

Abstract
During recent years, inborn errors of human IL-17 immunity have been demonstrated to underlie primary immunodeficiencies with chronic mucocutaneous candidiasis (CMC). Various defects in receptors responsible for sensing of Candida albicans or downstream signalling to IL-17 may lead to susceptibility to Candida infection. While CMC is common in patients with profound T cell immunodeficiencies, CMC is also recognised as part of other immunodeficiencies in syndromic CMC, or as relatively isolated CMC disease. We describe a 40-year-old woman with a clinical picture involving cutaneous bacterial abscesses, chronic oral candidiasis and extensive dermatophytic infection of the feet. By whole exome sequencing, we identified a STAT1-gain-of-function mutation. Moreover, the patient's peripheral blood mononuclear cells displayed severely impaired Th17 responses. The patient was treated with antifungals and prophylactic antibiotics, which led to resolution of the infection. We discuss the current knowledge within the field of Th17 deficiency and the pathogenesis and treatment of CMC.
Background
Chronic mucocutaneous candidiasis (CMC) is a hereditary primary immunodeficiency (PID) characterised by severe persistent or recurrent skin and mucosal infection by Candida albicans, dermatophytosis and onychomycosis.1 Since CMC may be part of other conditions ranging from very severe immunodeficiencies to more isolated and benign conditions, it has been suggested that CMC be divided into three different categories, including (1) CMC associated with profound primary T cell immunodeficiencies, (2) syndromic CMC and (3) isolated CMC disease.2 CMC disease was first described in sporadic cases in the 1960s, followed by the identification of inherited cases in consanguineous families in the 1970s; it is a rare condition with an estimated frequency of 1/100 000 individuals.3 The infectious phenotype typically presents in early infancy in otherwise healthy individuals with persistent or recurrent Candida infection.1 4–8 Some patients display bacterial infections of the respiratory tract and relatively mild staphylococcal infections of the skin,9 dermatophytosis6 and recurrent herpes virus disease.10 Moreover, autoimmune diseases, including thyroid autoimmunity and hepatitis, have been described.11–16 Finally, there is an associated risk of oral and oesophageal squamous cell carcinoma13 17 18 and cerebral aneurysms.19–21
Major progress has been made by the realisation that inborn errors of human IL-17 immunity underlie CMC, irrespective of the genetic cause.2 The main source of IL-17 is the Th17 subset of CD4+T lymphocytes, although CD8+T cells, natural killer (NK) T cells, innate lymphoid cells and γδ-T cells may also contribute.22 Differentiation of naïve T cells into Th17 subset of lymphocytes is a highly complex process driven by multiple cytokines, including IL-1β, IL-6, TGFβ, IL-21 and IL-23 in humans. Negative regulation of Th17 differentiation is exerted by IFNγ dependent Stat1-mediated responses.23 Th17 cells generate IL-17, IL-21 and IL-22, resulting in expression of proinflammatory cytokines (IL-1, IL-6, tumor necrosis factor-α (TNFα), G-CSF and GM-CSF), chemokines (CXCL1, CXCL5, IL-8, CCL2, CCL7) and antimicrobial peptides, from epithelia and fibroblasts.23 Accordingly, Th17 responses promote granulopoiesis and neutrophil recruitment to sites of infection, and mediate host defence against extracellular bacteria and fungi, particularly at mucosal surfaces.22–24 A critical role of the IL-17 pathway has been implicated in mucosal immunity to oral, dermal and disseminated candidiasis in mice.25
Given that defective Th17 responses underlie CMC, various defects in the normal pathway from sensing of C. albicans by pattern recognition receptors (PRR)s to downstream signalling molecules involved in generation and function of IL-17, IL-21 and IL-22, have been identified. First, CMC has been described as a consequence of rare mutations in PRRs, including Dectin1, Dectin2 and MINCLE, or the downstream adaptor CARD9, resulting in impaired cellular recognition of the Candida antigen by phagocytes and epithelial cells, therefore compromising differentiation of Th17 subsets.4 Moreover, CMC is frequently part of an autosomal dominant (AD) hyper-IgE syndrome (HIES) caused by STAT3-mutations resulting in insufficient responses to IL-6, IL-21 and IL-23, and defective generation of Th17 responses.26 27 CMC may also be present in autosomal recessive HIES caused by mutations in TYK2, DOCK8 and GMP3.28 In addition, CMC can present within the Mendelian susceptibility to mycobacteria (MSMD) syndrome caused by mutations in IL-12/23 and IL-12/23 receptor (IL-12R1b deficiency), ultimately resulting in decreased production of Th17 responses.29 30 CMC has also been described in patients with deficiency in ORAI1 or MST1.31 In the presence of STAT1 gain-of-function (GOF) mutations, impaired development of IL-17-producing cells has been suggested to be due to a role of Stat1-dependent IFNγ signalling in negative regulation of Th17 responses, although the precise mechanisms remain to be fully resolved.14 15 Direct interference with IL-17 function and responses can be caused by mutations in IL-17F and IL-17RA, respectively.32 Finally, CMC may be caused by the presence of autoantibodies directed against IL-17A, IL-17F and/or IL-22, abolishing the function of these cytokines.33 34
Case presentation
A 40-year-old woman presented to the International Center for Immunodeficiency Diseases at Aarhus University Hospital, for immunological evaluation. She had a history, since early childhood, of recurrent cutaneous abscesses mostly localised to the femora and nates regions, requiring repeated surgical incisions and antibiotics, as well as chronic oral candidiasis, and an unexplained condition involving excessive vegetative hyperkeratosis of her toes and toenails, with secondary bacterial and fungal infection. The previous medical history of the patient included several referrals to the department of dermato-venereology in our hospital. When the patient was 18 years of age, she developed vegetative hyperkeratosis on her toes and nails. Initial therapy was a topical antimycotic and corticosteroids with moderate and temporary effect. During her first pregnancy at 21 years of age, the hyperkeratosis progressed and became infected, requiring her to be admitted to hospital, with odorous macerated vegetative hyperkeratotic scales affecting the dorsal side of all toes, nail dystrophy and a sharply demarcated erythaematosus dermatitis suggesting dermatophytosis (figure 1A). Bacterial cultures from skin were negative while fungal cultures showed Trichophyton metagrophytes, and microscopy from the scales showed hyphae. The infection resolved with intensive use of topical silver sulfadiazine combined with standard doses of systemic dicloxacillin, metronidazole and terbinafine. In addition, the recurrent hyperkeratotic vegetations were treated with isotretinoin 40 mg daily. Two months after initiation of therapy, the patient's feet had improved significantly, but due to severe mucocutanous side effects, the isotretinoin was discontinued. At 38 years of age, the patient was referred with a similar clinical presentation. Due to suspicion of a psoriasiform skin pathology, she was treated with systemic methotrexate 15 mg weekly but failed to show improvement. Therefore, subcutaneous TNFα blockade with adalimumab was initiated. However, this resulted in a rapid and severe progression of the vegetative hyperkeratosis with secondary infection, and had to be discontinued (figure 1B). At this time, bacterial culture showed Pseudomonas aeruginosa and PCR identified Trichophyton mentagrophytes in the skin. The patient was treated with systemic ciprofloxacine and terbinafine, after which she experienced rapid improvement and an almost complete normalisation of the skin within 2 months (figure 1C). Punch biopsies from 1996 and 2013 showed massive epidermal hyperplasia with hyperkeratosis and psoriasiform hyperplasia, supporting the diagnosis of a hyperkeratotic genodermatosis suspected clinically (figure 2A, B). Fungae and bacteriae appeared in the hyperkeratosis with varying intensity in both biopsies (figure 2C). The extent of the epidermal changes and mixed infection was well documented in a final incisional biopsy performed in 2014. The patient is currently treated with daily prophylactic fluconazole 200 mg combined with cotrimoxazole 400/80 mg.
Figure 1.

Clinical photos. (A) Skin abnormalities of the patient's feet at presentation before treatment, (B) aggravation after weeks of treatment with TNF-α blockade with adalimumab, and (C) following 2 months of antifungal terbinafine treatment leading to almost complete resolution. (D–F) Feet of the father of the index patient (D) before, and (E and F) after treatment with terbinafine for 2 and 4 months, respectively. TNF-α, tumor necrosis factor-α.
Figure 2.
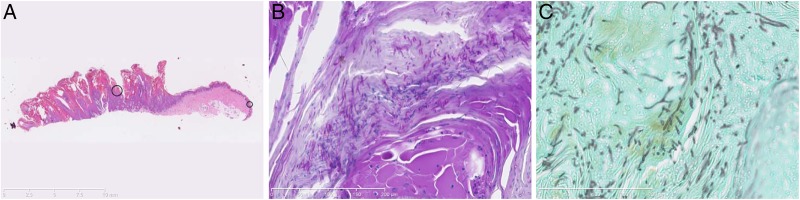
Histopathological sections from punch biopsies from the feet. (A) Massive epidermal hyperplasia with hyperkeratosis and psoriasiform hyperplasia after H&E staining and (B) Periodic-acid-Schiff staining. (C) Fungal hyphae in tissue, demonstrated by Grocott's methenamine silver staining.
Investigations
At 40 years of age, the patient had an immunological evaluation performed. She was found to have normal IgG, IgM and IgA, thereby excluding the presence of common variable immunodeficiency. In addition, IgE was within normal range, making a diagnosis of HIES less likely. She also had a normal distribution of lymphocytes, including total lymphocyte count of 1200 cells/µL, with 82% T cells, 8.9% B cells and 8.2% NK cells. Lymphocyte proliferation assay demonstrated an overall normal response to mitogens as measured by proliferation. In addition, a granulocyte oxidative burst was within normal range excluding chronic granulomatous disease. Complement activation was normal.
Since this routine immunological evaluation did not explain the infectious phenotype, we proceeded to perform whole exome sequencing (WES) with special attention to mutations in genes associated with Th17 defects. We identified a rare variant in STAT1 (NM_0073315.3) at nucleotide position 800 with a C to T substitution (c.800C>T) resulting in amino acid substitution from alanine to valine at position 267 (A267V) (figure 3). This variant is not reported in the ExAC database of 60.706 exomes. This mutation has been described to represent a GOF mutation causing increased Stat1 phosphorylation, decreased Th1 and Th17 responses and ultimately causing AD CMC.15 No homozygous, compound heterozygous, or de novo mutations were identified in any other genes with a known role in host immune responses, particularly not in the genes, including STAT3, DOCK8, TYK2 and GPM3, associated with HIES. The father of the index patient reported a similar medical history with recurrent hyperkeratotic and tender scaly feet and had also suffered from recurring oral, ocular and genital Candida infections. Sanger sequencing of the STAT1 gene was performed identifying the same A267V STAT1 GOF mutation, whereas the two healthy children of the index patient did not have the STAT1 variant. Finally, the grandfather of the index patient had a similar medical history but was deceased, thus precluding further analyses (figure 3A, B).
Figure 3.
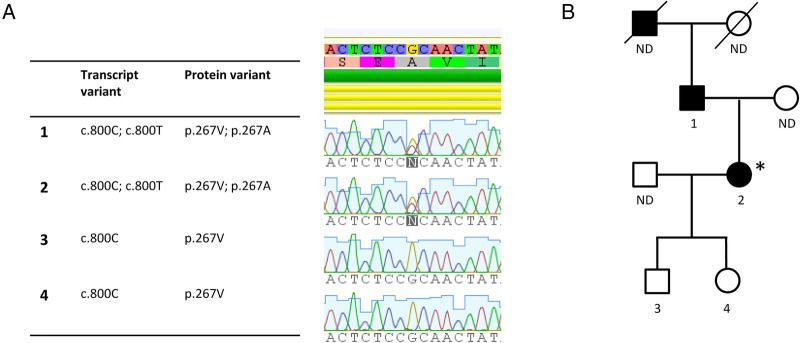
Genetics and pedigree. (A) WES was performed by TruSeq DNA sample preparation, targeting of exomes with SeqCap EZ Human Exome Library V.3.0 (Roche), sequencing on HiSeq, paired end 2×101 bp indexed. Reads were mapped to hg19 employing BWA and GATK. Single nucleotide polymorphisms were called employing HaplotypeCaller from the GATK package. VCF files were uploaded and analysed by Ingenuity Variant Analysis. The identified STAT1 GOF mutation at position c.800C>T results in amino acid substitution at position 267 (A267V) within the CC-domain of the STAT1 molecule. Confirmation by Sanger sequencing of the father (1), index patient (2, marked with *), son (3) and daughter (4). (B) Pedigree showing CMC disease in the grandfather, father (1) and index patient (2). CMC, chronic mucocutaneous candidiasis; GOF, gain-of-function; ND, not determined; VCF, variant call files; WES, whole exome sequencing.
To confirm our findings, we next evaluated Th17 function of the index patient by performing a Th17 assay measuring intracellular synthesis of IL-17 and IFNγ in response to PMA/ionomycin by flow cytometry (figure 4). In support of the diagnosis, we found a major reduction in percentages of cells producing IL-17 (figure 4B) as well as IFNγ (figure 4C) in patient peripheral blood mononuclear cells (PBMCs) compared to a healthy control.
Figure 4.
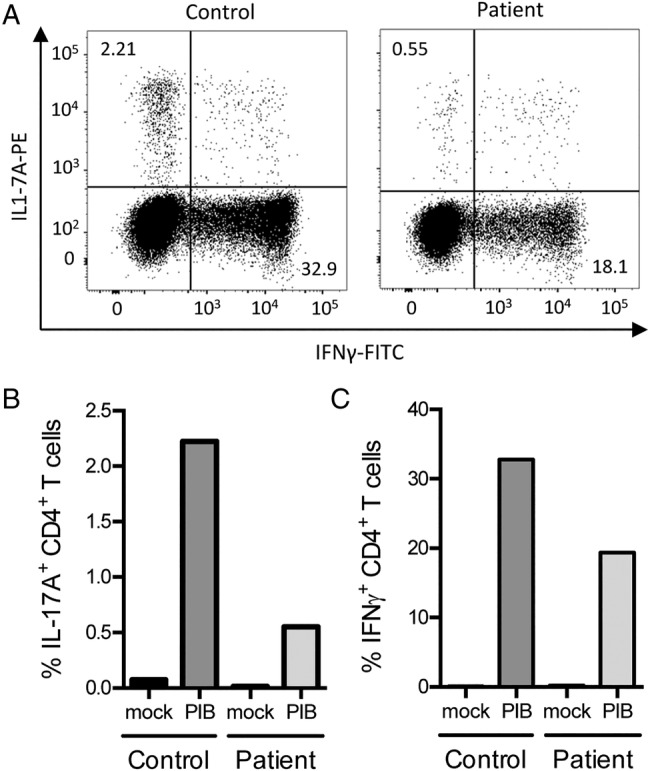
Th17 assay. PBMCs from the index patient and an age-matched and gender-matched control were isolated and stimulated with PMA (50 ng/mL), ionomycin (1 μg/mL) and brefeldin A (10 µg/mL) (PIB) for 4 h. (A) intracellular IL-17 and IFN-γ were measured by flow cytometry. The data demonstrated (B) a reduced fraction of IL-17 producing CD4+T cells in the index patient compared to control (2.21% vs 0.55%), and (C) reduced fraction of IFN-γ-producing CD4+ T cells in the index patient compared to control (32.9% vs 18.1%). The experiment was performed twice, each in duplicates. FITC, fluorescein isothiocyanate; PBMCs, peripheral blood mononuclear cells.
Discussion
Important reports appearing within the past few years have established that Th17 cytokines play an essential and non-redundant role in protection against C. albicans at mucocutaneous surfaces in humans. Although the common denominator appears to be defective IL-17 production or function in a broader sense, the genetic and molecular pathogenesis appears to be very diverse, and, accordingly, the infectious and autoimmune clinical pictures are heterogeneous and highly complex. We describe the clinical presentation of a 40-year-old woman with severe CMC involving oral Candida infection and dermatophytosis with hyperkeratosis of the feet as well as a predisposition to cutaneous abscesses. This clinical picture is in agreement with the CMCD entity.2 We identified a heterozygous STAT1 variant previously described as a GOF mutation and demonstrated impaired IL-17 and IFNγ production in patient PBMCs.15 The patient experienced an almost complete resolution of symptoms after 4 months of antifungal treatment, thereby supporting the diagnosis.
Two groups of investigators independently established heterozygous GOF mutations in STAT1 as a cause of Th17 deficiency and AD CMC. Van de Veerdonk et al15 analysed five different families, including 14 cases of AD CMC, and found defective production of IFNγ, IL-17 and IL-22 in response to Candida, and identified heterozygous mutations within conserved residues in exon 10 encoding the coiled-coil (CC) domain of STAT1. Simultaneously, Liu et al13 identified heterozygous variations in the STAT1 gene within the CC domain by WES. Functional analysis of the mutant alleles revealed GOF mutations by a mechanism involving impaired nuclear dephosphorylation of Stat1, resulting in enhanced responses to IFNα, IFNγ and IL-27, cytokines that are known to be antagonistic to Th17 development.35 36 These reports were further supported by Netea and colleagues, who described a functional mechanism whereby Stat1 hyperphosphorylation and defective IL-12R/IL-23R signalling underlie diminished Th1/Th17 responses in CMC.14 Subsequently, CMC caused by GOF mutations in exon 14 of the STAT1 DNA-binding domain has been described.37 More recently, the same group reported on two novel STAT1 GOF mutations resulting in impaired production of IL-17A and IL-22.38 Altogether, at least 30 different amino acid changes have been reported, of which 21 are in the CC domain and 9 are in the DNA-binding domain of STAT1.2 10 13–15 37 39 Overall, however, the mechanisms of defective IL-17 and IL-22 production remain to be fully understood. Interestingly, the STAT1 GOF mutation may also be directly involved in the pronounced psoriasiform hyperplasia observed in this patient and others.15 A recent study has shown that keratinocytes from psoriasis plaques have increased Stat1 phosphorylation compared to non-lesional skin from the same patients.40
The occurrence of severe pain and functional impairment of hands and feet together with secondary complications, such as squamous cell carcinoma, underscore the importance of diagnosis and prophylactic treatment of patients with CMC. As illustrated by the present case, diagnosis may be delayed but should be made by combining the clinical presentation of severe persistent or recurrent fungal infection of skin, mucous membranes and nails, with the presence of functionally impaired Th17 responses and genetic examination by WES or other genetic tools. Most authorities recommend antifungal prophylaxis with itraconazole or fluconazole. In the case presented here, terbinafine was initially chosen, before the specific diagnosis had been reached, to treat severe dermatophytosis, with very good results. In addition, since Th17 deficiency may also predispose to cutaneous abscesses with Staphylococcus aureus, in some cases, cotrimoxazole may be added.
This case illustrates the complex picture and spectrum of CMC. Moreover, this report supports previous experience in the field of WES being a useful approach in the identification of rare genetic aetiologies in individual patients with an unusual pathological clinical presentation. However, several aspects of CMC remain unravelled, including a better understanding of the precise molecular mechanism, whereby defective Th17 responses lead to a particular susceptibility to fungal infection. Intriguingly, other mutations at specific sites in STAT1 have been associated with other PIDs, including MSMD and herpes encephalitis, by mechanisms involving loss-of-function mutations.41 42 It will be interesting in the future to learn whether other genetic causes interfering with the generation of Th17 responses will be identified in patients with CMC. It therefore remains relevant and important to describe the specific clinical presentation and associated mutations in individual patients with CMC in order to gain more insight into this intriguing spectrum of diseases.
Learning points.
Chronic mucocutaneous candidiasis (CMC) may present as a complex clinical picture with a diverse genetic background.
CMC is characterised by defective Th17 responses; these responses are essential for antifungal immunity at mucosal surfaces.
Whole exome sequencing is a useful tool for the investigation and diagnosis of primary immunodeficiency (PIDs).
Genetic diagnosis is valuable in order to choose the correct treatment of PIDs, and may also have implications for genetic testing of family members and decisions on prophylactic treatment.
PIDs may teach us important lessons on basic immunology and protective immunity to different pathogens in humans.
Acknowledgments
The authors would like to thank the patient and her family for contributing material for the immunological evaluation and for giving permission to publish this case report.
Footnotes
Contributors: JN identified the patient, collected skin biopsies and took clinical pictures (figure 1), and also initiated treatment of the patient. ES provided histopathology stainings (figure 2). CSL was involved in identifying the patient and interpretation of data. MC performed the WES bioinformatical analysis and supplied figure 3. EKO performed the TH17 assay and interpretation of data (figure 4). THM was responsible for the immunological evaluation and treatment of the patient, suggested the diagnosis and genetic and immunological analyses, wrote the first draft of the manuscript, and was also responsible for finalising and revising the manuscript. All the authors read and approved the final version of the manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Kirkpatrick CH. Chronic mucocutaneous candidiasis. J Am Acad Dermatol 1994;31(3 Pt 2):S14–17. 10.1016/S0190-9622(08)81260-1 [DOI] [PubMed] [Google Scholar]
- 2.Puel A, Cypowyj S, Maródi L et al. Inborn errors of human IL-17 immunity underlie chronic mucocutaneous candidiasis. Curr Opin Allergy Clin Immunol 2012;12:616–22. 10.1097/ACI.0b013e328358cc0b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puel A, Picard C, Cypowyj S et al. Inborn errors of mucocutaneous immunity to Candida albicans in humans: a role for IL-17 cytokines? Curr Opin Immunol 2010;22:467–74. 10.1016/j.coi.2010.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glocker EO, Hennigs A, Nabavi M et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 2009;361:1727–35. 10.1056/NEJMoa0810719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Germain M, Gourdeau M, Hebert J. Case report: familial chronic mucocutaneous candidiasis complicated by deep candida infection. Am J Med Sci 1994;307:282–3. 10.1097/00000441-199404000-00008 [DOI] [PubMed] [Google Scholar]
- 6.Shama SK, Kirkpatrick CH. Dermatophytosis in patients with chronic mucocutaneous candidiasis. J Am Acad Dermatol 1980;2:285–94. 10.1016/S0190-9622(80)80040-5 [DOI] [PubMed] [Google Scholar]
- 7.Lilic D. Unravelling fungal immunity through primary immune deficiencies. Curr Opin Microbiol 2012;15:420–6. 10.1016/j.mib.2012.06.003 [DOI] [PubMed] [Google Scholar]
- 8.Eyerich K, Rombold S, Foerster S et al. Altered, but not diminished specific T cell response in chronic mucocutaneous candidiasis patients. Arch Dermatol Res 2007;299:475–81. 10.1007/s00403-007-0792-3 [DOI] [PubMed] [Google Scholar]
- 9.Chipps BE, Saulsbury FT, Hsu SH et al. Non-candidal infections in children with chronic mucocutaneous candidiasis. Johns Hopkins Med J 1979;144:175–9. [PubMed] [Google Scholar]
- 10.Tóth B, Méhes L, Taskó S et al. Herpes in STAT1 gain-of-function mutation [corrected]. Lancet 2012;379:2500 10.1016/S0140-6736(12)60365-1 [DOI] [PubMed] [Google Scholar]
- 11.Lilic D. New perspectives on the immunology of chronic mucocutaneous candidiasis. Curr Opin Infect Dis 2002;15:143–7. 10.1097/00001432-200204000-00007 [DOI] [PubMed] [Google Scholar]
- 12.Coleman R, Hay RJ. Chronic mucocutaneous candidosis associated with hypothyroidism: a distinct syndrome? Br J Dermatol 1997;136:24–9. 10.1111/j.1365-2133.1997.tb08741.x [DOI] [PubMed] [Google Scholar]
- 13.Liu L, Okada S, Kong XF et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 2011;208:1635–48. 10.1084/jem.20110958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smeekens SP, Plantinga TS, van de Veerdonk FL et al. STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS ONE 2011;6:e29248 10.1371/journal.pone.0029248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van de Veerdonk FL, Plantinga TS, Hoischen A et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med 2011;365:54–61. 10.1056/NEJMoa1100102 [DOI] [PubMed] [Google Scholar]
- 16.Hori T, Ohnishi H, Teramoto T et al. Autosomal-dominant chronic mucocutaneous candidiasis with STAT1-mutation can be complicated with chronic active hepatitis and hypothyroidism. J Clin Immunol 2012;32:1213–20. 10.1007/s10875-012-9744-6 [DOI] [PubMed] [Google Scholar]
- 17.Williamson DM. Chronic hyperplastic candidiasis and squamous carcinoma. Br J Dermatol 1969;81:125–7. 10.1111/j.1365-2133.1969.tb15992.x [DOI] [PubMed] [Google Scholar]
- 18.Koch D, Lilic D, Carmichael AJ. Autosomal dominant chronic mucocutaneous candidiasis and primary hypothyroidism complicated by oesophageal carcinoma. Clin Exp Dermatol 2009;34:e818–20. 10.1111/j.1365-2230.2009.03561.x [DOI] [PubMed] [Google Scholar]
- 19.Leroy D, Dompmartin A, Houtteville JP et al. Aneurysm associated with chronic mucocutaneous candidiasis during long-term therapy with ketoconazole. Dermatologica 1989;178:43–6. 10.1159/000248386 [DOI] [PubMed] [Google Scholar]
- 20.Grouhi M, Dalal I, Nisbet-Brown E et al. Cerebral vasculitis associated with chronic mucocutaneous candidiasis. J Pediatr 1998;133:571–4. 10.1016/S0022-3476(98)70072-1 [DOI] [PubMed] [Google Scholar]
- 21.Marazzi MG, Bondi E, Giannattasio A et al. Intracranial aneurysm associated with chronic mucocutaneous candidiasis. Eur J Pediatr 2008;167:461–3. 10.1007/s00431-007-0490-3 [DOI] [PubMed] [Google Scholar]
- 22.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol 2010;10:479–89. 10.1038/nri2800 [DOI] [PubMed] [Google Scholar]
- 23.McDonald DR. TH17 deficiency in human disease. J Allergy Clin Immunol 2012;129:1429–35. 10.1016/j.jaci.2012.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vautier S, Sousa MG, Brown GD. C-type lectins, fungi and Th17 responses. Cytokine Growth Factor Rev 2010;21:405–12. 10.1016/j.cytogfr.2010.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kagami S, Rizzo HL, Kurtz SE et al. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol 2010;185:5453–62. 10.4049/jimmunol.1001153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milner JD, Brenchley JM, Laurence A et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008;452:773–6. 10.1038/nature06764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mogensen TH. STAT3 and the Hyper-IgE syndrome: Clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. JAKSTAT 2013;2:e23435 10.4161/jkst.23435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Q, Davis JC, Lamborn IT et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med 2009;361:2046–55. 10.1056/NEJMoa0905506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Beaucoudrey L, Puel A, Filipe-Santos O et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med 2008;205:1543–50. 10.1084/jem.20080321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hambleton S, Salem S, Bustamante J et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 2011;365:127–38. 10.1056/NEJMoa1100066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feske S, Picard C, Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin Immunol 2010;135:169–82. 10.1016/j.clim.2010.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puel A, Cypowyj S, Bustamante J et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011;332:65–8. 10.1126/science.1200439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kisand K, Bøe Wolff AS, Podkrajsek KT et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med 2010;207:299–308. 10.1084/jem.20091669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puel A, Döffinger R, Natividad A et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 2010;207:291–7. 10.1084/jem.20091983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Diveu C, McGeachy MJ, Boniface K et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J Immunol 2009;182:5748–56. 10.4049/jimmunol.0801162 [DOI] [PubMed] [Google Scholar]
- 36.Ramgolam VS, Sha Y, Jin J et al. IFN-beta inhibits human Th17 cell differentiation. J Immunol 2009;183:5418–27. 10.4049/jimmunol.0803227 [DOI] [PubMed] [Google Scholar]
- 37.Takezaki S, Yamada M, Kato M et al. Chronic mucocutaneous candidiasis caused by a gain-of-function mutation in the STAT1 DNA-binding domain. J Immunol 2012;189:1521–6. 10.4049/jimmunol.1200926 [DOI] [PubMed] [Google Scholar]
- 38.Yamazaki Y, Yamada M, Kawai T et al. Two novel gain-of-function mutations of STAT1 responsible for chronic mucocutaneous candidiasis disease: impaired production of IL-17A and IL-22, and the presence of anti-IL-17F autoantibody. J Immunol 2014;193:4880–7. 10.4049/jimmunol.1401467 [DOI] [PubMed] [Google Scholar]
- 39.Boisson-Dupuis S, Kong XF, Okada S et al. Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr Opin Immunol 2012;24:364–78. 10.1016/j.coi.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hald A, Andrés RM, Salskov-Iversen ML et al. STAT1 expression and activation is increased in lesional psoriatic skin. Br J Dermatol 2013;168:302–10. 10.1111/bjd.12049 [DOI] [PubMed] [Google Scholar]
- 41.Dupuis S, Dargemont C, Fieschi C et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science 2001;293:300–3. 10.1126/science.1061154 [DOI] [PubMed] [Google Scholar]
- 42.Dupuis S, Jouanguy E, Al-Hajjar S et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet 2003;33:388–91. 10.1038/ng1097 [DOI] [PubMed] [Google Scholar]