

Abstract
Objective(s):
Rapid and accurate detection of Brucella abortus and Brucella melitensis from clinical samples is so important because antibiotic treatment has major side effects. This study reveals a new method in detection of clinical samples of brucellosis using real-time PCR and high-resolution melt (HRM) curve analysis.
Materials and Methods:
160 brucellosis suspicious samples with more than 1/80 serum antibody titers were collected and the results were compared with the RFLP method. In order to amplify the sequences for HRM analysis, vdcc, int-hyp and glk and for RFLP, omp2a and omp2b with PstI and Hinf1 restriction endonuclease were used. At last, the accuracy and specificity of the two methods were compared with each other.
Results:
Out of these 160 samples, multiplex real time PCR showed 108 positive samples (67.5%), including 56% B. melitensis and 44% B. abortus; whereas in PCR-RFLP 52 out of 160 samples were positive, where recognition of two species were accordant with HRM analysis, separation was based on the size of the amplified fragment. Using the designed primers and performing the assay, we confirmed this method to be much faster and have lower cost with more than 99% accuracy compared to methods such as RFLP.
Conclusion:
The present study showed that this technique, which scans gene segments and creates an analysis pattern for detection of clinical samples, is useful and more dominant compared with PCR-RFLP. Thus, this method can be used for brucellosis detection, and clinical and epidemiological research.
Keywords: Brucella, High-resolution melt analysis, Multiplex PCR, RFLP
Introduction
Pathogen members of the bacterial genus Brucella can infect mammals and humans (1). Infection in humans occurs during direct and indirect contact with infected animals or by using infected meat or dairy products. Also, direct contact with infected animal’s tissue and inhalation aerosolized droplets are ways of infection transmission (2). These small bacteria are facultative intracellular, aerobic, and gram-negative coccobacilli with 90% homology in nucleic acid sequence (3). The most prevalent species (Brucella melitensis, Brucella abortus) are responsible for over half a million human infections worldwide per year. Therefore, rapid detection and treatment of brucellosis is one of the health priorities (3-5).
Conventional laboratory tests which produce discordant results are based on chemical and antigenic features, metabolism differentiations (6, 7), CO2 need, stainability, phage sensitivity (8), H2S production, oxidative metabolism pattern, reaction with anti-serum for phenotype diagnosis and detection of isolates (7, 9). Recently, molecular methods such as genome sequencing, single nucleotide polymorphism (SNP) analysis (10, 11), Variable Number Tandem Repeat (VNTR) analysis (12) or microsatellite and real-time PCR are used to identify Brucella isolates (11, 13). High-resolution melt (HRM) analysis is a new technology that lets us scan genome pieces with high accuracy and analyze the results of nucleic acid fragments in comparison with each other, distinctively (14). It is done by brief and proper setting of the melting temperature profile that has special sensitivity to make a positive species determination (15).
In this study, we discuss a new method for detecting two common bacterial species causing brucellosis, which is to our knowledge the fastest and most accurate method to date. We improved a real-time PCR method which in combination with HRM analysis is useful for detecting and differentiating between B. melitensis and B. abortus (16). This is the first time that this technique is compared for separating the brucellosis species in human blood and clinical samples with RFLP. In this study we targeted three independent gene loci for amplifying gene pieces, then compared the curves and identified and differentiated Brucella genus members. This study can be useful for clinical samples and epidemiological and also veterinary studies for detecting Brucella species.
Materials and Methods
Collection and extraction of DNA samples
We prepared 160 Brucella isolates that were collected from suspicious patients with clinical history of brucellosis and positive serological test (Rose Bengal test and serum agglutination test) who had measurable antibody titers 1/80 from various parts of Iran including high frequency brucellosis areas (Kerman, South Khorasan, Khorasan Razavi).
Then blood samples were maintained at -70 °C until analysis. We followed the genome extraction protocol according to previous study (17).
Primer design
We used two sets of primers: Brucella genus diagnostic primers and B. melitensis, B. abortus strain determiner primers. First group of primers were used for detecting Brucella genus in samples. Also two sets of primers were used to amplify the omp2a and omp2b in PCR-RFLP (Table 1).
Table 1.
Oligonucleotide sequences for primers used to detect the Brucella spp. and PCR-RFLP
| Markers | Orientation | Primer (5’−3′) | Gene target | Amplicon size (bp) |
|---|---|---|---|---|
| Brucella spp. | Forward | 5’-GTGGCGATCTTGTCCG-3’ | vdcc | 67 bp |
| Reverse | 5’-ACGGCGATGGATTTCCG-3’ | |||
| B. melitensis | Forward | 5’-GTGGCGATCTTGTCCG-3’ | int-hyp | 125 bp |
| Reverse | 5’-ACGGCGATGGATTTCCG-3’ | |||
| B. abortus | Forward | 5’-GACCTCTTCGCCACCTATCTGG-3’ | glk | 164 bp |
| Reverse | 5’-CCTTGTGCGGGGCCTTGTCCT-3’ | |||
| omp2a | Forward | 5’-GGCTATTCAAAATTCTGGCG-3’ | 1100 bp | |
| omp2a | Reverse | 5’-ATCGATTCTCACGCTTTCGT-3’ | ||
| omp2b | Forward | 5’-CCTTCAGCCAAATCAGAATG-3’ | 1200 bp | |
| omp2b | Reverse | 5’-GGTCAGCATAAAAAGCAAGC-3’ |
Multiplex real-time PCR
The multiplex real time PCR assay was prepared using Eva Green (a double-stranded DNA intercalating dye) and all the positive samples that amplified 67 base pairs of the vdcc gene. For doing the tests the materials were transferred to 0.2 ml PCR microtubes.
Each PCR reaction mixture contained 5 μl)Solis BioDyne- Switzerland) master mix 4X (5x HOT FIREPol® EvaGreen® HRM Mix (no ROX)) that contained HOT FIREPol® DNA Polymerase, 5x EvaGreen® HRM buffer, 12.5 mM MgCl2, 2 μl template DNA (0.5 μg), 0.15 mM dNTP, 20 pmol of each forward and reverse primer and sterile distilled water up to 20 μl.
The optimized program to amplify target genes in Rotor-Gene (Corbett Rotor-Gene 6000 Qiagen, Valencia, CA) is as follows : 1 cycle of 95 °C for 15 min, 35 cycles of 95 °C for 10 sec and 60 °C for 40 sec, with data acquired at the 60 °C step in the green channel (14).
Studies of specificity
In order to evaluate the specificity of primers and the PCR assay, DNA of closely related non-Brucella species and human were employed in the test.
Sensitivity of real time PCR based on DNA concentration
To evaluate the sensitivity of real time PCR based on DNA concentration, the concentration of DNA extracted from each of the samples was measured as mentioned. Then, for each extracted DNA, different serials dilutions (10-1, 10-2, 10-3 and 10-4) were prepared and eventually, using the above-mentioned dilution (final protocol and the optimized thermal profile), the multiplex real time PCR reactions was performed.
HRM analysis of B. abortus and B. melitensis
In this method 5x HOT FIREPol® EvaGreen® HRM mix was used. We optimized the above program using HRM and through increasing the temperature from 83.5 by 0.1 degree steps up to 89. Then data were normalized according to available information.
Amplification of omp2a and omp2b
Cloackaert et al designed primers for amplification of omp2a and omp2b fragments; their sequences are shown in the following table (18). Each PCR reaction mixture contained; 1X PCR buffer, 2 mM MgCl2, 1 μl template DNA (0.5 μg), 0.15 mM dNTP, 2.5 U Taq DNA polymerase, 20 p mol of each forward and reverse primers and sterile distilled water up to 50 μl. PCR for omp2a was performed in a GenAmp PCR system (Eppendorf, German) as: pre-denaturation for 5 min at 94 °C followed by 35cycles each containing denaturation at 94 °C for 60 sec, annealing at 50 °C for 120 sec and extension at 72 °C for 180 sec, followed by final extension at 72 °C for 7 min. Also PCR for omp2b was performed according to the following program:
pre-denaturation for 5 min at 94 °C followed by 35 cycles, each containing denaturation at 94 °C for 45 sec, annealing at 58 °C for 60 sec and extension at 72 °C for 60 sec, followed by final extension at 72 °C for 7 min. Then, The PCR products were analyzed using the electrophoresis technique on 2% agarose gel for 1 hr at 25 mA, stained by SYBR-Green and visualized under UV transilluminator (Figure 1). Finally, amplification products were further evaluated by restriction digestion procedures.
Figure 1.
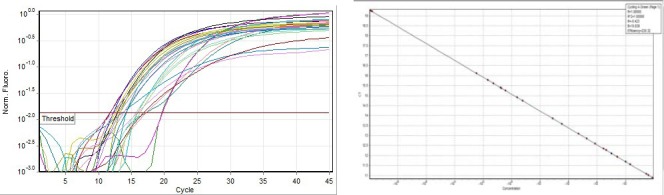
Isolated positive samples using real-time PCR amplification of the 67 bp. Negative samples not shown
Enzymatic digestion
To identify polymorphisms, the amplified products were subjected to restriction enzymes according to previous study, (18).
Results
Multiplex real-time PCR and multiplex PCR results
160 patients with brucellosis infection were tested. We detected and isolated positive samples using multiplex real-time PCR; 52 clinical samples were negative for all the targets in the real-time PCR and 108 for PCR-RFLP, including the control human gene, therefore, they were not considered in the analysis. 47 patients with B. abortus (brucellosis infection) and 61 patients with B. melitensis (brucellosis infection) were sampled for the HRM analysis method according to clinical criteria as described in materials and methods. The LLOD of the primers set for detection of B. melitensis and B. abortus, was 1.5 pg. For DNA template ranging (mean ± SD), result was shown in the crossing point cycles of DNA template ranging from 12.0 ± 0.25 to 15 ± 0.25, when it was performed in the amount ranging from 1 ng to 1.5 ng (Figure 1).
Figure 1 shows the amplification of the 67 base-pair sequence. Results show the specificities of primers which only paired with B. melitensis and B. abortus genome. Total extracted DNA from none of the microorganisms demonstrated any pairing with the designed primers at ∼2–4 ng concentration (Table 2). Since the concentration of the first DNA template for B. abortus and B. melitensis was 955 ng/μl, serial dilutions were prepared to calculate the pairing sensitivity, then PCR was performed. The lowest DNA copy number for detecting with this method was 10-4 copy number of the initial genome product, and the accurate results obtained in 10-2 copy number that mean the template concentration was 0. 955 ng/μl.
Table 2.
Performance of a real-time PCR assay for the detection of Brucella spp., Brucella abortus and Brucella melitensis (determine the specificity of PCR)
| Strain | PCR identification | Strain (from human) | PCR identification |
|---|---|---|---|
| Brucella spp. | 108/160 | Pseudomonas aeruginosa | 0/1 |
| B. abortus | 47/108 | Campylobacter spp. | 0/1 |
| B. melitensis | 61/108 | Klebsiella pneumoniae | 0/1 |
| Escherichia coli O157:H7 | 0/1 | Listeria monocytogenes | 0/1 |
| Agrobacterium tumefaciens PTCC 1654 | 0/1 | Proteus mirabilis | 0/1 |
| Vibrio cholerae PTCC 1611 | 0/1 | Salmonella enteritidis | 0/1 |
| Salmonella enterica ATCC:9270 | 0/1 | Staphylococcus aureus | 0/1 |
| Shigella flexneri ATCC:12022 | 0/1 | Streptococcus pneumoniae | 0/1 |
| Shigella sonnei ATCC:9290 | 0/1 | Staphylococcus epidermidis | 0/1 |
| Staphylococcus aureus ATCC:6538 | 0/1 | Escherichia coli | 0/1 |
| DNA Extraction from human Blood | 0/1 |
RFLP results
In our previous study with RFLP technique 52 cases obtained bands of 1100 bp for omp2a locus and 1200 bp for omp2b, which indicates that the samples were positive for Brucella (Figure 2-left) (17). According to Cloackaert et al 1995 (19) regarding enzymatic digestion of the amplified fragments, for 52 cases from digestion of omp2a fragment by Pst1 and Hinf1 enzymes, P3 and P2 patterns were obtained respectively, and for omp2b, P1 and P1 patterns were resulted, which indicates the B. melitensis biovar 1 (Figure 2-center) (18), and for 33 cases (44.5%) from digestion of omp2a fragment by Pst1 and Hinf1 enzymes, patterns of P2 and P2 and for omp2b patterns of P1 and P1 were resulted respectively, which indicates one of the B. abortus biovars 3, 5, 6 or 9 (Figure 2-right).
Figure 2.
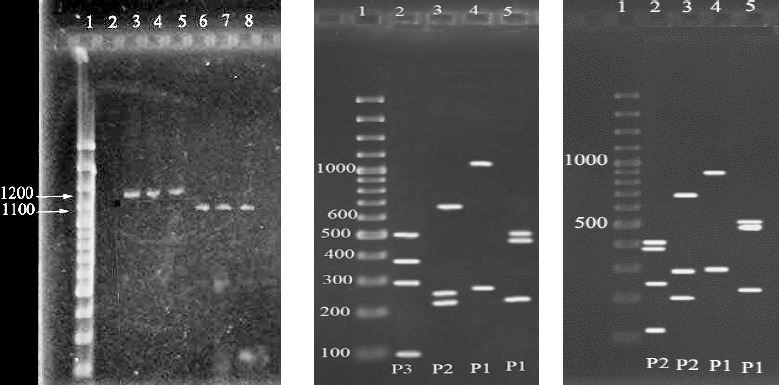
left: Agarose gel electrophoresis of PCR amplified products generated from DNA samples. Lane 1 shows DNA size marker (100bp DNA ladder, Fermentase). Lane 2 is negative control. Lanes 3 and 4 show amplified locus of omp2b. Lanes 6 and 7 show amplified locus of omp2a, Lanes 5 and 8 are positive controls. center: The patterns of enzymatic digestion for Brucella melitensis samples. Lane 1 is DNA size marker (100bp DNA ladder), Lanes 2 and 3 are digestion of omp2a fragment by Pst1 and Hinf1 and lanes 4 and 5 are digestion of omp2b fragment by Pst1 and Hinf1. right: The patterns of enzymatic digestion for B. abortus isolates. lane 1 is DNA size marker (100bp DNA ladder, Fermentase). Lanes 2 and 3 are digestion of omp2a fragment by Pst1 and Hinf1. Lanes 4 and 5 are digestion of omp2b fragment by Pst1 and Hinf1
Specificity of the HRM primers
To evaluate the specificity of the HRM primers the multiplex real-time PCR was used: 2 strains of the genus Brucella and 17 pathogenic bacteria of non-Brucella species and two DNA extractions from human Blood. DNA from these strains was used in the individual evaluation of each real-time PCR target assay, and in the assay for cross-reactivity at a 10 ng/μl concentration. No amplification signal was found for the other non-Brucella species (Table 2).
HRM analysis results
125 base pair of int-hyp target gene was amplified for Brucella and then HRM analysis curve was rechecked (Figure 3). As was expected B. melitensis species with mutation of guanine to thymine in amplified region was one of the recognized present species in the samples (Table 3). Glk target gene was amplified for Brucella species using the special primer with 164 bps. Figure 4 shows the analysis of HRM curve for separation of B. abortus with mutation of guanine to thymine; Table 3 shows the normalization region and genetic identity.
Figure 3.
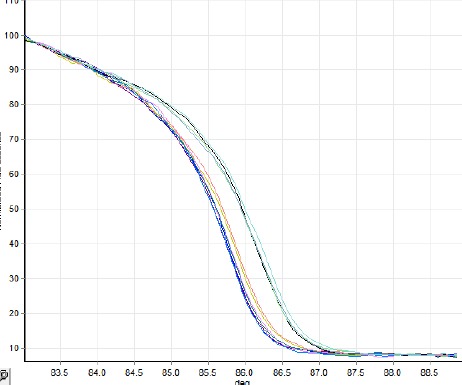
125 Base pair of int-hyp target gene was amplified for Brucella and then high-resolution melt analysis curve was rechecked; as was expected Brucella melitensis species with mutation of guanine to thymine was one of the recognized present species in the sample
Table 3.
Normalization region and genetic identity
| Markers | Normalization region | SNP or genetic identity | |
|---|---|---|---|
| First | Second | ||
| Brucella spp. | not applicable | not applicable | plus/minus |
| Brucella melitensis | 82.5–83.5 | 87.5–89 | T; B. melitensis G; other spp. |
| Brucella abortus | 83–83.5 | 87–89.5 | A; B. abortus G; other spp. |
Figure 4.
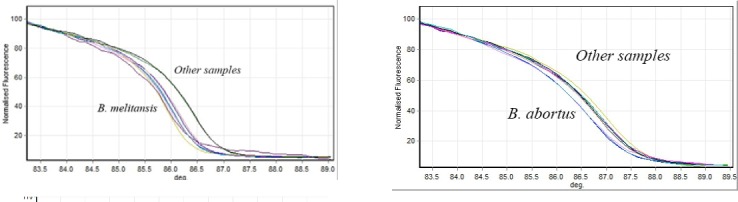
HRM assay separation curve of Brucella abortus from other species. glk target gene was amplified for the Brucella species using the special primer with 164 bps. Data shows the analysis of high-resolution melt curve for separation of B.abortus with mutation of guanine in 164 bps from glk gene
Discussion
HRM is a rapid, more convenient closed-tube method with novelty in bacterial genotyping which helps us to analyze the genetic mutations (20). This technique reveals distinctive details of the DNA double strain by temporal denaturation in order to found single nucleotide polymorphisms. The genome scanning action is the most important stage which makes possible detection of new varieties and isolation of each of the species in PCR products (21).
Detection and analysis of polymorphisms with RFLP is also used for identification and characterization of Brucella species with some advantages such as applicability, easy interpretation and could be used for numerous samples. This method uses the sets of omp2a, omp2b, omp25 and omp31 genes, to characterize all species of Brucella from each other and detect their biovars (19). The aim of this study was to investigate the efficiency and accuracy of both techniques and also design an examination for the separation of the two species by HRM analysis.
The results reveal that Brucella species (B. melitensis and B. abortus – unavailability of other Brucella species in Iran) could be detected by real-time PCR if accompanied by the post PCR step. The results were same as the PCR-RFLP method (17). The two stages of HRM were performed as closed-tube without any pollution transmission after running the PCR. In addition to rapidness and accuracy, being cost effective is another remarkable advantage of HRM. Inability to detect biovars in HRM is quiet mentionable.
Between 108 positive samples were detected by HRM analysis, 61 (56%) were B. melitensis and 47 (44.4%) were B. abortus. The results of this study for Brucella species rapid detection were the same as Winchell et al 2010 (14). In contrast with Winchell and colleagues’ study, our investigation was done on clinical samples therefore the presented HRM curves had a lower clarity.
Our modified HRM method compare with RFLP, reduced total period examination by optimizing and reducing the cycles. Also the used EvaGreen® color had lower toxicity than other florescence dyes (16).
The designed primers in this study were based on Gopaul et al in 2008 which had found SNP points by minor groove binding-based (genome scanning) method for Brucella species (11). Designed primers could determine each of B. melitensis and B. abortus in clinical trials.
Furthermore, to screen brucella genus with other microorganisms in clinical samples, we used a set of primers to amplify vdcc genome sequence. The 67 base-pair sequence separated positive samples for brucellosis from the total of suspicious samples with higher than 1.80 serum titer.
Conclusion
The goal of this study was to design an experiment to reveal admissible explanation in rapid detection of current brucella species with accuracy. The admissible curve results which are practical for human clinical blood samples should be obtained in 15 up to 30 cycles and it is recommended to compare the results with HRM sigmoid curve. For this aim, we used 1.5 ng/µl B. melitensis primer and performance of maximum number of cycles.
Acknowledgment
This study was supported by funds (grant number BMSU/MBRC -93-4) from the Molecular Biology Research Center, Baqiyatallah University of Medical Sciences, Tehran, Iran.
Footnotes
Conflict of interest
The authors declare that there is no conflict of interests.
References
- 1.Tiller RV, Gee JE, Frace MA, Taylor TK, Setubal JC, Hoffmaster AR, et al. Characterization of novel brucella strains originating from wild native rodent species in north queensland, Australia. Appl Environ Microbiol. 2010;76:5837–5845. doi: 10.1128/AEM.00620-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenberg T, Hamann H-P, Kaim U, Schlez K, Seeger H, Schauerte N, et al. Isolation of potentially novel brucella spp. from Frogs. Appl Environ Microbiol. 2012;78:3753–3755. doi: 10.1128/AEM.07509-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mirnejad R, Doust RH, Kachuei R, Mortazavi SM, Khoobdel M, Ahamadi A. Simultaneous detection and differentiates of Brucella abortus and Brucella melitensis by combinatorial PCR. Asian Pac J Trop. 2012;5:24–28. doi: 10.1016/S1995-7645(11)60239-3. [DOI] [PubMed] [Google Scholar]
- 4.Meisel S, Stöckel S, Elschner M, Melzer F, Rösch P, Popp J. Raman spectroscopy as a potential tool for detection of brucella spp. in Milk. Appl Environ Microbiol. 2012;78:5575–5583. doi: 10.1128/AEM.00637-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yagupsky P, Baron EJ. Laboratory exposures to brucellae and implications for bioterrorism. Emerg Infect Dis. 2005;11:1180–1185. doi: 10.3201/eid1108.041197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yagupsky P. Detection of Brucellae in blood cultures. J Clin Microbiol. 1999;37:3437–3442. doi: 10.1128/jcm.37.11.3437-3442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al Dahouk S, Scholz HC, Tomaso H, Bahn P, Gollner C, Karges W, et al. Differential phenotyping of Brucella species using a newly developed semi-automated metabolic system. BMC Microbiol. 2010;10:269. doi: 10.1186/1471-2180-10-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schofield DA, Sharp NJ, Westwater C. Phage-based platforms for the clinical detection of human bacterial pathogens. Bacteriophage. 2012;2:105–283. doi: 10.4161/bact.19274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Godfroid J, Nielsen K, Saegerman C. Diagnosis of brucellosis in livestock and wildlife. Croat Med J. 2010;51:296–305. doi: 10.3325/cmj.2010.51.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster J, Price L, Beckstrom-Sternberg S, Pearson T, Brown W, Kiesling D, et al. Genotyping of Brucella species using clade specific SNPs. BMC Microbiol. 2012;12:110. doi: 10.1186/1471-2180-12-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gopaul KK, Koylass MS, Smith CJ, Whatmore AM. Rapid identification of Brucella isolates to the species level by real time PCR based single nucleotide polymorphism (SNP) analysis. BMC Microbiol. 2008;8:86. doi: 10.1186/1471-2180-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wattiau P, Whatmore AM, Van Hessche M, Godfroid J, Fretin D. Nucleotide polymorphism-based single-tube test for robust molecular identification of all currently described brucella species. Appl Environ Microbiol. 2011;77:6674–6679. doi: 10.1128/AEM.00767-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higgins J, Stuber T, Quance C, Edwards WH, Tiller RV, Linfield T, et al. Molecular epidemiology of Brucella abortus isolates from cattle, elk, and bison in the United States 1998 to 2011. Appl Environ Microbiol. 2012;78:3674–3684. doi: 10.1128/AEM.00045-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winchell JM, Wolff BJ, Tiller R, Bowen MD, Hoffmaster AR. Rapid identification and discrimination of Brucella isolates by use of real-time PCR and high-resolution melt analysis. J Clin Microbiol. 2010;48:697–702. doi: 10.1128/JCM.02021-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zeinzinger J, Pietzka AT, Stöger A, Kornschober C, Kunert R, Allerberger F, et al. One-step triplex Hhgh-resolution melting analysis for rapid identification and simultaneous subtyping of frequently isolated salmonella serovars. Appl Environ Microbiol. 2012;78:3352–3360. doi: 10.1128/AEM.07668-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.White H, Potts G. Mutation scanning by high resolution melt analysis: evaluation of RotorGene 6000™ (Corbett Life Science), HR1™,and 384 well LightScanner™ (Idaho Technology) Wessex, United Kingdom: National Genetics Reference Laboratory; 2006. [Google Scholar]
- 17.Mirnejad R, Mohammadi M, Majdi A, Taghizoghi N, Piranfar V. Molecular typing of Brucella melitensis and B. abortus from human blood samples using PCR-RFLP Method. Jundishapur J Microbiol. 2013;6:e7197. [Google Scholar]
- 18.Cloeckaert A, Verger JM, Grayon M, Grepinet O. Restriction site polymorphism of the genes encoding the major 25 kDa and 36 kDa outer-membrane proteins of Brucella. Microbiol. 1995;141:2111–2121. doi: 10.1099/13500872-141-9-2111. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Goni I, Garcia-Yoldi D, Marin CM, de Miguel MJ, Munoz PM, Blasco JM, et al. Evaluation of a multiplex PCR assay (Bruce-ladder) for molecular typing of all Brucella species, including the vaccine strains. J Clin Microbiol. 2008;46:3484–3487. doi: 10.1128/JCM.00837-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merchant-Patel S, Blackall PJ, Templeton J, Price EP, Tong SYC, Huygens F, et al. Campylobacter jejuni and campylobacter coli genotyping by high-resolution melting analysis of a flaA fragment. Appl Environ Microbiol. 2010;76:493–499. doi: 10.1128/AEM.01164-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W, Raoult D, Fournier P-E. Bacterial strain typing in the genomic era. FEMS Microbiol Rev. 2009;33:892–916. doi: 10.1111/j.1574-6976.2009.00182.x. [DOI] [PubMed] [Google Scholar]