

Abstract
AIM: To study the genesis of neointima formation in pulmonary hypertension (PH), we investigated the role of caveolin-1 and related proteins.
METHODS: Male Sprague Dawley rats were given monocrotaline (M, 40 mg/kg) or subjected to hypobaric hypoxia (H) to induce PH. Another group was given M and subjected to H to accelerate the disease process (M + H). Right ventricular systolic pressure, right ventricular hypertrophy, lung histology for medial hypertrophy and the presence of neointimal lesions were examined at 2 and 4 wk. The expression of caveolin-1 and its regulatory protein peroxisome proliferator-activated receptor (PPAR) γ, caveolin-2, proliferative and anti-apoptotic factors (PY-STAT3, p-Erk, Bcl-xL), endothelial nitric oxide synthase (eNOS) and heat shock protein (HSP) 90 in the lungs were analyzed, and the results from M + H group were compared with the controls, M and H groups. Double immunofluorescence technique was used to identify the localization of caveolin-1 in pulmonary arteries in rat lungs and in human PH lung tissue.
RESULTS: In the M + H group, PH was more severe compared with M or H group. In the 4 wk M+H group, several arteries with reduced caveolin-1 expression in endothelial layer coupled with an increased expression in smooth muscle cells (SMC), exhibited neointimal lesions. Neointima was present only in the arteries exhibiting enhanced caveolin-1 expression in SMC. Lung tissue obtained from patients with PH also revealed neointimal lesions only in the arteries exhibiting endothelial caveolin-1 loss accompanied by an increased caveolin-1 expression in SMC. Reduction in eNOS and HSP90 expression was present in the M groups (2 and 4 wk), but not in the M + H groups. In both M groups and in the M + H group at 2 wk, endothelial caveolin-1 loss was accompanied by an increase in PPARγ expression. In the M + H group at 4 wk, increase in caveolin-1 expression was accompanied by a reduction in the PPARγ expression. In the H group, there was neither a loss of endothelial caveolin-1, eNOS or HSP90, nor an increase in SMC caveolin-1 expression; or any alteration in PPARγ expression. Proliferative pathways were activated in all experimental groups.
CONCLUSION: Enhanced caveolin-1 expression in SMC follows extensive endothelial caveolin-1 loss with subsequent neointima formation. Increased caveolin-1 expression in SMC, thus, may be a prelude to neointima formation.
Keywords: Endothelial cells, Neointima, Pulmonary hypertension, Smooth muscle cells
Core tip: Neointima in pulmonary hypertension (PH) is associated with poor prognosis. Caveolin-1, a cell membrane protein has a critical role in PH. We investigated the association of caveolin-1 and neointima formation in monocrotaline (MCT) + hypoxia-treated rats, and in human PH lung sections. The progressive caveolin-1 reduction in endothelial cells is followed by an increased caveolin-1 expression in smooth muscle cells (SMC). In human PH as well as in the MCT + hypoxia model, neointima was observed only in the arteries exhibiting an increased caveolin-1 expression in SMC. Thus, the increased caveolin-1 expression in SMC may in part, facilitate neointima formation.
INTRODUCTION
Pulmonary hypertension (PH) is a rare, but a progressive disease with a high morbidity and mortality rate. Although considerable progress has been made in the field; the pathogenesis of PH, however, is not yet fully understood, which makes the design of preventive and curative treatment a daunting challenge. The advances in therapeutic modalities have improved the life expectancy as well as the quality of life; the pulmonary vascular remodeling, however, remains progressive[1]. A number of diverse diseases can develop PH, and several PH-associated gene mutations are known to significantly increase the risk of familial PH[2,3]. Irrespective of the underlying disease, severe PH is typically characterized by endothelial dysfunction, impaired vasodilatation, increased vasoconstriction, cell proliferation, medial wall thickening, PH and right ventricular hypertrophy (RVH)[4]. The development of neointima and plexiform lesions in pulmonary arteries associate with poor outcomes although whether or not they are causative of disease or result from an abnormal hemodynamic milieu remains unclear in the human PH[5].
In the monocrotaline (MCT) model, endothelial caveolin-1 loss and the activation of proliferative and anti-apoptotic pathways are observed before PH becomes evident. Concurrent loss of several endothelial cell (EC) membrane proteins including PECAM-1, soluble guanylate cyclase and Tie2 is suggestive of an extensive EC membrane damage. At 2 wk post-MCT, PH and RVH are observed, accompanied by a further disruption of EC as indicated by the loss of cytosolic proteins such as heat shock protein (HSP) 90, Akt and IκB-α[6-8]. Importantly, preventive measures restore endothelial caveolin-1 resulting in the inhibition of proliferative pathways and attenuation of PH[9,10]. Caveolin-1 is a major scaffolding protein of caveolae (50-100 nm), a subset of lipid rafts in the plasma membrane of a number of different cell types including EC and smooth muscle cells (SMC). It plays a pivotal role in maintaining vascular homeostasis. It directly interacts with transducing molecules within caveolae and stabilizes them in an inactive form. It regulates cell proliferation, apoptosis, cell differentiation, cell cycle, and also eNOS function[11-13].
The presence of pulmonary arterial hypertension (PAH) in patients with CAV-1 mutation associated with reduced endothelial caveolin-1 expression, further supports a critical role of caveolin-1 in the lung vasculature[14,15]. Importantly, the loss of endothelial caveolin-1 and vWF accompanied by an increased caveolin-1 expression in SMC has recently been reported in children and adults with PAH associated with drug toxicity, congenital heart disease and idiopathic PAH (IPAH)[16-18]. Furthermore, pulmonary arterial SMC from the patients with IPAH revealed increased capacitative Ca2+ entry and DNA synthesis; both could be attenuated by silencing caveolin-1[18]. Thus, caveolin-1 switches from being an anti-proliferative to a pro-proliferative factor. Interestingly, the dual role of caveolin-1 is a known phenomenon in cancer[19].
Studies with rat models of PH using “VEGF receptor blocker (Sugen) + hypoxia”[20], MCT + pneumonectomy[21] and MCT + hypoxia[22] have shown severe PH with neointima and plexiform lesions, closely mimicking human PH. In these models, underlying EC damage is an important initial phase. We hypothesized that the extensive EC damage and/or loss might be a prerequisite for the increased caveolin-1 expression in SMC and subsequent development of neointima. To test this hypothesis, we treated rats with MCT and exposed them to hypobaric hypoxia (MCT + hypoxia) to accelerate the disease process. Hemodynamic data, lung histopathology, the expression of caveolin-1, and proliferative and anti-apoptotic factors, endothelial nitric oxide synthase (eNOS) and HSP90 proteins were examined. We evaluated the expression of caveolin-2 because it co-localizes with caveolin-1[23], and the expression of peroxisome proliferator-activated receptor (PPAR) γ, because it regulates caveolin-1 expression[24,25], and its loss is implicated in the pathogenesis of PH[26,27]. In addition, we examined caveolin-1 expression in the lung tissue from patients with IPAH and heritable PAH (HPAH).
MATERIALS AND METHODS
Male Sprague-Dawley rats (150-175 g, Charles River Wilmington, MA) were maintained at 22 °C on a 12 h light and dark cycle in the Animal Facility. They were allowed to acclimatize for 5 d, with free access to laboratory chow and water. The Protocols were approved by the Institutional Animal Care and Use Committee at New York Medical College (IACUC # 4-1-0113), and conform to the guiding principles for the use and care of laboratory animals of the American Physiological Society, and the National Institutes of Health. Rats were divided into 4 groups: Gr1, Control rats maintained in room air; Gr2, rats received MCT (40 mg/kg, sc), and kept in room air; Gr3, rats subjected to hypobaric hypoxia (atmospheric pressure 380 mmHg); and Gr4, rats received MCT 40 mg/kg and were subjected to hypobaric hypoxia starting on day 1. The hypoxia chamber was opened twice per week for 15 min to weigh the rats, replenish food and water, and to provide clean bedding similar to the other rats in room air. At the end of 2 and 4 wk, these rats were studied.
Human lung tissue was obtained from PAH patients at the time of post-mortem autopsy or lung transplantation; control tissue was obtained from healthy subjects who died due to traumatic injuries. Vanderbilt Pulmonary Hypertension Research Cohort study participants were recruited via the Vanderbilt Pulmonary Hypertension Center. The Vanderbilt University Medical Center Institutional Review Board approved all study protocols (IRB #9401). All participants, or their surrogate custodians as appropriate, gave informed written consent to participate in genetic and clinical studies. PAH was defined either by autopsy results showing plexogenic pulmonary arteriopathy in the absence of other causes such as congenital heart disease, or by clinical and cardiac catheterization criteria. These criteria included a mean pulmonary artery pressure ≥ 25 mmHg with a pulmonary capillary wedge or left atrial pressure ≤ 15 mmHg, and exclusion of other causes of PH in accordance with accepted international standards of diagnostic criteria[2]. HPAH was considered the type of PAH if a subject met one or both of the following criteria: (1) family history of two or more subjects with confirmed PAH according to international standards of diagnostic criteria; or (2) detection of a mutation in a PAH-specific gene, such as BMPR2. The majority of lung tissue specimens available for this study from PAH patients were from subjects deceased prior to the discovery of the BMPR2 gene and other genes that could be considered PAH-specific genes which are mutated in association with HPAH. Included in this study were 7 patients: 3 with IPAH and 4 with HPAH. The age ranged from 29 to 55 years except for one patient who was 6 years old diagnosed with HPAH.
Chemicals and antibodies
All chemicals including MCT were purchased from Sigma Aldrich, St Louis, MO. Antibodies: caveolin-1α (sc894), PPARγ (sc7273), HSP90 (sc13119) purchased from Santa Cruz laboratories, Santa Cruz, CA. PY-STAT3 (Tyr705, 9145), Bcl-xL (2764), p-Erk (Thr202/Tyr204, 4370), and Erk (4695) from Cell Signaling, Beverley, MA, β actin (A5441) and α-actin (C6198) from Sigma, caveolin-2 (610684), eNOS (610297) and STAT3 (610190) from BD Transduction, Palo Alto, CA.
Measurement of right ventricular systolic pressure
Rats anesthetized with pentobarbital (60 mg/kg, ip), were ventilated through a tracheostomy ( roughly equivalent to 70-80 breaths/min)[6]. A thoracotomy was performed; and right ventricular systolic pressure (RVSP) measured with a small needle attached to a tubing (PE50). After perfusing the lungs with normal saline, heart and lungs were removed. Right lung was frozen and stored at -80 °C. The heart and the left lung were kept in 10% buffered formaldehyde.
Estimation of right ventricular hypertrophy
The ratio of the right ventricle (RV) and the left ventricle including septum (LV) was used to assess right ventricular hypertrophy (RVH)[6,7]. In addition, the ratio of RV (mg)/final body weight (FBW, g) and the ratio of LV (mg)/FBW (g) were calculated.
Estimation of protein expression
Proteins (50-100 μg) from lung supernatants were used to examine the expression of proteins of interest[6,7]. The antibodies used were caveolin-1 (1:5000), Caveolin-2 (1:500), PPARγ (1:100), PY-STAT3 (1:200), Bcl-xl (1:200), p-Erk (1:2000), eNOS (1:400), or HSP90 (1: 3000). Loading protein was evaluated using β actin (1:10000), STAT3 (1: 2000) or Erk (1:2000) as appropriate. Protein bands visualized by chemiluminescence are expressed as % normal.
Lung histopathology and double immunofluorescence
Five to 6 μm lung sections were cut from the paraffin blocks, which were processed form the lung tissue preserved in 10% formaldehyde. Hematoxylin/eosin and elastic van Gieson stains were used for histopathological evaluation. Double immunofluorescence study (on all sections) was carried out at New York Medical College Facility, using caveolin-1 and α-actin antibodies as described previously[6,7]. Immunofluorescence was evaluated using a laser scanning confocal microscope.
Statistical analysis
The data are expressed as means ± SEM. Differences among multiple means were determined by one way Anova analysis using SPSS program. Specific differences were determined using Scheffe’s test with < 0.05 as significant.
RESULTS
Weight gain
At 2 wk (n = 5-8), the weight gain in the MCT and hypoxia groups was lower compared with the controls (controls, 63 ± 3 g; MCT, 38 ± 3 ga; hypoxia, 39 ± 2 ga). In the MCT + hypoxia group, there was a further reduction in the weight gain (6 ± 7 ga,c). There was no mortality in any of the groups. aP < 0.05 vs controls, cP < 0.05 vs MCT and hypoxia groups.
At 4 wk (n = 7-11), the mortality in the MCT and the MCT + hypoxia groups were 22% and 30% respectively, but none in the hypoxia alone group. Weight gain in the hypoxia group was comparable to the controls (97 ± 7 g vs hypoxia 94 ± 4 g, P = NS). The weight gain in the MCT group was significantly reduced (68 ± 7 ga) and a further reduction was noted in the MCT + hypoxia group (45 ± 5 ga,c). aP < 0.05 vs controls, cP < 0.05 vs MCT.
Hemodynamic data
At 2 wk, RVSP and RV/LV ratio were significantly higher in the MCT, hypoxia and MCT + hypoxia groups compared with the controls (Figure 1, top panel); with a further increase at 4 wk (Figure 1, bottom panel). The ratios of RV (mg)/FBW (g) confirmed increased RVH in the MCT + hypoxia groups at 2 and 4 wk compared with the MCT and hypoxia alone groups. RV (mg)/FBW (g) ratio: 2 wk; C, 0.5 ± 0.01, MCT, 1.02 ± 0.57a, Hypoxia, 1.19 ± 0.057a, MCT + Hypoxia, 1.54 ± 0.04a,c, 4 wk; C, 0.55 ± 0.019, MCT, 1.15 ± 0.56a, hypoxia, 1.05 ± 0.08a, MCT + hypoxia, 1.59 ± 0.01a,c. aP < 0.05 vs C, cP < 0.05 vs MCT or hypoxia group. The LV (mg)/FBW (g) ratio, however, was not different in any of the experimental groups compared with the controls (data not shown).
Figure 1.
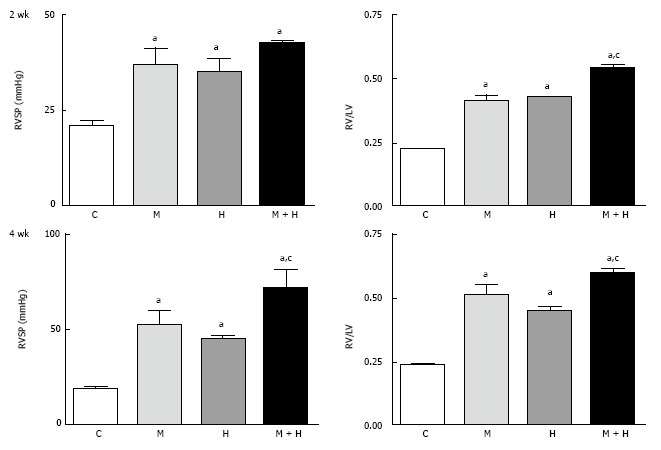
This figure depicts right ventricular systolic pressure and right ventricular hypertrophy in controls, monocrotaline, hypoxia and monocrotaline + hypoxia at 2 (n = 5-8) and 4 wk (n = 6-10). aP < 0.05 vs C, cP < 0.05 vs M and H. RVSP: Right ventricular systolic pressure; C: Controls; M: Monocrotaline; H: Hypoxia; M + H: Monocrotaline + hypoxia.
Histopathology
Experimental groups: Increased pulmonary arterial medial wall thickening is present in all the experimental groups at 2 and 4 wk (Figure 2, panels A and B). Figure 2C shows neointima in small arteries at 4 wk in the MCT + hypoxia group.
Figure 2.
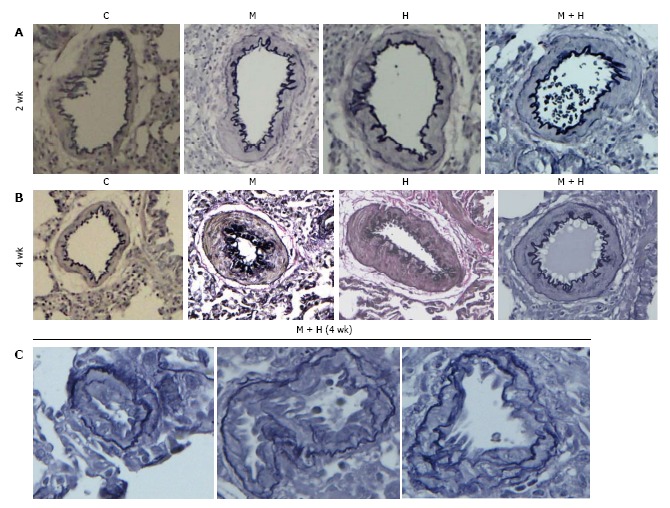
Pulmonary arteries (experimental groups). A and B: Pulmonary arteries (size 200-317 μm) from the controls and different experimental groups (elastic van Gieson stain): At 2 and 4 wk, arteries from MCT (M), hypoxia (H) and MCT + hypoxia (M + H) exhibit increased medial wall thickening compared with the control (C). Magnification = × 100; C: Arteries (size 100-155 μm) from 4 wk M + H group showing the presence of neointima. Fragmentation of internal elastic lamina can be seen in these arteries. Magnification = × 400.
Humans: Pulmonary arteries from IPAH and HPAH patients show varying degrees of medial wall thickening, neointima and luminal narrowing (Figure 3).
Figure 3.
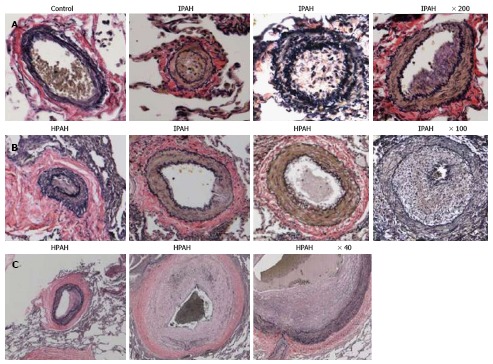
Pulmonary arteries (Human). A and B: Pulmonary arteries (size 134-323 μm) from a control, IPAH and HPAH patients. Control artery is thin walled. The arteries from patients exhibit varying degrees of muscular thickening, neointima and significant narrowing of the lumen; C: Larger arteries exhibiting vascular remodeling, extensive neointima formation and narrowing of the lumen.
Caveolin-1 and caveolin-2 expression
The expression of both caveolin-1 and caveolin-2 was significantly reduced in the MCT and MCT + hypoxia groups at 2 wk. In the hypoxia alone group, caveolin-1 expression was not reduced; however, the caveolin-2 expression was slightly but significantly reduced compared with the controls (Figure 4).
Figure 4.
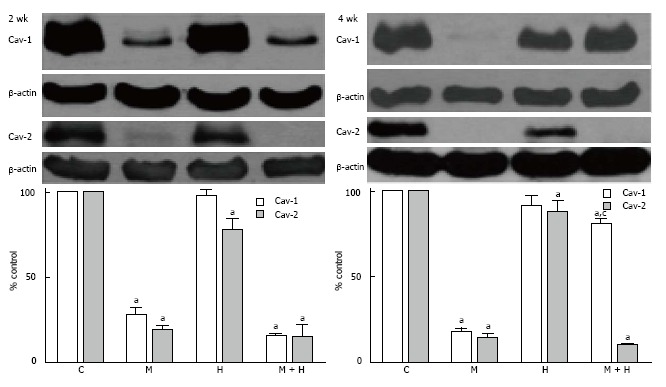
Western blots and bar graphs showing the expression of caveolin-1, caveolin-2 and β actin in controls, monocrotaline, hypoxia and monocrotaline + hypoxia at 2 (n = 3-6) and 4 wk (n = 5-8). aP < 0.05 vs C, cP < 0.05 vs M. C: Controls; M: Monocrotaline; H: Hypoxia; M + H: Monocrotaline + hypoxia.
At 4 wk, caveolin-1 and caveolin-2 were significantly reduced in the MCT group. In the hypoxia group, the expression of caveolin-1 was comparable to the controls; however, the expression of caveolin-2 was reduced, but not as low as seen in the MCT group. Importantly, in the MCT + hypoxia group, caveolin-1 expression was significantly increased compared with the MCT group (81% ± 3.9% vs 17% ± 3.6%, P < 0.05), although still low compared to the controls (81% ± 3.9% vs 100% ± 0%, P < 0.05). However, despite an increased caveolin-1 expression in this group, caveolin-2 showed a further reduction (Figure 4).
Localization of caveolin-1
Experimental groups: At 2 wk post-MCT, only 23% ± 0.87% of arteries exhibited the presence of endothelial caveolin-1. Consistent with previous observations[7]; in the current study, the endothelial caveolin-1 loss at 2 wk was not associated with an increased caveolin-1 expression in SMC. The MCT + hypoxia group showed a further reduction in the endothelial caveolin-1 expression (11% ± 1%). A few arteries displaying endothelial caveolin-1 loss exhibited increased expression of caveolin-1 in SMC (2.9% ± 0.25%). Expression of endothelial caveolin-1 in the hypoxia group, however, was not different compared with the controls (Figure 5, top panel).
Figure 5.
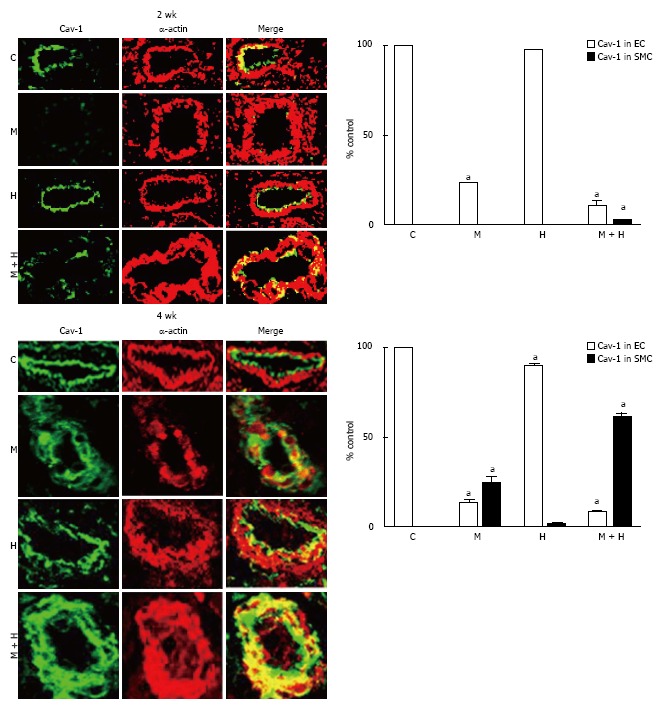
Immunofluorescence study depicting the expression of caveolin-1 (green) and smooth muscle α actin (red) in pulmonary arteries from controls, monocrotaline, hypoxia and monocrotaline + Hypoxia groups at 2 and 4 wk. The accompanying bar graphs (n = 4-5) shows the % arteries exhibiting the presence of caveolin-1 in endothelium (EC) and in smooth muscle layer (SMC). aP < 0.05 vs C. C: Controls; M: Monocrotaline; H: Hypoxia; M + H: Monocrotaline + hypoxia.
At 4 wk, in the MCT and MCT+ hypoxia groups, endothelial caveolin-1 was expressed in 13% ± 1.4% and 8% ± 0.79% of arteries respectively. In the MCT group, increased caveolin-1 expression in SMC was observed in 24% ± 3.5% of arteries. Importantly, in the MCT + hypoxia group, 61% ± 2% of arteries displayed increased caveolin-1 in SMC, consistent with the observed increase in total caveolin-1 expression in the lungs. However, the neointimal layer revealed scant expression of caveolin-1. Interestingly, in the hypoxia group, there were a few arteries with endothelial caveolin-1 loss (90% ± 0.89% vs C, 100% ± 0%, P < 0.05); and a smaller number of arteries (1.2% ± 0.58%) with endothelial caveolin-1 loss, exhibited an increased caveolin-1 expression in SMC (Figure 5, bottom panel).
Human lungs: The control pulmonary arteries showed well preserved caveolin-1 in the endothelial layer. Arteries from IPAH and HPAH patients showed varying degrees of alterations in caveolin-1 expression not unlike what was noted in the 4 wk MCT + hypoxia group, such as endothelial caveolin-1 loss, increased caveolin-1 expression in SMC and the presence of neointima (Figure 6).
Figure 6.
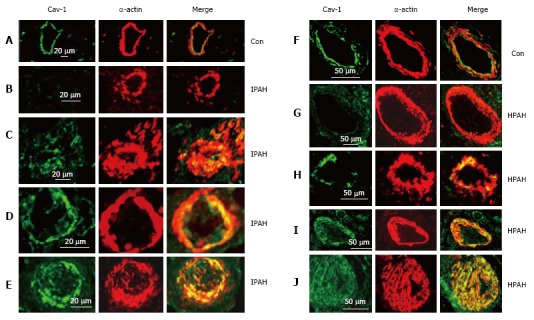
Immunofluorescence study showing the expression of caveolin-1 (green) and smooth muscle α-actin (red) in pulmonary arteries from the controls (A and F), and from the patients with idiopathic pulmonary arterial hypertension (B-E) and with heritable pulmonary arterial hypertension (G-J). In controls, endothelial caveolin-1 is well preserved and there is no enhanced expression of caveolin-1 in smooth muscle layer. Two arteries each from patients, IPAH (B and C), HPAH (G and F) show loss of endothelial caveolin-1 in B and G, and the appearance of increased expression of caveolin-1 in SMC in C and H. The next panels D, E, I and J from 4 different patients show loss of endothelial caveolin-1 and enhanced expression of caveolin-1 in SMC. PAH: Pulmonary arterial hypertension; IPAH: Idiopathic PAH; HPAH: Heritable PAH; SMC: Smooth muscle cells.
Caveolin-1 and PPARγ expression
At 2 wk, caveolin-1 loss in the MCT and MCT + hypoxia groups was accompanied by an increase in the expression of PPARγ (P < 0.05 vs controls, Figure 7). Since our previous studies had shown caveolin-1 loss at 48 h after MCT injection, we investigated the expression of PPARγ and caveolin-1 at 48 h (n = 4) and 1 wk (n = 4). At 48 h post-MCT, caveolin-1 expression was reduced to 56% ± 1.4% (P < 0.05 vs controls) associated with a PPARγ expression of 118% ± 9% (P = ns vs control). At 1 wk post-MCT, a further reduction in caveolin-1 (38% ± 1%) was associated with an increase in the expression of PPARγ (203% ± 22%, P < 0.05 vs control). No alterations were observed in the expression of PPARγ in the 2 wk hypoxia group (Figure 7).
Figure 7.
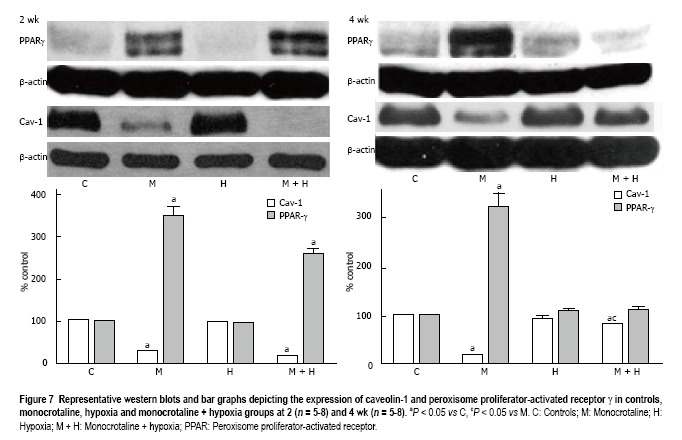
Representative western blots and bar graphs depicting the expression of caveolin-1 and peroxisome proliferator-activated receptor γ in controls, monocrotaline, hypoxia and monocrotaline + hypoxia groups at 2 (n = 5-8) and 4 wk (n = 5-8). aP < 0.05 vs C, cP < 0.05 vs M. C: Controls; M: Monocrotaline; H: Hypoxia; M + H: Monocrotaline + hypoxia; PPAR: Peroxisome proliferator-activated receptor.
At 4 wk, in the MCT group, a reciprocal increase in PPARγ expression accompanied the caveolin-1 loss. Importantly, in the MCT + hypoxia group, the increased total caveolin-1 expression in the lungs correlated with a reduction in the expression of PPARγ. In the hypoxia group, the expression of caveolin-1 was slightly decreased (90% ± 0.89%), and the PPARγ expression, however, was not altered (Figure 7).
Proliferative and anti-apoptotic pathways
As shown in Figure 8, both at 2 and 4 wk, the activation of p-Erk and PY-STAT3, and increased Bcl-xL expression were present in all experimental groups.
Figure 8.
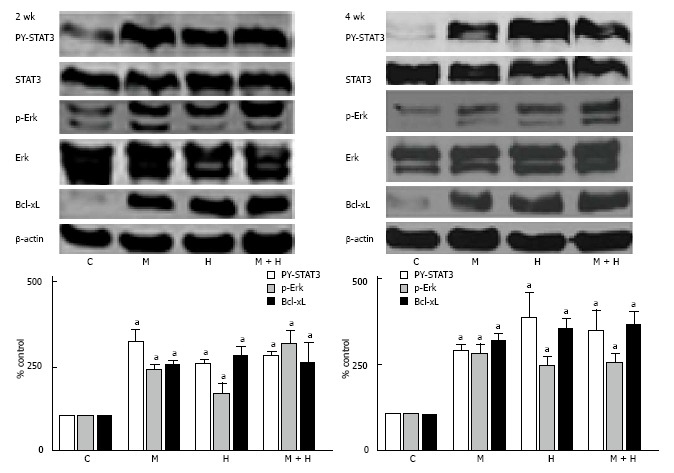
Representative western blots and bar graphs depicting the expression of PY-STAT3, p-Erk and Bcl-xL in controls, monocrotaline, hypoxia and monocrotaline + hypoxia at 2 (n = 4-7) and 4 wk (n = 5-8). STAT3, Erk and β-actin were used to assess the protein loading. aP < 0.05 vs C. C: Controls; M: Monocrotaline; H: Hypoxia; M + H: Monocrotaline + hypoxia.
eNOS and HSP90 expression
Although eNOS expression in the 2 wk-post MCT group was not significantly reduced compared with the controls, the expression of HSP90, however, was reduced (P < 0.05 vs controls). Expression of eNOS was increased in the hypoxia group, but the HSP90 expression was unaltered. In the MCT + hypoxia group, an increased eNOS expression, and a normal HSP90 expression were observed (Figure 9).
Figure 9.
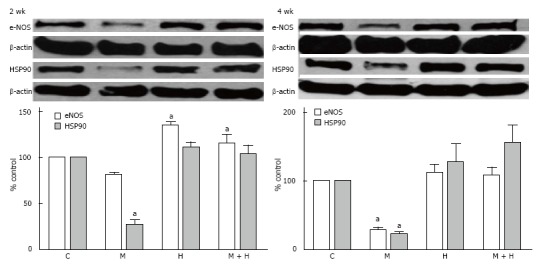
Representative western blots and bar graphs depicting the expression of endothelial nitric oxide synthase, HSP90 and β-actin in controls, monocrotaline, hypoxia and monocrotaline + hypoxia at 2 (n = 3-6) and 4 wk (n = 4-7). aP < 0.05 vs C. C: Controls; M: Monocrotaline; H: Hypoxia; M + H: Monocrotaline + hypoxia.
At 4 wk, in the MCT group, eNOS and HSP90 levels were reduced. In the hypoxia and MCT + hypoxia groups, eNOS and HSP90 levels were not altered (Figure 9).
DISCUSSION
The significant aspect of our study is the progressive disruption and loss of endothelial caveolin-1, activated proliferative pathways leading to PH in the MCT model. By 4 wk, a further reduction in endothelial caveolin-1 is accompanied by an increased caveolin-1 expression in SMC, observed in 24% of the arteries. The total caveolin-1 expression, however, remained significantly low. Exposure of MCT-treated rats to hypoxia accelerated the disease process. An increased number of arteries exhibited augmented caveolin-1 expression in SMC associated with an increase in total caveolin-1 expression. Importantly, some of the arteries exhibiting an increased caveolin-1 expression in SMC displayed neointima with scant caveolin-1. Furthermore, lung sections from patients with IPAH as well as HPAH showed similar changes, i.e., endothelial caveolin-1 loss, increased caveolin-1 in SMC. Neointimal lesions were seen only in arteries with increased caveolin-1 expression in SMC.
Neointima and plexiform lesions have been described in rodent PH models such as Sugen + hypoxia and pneumonectomy + MCT[20,21,28]. In the Sugen + hypoxia model, the initial EC apoptosis is followed by cellular proliferation and angiogenesis deregulation resulting in plexiform lesions with significantly reduced caveolin-1 expression[29,30]. The reduced expression of caveolin-1 in plexiform lesion is supported by the electron microscopic examination showing a lack of caveolae[31]; the total caveolin-1 protein levels in the lungs, however, are not decreased[32]. In-vitro studies have shown that in response to cyclic stretch, caveolin-1 in SMC shifts to non-caveolar sites, mediates Erk activation and participates in cell proliferation. Interestingly, SMC not expressing caveolin-1 fail to proliferate when subjected to cyclic stretch[33,34]. It is likely, that the extensive damage and/or loss of EC, leads to the exposure of SMC to direct shear stress and pressure, resulting in the caveolin-1 shift from caveolae to non-caveolar sites, thus altering caveolin-1 function.
In the hypoxia group, at 2 wk, there was no endothelial caveolin-1 loss, indicating that there was no physical disruption of EC. During hypoxia, caveolin-1 forms a tight complex with eNOS[19,35], leading to the dysfunction of both factors. Removal of hypoxia[36,37] or eNOS/caveolin-1 complex disruption attenuates PH[38]. At 4 wk, the total caveolin-1 expression in the lungs was not altered, but immunofluorescence studies revealed a small loss in endothelial caveolin-1 accompanied by 1.2% of arteries exhibiting increased caveolin-1 expression in SMC. It is noteworthy that in infants with respiratory distress syndrome or bronchopulmonary dysplasia, PH in the absence of EC disruption, does not lead to endothelial caveolin-1 loss or increased caveolin-1 expression in SMC. However, accompanying inflammation results in endothelial cell membrane disruption and endothelial caveolin-1 loss with subsequent increased caveolin-1 expression in SMC[16]. These studies suggest that the endothelial disruption and the endothelial caveolin-1 loss may be necessary for the increased caveolin-1 expression in SMC.
Caveolin-2 loss concomitant with caveolin-1 loss has been shown in the experimental models of PH, and the rescue of caveolin-1 restores caveolin-2 expression[10,39]. Caveolin-2 is expressed in a number of cell types including EC and SMC, and it colocalizes with caveolin-1 and necessitates caveolin-1 for its transport to caveolae[23]. However, caveolin-2 is not necessary for caveolar localization of caveolin-1; but the co-expression of caveolin-1 and 2 results in a more efficient formation of caveolae[40,41]. In the present study, MCT-treated rats exhibited a significant loss of caveolin-2 concomitant with the loss of caveolin-1. In the MCT + hypoxia group at 4 wk, despite an increase in the total caveolin-1 expression, a significant loss of caveolin-2 was present, which supports the view that the major part of caveolin-1 in SMC may not be localized in caveolae. In the hypoxia group, despite the presence of caveolin-1, some loss of caveolin-2 was observed, suggesting that a part of caveolin-1 may not be available for caveolin-2 localization.
All experimental groups (MCT, hypoxia and MCT + hypoxia) at 2 and 4 wk revealed the activation of PY-STAT3, pERK1/2 and Bcl-xL. Caveolin-1 is a well known inhibitor of pro-proliferative and anti-apoptotic factors[11,42]; and the rescue of caveolin-1 as a preventive measure in the MCT model, inhibits the activation of proliferative pathways and attenuates PH[9,10]. Interestingly, in the presence of caveolin-1 in hypoxia groups and MCT + hypoxia group at 4 wk, proliferative pathways were activated; which strongly suggest that caveolin-1 is dysfunctional in these groups.
In the 4 wk MCT group, the expression of eNOS and HSP90 was significantly reduced, but was normal in the MCT + hypoxia groups. In addition, caveolin-1 expression in native EC and in neointimal cells was sparse in the latter group. Strong eNOS expression and low caveolin-1 expression have been reported in the plexiform lesions[39,43], besides, oxidant stress is a critical feature in patients with IPAH[44]. The major cause of PH in caveolin-1 knockout mice is thought to be eNOS uncoupling and subsequent oxidative and nitrosative stress; and PH is attenuated by caveolin-1 re-expression, eNOS inhibition or treatment with superoxide dismutase mimetic[45,46]. Furthermore, EC from patients with IPAH show caveolin-1 degradation induced by sustained eNOS and Src signaling[47]. It is important to note, that caveolin-1 regulates eNOS-derived NO and superoxide, and NOX activity. Caveolin-1 sequestrates uncoupled eNOS, inhibits superoxide formation and prevents eNOS oxidase activity[48,49]. These observations support a pivotal role for caveolin-1 in preventing oxidative and nitrosative stress.
Protein and mRNA expression of PPARγ is described to be low in IPAH, Sugen + hypoxia[26] and the shunt[27] models of PH, but not in chronic obstructive pulmonary disease patients[26]. PPARγ, a ligand-activated transcription factor belongs to the nuclear hormone superfamily. In several cell systems, PPARγ has been shown to upregulate caveolin-1 expression[24,25,50]. In the present study, PPARγ levels revealed an inverse relationship with caveolin-1 in the MCT groups; initial low endothelial and total ceveolin-1 levels were associated with increased PPARγ levels. At 4 wk in the MCT + hypoxia group, an increase in total caveolin-1 was associated with a decrease in PPARγ levels. The increased expression of PPARγ may be a compensatory mechanism to upregulate the caveolin-1 expression during the initial phase of PH associated with significantly reduced caveolin-1 levels. In the hypoxia group, however, the PPARγ levels were not altered. A thiazolidinedione (TZD) compound (PPARγ activator) has been reported to attenuate hypoxia-induced PH[51]. Some of the TZD compounds are reported to have cholesterol disruptive function independent of PPARγ[52]. Interestingly, cholesterol lowering statins in the hypoxia model of PH has been shown to disrupt the tight complex of eNOS and caveolin-1 resulting in the restoration of eNOS function and the attenuation of PH[38]. Recent studies have shown that increased PPARγ expression portends poor prognosis in some forms of cancer[53,54]. In view of these observations, increasing PPARγ levels as a therapeutic measure in PH is of some concern. It is possible that PPARγ activation may be beneficial in some forms of PH or at some stage during the disease; or a selective increase in PPARγ expression in EC may be useful. In any case, further studies are necessary to ascertain the roles of PPARγ and caveolin-1, and their interrelationship in PH.
In conclusion, addition of hypoxia to MCT-treated rats results in an acceleration of the disease process. Extensive endothelial damage, progressive endothelial caveolin-1 loss, and increased caveolin-1 expression in SMC accompanied by an augmented total caveolin-1 protein expression in lungs is followed by neointima formation. In addition, caveolin-1 and PPARγ revealed an inverse relationship (Figure 10). Importantly, lung sections from IPAH and HPAH patients showed similar alterations in caveolin-1 expression, i.e., endothelial caveolin-1 loss and increased caveolin-1 expression in SMC. Both in humans and the MCT + hypoxia group, neointimal lesions were observed only in the arteries exhibiting increased caveolin-1 expression in SMC. Since increased caveolin-1 expression in SMC has been shown to be actively pro-proliferative, this alteration in caveolin-1 expression may be a prelude to neointima formation. In the hypoxia group, in the absence of endothelial disruption or the endothelial caveolin-1 loss, there was neither an increased expression of caveolin-1 in SMC nor neointima. These results suggest that the endothelial cell integrity may be an important factor that determines the course of the disease.
Figure 10.
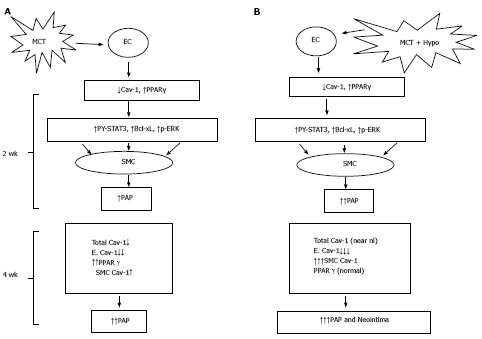
Monocrotaline injury to endothelial cells resulting in the loss of caveolin-1 and the activation of the proliferative pathways (PY-STAT3, Bcl-xL, p-ERK) leading to PH at 2 wk; and a reciprocal relationship between caveolin-1 and peroxisome proliferator-activated receptorγ expression (A) and MCT + hypoxia (MCT + Hypo) accelerates the disease process (B). At 4 wk, there is a further loss of endothelial caveolin-1 (E. Cav-1) and enhanced expression of cav-1 in smooth muscle cells (SMC), however, the total cav-1 levels remain low (17% vs C, 100%). These alterations are accompanied by a further increase in pulmonary artery pressure. Panel B shows MCT + hypoxia (MCT + Hypo) accelerates the disease process. At 2 wk, extensive endothelial caveolin-1 is accompanied by the activation of proliferative pathways and PH (higher pulmonary artery pressure compared with MCT alone group). At 4 wk, a further loss of E. Cav-1 is accompanied by significantly increased expression of caveolin-1 in SMC compared with MCT alone group. At this stage total caveolin-1 level is closer to normal (81% vs C, 100%), and neointimal lesions can be seen. MCT: Monocrotaline; EC: Endothelial cells; PPAR: Peroxisome proliferator-activated receptor.
ACKNOWLEDGEMENTS
We thank the patients for their valuable contributions, and acknowledge Lisa Wheeler (Vanderbilt University, Nashville) who coordinated the study enrollment and sample acquisition for human studies.
COMMENTS
Background
Neointima formation in pulmonary hypertension (PH) portends poor prognosis. Despite major advances in the field, the mechanism/s of pathogenesis is not yet clear, which makes the therapeutic measures a challenge.
Research frontiers
Caveolin-1, a membrane protein plays a significant role in pulmonary vascular homeostasis and in the pathogenesis of PH.
Innovation and breakthroughs
An important aspect of the study is that caveolin-1 plays a dual role in the pathogenesis of PH, i.e., as an anti-proliferative and a pro-proliferative factor. This change in function of caveolin-1 is similar to what has been reported in cancer. Loss of endothelial caveolin-1 leads to the activation of proliferative pathways, vascular remodeling and PH. As the disease progresses, the resulting extensive endothelial caveolin-1 loss associated with endothelial cell damage is followed by an enhanced expression of caveolin-1 in smooth muscle cells (SMC). The authors have shown that by subjecting monocrotaline-treated rats to hypoxia accelerates the disease process; by 4 wk, a large number of arteries exhibit enhanced expression of caveolin-1 in SMC. This caveolin-1 becomes pro-proliferative and facilitates cell proliferation, cell migration and neointima formation. In the experimental models and humans, neointima is observed only in the arteries exhibiting extensive endothelial damage and enhanced expression of caveolin-1 in SMC.
Application
The results would lead to further research in the role of caveolin-1 in SMC in PH, and in assessing the effects of modulation of caveolin-1 expression.
Peer-review
The present investigation is well written and interesting.
Footnotes
P- Reviewer: Izawa KP, Lazzeri C S- Editor: Ji FF L- Editor: A E- Editor: Lu YJ
Supported by Funds from NYMC Research Endowment Fund under the College’s intramural support program (RM); the National Institutes of Health (JEL, R01 HL111259); and K23 HL098743 (EDA).
Institutional review board statement: The study was reviewed and approved by the Vanderbilt University.
Institutional animal care and use committee statement: All procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee (New York Medical College) (IACUC procol # IACUC#4-1-0113), and conform to the guiding principles for the use and care of laboratory animals of the American Physiological Society, and the National Institutes of Health.
Conflict-of-interest statement: None.
Data sharing statement: No additional data are available.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 3, 2015
First decision: June 9, 2015
Article in press: September 8, 2015
References
- 1.Pogoriler JE, Rich S, Archer SL, Husain AN. Persistence of complex vascular lesions despite prolonged prostacyclin therapy of pulmonary arterial hypertension. Histopathology. 2012;61:597–609. doi: 10.1111/j.1365-2559.2012.04246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, Gomez Sanchez MA, Krishna Kumar R, Landzberg M, Machado RF, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D34–D41. doi: 10.1016/j.jacc.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 3.Soubrier F, Chung WK, Machado R, Grünig E, Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62:D13–D21. doi: 10.1016/j.jacc.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 4.Tuder RM, Archer SL, Dorfmüller P, Erzurum SC, Guignabert C, Michelakis E, Rabinovitch M, Schermuly R, Stenmark KR, Morrell NW. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D4–12. doi: 10.1016/j.jacc.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jonigk D, Golpon H, Bockmeyer CL, Maegel L, Hoeper MM, Gottlieb J, Nickel N, Hussein K, Maus U, Lehmann U, et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am J Pathol. 2011;179:167–179. doi: 10.1016/j.ajpath.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang J, Wolk JH, Gewitz MH, Mathew R. Progressive endothelial cell damage in an inflammatory model of pulmonary hypertension. Exp Lung Res. 2010;36:57–66. doi: 10.3109/01902140903104793. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Wolk JH, Gewitz MH, Mathew R. Caveolin-1 expression during the progression of pulmonary hypertension. Exp Biol Med (Maywood) 2012;237:956–965. doi: 10.1258/ebm.2012.011382. [DOI] [PubMed] [Google Scholar]
- 8.Mathew R, Huang J, Shah M, Patel K, Gewitz M, Sehgal PB. Disruption of endothelial-cell caveolin-1alpha/raft scaffolding during development of monocrotaline-induced pulmonary hypertension. Circulation. 2004;110:1499–1506. doi: 10.1161/01.CIR.0000141576.39579.23. [DOI] [PubMed] [Google Scholar]
- 9.Huang J, Kaminski PM, Edwards JG, Yeh A, Wolin MS, Frishman WH, Gewitz MH, Mathew R. Pyrrolidine dithiocarbamate restores endothelial cell membrane integrity and attenuates monocrotaline-induced pulmonary artery hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1250–L1259. doi: 10.1152/ajplung.00069.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jasmin JF, Mercier I, Dupuis J, Tanowitz HB, Lisanti MP. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Circulation. 2006;114:912–920. doi: 10.1161/CIRCULATIONAHA.106.634709. [DOI] [PubMed] [Google Scholar]
- 11.Krajewska WM, Masłowska I. Caveolins: structure and function in signal transduction. Cell Mol Biol Lett. 2004;9:195–220. [PubMed] [Google Scholar]
- 12.Maniatis NA, Chernaya O, Shinin V, Minshall RD. Caveolins and lung function. Adv Exp Med Biol. 2012;729:157–179. doi: 10.1007/978-1-4614-1222-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathew R. Pathogenesis of pulmonary hypertension: a case for caveolin-1 and cell membrane integrity. Am J Physiol Heart Circ Physiol. 2014;306:H15–H25. doi: 10.1152/ajpheart.00266.2013. [DOI] [PubMed] [Google Scholar]
- 14.Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA, Palomero T, Sumazin P, Kim HR, Talati MH, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5:336–343. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Best DH, Austin ED, Chung WK, Elliott CG. Genetics of pulmonary hypertension. Curr Opin Cardiol. 2014;29:520–527. doi: 10.1097/HCO.0000000000000105. [DOI] [PubMed] [Google Scholar]
- 16.Dereddy N, Huang J, Erb M, Guzel S, Wolk JH, Sett SS, Gewitz MH, Mathew R. Associated inflammation or increased flow-mediated shear stress, but not pressure alone, disrupts endothelial caveolin-1 in infants with pulmonary hypertension. Pulm Circ. 2012;2:492–500. doi: 10.4103/2045-8932.105038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathew R, Huang J, Katta US, Krishnan U, Sandoval C, Gewitz MH. Immunosuppressant-induced endothelial damage and pulmonary arterial hypertension. J Pediatr Hematol Oncol. 2011;33:55–58. doi: 10.1097/MPH.0b013e3181ec0ede. [DOI] [PubMed] [Google Scholar]
- 18.Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, Swaney JS, Niesman IR, Schermuly RT, Pullamsetti SS, et al. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. FASEB J. 2007;21:2970–2979. doi: 10.1096/fj.07-8424com. [DOI] [PubMed] [Google Scholar]
- 19.Mathew R. Cell-specific dual role of caveolin-1 in pulmonary hypertension. Pulm Med. 2011;2011:573432. doi: 10.1155/2011/573432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD, Voelkel NF, McMurtry IF, Oka M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747–2754. doi: 10.1161/CIRCULATIONAHA.109.927681. [DOI] [PubMed] [Google Scholar]
- 21.Okada K, Tanaka Y, Bernstein M, Zhang W, Patterson GA, Botney MD. Pulmonary hemodynamics modify the rat pulmonary artery response to injury. A neointimal model of pulmonary hypertension. Am J Pathol. 1997;151:1019–1025. [PMC free article] [PubMed] [Google Scholar]
- 22.Morimatsu Y, Sakashita N, Komohara Y, Ohnishi K, Masuda H, Dahan D, Takeya M, Guibert C, Marthan R. Development and characterization of an animal model of severe pulmonary arterial hypertension. J Vasc Res. 2012;49:33–42. doi: 10.1159/000329594. [DOI] [PubMed] [Google Scholar]
- 23.Parolini I, Sargiacomo M, Galbiati F, Rizzo G, Grignani F, Engelman JA, Okamoto T, Ikezu T, Scherer PE, Mora R, et al. Expression of caveolin-1 is required for the transport of caveolin-2 to the plasma membrane. Retention of caveolin-2 at the level of the golgi complex. J Biol Chem. 1999;274:25718–25725. doi: 10.1074/jbc.274.36.25718. [DOI] [PubMed] [Google Scholar]
- 24.Burgermeister E, Tencer L, Liscovitch M. Peroxisome proliferator-activated receptor-gamma upregulates caveolin-1 and caveolin-2 expression in human carcinoma cells. Oncogene. 2003;22:3888–3900. doi: 10.1038/sj.onc.1206625. [DOI] [PubMed] [Google Scholar]
- 25.Hu Q, Zhang XJ, Liu CX, Wang XP, Zhang Y. PPARgamma1-induced caveolin-1 enhances cholesterol efflux and attenuates atherosclerosis in apolipoprotein E-deficient mice. J Vasc Res. 2010;47:69–79. doi: 10.1159/000235927. [DOI] [PubMed] [Google Scholar]
- 26.Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, Wick M, Nemenoff RA, Geraci MW, Voelkel NF. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92:1162–1169. doi: 10.1161/01.RES.0000073585.50092.14. [DOI] [PubMed] [Google Scholar]
- 27.Tian J, Smith A, Nechtman J, Podolsky R, Aggarwal S, Snead C, Kumar S, Elgaish M, Oishi P, Göerlach A, et al. Effect of PPARgamma inhibition on pulmonary endothelial cell gene expression: gene profiling in pulmonary hypertension. Physiol Genomics. 2009;40:48–60. doi: 10.1152/physiolgenomics.00094.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White RJ, Meoli DF, Swarthout RF, Kallop DY, Galaria II, Harvey JL, Miller CM, Blaxall BC, Hall CM, Pierce RA, et al. Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L583–L590. doi: 10.1152/ajplung.00321.2006. [DOI] [PubMed] [Google Scholar]
- 29.Sakao S, Taraseviciene-Stewart L, Wood K, Cool CD, Voelkel NF. Apoptosis of pulmonary microvascular endothelial cells stimulates vascular smooth muscle cell growth. Am J Physiol Lung Cell Mol Physiol. 2006;291:L362–L368. doi: 10.1152/ajplung.00111.2005. [DOI] [PubMed] [Google Scholar]
- 30.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF, Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001;15:427–438. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 31.Smith P, Heath D. Electron microscopy of the plexiform lesion. Thorax. 1979;34:177–186. doi: 10.1136/thx.34.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Achcar RO, Demura Y, Rai PR, Taraseviciene-Stewart L, Kasper M, Voelkel NF, Cool CD. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest. 2006;129:696–705. doi: 10.1378/chest.129.3.696. [DOI] [PubMed] [Google Scholar]
- 33.Kawabe J, Okumura S, Lee MC, Sadoshima J, Ishikawa Y. Translocation of caveolin regulates stretch-induced ERK activity in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2004;286:H1845–H1852. doi: 10.1152/ajpheart.00593.2003. [DOI] [PubMed] [Google Scholar]
- 34.Sedding DG, Braun-Dullaeus RC. Caveolin-1: dual role for proliferation of vascular smooth muscle cells. Trends Cardiovasc Med. 2006;16:50–55. doi: 10.1016/j.tcm.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Murata T, Sato K, Hori M, Ozaki H, Karaki H. Decreased endothelial nitric-oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia-induced pulmonary hypertension. J Biol Chem. 2002;277:44085–44092. doi: 10.1074/jbc.M205934200. [DOI] [PubMed] [Google Scholar]
- 36.Burke DL, Frid MG, Kunrath CL, Karoor V, Anwar A, Wagner BD, Strassheim D, Stenmark KR. Sustained hypoxia promotes the development of a pulmonary artery-specific chronic inflammatory microenvironment. Am J Physiol Lung Cell Mol Physiol. 2009;297:L238–L250. doi: 10.1152/ajplung.90591.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sluiter I, van Heijst A, Haasdijk R, Kempen MB, Boerema-de Munck A, Reiss I, Tibboel D, Rottier RJ. Reversal of pulmonary vascular remodeling in pulmonary hypertensive rats. Exp Mol Pathol. 2012;93:66–73. doi: 10.1016/j.yexmp.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 38.Murata T, Kinoshita K, Hori M, Kuwahara M, Tsubone H, Karaki H, Ozaki H. Statin protects endothelial nitric oxide synthase activity in hypoxia-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2005;25:2335–2342. doi: 10.1161/01.ATV.0000186184.33537.48. [DOI] [PubMed] [Google Scholar]
- 39.Taraseviciene-Stewart L, Scerbavicius R, Choe KH, Cool C, Wood K, Tuder RM, Burns N, Kasper M, Voelkel NF. Simvastatin causes endothelial cell apoptosis and attenuates severe pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;291:L668–L676. doi: 10.1152/ajplung.00491.2005. [DOI] [PubMed] [Google Scholar]
- 40.Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, Kneitz B, Hou H, Christ GJ, Edelmann W, et al. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol. 2002;22:2329–2344. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujimoto T, Kogo H, Nomura R, Une T. Isoforms of caveolin-1 and caveolar structure. J Cell Sci. 2000;113 Pt 19:3509–3517. doi: 10.1242/jcs.113.19.3509. [DOI] [PubMed] [Google Scholar]
- 42.Hassan GS, Williams TM, Frank PG, Lisanti MP. Caveolin-1-deficient aortic smooth muscle cells show cell autonomous abnormalities in proliferation, migration, and endothelin-based signal transduction. Am J Physiol Heart Circ Physiol. 2006;290:H2393–H2401. doi: 10.1152/ajpheart.01161.2005. [DOI] [PubMed] [Google Scholar]
- 43.Mason NA, Springall DR, Burke M, Pollock J, Mikhail G, Yacoub MH, Polak JM. High expression of endothelial nitric oxide synthase in plexiform lesions of pulmonary hypertension. J Pathol. 1998;185:313–318. doi: 10.1002/(SICI)1096-9896(199807)185:3<313::AID-PATH93>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 44.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–769. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 45.Murata T, Lin MI, Huang Y, Yu J, Bauer PM, Giordano FJ, Sessa WC. Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J Exp Med. 2007;204:2373–2382. doi: 10.1084/jem.20062340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, Brovkovych V, Yuan JX, Wharton J, Malik AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009;119:2009–2018. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bakhshi FR, Mao M, Shajahan AN, Piegeler T, Chen Z, Chernaya O, Sharma T, Elliott WM, Szulcek R, Bogaard HJ, et al. Nitrosation-dependent caveolin 1 phosphorylation, ubiquitination, and degradation and its association with idiopathic pulmonary arterial hypertension. Pulm Circ. 2013;3:816–830. doi: 10.1086/674753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen F, Barman S, Yu Y, Haigh S, Wang Y, Black SM, Rafikov R, Dou H, Bagi Z, Han W, et al. Caveolin-1 is a negative regulator of NADPH oxidase-derived reactive oxygen species. Free Radic Biol Med. 2014;73:201–213. doi: 10.1016/j.freeradbiomed.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karuppiah K, Druhan LJ, Chen CA, Smith T, Zweier JL, Sessa WC, Cardounel AJ. Suppression of eNOS-derived superoxide by caveolin-1: a biopterin-dependent mechanism. Am J Physiol Heart Circ Physiol. 2011;301:H903–H911. doi: 10.1152/ajpheart.00936.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Llaverias G, Vázquez-Carrera M, Sánchez RM, Noé V, Ciudad CJ, Laguna JC, Alegret M. Rosiglitazone upregulates caveolin-1 expression in THP-1 cells through a PPAR-dependent mechanism. J Lipid Res. 2004;45:2015–2024. doi: 10.1194/jlr.M400049-JLR200. [DOI] [PubMed] [Google Scholar]
- 51.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, Hart CM. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol. 2010;42:482–490. doi: 10.1165/rcmb.2008-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang M, Wise SC, Leff T, Su TZ. Troglitazone, an antidiabetic agent, inhibits cholesterol biosynthesis through a mechanism independent of peroxisome proliferator-activated receptor-gamma. Diabetes. 1999;48:254–260. doi: 10.2337/diabetes.48.2.254. [DOI] [PubMed] [Google Scholar]
- 53.Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, Briggs M, Heyman R, Auwerx J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat Med. 1998;4:1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- 54.Zaytseva YY, Wallis NK, Southard RC, Kilgore MW. The PPARgamma antagonist T0070907 suppresses breast cancer cell proliferation and motility via both PPARgamma-dependent and -independent mechanisms. Anticancer Res. 2011;31:813–823. [PubMed] [Google Scholar]