J, Wüllner U. Inhibition of thioredoxin reductase induces apoptosis in neuronal cell lines: role of glutathione and the MKK4/JNK pathway. Biochem Biophys Res Commun. 2007;359: 759–64. [DOI] [PubMed] [Google Scholar]
- 8. Shi Y, Nikulenkov F, Zawacka-Pankau J, Li H, Gabdoulline R, Xu J, et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. Macmillan Publishers Limited; 2014;21: 612–23. 10.1038/cdd.2013.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Droge W. Free Radicals in the Physiological Control of Cell Function. Physiol Rev. 2002;82: 47–95. [DOI] [PubMed] [Google Scholar]
- 10. McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8: 1775–89. [DOI] [PubMed] [Google Scholar]
- 11. Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev. 2011;63: 218–42. 10.1124/pr.110.002980 [DOI] [PubMed] [Google Scholar]
- 12. Jia Y-T, Wei W, Ma B, Xu Y, Liu W-J, Wang Y, et al. Activation of p38 MAPK by reactive oxygen species is essential in a rat model of stress-induced gastric mucosal injury. J Immunol. American Association of Immunologists; 2007;179: 7808–19. [DOI] [PubMed] [Google Scholar]
- 13. Sato A, Okada M, Shibuya K, Watanabe E, Seino S, Narita Y, et al. Pivotal role for ROS activation of p38 MAPK in the control of differentiation and tumor-initiating capacity of glioma-initiating cells. Stem Cell Res. 2014;12: 119–31. 10.1016/j.scr.2013.09.012 [DOI] [PubMed] [Google Scholar]
- 14. Kanaji N, Nelson A, Wang X, Sato T, Nakanishi M, Gunji Y, et al. Differential roles of JNK, ERK1/2, and p38 mitogen-activated protein kinases on endothelial cell tissue repair functions in response to tumor necrosis factor-α. J Vasc Res. 2013;50: 145–56. 10.1159/000345525 [DOI] [PubMed] [Google Scholar]
- 15. Sharma G-D, He J, Bazan HEP. p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: evidence of cross-talk activation between MAP kinase cascades. J Biol Chem. 2003;278: 21989–97. [DOI] [PubMed] [Google Scholar]
- 16. Karkali K, Panayotou G. The Drosophila DUSP puckered is phosphorylated by JNK and p38 in response to arsenite-induced oxidative stress. Biochem Biophys Res Commun. 2012;418: 301–6. 10.1016/j.bbrc.2012.01.015 [DOI] [PubMed] [Google Scholar]
- 17. Seisenbacher G, Hafen E, Stocker H. MK2-dependent p38b signalling protects Drosophila hindgut enterocytes against JNK-induced apoptosis under chronic stress. PLoS Genet. 2011;7: e1002168 10.1371/journal.pgen.1002168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Craig CR, Fink JL, Yagi Y, Ip YT, Cagan RL. A Drosophila p38 orthologue is required for environmental stress responses. EMBO Rep. 2004;5: 1058–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461: 537–41. 10.1038/nature08313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7: 491–501. [DOI] [PubMed] [Google Scholar]
- 21. Bosch M, Serras F, Martin-Blanco E, Baguna J. JNK signaling pathway required for wound healing in regenerating Drosophila wing imaginal discs. Dev Biol. 2005;280: 73–86. [DOI] [PubMed] [Google Scholar]
- 22. Bosch M, Baguna J, Serras F. Origin and proliferation of blastema cells during regeneration of Drosophila wing imaginal discs. Int J Dev Biol. 2008;52: 1043–1050. 10.1387/ijdb.082608mb [DOI] [PubMed] [Google Scholar]
- 23. Bergantiños C, Corominas M, Serras F. Cell death-induced regeneration in wing imaginal discs requires JNK signalling. Development. 2010;137: 1169–1179. 10.1242/dev.045559 [DOI] [PubMed] [Google Scholar]
- 24. Smith-Bolton RK, Worley MI, Kanda H, Hariharan IK. Regenerative growth in Drosophila imaginal discs is regulated by Wingless and Myc. Dev Cell. 2009;16: 797–809. 10.1016/j.devcel.2009.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mattila J, Omelyanchuk L, Kyttala S, Turunen H, Nokkala S. Role of Jun N-terminal Kinase (JNK) signaling in the wound healing and regeneration of a Drosophila melanogaster wing imaginal disc. Int J Dev Biol. 2005;49: 391–399. [DOI] [PubMed] [Google Scholar]
- 26. Lee N, Maurange C, Ringrose L, Paro R. Suppression of Polycomb group proteins by JNK signalling induces transdetermination in Drosophila imaginal discs. Nature. 2005;438: 234–237. [DOI] [PubMed] [Google Scholar]
- 27. Fan Y, Wang S, Hernandez J, Yenigun VB, Hertlein G, Fogarty CE, et al. Genetic models of apoptosis-induced proliferation decipher activation of JNK and identify a requirement of EGFR signaling for tissue regenerative responses in Drosophila. PLoS Genet. Public Library of Science; 2014;10: e1004131 10.1371/journal.pgen.1004131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pastor-Pareja JC, Wu M, Xu T. An innate immune response of blood cells to tumors and tissue damage in Drosophila. Dis Model Mech. 2008;1: 144–154. 10.1242/dmm.000950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shlevkov E, Morata G. A dp53/JNK-dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ. 2012;19: 451–60. 10.1038/cdd.2011.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kondo S, Senoo-Matsuda N, Hiromi Y, Miura M. DRONC coordinates cell death and compensatory proliferation. Mol Cell Biol. 2006;26: 7258–7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wells BS, Yoshida E, Johnston LA. Compensatory proliferation in Drosophila imaginal discs requires Dronc-dependent p53 activity. Curr Biol. 2006;16: 1606–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131: 5591–5598. [DOI] [PubMed] [Google Scholar]
- 33. Perez-Garijo A, Shlevkov E, Morata G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development. 2009;136: 1169–1177. 10.1242/dev.034017 [DOI] [PubMed] [Google Scholar]
- 34. Wu M, Pastor-Pareja JC, Xu T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature. 2010;463: 545–548. 10.1038/nature08702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Álvarez-Fernández C, Tamirisa S, Prada F, Chernomoretz A, Podhajcer O, Blanco E, et al. Identification and functional analysis of healing regulators in Drosophila. PLoS Genet. 2015;11: e1004965 10.1371/journal.pgen.1004965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, Edgar BA. Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell. 2009;137: 1343–55. 10.1016/j.cell.2009.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147: 742–58. 10.1016/j.cell.2011.10.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. King RS, Newmark PA. The cell biology of regeneration. J Cell Biol. 2012;196: 553–62. 10.1083/jcb.201105099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vriz S, Reiter S, Galliot B. Cell death: a program to regenerate. Curr Top Dev Biol. 2014;108: 121–51. 10.1016/B978-0-12-391498-9.00002-4 [DOI] [PubMed] [Google Scholar]
- 40. Nishina T, Komazawa-Sakon S, Yanaka S, Piao X, Zheng D-M, Piao J-H, et al. Interleukin-11 links oxidative stress and compensatory proliferation. Sci Signal. 2012;5: ra5 10.1126/scisignal.2002056 [DOI] [PubMed] [Google Scholar]
- 41. Worley MI, Setiawan L, Hariharan IK. Regeneration and transdetermination in Drosophila imaginal discs. Annu Rev Genet. 2012;46: 289–310. 10.1146/annurev-genet-110711-155637 [DOI] [PubMed] [Google Scholar]
- 42. Bergantiños C, Vilana X, Corominas M, Serras F. Imaginal discs: Renaissance of a model for regenerative biology. Bioessays. 2010;32: 207–217. 10.1002/bies.200900105 [DOI] [PubMed] [Google Scholar]
- 43. Yagi R, Mayer F, Basler K. Refined LexA transactivators and their use in combination with the Drosophila Gal4 system. Proc Natl Acad Sci U S A. 2010;107: 16166–16171. 10.1073/pnas.1005957107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 1998;12: 557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chatterjee N, Bohmann D. A versatile ΦC31 based reporter system for measuring AP-1 and Nrf2 signaling in Drosophila and in tissue culture. Jennings B, editor. PLoS One. Public Library of Science; 2012;7: e34063 10.1371/journal.pone.0034063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kelly GS. Clinical applications of N-acetylcysteine. Altern Med Rev. 1998;3: 114–127. [PubMed] [Google Scholar]
- 47. Cully M, Genevet A, Warne P, Treins C, Liu T, Bastien J, et al. A role for p38 stress-activated protein kinase in regulation of cell growth via TORC1. Mol Cell Biol. 2010;30: 481–95. 10.1128/MCB.00688-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suzanne M, Irie K, Glise B, Agnès F, Mori E, Matsumoto K, et al. The Drosophila p38 MAPK pathway is required during oogenesis for egg asymmetric development. Genes Dev. 1999;13: 1464–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Seong K-H, Li D, Shimizu H, Nakamura R, Ishii S. Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell. 2011;145: 1049–61. 10.1016/j.cell.2011.05.029 [DOI] [PubMed] [Google Scholar]
- 50. Frantz B, Klatt T, Pang M, Parsons J, Rolando A, Williams H, et al. The activation state of p38 mitogen-activated protein kinase determines the efficiency of ATP competition for pyridinylimidazole inhibitor binding. Biochemistry. 1998;37: 13846–53. [DOI] [PubMed] [Google Scholar]
- 51. Galko MJ, Krasnow MA. Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol. 2004;2: E239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bunker BD, Nellimoottil TT, Boileau RM, Classen AK, Bilder D. The transcriptional response to tumorigenic polarity loss in Drosophila. Elife. 2015;4 10.7554/eLife.03189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Amoyel M, Anderson AM, Bach EA. JAK/STAT pathway dysregulation in tumors: a Drosophila perspective. Semin Cell Dev Biol. 2014;28: 96–103. 10.1016/j.semcdb.2014.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dobens LL, Martin-Blanco E, Martinez-Arias A, Kafatos FC, Raftery LA. Drosophila puckered regulates Fos/Jun levels during follicle cell morphogenesis. Development. 2001;128: 1845–1856. [DOI] [PubMed] [Google Scholar]
- 55. Lesch C, Jo J, Wu Y, Fish GS, Galko MJ. A targeted UAS-RNAi screen in Drosophila larvae identifies wound closure genes regulating distinct cellular processes. Genetics. 2010;186: 943–57. 10.1534/genetics.110.121822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ramet M, Lanot R, Zachary D, Manfruelli P. JNK signaling pathway is required for efficient wound healing in Drosophila. Dev Biol. 2002;241: 145–156. [DOI] [PubMed] [Google Scholar]
- 57. Freel CD, Richardson DA, Thomenius MJ, Gan EC, Horn SR, Olson MR, et al. Mitochondrial localization of Reaper to promote inhibitors of apoptosis protein degradation conferred by GH3 domain-lipid interactions. J Biol Chem. 2008;283: 367–79. [DOI] [PubMed] [Google Scholar]
- 58. Sandu C, Ryoo HD, Steller H. Drosophila IAP antagonists form multimeric complexes to promote cell death. J Cell Biol. 2010;190: 1039–52. 10.1083/jcb.201004086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Varkey J, Chen P, Jemmerson R, Abrams JM. Altered cytochrome c display precedes apoptotic cell death in Drosophila. J Cell Biol. 1999;144: 701–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Abdelwahid E, Yokokura T, Krieser RJ, Balasundaram S, Fowle WH, White K. Mitochondrial disruption in Drosophila apoptosis. Dev Cell. 2007;12: 793–806. [DOI] [PubMed] [Google Scholar]
- 61. Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem. 1996;271: 15703–7. [DOI] [PubMed] [Google Scholar]
- 62. Ilmarinen P, Moilanen E, Kankaanranta H. Mitochondria in the center of human eosinophil apoptosis and survival. Int J Mol Sci. Multidisciplinary Digital Publishing Institute; 2014;15: 3952–69. 10.3390/ijms15033952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol. 2004;14: 1262–1266. [DOI] [PubMed] [Google Scholar]
- 64. Dichtel-Danjoy M-L, Ma D, Dourlen P, Chatelain G, Napoletano F, Robin M, et al. Drosophila p53 isoforms differentially regulate apoptosis and apoptosis-induced proliferation. Cell Death Differ. 2013;20: 108–16. 10.1038/cdd.2012.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wells BS, Johnston LA. Maintenance of imaginal disc plasticity and regenerative potential in Drosophila by p53. Dev Biol. Elsevier Inc; 2012;361: 263–276. 10.1016/j.ydbio.2011.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7: 491–501. [DOI] [PubMed] [Google Scholar]
- 67. Fan Y, Bergmann A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell. 2008;14: 399–410. 10.1016/j.devcel.2008.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Herrera SC, Martin R, Morata G. Tissue homeostasis in the wing disc of Drosophila melanogaster: immediate response to massive damage during development. PLoS Genet. 2013;9: e1003446 10.1371/journal.pgen.1003446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Autsavapromporn N, de Toledo SM, Little JB, Jay-Gerin J- P, Harris AL, Azzam EI. The role of gap junction communication and oxidative stress in the propagation of toxic effects among high-dose α-particle-irradiated human cells. Radiat Res. 2011;175: 347–57. 10.1667/RR2372.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Feine I, Pinkas I, Salomon Y, Scherz A. Local oxidative stress expansion through endothelial cells—a key role for gap junction intercellular communication. Ushio-Fukai M, editor. PLoS One. Public Library of Science; 2012;7: e41633 10.1371/journal.pone.0041633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochim Biophys Acta. 2006;1758: 994–1003. [DOI] [PubMed] [Google Scholar]
- 72. Bienert GP, Møller ALB, Kristiansen KA, Schulz A, Møller IM, Schjoerring JK, et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem. 2007;282: 1183–92. [DOI] [PubMed] [Google Scholar]
- 73. Son Y, Kim S, Chung H-T, Pae H-O. Reactive oxygen species in the activation of MAP kinases. Methods Enzymol. 2013;528: 27–48. 10.1016/B978-0-12-405881-1.00002-1 [DOI] [PubMed] [Google Scholar]
- 74. Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2: 222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chakrabarti S, Poidevin M, Lemaitre B. The Drosophila MAPK p38c regulates oxidative stress and lipid homeostasis in the intestine. PLoS Genet. Public Library of Science; 2014;10: e1004659 10.1371/journal.pgen.1004659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Razzell W, Evans IR, Martin P, Wood W. Calcium Flashes Orchestrate the Wound Inflammatory Response through DUOX Activation and Hydrogen Peroxide Release. Curr Biol. 2013;23: 424–429. 10.1016/j.cub.2013.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Muliyil S, Narasimha M. Mitochondrial ROS regulates cytoskeletal and mitochondrial remodeling to tune cell and tissue dynamics in a model for wound healing. Dev Cell. 2014;28: 239–52. 10.1016/j.devcel.2013.12.019 [DOI] [PubMed] [Google Scholar]
- 78. Hinz U, Giebel B, Campos-Ortega JA. The basic-helix-loop-helix domain of Drosophila lethal of scute protein is sufficient for proneural function and activates neurogenic genes. Cell. 1994;76: 77–87. [DOI] [PubMed] [Google Scholar]
- 79. McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL. Spatiotemporal rescue of memory dysfunction in Drosophila. Science (80-). 2003;302: 1765–1768. [DOI] [PubMed] [Google Scholar]
- 80. Wing JP, Zhou L, Schwartz LM, Nambu JR. Distinct cell killing properties of the Drosophila reaper, head involution defective, and grim genes. Cell Death Differ. 1998;5: 930–939. [DOI] [PubMed] [Google Scholar]
- 81. Martin FA, Morata G. Compartments and the control of growth in the Drosophila wing imaginal disc. Development. 2006;133: 4421–4426. [DOI] [PubMed] [Google Scholar]
- 82. Baena-Lopez LA, García-Bellido A. Control of growth and positional information by the graded vestigial expression pattern in the wing of Drosophila melanogaster. Proc Natl Acad Sci U S A. 2006;103: 13734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Barrio R, de Celis JF. Regulation of spalt expression in the Drosophila wing blade in response to the Decapentaplegic signaling pathway. Proc Natl Acad Sci U S A. 2004;101: 6021–6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pastor-Pareja JC, Grawe F, Martin-Blanco E, Garcia-Bellido A. Invasive cell behavior during Drosophila imaginal disc eversion is mediated by the JNK signaling cascade. Dev Cell. 2004;7: 387–399. [DOI] [PubMed] [Google Scholar]
- 85. Harrison DA, Binari R, Nahreini TS, Gilman M, Perrimon N. Activation of a Drosophila Janus kinase (JAK) causes hematopoietic neoplasia and developmental defects. Embo J. 1995;14: 2857–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bach EA, Ekas LA, Ayala-Camargo A, Flaherty MS, Lee H, Perrimon N, et al. GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr Patterns. 2007;7: 323–331. [DOI] [PubMed] [Google Scholar]
- 87. Hou XS, Melnick MB, Perrimon N. Marelle acts downstream of the Drosophila HOP/JAK kinase and encodes a protein similar to the mammalian STATs. Cell. 1996;84: 411–419. [DOI] [PubMed] [Google Scholar]
- 88. Glise B, Bourbon H, Noselli S. hemipterous encodes a novel Drosophila MAP kinase kinase, required for epithelial cell sheet movement. Cell. 1995;83: 451–461. [DOI] [PubMed] [Google Scholar]
- 89. Lai S-L, Lee T. Genetic mosaic with dual binary transcriptional systems in Drosophila. Nat Neurosci. 2006;9: 703–9. [DOI] [PubMed] [Google Scholar]
- 90. Repiso A, Bergantinos C, Serras F. Cell fate respecification and cell division orientation drive intercalary regeneration in Drosophila wing discs. Development. 2013;140: 3541–3551. 10.1242/dev.095760 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Propagation of ROS labeled with CellROX Green towards the adjacent tissue during the first 15’ after injury. Thermal scale corresponds to the same as in Fig 1B. White line indicates cut edge. (B) ROS detected with H2DCFDA after ptc>rpr. ROS are found in dead cells and in adjacent living cells. TP-3: TO-PRO-3. (C, D) ROS detected with H2DCFDA after physical injury (white wedge).
(TIF)
(A) Ex vivo analysis of cut imaginal discs cultured in Schneider’s medium, incubated with NAC, Trolox or VitC. Top row shows images of control discs (no antioxidant). Lower row shows images of discs incubated with the indicated antioxidant. Dotted lines indicate zones used as ROI for pixel intensity measurements (below). White wedges indicate the position of the cut. *P<0.05 **P<0.01. (B) Examples of control wings kept at 17°C (sal EPv >rpr OFF) that grew in food supplemented with antioxidant. All cases, showed normal set of interveins and veins. (C) Examples of control wings kept at 17°C (sal EPv >rpr OFF) that grew from the indicated genotypes. (D) Controls for transgenes of Fig 2F. Activation of transgenes (nub>Cat; nub>Sod; nub>Sod:Cat) in the absence of cell death results in normal wings.
(TIF)
(A) Endogenous expression of the TRE-red and puc-lacZ reporters. Note that both reporters are expressed only at the tip of the notum (n: notum; wp: wing pouch). (B) TRE-red and puc>GFP expression after physical injury. Note that TRE-red expression is activated earlier and more extensive than puc>GFP. White wedges indicate the position of the cut. Dotted line indicates the edges of the disc. (C) The JNK Inhibitor IX eliminates TRE-red activity and upd expression. Top row: rpr–ablated disc from larvae fed with standard food stained for nuclei (TP3: TO-PRO-3), TRE-red reporter, and anti-Upd. Bottom row: rpr–ablated disc from larvae supplemented with JNK Inhibitor IX. (D) JNK Inhibitor IX inhibits regeneration. Quantification of regenerated sal EPv >rpr wings after feeding with standard food or JNK Inhibitor IX supplemented.
(TIF)
To test whether P-p38 is activated after an independent mechanism of oxidative stress in the absence of damage, larvae were fed with 1% H2O2 for 2 h before processed for imaging. (A) Live imaging showing high ROS in the entire disc. (B) Fixed disc stained with P-p38.
(TIF)
(A) Inhibition of p38 with RNAi constructs prevents tissue repair. Ectopic expression of p38 RNAis under the control of ci-Gal4 and simultaneous cell death induction with sal E/Pv -LHG LexO-rpr when shifted to 29°C for 11 h. Adult wing size was measured after ectopic expression of the indicated RNAi transgenes (red). The experiments with rpr-ablation are indicated in blue. (B) Examples of control wings of Fig 5B, in which no rpr-ablation was induced (kept at 17°C) for the indicated concentrations of the p38 inhibitor SB202190. (C) Test for the reliability of the SB202190. Discs were dissected from rpr-ablated larvae that were fed with 5 μM SB202190, fixed and imaged. SB202190 intake reduces P-p38 activation after cell death as measured from the Mean Pixel Intensity in comparison to DMSO fed larvae. TRE-red in individuals fed with 5 μM SB202190 is active. Right: Mean Pixel Intensities for both experiments. For p38: control 48.77 ± 30.82 (S.D.); SB202190 10.52 ± 8.09 (S.D.). For TRE-red: control 136.46 ± 44.5 (S.D.); SB202190 93.5 ± 23.19 (S.D.). ***P<0.001 for the P-p38 and P = 0,15 n.s. for TRE-red.
(TIF)
(A) RNA interference of MKK lic inhibits p38 phosphorilation after injury. The UAS-RNAi lic was activated in the posterior compartment together with UAS-GFP (white). Two injuries were inflected with tungsten needles, one in the anterior and one in the posterior compartment. The cuts were performed in Schneider’s medium, and fixation for immunostaining 20’ after injury. P-p38 activation was localized in the anterior compartment and almost absent around the posterior cut. (B) Blocking JNK with JNK Inhibitor IX does not affect P-p38 after rpr-ablation. The domain of dead cells is outlined in white.
(TIF)
Percentages of regenerated wings for the indicated genetic background after sal E/Pv >rpr ablation. Right wings: sal E/Pv >rpr OFF column: wings of those genetic backgrounds without cell death (kept at 17°C). All wings raised in those conditions contain the normal set of veins and interveins. sal E/Pv >rpr ON column: top (wt) is an example of fully regenerated wing. The rest of wings are examples of non-regenerated or incomplete regeneration in the heterozygous condition indicated.
(TIF)
Quantification of in situ hybridizations of upd mRNA (A) and upd3 mRNA (B) and antibody localization for Upd (C). Regions of interest were determined around the wound edges (as in S2 Fig) of wild type discs (wt) and p38a 1-/- mutants. Images in Fig 8A–8C are examples of the quantification shown here. ***P<0.001 **P<0.01. Bars indicate standard deviation.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.