

Abstract
Objective(s):
Melatonin is known as an anti-inflammatory agent, and it has been proven to exert neuroprotection through inhibition of cell death (apoptosis) in several models of brain injury. Secondary injury following the primary traumatic brain injury (TBI) results in glial cells activation, especially astrocytes. In fact, astrocyte activation causes the production of pro-inflammatory cytokines that may lead to secondary injury. Since most TBI research studies have focused on injured neurons and paid little attention to glial cells, the aim of current study was to investigate the effects of melatonin against astrocytes activation (astrogliosis), as well as inhibition of apoptosis in brain tissue of male rats after TBI.
Materials and Methods:
The animals were randomly allocated into five groups: sham group, TBI+ vehicle group (1% ethanol in saline) and TBI+ melatonin groups (5 mg/kg, 10 mg/kg and 20 mg/kg). All rats were intubated and then exposed to diffuse TBI, except for the sham group. Immunohistochemical methods were conducted using glial fibrillary acidic protein (GFAP) marker and TUNEL assay to evaluate astrocyte reactivity and cell death, respectively.
Results:
The results showed that based on the number of GFAP positive astrocytes in brain cortex, astrogliosis was reduced significantly (P<0.05) in melatonin- treated groups (no dose dependent) compared to the vehicle group. Furthermore, based on TUNEL results, melatonin treatment considerably reduced the number of apoptotic cells (P<0.05).
Conclusion:
In total, the present findings suggest that melatonin treatment following TBI diminishes astrocyte reactivity and neuronal cells apoptosis in brain cortex in the rat model.
Keywords: Apoptosis, Astrogliosis, GFAP, Melatonin, Traumatic brain injury
Introduction
Traumatic brain injury (TBI) is the leading cause of death and disability among the youth in developed countries. Annually, 1.5 million people lose their lives due to TBI worldwide (1). Based on an epidemiological study, accidents are the second leading cause of mortality and morbidity in Iran (2). TBI following motorcycle accidents is the most common cause of permanent disability of individuals in Iran (3). TBI is known to cause primary mechanical injury of neurons; however, the secondary damage following injury is complex with a broad spectrum of symptoms. The impact can be devastating on person’s life (4). Neural inflammation and oxidative stress are the main pathological mechanisms of neuronal cell apoptosis after TBI (5, 6). The role of inflammatory cascade activated after TBI is very important in brain tissue homeostasis, mediated by the release of pro- and anti-inflammatory cytokines and chemokine (Interleukin, TNF-α, Fas Ligand), which are normally scarcely detectable in healthy brain tissue, but quickly upregulated in response to stressful or pathological conditions (7).
Different strategies such as melatonin treatment can be adopted to reduce the complications of TBI, using varieties of antioxidant compounds (8). Melatonin (N-acetyl 5-methoxytryptamine), originally identified by Lerner in 1958, is a tryptophan derivatives that is produced mainly in the pineal gland. Control of circadian rhythm and sleep induction are the main physiological functions of melatonin (9, 10). There is some evidence showing the reduction of melatonin secretion after TBI (11); hence, some scientists tried to use the exogenous treatment of melatonin to evaluate its neuroprotective effects on different neural cell injuries. For instance, administration of melatonin after TBI in an animal contusion model facilitates neurobehavioral recovery (3, 4). Besides, experimental and clinical data confirm that melatonin has an important role in the reduction of adhesion molecules and pro-inflammatory cytokines. Moreover, experimental evidence supports its function as a direct and indirect antioxidant, free radicals scavenger and antioxidant enzymes stimulating agent (12).
Most TBI research studies have focused on injured neurons, while little attention has been paid to glial cells. Glial cells, particularly astrocytes become activated after TBI and have an important role in additional injury (13-15). Activated astrocytes are one of the main sources of pro-inflammatory cytokines, which are the main cause of prominent histological brain injury (16). Nevertheless, not enough attention has been paid to the effects of melatonin on astrocytes function and activation. Since brain tissue is susceptible to inflammation and free radicals (17), we conducted a study to examine the neuroprotective effects of melatonin on the prevention of neuronal cells apoptosis and astrocytes activation in male rats after TBI.
Materials and Methods
Animals and experimental protocols
All experiments of this study were performed in accordance with the animal experimental protocols approved by the Ethics Committee of Kerman University of Medical Sciences (EC/KNRC/90-2). Animals (NMRI adult male rats weighing 250–300 g) maintained in a climate controlled room (22–25 °C), with a 12-hr light/dark cycle and free access to laboratory chow and water.
A total of 40 adult male rats were randomly divided into five groups before TBI induction; sham (intact group); a vehicle-treated TBI group that received an injection of vehicle (1% ethanol in saline) after TBI at the corresponding points in time; and melatonin-treated TBI groups (TBI+M5, TBI M10, TBI+M20) that were exposed to brain trauma and received 5 mg/kg, 10 mg/kg, and 20 mg/kg melatonin IP, respectively (Sigma, St. Louis, MO) at 1, 24, 48 and 72 hr post-TBI (18). Eventually, the brain tissues of animals from each group were processed for histological examinations.
Induction of TBI
All rats were intubated and then exposed to diffuse TBI (moderate type). As previously described by marmarou (19), in this method a 250 g weight is dropped onto the head of the anesthetized male rat (from free-falling tube with a height of 2 m), while a 3 mm thick steel’s disc with a diameter of 10 mm was attached to the animal’s skull. After induction of brain trauma, the animals were connected to a respiratory pump (Germany, TSA animal respiratory compact), and following spontaneous breathing and recovery, they were returned to the individual cages (20).
Preparing the brain tissue
For the histological assessment of brain tissue, animals were deeply anesthetized with chloral hydrate (400 mg/kg, IP, Merck) 72 hr after TBI, and sacrificed by cardiac perfusion with 140–180 ml of 0.9% heparinized normal saline followed by 140 to 180 ml of cold 4% paraformaldehyde in phosphate buffered saline (PBS, pH of 7.5). The perfusion lasted for 12 to 20 min (until the lungs and liver were clear of blood). Then, the brains were carefully removed and immediately immersed in an aldehyde fixative (10% formaldehyde in 0.1 M sodium phosphate-buffered) and kept overnight at 4 °C. They were then dehydrated in alcohol solution and eventually embedded in paraffin (21, 22).
Immunohistochemistry
Coronal serial sections (5 μm) of the brain tissue including the cortex and underlying white matter were cut on a microtome (using stereotaxic coordinates of the Bregma AP +3.22 to — +4.78 mm, with 1-mm interval), mounted on poly-l-lysine-coated slides (St Louis, MO, Sigma Chemical Co), and dried at room temperature overnight (23). Three sets of sections were obtained from each brain. One set of the sections was stained with Hemotoxylin and Eosin for morphologic analysis, and the other two sets were subjected to immunohistochemistry (IHC) by using glial fibrillary acidic protein (GFAP) antibody and TUNEL staining.
Mouse monoclonal anti-GFAP antibody (1:500 dilutions, Dako A/S Denmark) was used to assess astrocytic reactivity. For this purpose, sections were deparaffinized in a microwave oven (65 °C) for 15 min and then in xylene (5 min). After rinsing in PBS, the sections were put in the citrate buffer (10 mM, pH of 6 and temperature of 90°C) for 1 hr and then kept in hydrogen peroxide (0.3% H2O2 in 60% methanol/PBS) for 15 min to inhibit endogenous peroxidase activity. The sections were incubated with primary antibody and stored overnight at 4 °C having been rinsed three times in PBS, and were incubated for 1.5 hr at 25 °C with a 1:400 diluted secondary antibody (rabbit anti-mouse antibody). To detect antibody-antigen complex, 0.1% diaminobenzidine (24) was used in the presence of 0.03% H2O2 (25).
The TUNEL assay was performed on the cerebral sections to detect the cell death (using the Kit POD; Indianapolis, Roche, IN). After incubation with Diamino Benzidine DAB and hydrogen peroxide, a dark brown color revealed damage to neuronal perikarya (26).
Cell counting
Five coronal sections were chosen for cell counting (between the Bregma AP —0.32 and —0.68 mm). The number of surviving and degenerating neurons was counted in five random and non-overlapping regions (at a magnification of X200). Percent of neuronal damage was calculated based on the ratio of the number of degenerated neurons to that of both surviving and degenerated neurons. In addition, the number of astrocytes were counted in five random and separated regions of the cortex (at the level between Bregma AP- 0.12 and +0.28) (23).
Statistical analyses
The data were expressed as mean±SEM. One way ANOVA and Tukey-Kramer multiple post hoc test were used to evaluate the differences between the groups, and P<0.05 was designated for statistical significance.
Results
Apoptotic cells in brain cortex
Histological studies in the vehicle group animals showed severe morphological changes including extensively dark pyknotic nuclei and shrunken cytoplasm in the brain cortex following TBI. However, in melatonin treatment groups, the severity of degenerative changes in the nucleus and cytoplasm was lower than those in the vehicle and sham groups (P<0.05) (Table 1). In addition, there were few TUNEL positive cells of brain cortex in the sham group, while many TUNEL positive cells were detected in the vehicle group (Figure 1).
Table 1.
The effect of melatonin on cortical neuronal death following TBI
| (DC/CCx 1oo)** | DC** | CC* | Groups |
|---|---|---|---|
| 11.83## | 42 | 355 | Sham |
| 69.69 | 253 | 363 | Vehicle |
| 38.10 # | 133 | 349 | M.5 mg/kg |
| 35.42 # | 124 | 350 | M.10 mg/kg |
| 33.91 # | 116 | 342 | M.20 mg/kg |
counted cells
Average number of the degenerated cells (DC)
Average percentage of dead cells
P<0.05 for melatonin (5, 10 and 20 mg/kg)
P<0.01(sham vs Vehicle)
Figure 1.
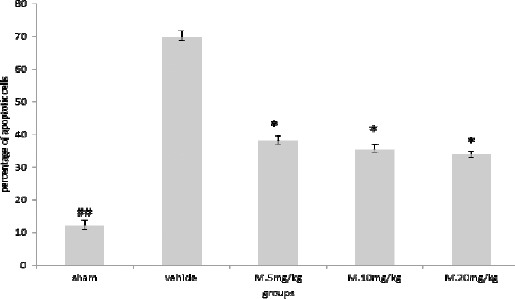
Melatonin treatment reduced apoptosis of neurons induced by TBI. Results are expressed as mean±SEM and data were analyzed by one-way ANOVA followed by Tukey Kramer multiple comparisons test
* Significantly different from the vehicle group (P<0.05)
## Significantly different from the vehicle group (P<0.01)
The results of neuronal cell counting showed significant difference between TBI+ 5 mg/kg melatonin (P<0.05), TBI+ 10 mg/kg melatonin (P<0.05), and TBI+20 mg/kg melatonin (P<0.05) groups, compared to the vehicle group (Figure 2).
Figure 2.
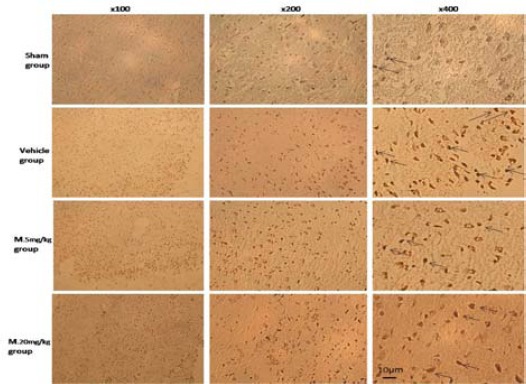
Immunohistochemical analysis of TUNEL in brain cortex of rat. Arrows show the apoptotic cells. Bar=10 µm
Activated astrocytes numbers
Our findings showed that TBI causes a dramatic increase in the number of activated astrocytes in brain cortex. But, melatonin treatment significantly decreased the number of GFAP positive astrocytes (P<0.05) that were not dose dependent (Figure 3 and 4).
Figure 3.
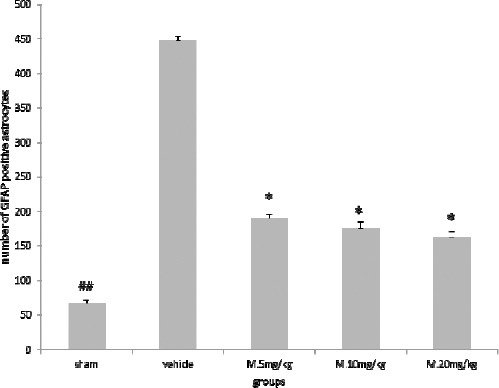
Melatonin treatment reduced the number of glial fibrillary acidic protein (GFAP) positive astrocytes. Results are expressed as mean±SEM, and data were analyzed by One-way ANOVA followed by Tukey-Kramer multiple comparisons test
*Significantly different from the vehicle group (P<0.05)
## Significantly different from the vehicle group (P<0.01)
Figure 4.
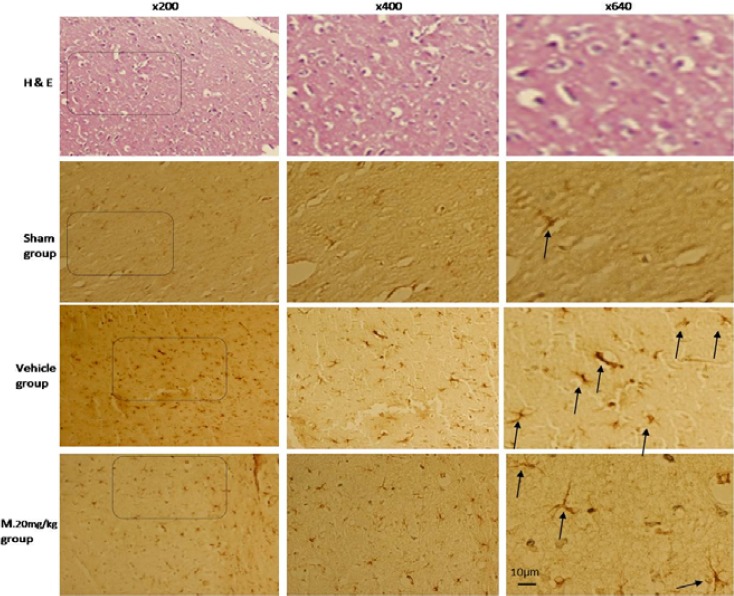
Immunohistochemical analysis of glial fibrillary acidic protein (GFAP) positive cells in brain cortex of rat. Arrows show the GFAP positive cells. Bar=10 µm
Discussion
The results of this study have shown the neuroprotective effect of melatonin after TBI in male rat. Our findings indicate that TBI causes a dramatic increase in neuronal cell death in brain tissue, and based on TUNEL assay, the percentage of apoptotic cells significantly decreased in the melatonin treatment groups. It seems that melatonin plays a great role in antiapoptotic activities via the inhibition of intrinsic apoptotic pathways, as well as, the activation of several associated signal molecules in various brain regions in TBI model (27). In an experimental study, it was evaluated that treatment with melatonin (5 and 10mg/kg, IP) improved the survival rate in a stroke model of mice (28). Melatonin has been evaluated as effective in TBI, through increasing superoxide dismutase (SOD) and glutathione peroxidase (GPx) activities (29).
The result of the present study does not show any significant difference between three different doses of melatonin in our animal groups. A previous study has also shown that melatonin significantly attenuated neuronal cell death in hippocampal CA1 and CA3 regions and dentate gyrus of immature rats after head trauma, which was equally effective at doses of 5 and 20 mg/kg (30). In another experimental animal model, melatonin treatment led to glial cell death reduction in white matter in mid-gestation fetal sheep following umbilical cord occlusion (31).
The anti-inflammatory effect of melatonin has been introduced as a chief protective mechanism against brain injury (32, 33). However, the modulation behaviors of melatonin to suppress activated astrocytes have not been particularly investigated in a research with respect to TBI. Astrocytes activation occurs in response to many CNS pathologies, such as trauma, tumor formation, and neurodegenerative disease. The process of astrocyte activation results in so-called “reactive astrogliosis”, which is a reaction with specific structural and functional characteristics including hypertrophy of cellular process of astrocytes (34). Up-regulation of intermediate filament proteins, particularly GFAP by reactive astrocytes may be applied to distinguish reactive astrogliosis. Hence, the present study has focused on reactive astrogliosis based on GFAP immunoreactivity (35). In fact, astrocytes, as the largest and the most abundant glial cells in CNS, may be the target of melatonin. We have demonstrated that the number of GFAP positive cells is reduced in the melatonin treatment groups, which indicates the alleviation of astrogliosis process induced by TBI. Melatonin has been shown to modify immunity and the stress response, and has an antioxidant activity (scavenge free radicals) (31). Melatonin has a greater antioxidant effect than vitamin E, and can indirectly increase the expression of other endogenous antioxidant enzymes (36). Ananth and colleagues demonstrated that domoic acid-induced astrocyte activation is attenuated significantly by the intravenous administration of melatonin in the hippocampus of adult rats (37). Also, studies have shown that among the pathological changes occurred in the acute post-traumatic period, impairment of the blood–brain barrier (BBB) has a major role and allows the entry of circulating lymphocytes, monocytes, and neutrophils into the injured site, which directly affects inflammation and glial cell reactivities that may lead to neural cell death (38, 39).
In addition, it was shown that melatonin administration significantly up regulated STAT1 DNA-binding activity, and led to the reduction of pro-inflammatory cytokines such as IL-6β, and Nitric Oxide Synthase NOS (33). However, the modulation behaviors of melatonin to astrocyte, an important source of pro-inflammatory cytokines, have not yet been extensively explored. Proliferation of astrocytes and their migration toward the lesion site can be recorded by GFAP immunoreactivity (40).
Also, we showed the importance of the therapeutic time-window after the initial insult of TBI. It is critical to start interventions as soon as possible upon TBI (41). In our experiment, the treatment started one hr after TBI and continued until 72 hr, which is a critical time period for histological examination.
It would be necessary to mention that there are several limitations to our study. Firstly, the mechanisms by which melatonin reduces GFAP positive astrocytes are not explained. In addition, this study only reveals the neuroprotective effects of melatonin after a short time period following TBI. Therefore, further investigations need to be performed to clarify the molecular mechanisem of melatonin in suppression of astrocyte reactivity.
Conclusion
Findings support that melatonin is highly protective of both neurons and glial cells following TBI in our animal model. In general, the present findings suggest that melatonin treatment of TBI markedly diminish astrocyte reactivity (astrogliosis), as well as the number of apoptotic neurons in brain cortex of traumatic brain injury in rat model.
Acknowledgment
The present study was financially supported by Neuroscience Research Center, Institute of Neuro-pharmacology, Kerman University of Medical Sciences, Kerman, Iran. This work was part of MSc thesis of Mr Babaee at the Department of Anatomical Sciences. The authors have no conflicts of interest to declare.
References
- 1.Chua KSG NY, Yap SGM, Bok C. A brief review of traumatic brain injury rehabilitation. Ann Acad Med Singapore. 2007;36:31–42. [PubMed] [Google Scholar]
- 2.Khorasani-Zavareh D, Mohammadi R, Khankeh HR, Laflamme L, Bikmoradi A, Haglund BJ. The requirements and challenges in preventing of road traffic injury in Iran. A qualitative study. BMC Public Health. 2009;9:486. doi: 10.1186/1471-2458-9-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aghakhani N, Azami M, Jasemi M, Khoshsima M, Eghtedar S, Rahbar N. Epidemiology of traumatic brain injury in urmia, iran. Iran Red Crescent Med J. 2013;15:173. doi: 10.5812/ircmj.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, et al. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. doi: 10.1016/j.freeradbiomed.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 6.Bayir H, Kochanek PM, Clark RS. Traumatic brain injury in infants and children: mechanisms of secondary damage and treatment in the intensive care unit. Crit Care Clin. 2003;19:529–549. doi: 10.1016/s0749-0704(03)00014-9. [DOI] [PubMed] [Google Scholar]
- 7.Kunz A, Dirnagl U, Merqenthaler P. Acute pathophysiological processes after ischaemic and traumatic brain injury. Best Pract Res Clin Anaesthesiol. 2010;24:495–509. doi: 10.1016/j.bpa.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Hall ED, Vaishnav RA, Mustafa AG. Antioxidant therapies for traumatic brain injury. Neurothera-peutics. 2010;7:51–61. doi: 10.1016/j.nurt.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reiter RJ. Melatonin: the chemical expression of darkness. Mol Cell Endocrinol. 1991;79:C153–158. doi: 10.1016/0303-7207(91)90087-9. [DOI] [PubMed] [Google Scholar]
- 10.Claustrat B, Brun J, Chazot G. The basic physiology and pathophysiology of melatonin. Sleep Med Rev. 2005;9:11–24. doi: 10.1016/j.smrv.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Seifman MA, Gomes K, Nguyen PN, Bailey M, Rosenfeld JV, Cooper DJ, et al. Measurement of serum melatonin in intensive care unit patients: changes in traumatic brain injury, trauma, and medical conditions. Front Neurol. 2014;5:273. doi: 10.3389/fneur.2014.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmadiasl N, Shokofeh B, Alireza A. Combination Antioxidant Effect of erythropoietin and melatonin on renal ischemia-reperfusion injury in rats. Iran J Basic Med Sci. 2013;16:1209–1211. [PMC free article] [PubMed] [Google Scholar]
- 13.Pineau I, Sun L, Bastien D, Lacroix S. Astrocytes initiate inflammation in the injured mouse spinal cord by promoting the entry of neutrophils and inflammatory monocytes in an IL-1 receptor/MyD88-dependent fashion. Brain Behav Immun. 2010;24:540–553. doi: 10.1016/j.bbi.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Barreto GE, Gonzalez J, Torres Y, Morales L. Astrocytic-neuronal crosstalk: Implications for neuroprotection from brain injury. Neurosci Res. 2011;71:107–113. doi: 10.1016/j.neures.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Cameron B, Landreth GE. Inflammation, microglia, and alzheimer’s disease. Neurobiol Dis. 2010;37:503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Candace LF, Bruce GL. Astroglia: Important mediators of traumatic brain injury. Prog Brain Res. 2007:161. doi: 10.1016/S0079-6123(06)61005-4. [DOI] [PubMed] [Google Scholar]
- 17.Skaper SD, Floreani M, Ceccon M, Facci L, Giusti P. Excitotoxicity, oxidative stress, and the neuroprotective potential of melatonin. Ann N Y Acad Sci. 1999;890:107–118. doi: 10.1111/j.1749-6632.1999.tb07985.x. [DOI] [PubMed] [Google Scholar]
- 18.Dehghan F, Hadad MK, Asadikram G, Najafipour H, Shahrokhi N. Effect of melatonin on intracranial pressure and brain edema following traumatic brain injury: role of oxidative stresses. Arch Med Res. 2013;44:251–258. doi: 10.1016/j.arcmed.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Marmarou A, Foda MAAE, Brink W, Campbell J. A new model of diffuse brain injury in rats: Part I: Pathophysiology and biomechanics. J Neurosurg. 1994;80:291–300. doi: 10.3171/jns.1994.80.2.0291. [DOI] [PubMed] [Google Scholar]
- 20.Keshavarzi Z, Khaksari M, Shahrokhi N. The effects of cyclooxygenase inhibitors on the gastric emptying and small intestine transit in the male rats following traumatic brain injury. Iran J Basic Med Sci. 2014;17:406–410. [PMC free article] [PubMed] [Google Scholar]
- 21.Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007;446:1091–1095. doi: 10.1038/nature05704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, et al. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. doi: 10.1016/j.freeradbiomed.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 23.Lee MY, Kuan YH, Chen HY, Chen TY, Chen ST, Huang CC, et al. Intravenous administration of melatonin reduces the intracerebral cellular inflammatory response following transient focal cerebral ischemia in rats. J Pineal Res. 2007;42:297–309. doi: 10.1111/j.1600-079X.2007.00420.x. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad molai Gh, Dabiri Sh, Asadi karam GM, Shahrokhi N. Comparision of the effect of progestrone, allopregnanolone and gender on suppressing edema formation after traumatic brain injury in rat. Kerman Univ Med Sci. 2008;15:47–59. [Google Scholar]
- 25.Baydas G, Reiter RJ, Yasar A, Tuzcu M, Akdemir I, Nedzvetskii VS. Melatonin reduces glial reactivity in the hippocampus, cortex, and cerebellum of streptozotocin-induced diabetic rats. Free Radic Biol Med. 2003;35:797–804. doi: 10.1016/s0891-5849(03)00408-8. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Chopp M, Jiang N, Zhang ZG, Zaloga C. Induction of DNA fragmentation after 10 to 120 minutes of focal cerebral ischemia in rats. Stroke. 1995;26:1252–1258. doi: 10.1161/01.str.26.7.1252. [DOI] [PubMed] [Google Scholar]
- 27.Kabadi SV, Maher TJ. Posttreatment with uridine and melatonin following traumatic brain injury reduces edema in various brain regions in rats. Ann N Y Acad Sci. 2010;1199:105–113. doi: 10.1111/j.1749-6632.2009.05352.x. [DOI] [PubMed] [Google Scholar]
- 28.Chern CM, Liao JF, Wang YH, Shen YC. Melatonin ameliorates neural function by promoting endogenous neurogenesis through the MT2 amelatonin receptor in ischemic-stroke mice. Free Radic Biol Med. 2012;52:1634–1647. doi: 10.1016/j.freeradbiomed.2012.01.030. [DOI] [PubMed] [Google Scholar]
- 29.Dehghan F, Khaksari Hadad M, Asadikram G, Najafipour H, Shahrokhi N. Effect of melatonin on intracranial pressure and brain edema following traumatic brain injury: role of oxidative stresses. Arch Med Res. 2013;44:251–258. doi: 10.1016/j.arcmed.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Ozdemir D, Uysal N, Gonenc S, Acikgoz O, Sonmez A, Topcu A, et al. Effect of melatonin on brain oxidative damage induced by traumatic brain injury in immature rats. Physiol Res. 2005;54:631–637. [PubMed] [Google Scholar]
- 31.Welin AK, Svedin P, Lapatto R, Sultan B, Hagberg H, Gressens P, et al. Melatonin reduces inflammation and cell death in white matter in the mid-gestation fetal sheep following umbilical cord occlusion. Pediatr Res. 2007;61:153–158. doi: 10.1203/01.pdr.0000252546.20451.1a. [DOI] [PubMed] [Google Scholar]
- 32.Keskin I, Kaplan S, Kalkan S, Sutcu M, Ulkay MB, Esener OB. Evaluation of neuroprotection by melatonin against adverse effects of prenatal exposure to a nonsteroidal anti-inflammatory drug during peripheral nerve development. Int J Dev Neurosci. 2015;41:1–7. doi: 10.1016/j.ijdevneu.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Tsai MC, Chen WJ, Tsai MS, Ching CH, Chuang JI. Melatonin attenuates brain contusion-induced oxidative insult, inactivation of signal transducers and activators of transcription 1, and upregulation of suppressor of cytokine signaling-3 in rats. J Pineal Res. 2011;51:233–245. doi: 10.1111/j.1600-079X.2011.00885.x. [DOI] [PubMed] [Google Scholar]
- 34.Marmarou CR, Liang X, Abidi NH, Parveen S, Taya K, Henderson SC, et al. Selective vasopressin-1a receptor antagonist prevents brain edema, reduces astrocytic cell swelling and GFAP V1aR and AQP4 expression after focal traumatic brain injury. Brain Res. 2014;1581:89–102. doi: 10.1016/j.brainres.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 36.Babaei-Balderlou F, Zare S, Heidari R, Farrokhi F. Effects of melatonin and vitamin E on peripheral neuropathic pain in streptozotocin-induced diabetic rats. Iran J Basic Med Sci. 2010;13:1–8. [Google Scholar]
- 37.Ananth C, Gopalakrishnakone P, Kaur C. Protective role of melatonin in domoic acid-induced neuronal damage in the hippocampus of adult rats. Hippocampus. 2003;13:375–387. doi: 10.1002/hipo.10090. [DOI] [PubMed] [Google Scholar]
- 38.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 39.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Domowicz MS, Henry JG, Wadlington N, Navarro A, Kraig RP. Astrocyte precursor response to embryonic brain injury. Brain Res. 2011;1389:35–49. doi: 10.1016/j.brainres.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding K, Wang H, Xu J, Lu X, Zhang L, Zhu L. Melatonin reduced microglial activation and alleviated neuroinflammation induced neuron degeneration in experimental traumatic brain injury: Possible involvement of mTOR pathway. Neurochem Int. 2014;76:23–31. doi: 10.1016/j.neuint.2014.06.015. [DOI] [PubMed] [Google Scholar]