

Abstract
Pluripotency of embryonic stem cells (ESCs) and induced pluripotent stem cells is regulated by a well characterized gene transcription circuitry. The circuitry is assembled by ESC specific transcription factors, signal transducing molecules and epigenetic regulators. Growing understanding of stem-like cells, albeit of more complex phenotypes, present in tumors (cancer stem cells), provides a common conceptual and research framework for basic and applied stem cell biology. In this review, we highlight current results on biomarkers, gene signatures, signaling pathways and epigenetic regulators that are common in embryonic and cancer stem cells. We discuss their role in determining the cell phenotype and finally, their potential use to design next generation biological and pharmaceutical approaches for regenerative medicine and cancer therapies.
Keywords: Embryonic stem cells, Cancer stem cells, Pluripotency, Self-renewal, Tumorigenicity
Core tip: Accumulating experimental evidence has revealed the existence of common stemness regulators for embryonic and cancer stem cells. In this review, we highlight current results on biomarkers, gene signatures, signaling pathways and epigenetic regulators that determine the phenotype of these two types of stem cells. We also discuss how this knowledge may promote the design of next generation biological and pharmaceutical tools for regenerative medicine and cancer therapies.
INTRODUCTION
Embryonic stem cells (ESCs) have two unique properties, self-renewal and pluripotency[1]. Mouse ESCs (mESCs) are isolated from day 3.5 blastocyst and possess ground state pluripotency whereas human ESCs (hESCs) are isolated from late blastocyst and correspond to the epiblast stem cells of the mouse[2,3]. The pluripotency of ESCs is determined by the concerted action of signaling pathways that respond to external stimuli, intrinsically expressed transcription factors and complexes that govern the epigenetic state. The extended transcriptional network of ESCs is centered on the triad of master regulators of pluripotency Oct4, Sox2 and Nanog[4]. In the last decade the introduction in somatic cells of transcription factors (Oct4, Sox2, Klf4, c-Myc), microRNAs and small molecules allowed the generation of induced pluripotent stem (iPS) cells[5]. Due to their ability to give rise to any type of differentiated cells and tissues, both ES and iPS cells offer many opportunities for modeling human diseases and development of regenerative medicine[6].
ESCs and tumor cells share many common properties exemplified by rapid proliferation, similar metabolic requirements and inhibition of differentiation. Pluripotent ESCs have inherent tumorigenic potential and they generate benign tumors and teratomas when injected in immunodeficient mice[7]. Reprogramming of somatic cells into pluripotency by oncogenes like Myc and Klf4 suggest a strong link between pluripotency and tumorigenicity[8,9]. Currently, growing experimental evidence has revealed that tumors contain a variable number of cells that have self-renewal and partial differentiation capacities[10-12]. Because these cells share these properties with the adult tissue stem cells from which they are likely derived they were termed cancer stem cells (CSCs)[10-12]. The procedures of somatic cell reprogramming and CSC establishment are both dependent on transitions between epithelial and mesenchymal states (EMT/MET)[13,14]. In addition, CSCs from epithelial tumors also exhibit ESC-like signatures[15,16] that include the oncogene c-Myc and factors important for pluripotency such as Sox2, Dnmt1, Cbx3 and HDAC1[16].
The CSC model[10-12] ultimately links Cancer with Stem cell biology and provides a common framework that is proposed to account for all the properties of stem cells, regardless of their early or late developmental origin in normal or pathological states. In this review, we analyze common regulatory mechanisms of embryonic and CSCs focusing on biomarkers, signaling pathways, transcription factors and epigenetic complexes. This information can elucidate the risks stemming from the tumorigenic potential of pluripotent ESC and iPS cells used in tissue regeneration therapies. Additionally, knowledge about their stem cell properties is valuable for the eradication of CSCs that are responsible for therapy resistance, tumor invasion and metastasis.
GENERAL PROPERTIES AND MARKERS FOR EMBRYONIC/PLURIPOTENT AND CSC
Three types of markers for the identification of either ESCs or CSCs are utilized: cell surface molecules, signaling pathway markers and transcription factors[17]. However, cell surface molecules are mostly used as biomarkers since they can be assessed on intact living cells.
ESC biomarkers
All pluripotent stem cells express on their surface glycan epitopes that show species-specific and differentiation stage-specific expression, the stage specific embryonic antigens (SSEA-1, 3 and 4). SSEA-1 (CD15/Lewis x) is expressed on mESCs and embryonic carcinoma cells (ECCs) but is absent from hESCs[18,19]. Upon mESC differentiation SSEA-1 expression decreases whereas SSEA-4 expression is induced. Undifferentiated hESCs express SSEA-3/4 but following differentiation they are both silenced and SSEA-1 is induced[20-22] (Table 1).
Table 1.
Biomarkers of pluripotency in mouse and human embryonic stem cells
| Biomarker | Role in mESC | Role in hESC |
| Oct4 | Pluripotency[200] | Pluripotency[4] |
| Sox2 | Pluripotency[215] | Pluripotency[4] |
| Nanog | Pluripotency[227] | Pluripotency[4] |
| Klf4 | Pluripotency[253,255] | Pluripotency[253] |
| c-Myc | Pluripotency[190] | Pluripotency[190] |
| SSEA1/CD15 | Pluripotency[18,19] | Not expressed |
| SSEA 3, 4 | Not expressed | Pluripotency[20,22] |
| TRA-1-68 | Not expressed | Pluripotency[24] |
| TRA-1-81 | Not expressed | Pluripotency[24] |
| Cripto-1 | Pluripotency[28,474] | Pluripotency[28,474] |
hESC: Human embryonic stem cell; mESCs: Mouse embryonic stem cells; SSEA: Stage specific embryonic antigens; TRA: Tumor rejection antigens.
Human ESC, embryonic carcinoma and germ tumors are characterized by the expression of tumor rejection antigens (TRA-1-60 and TRA-1-81). These are proteoglycan epitopes that reside on the 200 kDa form of podocalyxin (SC-podocalyxin)[23,24]. Because they are not expressed on somatic cells, TRAs are useful markers for the isolation of human iPS cells during reprogramming[25].
Cluster of differentiation antigens (CDs) are membrane proteins that function in diverse processes such as cell adhesion, communication and differentiation. Various family members are expressed in mESCs, hESCs and ECCs. Their expression usually changes following differentiation[26]. CD324 (E-cadherin), CD31 (PECAM-1), CD24, CD90 (Thy-1), CD9, CD59, CD133 and CD326 (EpCAM) are present on the membrane of ESCs and ECCs although their expression levels can vary depending on the cell line and culture conditions[17]. Among CDs the epithelial marker CD326 (EpCAM) is the more closely correlated with the undifferentiated state and is rapidly lost upon differentiation[27] (Table 1). Interestingly, some of the above CDs are also used as CSC markers, as will be discussed below.
Among these markers of stemness, Cripto-1 (CR1, TDGF-1) represents an important component of a critical core pathway that is used by ESCs (Table 1). This growth factor acts during embryogenesis as a TGF-β ligand, co-receptor and as an oncogene. Moreover, Cripto-1 is involved in PI3K/Akt and MAPΚ pathways in a SMAD-independent manner and it enhances the Wnt and Notch pathways acting as a chaperone for low-density lipoprotein receptor-related protein 5 (LRP5) and Notch respectively[28]. Cripto-1 is crucial for early embryonic development and is expressed in both mouse and human ESCs resulting in maintenance of stem cell pluripotency. Additionally, Cripto-1 regulates ESC fate choices by repressing the neural and enhancing the cardiomyocytic differentiation[29]. Recently, it was shown that Cripto-1 performs an essential role in the etiology and progression of several types of human tumors, where it is expressed in a CSC subpopulation and facilitates epithelial-mesenchymal transition (EMT)[28]. Intracellular transcription factors such as Oct4, Sox2, Nanog, Klf4 that are specifically expressed in undifferentiated stem cells are also important biomarkers. Their importance for pluripotency is examined in detail below.
CSC biomarkers in solid tumors
Breast cancer was the first type of solid tumor where CSCs were identified. Cells exhibiting the EPCAM+ ESA+CD44+CD24-/lowLin- phenotype were able to propagate breast tumors when injected even in a low concentration into the mammary fat pad of immunodeficient mice[30]. Further studies identified a subpopulation of CD44+CD24- cells expressing Aldehyde dehydrogenase (ALDH) capable of propagating tumor, by injection of as little as 20 cells in immunodeficientmice[31]. CD44 is a cell membrane protein that binds hyaluronan (HA) and has a role in cell-cell and cell-extracellular matrix (ECM) interactions. Studies have implicated CD44 in breast cancer cell adhesion, migration, invasion and metastasis[32]. Additionally, it protects cells from apoptosis which is an important characteristic of CSCs[33]. HA-CD44 interaction promotes multiple cascades, activating gene transcription of stem cell-related factors in many different tumors, such as ovarian, breast, head and neck cancer[33]. Like CD44, CD24 is a widely expressed glycosylated cellular adhesion protein. Low expression of CD24 in breast CSCs was shown to enhance their growth ability and metastatic potential, through a chemokine receptor CXCR4 response[34]. Additional surface markers for breast CSCs are: CD13, also a marker for brain and colon CSCs, a6-integrin, CD61, CD29 and CD49.
The ALDH family of enzymes catalyzes the oxidation of aldehydes into carboxylic acids in a NADP+ dependent manner[35]. ALDHs play a crucial role in retinoic acid biosynthesis, metabolism of cyclophosphamides and clearing toxic byproducts of reactive oxygen species[36]. High ALDH activity was measured in human adult hematopoietic and breast stem cells, murine neural stem cells, as well as leukemia, breast, colon, head and neck CSCs[35]. Emerging evidence supports the significance of ALDH as a biomarker for adult and CSCs of different origin, including pancreatic, prostate, ovarian, lung, head and neck squamous cell carcinoma[37].
The first evidence for the existence of CSCs in brain tumors was provided by Singh et al[38]. Brain CSCs express the cell surface marker CD133 and lack the expression of neural differentiation markers. CD133 is a transmembrane glycoprotein expressed in mouse and human ESCs, as well as in different types of adult stem/progenitor cells, including hematopoietic and neural stem cells, endothelial precursors, mesenchymal progenitors, kidney, mammary glands and pancreatic, colorectal, testis, prostate, ovarian, lung and melanoma CSCs[39]. The exact function of this protein remains unknown, but it was proposed to act as an organizer of the cell membrane topology[40]. Limitations on the exclusive use of CD133 as a brain CSC marker arise from a study by Beier et al[41] who have reported that CD133 negative cells from glioblastoma sphere cultures are able to propagate tumors in immunodeficient mice. Moreover, CD133 expression was shown to depend on culture conditions and hypoxia levels. Additional markers shared by normal and cancer brain stem cells are nestin, Sox2, Musashi-1 and Bmi-1[42]. Several studies have indicated that multidrug resistance transporters (MDR) such as ATP binding cassette (ABC) transporters and breast cancer resistance protein (BCRP/ABCG2) increase drug efflux from the cells[43,44].
Stage-specific embryonic antigen 1 (SSEA1), also known as CD15, is a well characterized marker of undifferentiated mouse and differentiated human ESCs. It was first identified in neural embryonic progenitors and represents a putative brain CSC marker. SSEA1 expression correlates with increased cell proliferation, decreased differentiation and apoptosis[45]. CD15+ cells exhibit high tumorigenicity. Another potential brain CSC marker is Nestin, an intermediate filament protein that was first identified as a neural stem cell marker[46]. It is expressed in many different brain tumors and is involved in stemness, cell growth, invasion and migration[47]. Finally, brain, like other CSCs from breast and lung are resistant to chemotherapy and radiotherapy because of the expression of MDR1 transporter on their cell surface[48].
Colorectal CSCs also express CD133[49] and the surface molecule Epithelial Specific Antigen (ESA/EpCAM), while lacking expression of intestinal differentiation markers such as cytokeratin 20 (CK20). EpCAM is an epithelial adhesion molecule involved in proliferation, differentiation, migration and signaling[50,51]. Another adhesion protein, CD166, was proposed as a biomarker in colorectal CSCs[51]. CD166 is present in a wide variety of normal tissues, as well as in different cancers including breast, lung, prostate and melanoma[52]. CD166 is used as a positive prognostic marker for survival in colorectal cancer despite the contradictory studies regarding its timeframe of expression during tumorigenesis. Other recently identified potential markers include CD24, CD29 and Lgr5. CD29 (β1-integrin), a transmembrane receptor for extracellular proteins activates signaling cascades responsible for proliferation, differentiation, migration, survival or death[53]. Finally, Leucine-rich repeat-containing G protein coupled receptor 5 (Lgr5), a receptor for R-spondins is characterized by a large extracellular and seven transmembrane domains. It binds R-spondin proteins which activate Wnt/β-catenin signaling as a Wnt pathway co-receptor[54,55]. Lgr5, a Wnt pathway target itself, is expressed in various stem cell types. High Lgr5 expression levels are associated with high vimentin and low miR-200c expression followed by increased invasiveness and lymph node metastasis[56].
Pancreatic CSCs are characterized as CD44+CD24+EpCAM+[57], like CSCs from other solid tumors, such as ovarian cancer[46]. On the contrary, breast CSCs exhibit low CD24 expression. Tyrosine kinase c-Met and CD133 have emerged as additional pancreatic CSC markers[58,59]. Indeed, solely CD133+ cells, were shown to induce tumor formation in high frequency[59]. Moreover, concomitant expression of CXCR4 promotes metastasis and represents a useful target for antitumor drugs[59,60].
Prostate CSCs are characterized by high expression of CD44 and CD133, as well as the existence of ALDH1A1 and ABCG2 transporter on their cell surface, which confer chemoresistance. Α2β1 integrin was also proposed as a prostate CSC marker, in addition to the lack of differentiation markers, such as PSA[61].
Ovarian CSC markers are CD133, CD44 and CD24[62]. Additional markers are EpCAM, ALDH1 and CD117 or c-kit proto-oncogene[62]. The c-KIT receptor is activated by autophosphorylation upon stem cell factor (SCF) binding and is involved in cell proliferation, apoptosis, differentiation and adhesion[46]. Moreover, CD117 was associated with chemotherapy resistance[63]. Finally, other molecules that are significant for ovarian CSCs are MyD88 for chemoresistance, Lin28 and Oct4 for cancer stemness and dedifferentiation[35].
Melanoma CSCs were recently found to be positive for CD20, CD133 and the ABC transporters, ABCG2 and ABCB5[64]. Additional markers are CD133 and CD166[64]. CD133+ melanoma cells, which overexpress CD166 and nestin, exhibit tumor-propagating ability and high expression levels of genes responsible for tumor initiation and metastasis[65]. CD271, which was proven to be important for maintenance of stem-like properties and tumorigenicity of melanoma cells[66] is considered as the most convincing biomarker for melanoma CSCs. In addition, CXCR6 is implicated in their asymmetric division[67] and Oct4 that is induced upon hypoxia is capable of promoting melanoma cell dedifferentiation into CSCs[68].
Lung CSCs express CD133 as well as CD24, CD34, CD44, CD87 and ALDH1[35,69]. CD133+ cells show increased expression of Oct4 protein and the ABCG2 transporter[70]. Finally, Bmi-1 which is expressed in human small cell lung cancers could be applied as a potential lung CSC marker due to its role in self-renewal[71].
Hepatocellular CSCs express CD133[72]. CD133+ cells exhibited increased expression of Oct4, Notch, Wnt/β-catenin, Hedgehog and Bmi-1, genes implicated in self-renewal, pluripotency, proliferation and differentiation[73]. EpCAM, CD90 (Thy-1), CD44, CD13, ALDH1, ABCG2, CD117 and AFP represent additional hepatic CSC markers[74].
The head and neck squamous cell carcinoma share the same biomarkers as most of the above described CSCs, including CD44, CD133, ABCG2, ALDH1, c-Met, Bmi-1 and Lgr5[75-77].
In conclusion, putative CSCs have been characterized and enriched from many types of solid tumors using various cell surface markers. These CSC biomarkers offer important biological diagnostic and therapeutic tools (Table 2).
Table 2.
Biomarkers of cancer stem cells
| Cancer type | Biomarkers |
| Breast | CD44[32,33] ALDH[31,35] EpCAM[30] CD13[35] A6-integrin[35] CD61[35] CD29[35] CD49[35] ABCG2[43,44] Nanog[204] Klf4[263,264] |
| Brain | CD133[39] Nestin[42] Sox2[42] Musashi-1[42] Bmi-1[42] ABCB1[48] ABCG2[48] ABCC1[48] SSEA1[45] Nanog[236] Sox2[42,225] Klf4[269] |
| Colorectal | CD133[49] EpCAM[50,51] CD166[51] CD24[51] CD29[51] Lgr5[56] Klf4[266] |
| Pancreatic | CD44[57] CD24[57] EpCAM[57] c-Met[58] CD133[59] CXCR4[59,60] Sox2[221] |
| Prostate | CD44[61] CD133[61] ALDH1A1[61] ABCG2[61] A2β1[61] Sox2[61] |
| Ovarian | CD133[62] CD44[62] CD24[62] EpCAM[62] ALDH1[62] CD117[62] c-kit[62] CD117[63] MyD88[35] Lin28[35] Oct4[35] Nanog[232,238] Sox2[219] |
| Melanoma | CD20[64] CD133[64] ABCG2[64] ABCB5[64] CD133[64] CD166[64] CD271[66] CXCR6[67] Oct4[68] Sox2[224] |
| Lung | CD133[69] CD24[35] CD34[35] CD44[35] CD87[35] ALDH1[35] ABCG2[70] Bmi-1[71] Nanog[234] Sox2[223] |
| Hepatocellular | CD133[72] EpCAM[74] CD90[74] CD44[74] CC13[74] ALDH1[74] ABCG2[74] CD117[74] AFP[74] |
| Head and neck (HNSCC) | CD44[75] CD133[76] ABCG2[76,77] ALDH1[77] c-Met[77] Bmi-1[77] Lgr5[77] |
CONVERGENCE OF SIGNALING PATHWAYS IN EMBRYONIC AND CSC
Pluripotency of ESCs is regulated by core transcription factors as well as key signaling pathways, including LIF/Stat3 in the case of mESCs, Wnt, Hedgehog, Notch, FGF and TGF-β for both mESCs and hESCs[78]. One hallmark of CSCs is their self-renewal capacity driven by developmental pathways[13,79,80]. Below, we will outline the common signaling mechanisms in self-renewal, differentiation and pluripotency of mES, hES and CS cells in solid tumors.
Jak/Stat signaling
Binding of cytokines to their cognate receptors induces the activation of Jak kinases and the phosphorylation, dimerization and nuclear shuttling of signal transducers and activators of transcription (STATs). The Jak/Stat signaling pathway is activated by the cytokine LIF (leukemia inhibitory factor) in mESCs and is required for their self-renewal and pluripotency[81]. In contrast, LIF does not support the pluripotency of human ESC.
Numerous studies have shown the ability of STAT3 to promote tumorigenesis when it is aberrantly activated[82]. Most importantly Stat3 has been recently shown to regulate the survival and proliferation of colon[83] prostate[84] and breast[85] CSCs.
Wnt/β-catenin signaling
The Wnt/β-catenin branch of Wnt signaling-also referred to as the “canonical” Wnt-pathway- is important for proper embryonic development and adult tissue homeostasis. There are more than 30 extracellular Wnt-ligands, which bind to the receptor complex Frizzled and LRP5/6 (member of the LDL receptor family)[86]. In the absence of Wnt signals, the scaffolding proteins Axin and adenomatous polyposis coli (APC) anchor the intracellular signaling protein β-catenin to a “destruction” complex involving Glycogen-activated kinase-3 (GSK-3). Wnt ligand-receptor binding initiates a series of events resulting in inhibition of the destruction complex and β-catenin cytoplasmic accumulation[87]. Its concentration-dependent nuclear translocation and interaction with the T-cell factor/lymphocyte enhancer binding factor (Tcf/Lef) family leads to transcription of proliferative genes such as c-Myc and cyclinD1[88,89].
Many members of the Wnt signaling pathway are implicated in stem-cell proliferation and activity. In mESCs, Wnt promotes self-renewal[90]. Studies using small-molecule inhibitors have highlighted GSK-3 as a critical regulator of pluripotency for both mouse and human ESCs[91]. β-catenin is dispensable for mESC maintenance, however, in its absence, the positive effect of GSK3 inhibition on self-renewal is abolished[92] and β-catenin mutant mice display embryonic neural progenitor defects[93]. Double mutants of GSK3 and β-catenin promote exit from pluripotency and induction of neuroectoderm differentiation[94]. From the mechanistic point of view, accumulating evidence indicates that the key pathway effector Tcf3 is acting as a repressor of Oct3/4, Sox2 and Nanog, whereas β-catenin inhibits this repression by converting Tcf3 into activator[95]. Additionally, active β-catenin interacts with Oct4 and enhances its activity in a Tcf-3 independent manner[96]. In hESCs, there is an ongoing debate about the contribution of Wnt signaling in self-renewal or differentiation[91,97]. Long-term inhibition of Wnt did not affect survival of hESCs, whereas activation of Wnt signaling resulted in activation of mesoderm differentiation. In fact, differential activity of Wnt signaling associates with distinct lineage-specific differentiation potential of hESCs[98].
Wnt signaling also regulates the activity of somatic SCs including those in the skin, blood, brain and the mammary gland, whereas aberrant signaling results in neoplasia[94]. Mutations in key mediators of the Wnt pathway have been observed in approximately 90% of all colon cancers[78]. Wnt activity was found to be enhanced in the CD133+ stem-like cell population of colorectal cancer and in Lgr5+ (a Wnt family member) intestinal crypt stem cells, which were identified as the origin of adenocarcinomas[94,99]. Following transplantation in a mouse model for lineage tracing, Lgr5+ cells expand clonally repopulating all other adenomas[100]. Myofibroblast-secreted factors were linked through β-catenin-dependent transcription to colon CSC clonogenicity and were shown to restore the CSC phenotype in more differentiated tumor cells, both in vitro and in vivo[101]. These findings indicate a Wnt-dependent role of the tumor microenvironment in colon carcinogenesis.
Wnt is also involved in mammary gland tumorigenesis, since Wnt signaling expands mammary gland stem cells in early tumorigenic lesions of MMTV-Wnt1 transgenic mice[102]. The mammary gland CSCs pool is sustained through recruitment of Wnt ligands by periostin, a component of the extracellular matrix of fibroblasts. Infiltrating tumor cells induce its expression in the stroma of secondary organs, such as lung, to allow colonization. Inhibition of its function prevents metastasis[103].
Finally, although the role of Wnt in brain CSCs is still unclear, autocrine activation of Wnt/β-catenin signaling by the orphan nuclear receptor Tlx, that induces glioma formation when overexpressed, was shown to enhance the proliferation of murine neural stem cells[104]. Moreover, a recent study linked low oxygen levels in the hippocampus with neurogenesis through HIF-1α enhanced Wnt signaling[105]. Interestingly, brain tumor formation is promoted by hypoxia[94] and HIFs were identified as key players in stemness and malignancy maintenance of colon cancer cells[106].
Hedgehog and Notch signaling
Hedgehog (Hh) signaling is implicated in many developmental processes, such as in proliferation and cell-fate specification of neural stem and neural crest stem cells. In lack of Hh pathway activity, many target genes like the Hh receptor Patched (PTC) and the Gli family of transcription factors are actively repressed. Secreted Hh-transmembrane PTC binding results in releasing PTC-repression on Smoothened (SMO), a G-protein-coupled receptor that activates downstream intracellular components[107]. Despite the presence of functional pathway components, SHh (Sonic Hedgehog) ligand appears to be dispensable for maintaining pluripotency and promoting proliferation of undifferentiated hESCs. However, addition of exogenous SHh to embryoid bodies generated by hESCs promotes differentiation towards neuroectodermal[108]. On the contrary, SHh was reported to stimulate mESC proliferation through the concerted effect of activated Gli1, increased intracellular Ca2+ levels, PKC (protein kinase C) and epidermal growth factor receptor (EGFR) activation[109].
In adults the Hh pathway is mainly inactive, but it has been reported to participate in tissue maintenance and repair. Its deregulated activity has been linked to cancer development, however little is known about its role in CSCs[110]. Hh signaling downstream effectors Gli1/2 and Bmi-1, a transcriptional repressor of the polycomb group and central regulator of self-renewal in normal stem cells, were recently shown to control proliferation and pluripotency of breast CSCs and normal human mammary stem/progenitor cells[111]. Active Hh signaling has also been identified in glioblastoma CSCs. Inhibition of the pathway by chemical molecules or siRNAs leads to loss of their tumorigenic potential[112,113]. Hh signaling is furthermore preferentially activated in colon carcinoma CSCs derived from primary clinical specimens, whereas its hindrance negatively regulates cancer cell proliferation and induced apoptosis of colon CSCs[114]. Finally, a number of recent studies have implicated Hh signaling in EMT and metastasis, for example in pancreatic cancer cell lines inhibition of Hh results in EMT-inhibition and blocks metastasis[115].
Notch signaling is a crucial regulator of cell to cell communication during embryogenesis, cellular proliferation, differentiation, and apoptosis[79]. The Notch pathway consists of a membrane-tethered receptor (Notch 1-4), that undergoes proteolytic cleavage by ADAM-type metalloproteases and γ-secretase upon ligand binding (membrane-associated Delta-like, Jagged in mammals). Following cleavage, the released intracellular Notch domain shuttles to the nucleus to form a transcriptional complex with recombining binding protein suppressor of hairless (RBPJ, also known as CBF1) and mastermind-like proteins, ultimately activating genes of the hairy and enhancer of split-related family[78,79].
Despite the presence of functional pathway components during early embryonic development, Notch signaling is dispensable for the maintenance of hESC or mESC pluripotency and constitutively activated Notch does not alter the stem cell phenotype[116-118]. Inhibition of pathway components by siRNAs or by a chemical compound against γ-secretase activity (GSI) interfered with hESC proliferation without driving their differentiation[119]. However, a number of studies indicate a decisive role for Notch in ESC fate determination. Activated Notch signaling was shown to promote neural commitment of both hESCs and mESCs[117]. In undifferentiated hESCs it is required to form the progeny of all three embryonic germ layers except for trophoblast cells[120]. In mESCs, there appears to be no in vivo requirement for the Notch pathway, until after all three germ layers have formed[121].
Notch signaling is important for tissue maintenance in many organs, including the skin, blood, intestine, liver, kidney, central nervous system, bone and muscle[121]. It promotes the maintenance of the neural, myogenic and intestinal stem cell pool in both Drosophila and mouse[122].
Deregulation of Notch has been reported in several cancer types and is progressively linked to CSC self-renewal[78]. Notch pathway components are characterized by higher expression level in pancreatic CSCs. Their inhibition using either GSI or Hes1 shRNA reduced CSC numbers and tumorsphere formation. Conversely, Notch activation increased pancreatic CSC self-renewal. In vivo treatment of orthotopic pancreatic tumors in NOD/SCID mice with GSI blocked tumor proliferation and reduced the CSC population[123]. Notch signaling is also activated and plays a crucial role in promoting CSC survival, proliferation and tumor initiation (but not progression) in colon cancer. An antibody against Notch ligand DLL4 inhibited tumor growth in a xenograft mouse model[124,125].
In medulloblastoma, increased Notch and Hh signaling have been linked to the maintenance of a stem-like cell population. Pharmacological depletion of Notch signaling inhibits medulloblastoma growth in mouse xenografts[126]. In this context, Notch was proposed to interact with Hh signaling to promote oncogenesis[127]. Additional pathway interactions were found in human breast epithelial cells, where oncogenic conversion is driven by increased Wnt signaling via Notch-dependent mechanism[128]. Deregulation of Notch signaling is an early event in pre-invasive ductal carcinomas. Reduced mammosphere forming efficiency of in situ ductal carcinoma in the presence of Notch inhibitors suggested that Notch regulates breast CSC self-renewal[129]. In normal breast tissue, Notch1 was proposed to regulate progenitor-to-luminal differentiation, whereas Notch4 stem-to-progenitor cell transitions. Interestingly, inhibition of Notch4 and, to a lesser extent, Notch1 signaling results in decrease of the stem-like cell population and of tumorsphere formation in primary breast cancer samples and cell lines and in limited tumor formation in vivo[130]. Furthermore, Notch4 (but not Notch1) activation inhibits mammary epithelial cell differentiation and promotes mammary carcinogenesis in mice[80,131].
TGF-β signaling
The TGF-β pathway plays a major role in development. Depending on the downstream effector molecules, it can be classified into the Smad1/5/8, the Smad2/3 and the Tab/Tak pathways. Secreted TGF-β ligands bind to the extracellular domain of Ser/Thr kinase type I and type II TGF-β trans-membrane receptors (TGF-βR) thereby phosphorylating and activating latent cytoplasmic SMAD transcription factors[132]. Among 42 known ligands in humans, bone morphogenetic proteins (BMPs) and growth differentiating factors bind type I receptors (GDFs) activate Smad1/5. Activin and Nodal trigger phosphorylation of Smad2/3 through TGF-βR I/II. Activated Smads form a higher-order protein complex with Smad4, which then translocates to the nucleus to modify gene transcription. Inhibitory cytoplasmic Smad6/7, as well as molecules secreted by neighboring cells like Lefty, further increase the regulatory complexity of the pathway[78].
During embryonic development cell fate determination, such as mesoderm and primitive streak formation in the mouse, as well as neural induction and mesoderm specification in Xenopus are affected by the TGF-β pathway[133]. Both the Smad1/5/8 and the Smad2/3 branches are involved in ESC pluripotency/differentiation. Activin/Nodal/Smad2/3 signaling is important for sustaining self-renewal and pluripotency of mouse and human ESCs[134-136], whereas BMP/Smad1/5/8 signaling promotes self-renewal in mESCs[137,138] and differentiation in hESCs[139-141]. The partly divergent signaling outcomes observed in mouse vs human ESCs are most likely due to the different developmental stages from which they are derived, hESCs being more similar to mouse epiblast stem cells (EpiSCs)[3,142].
In mESC culture, concerted BMP/LIF signaling sustains pluripotency through the induction of inhibitor of differentiation (Id) proteins, and by inhibiting two major differentiation pathways, namely extracellular receptor kinase (ERK) and p38 mitogen-activated protein kinase (MAPK) at the same time[137,138,143]. Furthermore, it was recently reported that mESC self-renewal is endogenously activated by autocrine loops of Activin/Nodal[135].
In hESC culture, Activin A, which is secreted by mouse embryonic fibroblast feeder layers, suppresses BMP signaling and hESC differentiation, while stimulating the expression of pluripotency factors (e.g., NANOG, OCT4, FGF2/8, NODAL)[144,145]. Nodal is secreted by hESCs themselves, reinforcing their pluripotent state by an autocrine mechanism. A regulatory loop is formed by simultaneous secretion of the Nodal inhibitor Lefty. Upon differentiation Nodal expression is rapidly down-regulated[134,136,146]. Pluripotency is further sustained by downstream pathway effectors Smad2/3 which bind to and trans-activate Nanog expression in undifferentiated hESCs[147,148]. Smad2/3 phosphorylation levels decrease upon early hESC differentiation[134]. Smad3 alone was also shown to form a complex with Oct4 and directly regulate many Oct4 targets[149]. On the contrary, SMAD1/5 phosphorylation levels increase upon hESC differentiation[134,150], a finding consistent with the ability of BMP4 to initiate differentiation to trophoblasts in vitro[139].
The TGF-β pathway is also involved in EMT during embryonic morphogenesis. Interestingly, some epithelial cells acquire thereby self-renewing characteristics reminiscent of stem cells[151]. In carcinomas, EMT can lead to metastasis and high-grade malignancy[152].
TGF-β signaling plays a complex, context-dependent and tissue-specific role in cancer development and CSC proliferation. While inhibiting the onset of cancinogenesis the pathway may promote invasion and metastasis at later disease stages[153].
Skin epithelia lacking TGF-βR II exhibiting enhanced integrin/focal adhesion kinase (FAK) signaling were prone to age-dependent squamous cell carcinoma development[154]. Moreover, tumor regression occurred in cancer cells lacking integrin/FAK signaling[155]. CSCs isolated from the tumor/stromal interface of TGF-βR II null squamous carcinoma formed less differentiated, highly aggressive and metastatic skin cancers. Interestingly, FAK depletion counterbalanced the TGF-βR II-null phenotype[156]. These data support an important role for TGF-β in counterbalancing the integrin/FAK-dependent tumorigenic effects in squamous cell carcinoma by down-regulating CSC proliferation and expansion[133].
Breast cancer cells respond to TGF-β by exhibiting stem-like properties. A recent study revealed that chemotherapy relapse of triple-negative breast cancer (TNBC) might involve expansion of CSCs caused by activated TGF-β and IL-8 signaling in The resistant CSCs are prone to TGF-β pathway inhibitors[157].
In malignant gliomas, TGF-β signaling also appears to exert agonistic effects on tumorigenesis through the Nodal/Activin branch of the pathway, increasing self-renewal of glioma stem cells (GSCs) by enhancing LIF/STAT signaling[158]. Furthermore, TGFβ-Sox4-Sox2 signaling appears to be important for the maintenance of stemness of GSCs[159]. On the other hand, the activation of the BMP branch of the pathway by BMP4, initiated neural differentiation and blocked tumor growth in a mouse xenograft model[160]. In addition, epigenetically silenced BMP signaling was proposed to desensitize glioblastoma stem-like cells to normal differentiation cues and to promote their proliferation[161].
The differential CSC responses to TGF-β cues underlie a serious dilemma over the clinical use of TGF-β agonists/antagonists[133].
Fibroblast growth factor signaling
Most fibroblast growth factor ligands (FGF1-22) function in a classical autocrine or paracrine manner. Ligand-receptor binding results in autophosphorylation and dimerization of the intracellular region of a tyrosine kinase trans-membrane receptor (FGFR1-4)[162]. The signal is further relayed through four main pathways: RAS-RAF-MAPK (ERK), PLCγ-PKC, PI3K-AKT and JAK/STAT. Emerging evidence suggests that FGFRs also traffic to the nucleus, activating entirely different downstream molecules[163].
Mutations of FGF pathway components result in pri-implantation lethality of the early mouse embryo[162]. Autocrine FGF-induced ERK1/2 signaling is dispensable for mESC pluripotency but requisite for their differentiation into neural and mesendodermal lineages[164,165]. Ying et al[166] further suggested that addition of LIF and BMP to culture media promotes mESC self-renewal solely by compensating for the pro-differentiation effects of FGF4. mESCs are comprised of heterogeneous populations. Cells primed for differentiation towards the primitive endoderm express FGF5 and Brachyury. FGF4 signaling on the other hand additionally maintains the primed state towards germ layer differentiation[162,167].
In stark contrast to mESCs, hESCs require exogenous FGF2 to sustain self-renewal and the capacity to give rise to somatic lineages[168,169]. The combined use of FGF2 and Activin is the most effective in maintaining hESCs and EpiSCs self-renewal. Recent studies suggest that spontaneous extra-embryonic differentiation, to which both hESCs and EpiSCs are prone, may be blocked by FGF[3,162]. FGF2 seems to influence the pluripotent state of hESCs on several levels. It activates NANOG expression in cooperation with Activin signaling through SMAD2/3[147] and synergizes with Noggin to repress trophoblast-inducing BMP signaling[140,170]. FGF/ERK signaling leads to phosphorylation of c-Myc, c-Jun and c-Fos. Its inhibition leads to a decrease in the expression of core pluripotency factors Nanog, and Oct4[171-173]. Interestingly, ERK and GSK-3 inhibition have successfully supported the reprogramming procedure for the generation of human iPS cells[174]. Furthermore, conversion of hESCs to an mESC-like phenotype was achieved by the concerted action ofERK and p38 inhibitors with LIF[175]. A number of recent studies raise the possibility of differential and sometimes opposing functions of FGF in hESCs, depending on the downstream effector signaling cascades[172,176].
Exogenous FGF2 is also required for growth and maintenance of CSCs isolated from different human carcinomas (e.g., brain, breast) in tumorspheres, but the mechanisms of FGF action remain to be elucidated[177,178]. A recent study revealed that the expansion of a functional breast CSC pool in response to estrogens is induced through a paracrine FGF/FGFR/Tbx3 signaling cascade, which is also functional in epithelialstem cells from the normal mammary gland[179]. Guthridge and colleagues transformed NIH3T3 cells with FGF4 and found out, that HSP-90, p63, LAMP-1 and CyclinD1 were massively activated[180]. The obtained results suggest a link between FGF4, CSC expansion and tumorigenesis[181]. Indeed, Cyclin D1 is considered to be a marker for cancer onset and progression, while in stratified epithelial tissues p63 is thought to regulate stem cell characteristics[182,183].
In conclusion, emerging evidence indicates a complex crosstalk between signaling pathways in development, adult tissue homeostasis and cancer. A better understanding of the pathway interplay and how it controls the biology of ESCs, SCs and CSCs will be essential for the advance of regenerative medicine and for developing effective cancer therapies.
COMMON TRANSCRIPTIONAL REGULATORS OF EMBRYONIC/PLURIPOTENT AND CSC
Regulatory networks in pluripotency
Transcription factors Oct3/4 (Pou5f1), Sox2 and Nanog constitute the “core pluripotency network” that regulates pluripotency of both mouse and human ESCs. They bind synergistically to their own promoter/enhancer elements establishing an auto-regulatory circuit[4,184]. This master pluripotency network, including additional factors such as Sall4, Klf4, and Stat3 binds to and regulates the expression of two distinct groups of genes in ESCs: genes related to self-renewal (active) and genes related to differentiation (silenced)[185]. The latter group of genes is co-occupied by the epigenetic silencing complexes Polycomb (PRC1 and PRC2)[186]. A second important multi-protein complex is centered on the oncoprotein Myc (Myc network). Both networks are mutually regulating each other and interact physically and functionally with chromatin remodeling and modification complexes[187,188]. The Myc complex binds near the transcription start site (TSS), whereas the core pluripotency complex binds to upstream promoters and enhancers[189-191]. The master pluripotency factors (Oct4, Sox2, Nanog) form super-enhancers, clusters of enhancer elements that recruit Mediator and determine cell identity[192,193].
Oct4, Sox2, Klf4 and Myc were the initial factors with the potential to reprogram somatic cells into pluripotency[188,194]. The individual role of these factors in pluripotent and CSCs will be examined below.Oct4 in ESCs and CSCs Oct4 belongs to the POU family of homeodomain proteins and is encoded by the Pou5f1 gene. Its expression has been identified in undifferentiated ESCs, embryonic carcinoma cells (ECCs), pluripotent epiblast and embryonic germ cells (EGCs)[195-197]. Nichols et al[198] reported that Oct4 expression is essential for the maintenance of ESC properties. They showed that Oct4-deficient embryos did not form a pluripotent inner cell mass and differentiated to trophectoderm[199]. Moreover, inhibition of Oct4 in mESCs led to the upregulation of trophectoderm genes (Cdx2), while its overexpression caused differentiation into primitive endoderm and mesoderm[200]. Under serum free culture conditions Oct4 overexpression in ESCs promoted neuroectoderm formation and subsequent neuronal differentiation[201]. Oct4 cooperates with Sox2 to regulate the expression level of genes important for self-renewal and pluripotent phenotype of ESCs (e.g., Nanog). On the other hand, when the Oct4 or Sox17 expression levels increased, Sox2 was replaced by Sox17 and targeted genes that trigger the endodermal expression program[202]. These results indicated that the precise levels of Oct4 determined the ESC fate and that Oct4 is a master player in sustaining stem cell self-renewal.
Numerous studies have indicated that Oct4 plays a crucial role in tumorigenesis and tumor metastasis. It was shown to be upregulated in many human cancers such as bladder, seminoma, prostate and breast cancer[15,203-206]. Hu et al[207] reported that in murine lung carcinoma cells and human breast cancer MCF7 cells, ablation of Oct4 expression leads to apoptosis of CSC-like cells through the Oct4/Tcl1/Akt1 pathway and inhibition of tumor growth. Another study confirmed that the reduction of Oct4 in lung cancer cells blocked the clonogenicity and tumor invasion[70]. Chiou et al[208] enriched oral CSCs by sphere formation and concluded that these cells highly expressed Oct4 and had similar characteristics of stem cells and malignant tumors. A year later, Rentala et al[209] reported that the expression of Oct4 in prostate CSCs maintained their stem cells properties. Moreover, it has been revealed that ectopic expression of Oct4 into normal primary breast epithelial preparations generated cell lines which form triple-negative breast carcinomas in nude mice[210]. Recently, Wang et al[211] demonstrated that cervical cancer cells expressed higher Oct4 levels than normal cervix cells. They proposed that Oct4 promotes tumor formation in vivo and inhibits apoptosis by the activation of miR-125b expression[211]. In addition, Oct4 has been suggested to regulate stemness of head and neck squamous carcinoma CSCs. The overexpression of Oct4 activated Cyclin E leading to tumor growth and tumor invasion through slug expression[212].
Sox2 in ESCs and CSCs
Sox2 is a member of the Sox (SRY-related HMG box) family that consists of transcription factors with a single high-mobility group box DNA-binding domain and also belongs to the SOXB1 subgroup[213]. Sox2 is expressed in the inner cell mass (ICM) and extraembryonic ectoderm of pre-implantation blastocysts[214]. Sox2 deficient blastocysts could not form a pluripotent ICM. Moreover, Sox2-deficient mESCs differentiated primarily into trophectoderm, while the Oct4 overexpression rescued the pluripotency of Sox2-null mESCs[215]. As a result, Sox2 is critical for the maintenance of Oct4 expression and hence the stem cells’ properties. Furthermore, Masui et al[215] identified a synergistic function of Sox2 and Oct4 for the activation of Oct-Sox enhancers, leading to the regulation of various pluripotency genes, including Nanog, Oct4 and Sox2. Overexpression of Sox2 in ESCs led to their differentiation[216,217]. This effect was due to the repression of pluripotency genes Sox2, Oct4, Nanog, Fgf4 and Utf1[216] and the induction of neuroectoderm, mesoderm and trophectoderm[217].
To date, many reports have demonstrated the involvement of Sox2 in cancer biology and especially in CSCs. Sox2 is critical for osteosarcoma cell self-renewal and antagonizes the pro-differentiation Wnt pathway which can also affect negatively the expression of Sox2[218]. In addition, it is implicated in the promotion of cell migration and invasion in ovarian cancer, through regulating fibronectin 1[219]. Studies in gastric cancer showed that inhibition of Sox2 results in reduction of spheres formation and in increase of apoptotic sphere cells[220]. The contribution of Sox2 in pancreatic CSCs was suggested by the fact that it regulates stemness via the control of genes of G1/S transition and EMT[221]. In prostate CSCs, the inhibition of EGFR signaling led to the decrease of Sox2 expression and self-renewal of prostate CSCs. Moreover, knockdown of Sox2 reduces the ability of prostate CSCs to grow under anchorage-independent conditions[222]. Similar findings have been extracted from non-small cell lung cancer studies. Singh and colleagues inhibited the expression of Sox2 and noticed a 2.5-fold reduction in sphere formation[223]. Additionally, EGFR/Src/Akt signaling influenced Sox2 protein expression, due to the decreased levels of Sox2 during the EGFR or SRC inhibition[223]. In melanoma CSCs, Sox2 is highly expressed and interact with Hedgehog-GLI (HH-GLI) signaling[224]. In more detail, Santini et al[224] showed that knockdown of Sox2 decreases the melanoma sphere formation and self-renewal of melanoma CSCs. Two HH-GLI signaling transcription factors, GLI1 and GLI2, have the ability to bind the proximal promoter of Sox2 and thus the HH-GLI signaling regulates Sox2. Finally, Favaro et al[225] proved that Sox2 is required for CSC maintenance in a high-grade oligodendroglioma mouse model.
Nanog in ESCs and CSCs
Nanog is the third member of the core pluripotency network in undifferentiated ESCs[226-228]. It is a homeodomain containing transcription factor, which was discovered through a functional screening for pluripotency factors which allowed the maintenance of ESC properties, in the absence of the LIF-STAT3 pathway[227,228]. Chambers et al[228] also added that Nanog expression is high in Oct4-null embryos, whereas its overexpression does not counteract the differentiation program of ESCs prompted by Oct4 deletion. In the absence of Nanog, embryos do not form a pluripotent ICM[227,229], although Nanog-null mESCs can be established[227,228]. Intriguingly, these Nanog-deficient mESCs although disposed to differentiation, could still be maintained in the undifferentiated state[227,228]. In 2005, Hyslop et al[230] showed that Nanog down-regulation in human ESCs promotes differentiation towards extraembryonic lineage, as shown by the upregulation of endodermal- and trophectodermal-characteristic genes. This suggests a pivotal role for Nanog in the maintenance of pluripotency in human embryonic development. Oct4/Sox2 heterodimers bind to the octamer/Sox elements within the Nanog proximal promoter and regulate Nanog expression in ESCs[231]. Moreover, Nanog, Oct4 and Sox2 cooperate with signaling pathways mediators resulting in delivering signals directly to the genes regulated by the core factors[191]. Sites co-occupied by the three core regulators generally have enhancer activity, while the transcription of the respective genes requires the recruitment of at least one of the trio[191].
Nanog expression was investigated in several types of cancer, including lung, breast, oral, kidney, gastric, cervix, brain, ovarian and prostate cancer[232-237]. In particular, high expression levels of Nanog are related to a poor prognosis for ovarian serous carcinoma, colorectal, and breast cancer patients[238-240]. In oral squamous cell and lung adenocarcinoma, Nanog and Oct4 high levels were linked to advanced cancer stage and shorter patient survival[208,234]. Several groups demonstrated that Nanog expression is much higher in CSCs than in non-stem cancer cells in many types of cancer[233,241-246]. In colorectal cancer, Nanog-positive CSCs constitute approximately 2% of the total cancer cell population[241]. In addition, a direct connection between the surface markers of CSC and Nanog has not been clarified yet, but there are many studies which demonstrated that cancer cells expressing these markers would have higher levels of pluripotency genes[247]. For instance, CD133- or CD44- cancer cells express significant lower levels of Nanog compared to CD133+ or CD44+ cells, respectively[248,249]. Moreover, functional studies in various cancer types showed that Nanog induces CSC-like characteristics. Jeter et al[242] demonstrated that NANOGP8 overexpression in prostate cancer increased clonogenicity and tumor regenerative ability[247]. Nanog activation leads a small population of colorectal cancer cells to acquire a stem-cell like phenotype[247]. Furthermore, Han et al[250] proved that Nanog binds to Cyclin D1 promoter region and regulates proliferation and cell cycle of breast cancer cells[247,250]. Recently, Siu et al[251] reported that increased expression of Nanog in ovarian cancer controls cell proliferation, migration and invasion through E-cadherin and FoxJ1 deregulation[247,251]. These results propose that Nanog may constitute a CSC marker and play a vital role in cancer progression[251].
Klf4 in ESCs and CSCs
Following the identification of KLF4 as a critical transcription factor for reprogramming, more attention was given to its actions. Klf4 belongs to the Kruppel-like transcription factor family and has a central role in cell cycle regulation, somatic cell reprogramming and pluripotency[194,252-254]. Klf4 is highly expressed in mESCs and its expression decreases strongly upon differentiation[255]. The inhibition of Klf4, using RNAi, leads to the differentiation of ESCs[252,254], while Klf4 ectopic expression postpones differentiation, enhances the expression of Oct4 and promotes self-renewal[256]. Klf4, in conjunction with Oct4 and Sox2 drives the expression of Lefty1[257] and Nanog[258]. In addition, the expression of Klf4 is regulated by STAT3 and Nanog[252] being a direct target of both transcription factors[252,259]. In a recent report, Aksoy et al[260] described that Klf4 reduction induces differentiation towards visceral and definitive endoderm, concluding that Klf4 inhibits endoderm differentiation in mESCs.
It is not surprising that Klf4 plays a key role in maintaining CSC populations. It is known that telomerase activity is sustained by Klf4 via telomerase reverse transcriptase in both CSCs and hESCs, suggesting that Klf4 is important for the long-term proliferative potential of these cells[261]. Moreover, Hoffmeyer et al[262] reported that β-catenin regulates Tert expression via the interaction with Klf4, supporting a connection between stem cells and oncogenesis. In 2011, Yu et al[263] reported for the first time that Klf4 is crucial in maintaining breast CSCs and inducing cell migration and invasion. Klf4 is expressed at high levels in CSC populations in mouse primary mammary tumor and human breast cancer cell lines. The inhibition of Klf4 in MCF-7 and MDA-MB-231 decreases the ability of breast CSCs to self-renew and form mammospheres and tumors in vivo[264]. On the other hand, it was shown that Klf4 suppresses metastasis in MDA-MB-231 cells by maintaining the expression of E-cadherin and inhibiting EMT[265]. Klf4 is highly expressed in colorectal CSCs and its knockdown leads to the decrease of spheres formation, migration, invasion and EMT. These results prove the essential role of Klf4 for maintaining colorectal CSCs[266]. Furthermore, Wellner et al[267] reported that Klf4 is induced by ZEB1 through the repression of stemness inhibitor miR-203, and controls/enhances pancreatic and colorectal cancer cells ability to initiate tumor development. Moreover, repression of Klf4 by miR-7 inhibits metastasis of human breast CSCs in nude mice[268] whereas inhibition of Klf4 by mir-152 suppresses the generation of glioblastoma SCs[269].
These results propose that Klf4 has important roles not only in stem cell self-renewal and cell motility, but also in CSC and carcinoma cell invasion and metastasis.
Myc in ESCs and CSCs
Myc family which includes three important members - c-Myc, N-Myc and L-Myc - acts as an essential regulator in cell growth, proliferation, differentiation and apoptosis and it is thought to be crucial for stem cell pluripotency and proliferation[270-272]. Myc is directly regulated by LIF/STAT3 signaling and its constitutive activity renders ESC self-renewal independent of LIF. In contrast, the overexpression of Myc dominant negative form induces differentiation[273]. Although the individual inactivation of c-Myc and N-Myc has no effect on pluripotency, their simultaneous deletion destabilizes the pluripotent state leading to primitive endoderm and mesoderm differentiation[274]. Moreover, the overexpression of either c-Myc or N-Myc restore pluripotency of ESCs[274], supporting the idea that c-Myc and N-Myc perform redundant roles in maintaining pluripotent stem cell identity. Recently, Chappell et al[275] showed that Myc represses MAPK signaling and results in inhibiting differentiation.
Several genome-wide analyses have been performed in order to determine how Myc regulates ESC pluripotency. These studies showed that Myc binds to and possibly regulates the transcription of at least 8000 genes in ESCs[191,276-278]. The Myc-centered complex in ESCs is binding to the TSS and includes also E2F Max and NuA4 HAT complex[189].
Myc deregulation and elevation have been pobserved in a wide range of human malignancies, associated with aggressive and poorly differentiated tumors[279]. It is well-known that Myc is involved in the regulation of 15% of genes in the human genome[280] and regulates important pro-tumorigenic factors including KRAS and AKT, and tumor-suppressors PTEN and p53[281,282]. Some reports also showed that Myc-centered protein interaction networks in ESCs are enriched in some cancers, especially in the CSCs, conferring metastatic potential and poor outcome[16,183]. These findings suggest that the Myc network is responsible for the similarities between ESCs and cancer cells. Wang et al[283] determined that glioma CSCs expressed high levels of Myc, which is crucial for growth, proliferation and survival. Furthermore, glioma CSCs with low levels of Myc did not generate neurospheres in vitro or tumors after xenotransplantation in the brains of iimmunodeficient mice[283]. Salcido et al[284] found that Myc is expressed at high levels in CSC population of small-cell lung cancer. Additionally, it has been demonstrated that Myc expression is significantly upregulated in tumor spheres formed by rhabdomyosarcoma cell lines[285]. In a recent study, it has been indicated that silencing of Myc using promoter targeting siRNA, decreased prostate CSC maintenance and tumorigenicity and induced senescence in the prostate CSC subpopulation[286]. The clarification of the role of Myc in hepatic CSCs biology came from Akita work in 2014. They revealed a direct link between c-Myc expression levels and CSC properties and they have further mechanistically demonstrated that c-Myc modulates the hepatic CSC phenotype in a p53-dependent manner[287].
Collectively the result of accumulating research suggests that ESCs and CSCs share critical transcription factors (Table 3). It is clear that pluripotency factors Oct4, Nanog, Sox2, Klf4 and Myc play a crucial role in cancer development and contribute to cancer treatment. However, further investigation of their role in determining the CSC phenotypes, will provide the exact regulatory mechanisms and possibly new regulatory factors relating to tumorigenesis and metastasis.
Table 3.
Common signaling pathways, transcription factors, non-coding RNAs and epigenetic regulators of embryonic stem cells and cancer stem cells in solid tumors
| ESCs | CSC type | |
| Signaling pathways | ||
| Wnt/β-catenin | Self-renewal in mESCs/hESCs[91] Differentiation in hESCs[98] | Brain[104] Breast[103] Colon[99] Lung[475] Prostate[476] |
| Hedgehog | Self-renewal in mESCs[109] Differentiation in hESCs[108] | Brain[112] Breast[111] Pancreas[115] |
| Notch | Differentiation in mESCs and hESCs[117] | Brain[126] Breast[128] Colon[125] Pancreas[123] |
| TGF-β | Activin/Nodal promote self-renewal in mESCs and hESCs[134,135] BMP promotes self-renewal in mESCs[138] and differentiation in hESCs[141] | Brain[158] Breast[157] Skin[133] |
| FGF | Differentiation in mESCs[164] Self-renewal in hESCs[169] | Bladder[477] Brain[177] Breast[178] |
| Transcription factors | ||
| 4-Oct | Self-renewal and pluripotency[198] | Breast[207,210] Lung[70,207] Oral[208] Prostate[209] Cervical[211] Head and neck[212] |
| Sox2 | Self-renewal and pluripotency[215] | Osteosarcoma[218] Ovarian[219] Gastric[220] Pancreatic[221] Prostate[222] Lung[223] Melanoma[224] |
| Nanog | Self-renewal and pluripotency[226-228] | Breast[250] Oral[208] Lung[234] Colorectal[238,241] Prostate[242] Ovarian[251] |
| Klf4 | Self-renewal and pluripotency[254,256] | Breast[263-265,268] Colorectal[266,267] Pancreatic[267] Brain[269] |
| c-Myc | Self-renewal and pluripotency[189,191,270] | Brain[283] Lung[284] Rhabdomyosarcoma[285] Prostate[286] Hepatic[287] |
| DNA methylation regulators | ||
| DNMT1 | Differentiation[293] | Colon[336] Breast[338] |
| TET2 | Differentiation[315,322] | Breast[344] |
| Chromatin modifications regulators | ||
| EZH2 | Self-renewal and pluripotency[186] | Breast[393] Pancreas[393] Brain[395] Prostate[396] Bone[397] |
| BMI-1 | Self-renewal and pluripotency[186] | Prostate[400] Esophageal[401] Head and neck[402] Cervical[403] Colorectal[404] Laryngeal[405] Ovarian[406] Salivary adenoid cystic carcinoma[399] |
| Suz12 | Self-renewal and pluripotency[186] | Breast[407] Colon[408] |
| MLL1 | Self-renewal and pluripotency[186] | Brain[409] |
| MicroRNAs | ||
| Let-7 | Differentiation[430,431] | Breast[434] Prostate[448] |
| MiR-200 family | Differentiation[267] | Breast[437] |
| MiR-34a | Differentiation[429] | Brain[444] Prostate[448] Pancreatic[451] Gastric[450] Colon[452] |
| MiR-145 | Differentiation[424] | Brain[445] Breast[440] |
| Long non-coding RNAs | ||
| LncRNA-RoR | Self-renewal[418] | Breast[461] |
CSC: Cancer stem cell; ESCs: Embryonic stem cells; mESCs: Mouse ESCs; hESCs: Human ESCs.
COMMON EPIGENETIC REGULATORS IN ESCS AND CSC
Cell epigenetic state has been recognized as an important factor in diverse developmental and differentiation processes via global or gene specific regulatory mechanisms. Genome wide analyses and knockout studies provide new information about the role of epigenetic processes in self-renewal and cancer initiation and permit the development of “epigenetic’’ therapy as a cancer treatment option[288,289].
This part of the review highlights our current view of the most important common epigenetic regulators associated with DNA methylation, histone modifications as well as long-non coding RNAs and miRNAs in ESCs and CSCs, their significance in normal development and their deregulation in tumorigenesis.
DNA methylation regulators in ESCs and CSCs
DNA methylation is generated by DNA methyltransferases (DNMTs), enzymes that add methyl groups on cytosines. The most studied members of the mammalian DNMT family include DNMT1, DNMT3a and DNMT3b. DNMT1 is thought to be responsible for the replicative maintenance of the DNA methylation, while DNMT3A and DNMT3B function as de novo methyltransferases[290]. Nevertheless, recent evidence shows that DNMT1 may also be required for de novo DNA methylation[291] and that DNMT3a and DNMT3b can also participate in the maintenance of the DNA methylome[292]. These findings emphasize the need to clarify the exact role of each DNMT and their potential crosstalk.
Restriction enzyme digestion-mediated and Methylated DNA Immunoprecipitation-ChIP (MeDIP-ChIP) analyses of global DNA methylation show that the DNA methylation levels are reduced in mouse ESC compared to the somatic cells and that methylations on promoter regions lie primarily outside of CpG islands[293-295]. A methylation analysis of CpGs by Bibikova et al[296] reported that the methylation levels of over 370 genes in 14 hESC lines were lower than those of mESCs[294].
Two groups, Lister et al[297] and Laurent et al[298] have compared the methylation maps of hESCs and human fibroblast cell lines and observed significantly higher levels of non-CpG methylation present in hESCs that may be due to differences in methylation regulatory mechanisms between un- and differentiated cell types. Laurent et al[298] observed that CpA methylation was the most frequent type of non-CpG methylation in hESCs, and thatthis modification was lost upon differentiation. Non-CpG methylation is also present in mESCs andis reduced from 8% to 4.3%, six days after induction of differentiation[299]. This non-CpG methylation is more abundant within gene bodies than promoter regions and is catalyzed by DNMT3a and DNMT3b requiring also the presence of DNMT3L[300]. Methylation profiles of iPS cells are highly similar to the ones of ESC, showing that this is a unique characteristic of pluripotent cells[297,301]. However its functional role remains unclear.
Deletion of Dnmt1 or 3b in mice results in embryonic lethality[302], while Dnmt3a-/- mice die within 4 wk after birth showing that these enzymes are essential for normal development[303]. DNMT1 overexpressing ESCs when injected in blastocysts resulted in embryonic lethality, resembling the effect of DNMT1 deficiency[304]. DNMT1, DNMT3a and DNMT3b are expressed in hESCs[305]. Mouse knockout experiments have shown that deletion of DNMTs does not affect ESCs self-renewal but deregulates cell specification, suggesting that global methylation may be dispensable for the undifferentiated state but is critical for differentiation. More specifically, DNMT1-/- EBs contain a large number of Oct4 positive pluripotent cells indicating that methylation is required for proper cell differentiation[293]. In Dnmt3a-/- Dnmt3b-/- mouse ESCs, only 0.6% of CpGs are demethylated[293], suggesting that these molecules have limited contribution on global DNA methylation. Furthermore, Pawlak et al[306] showed that Dnmt3a and Dnmt3b are dispensable for nuclear reprogramming.
Although many observations implied the reversibility of DNA methylation, only recently were identified the TET (ten eleven translocation) enzymes that actively demethylate DNA. The mammalian TET family has three members, TET1, TET2 and TET3 that catalyze 5mC oxidation and generate the 5mC derivatives 5hmC, 5fC or 5caC (5-hydroxymethylcytosine, 5-formylcytosine, 5-carboxylcytosine)[307-310]. 5hmC is the first intermediate toward DNA demethylation and its variable amounts in different cells and tissues implies a distinct regulatory role[311]. Recent reports, studying the genomic distribution of 5hmC in mouse and human ESCs, provide evidence that this modification may function as a specific epigenetic mark in gene expression regulation[312-314].
The expression levels of TET1 and 2 are high in undifferentiated ESCs and decline upon differentiation, in parallel with an increase of TET3[315]. Further studies have shown that TET1 adds 5hmC on promoter regions and TSSs whereas TET2 activity is detected in the coding regions[316]. TET1 activity in ESCs is associated with the demethylation and expression of pluripotency related genes as well as repression of Polycomb targeted developmental regulators[317]. Moreover, TET proteins were found to participate in various gene expression regulatory complexes via interaction with Sin3A co-repressor[318], Polycomb Repressing Complex 2 (PRC2)[317] and the O-linked N-acetylglucosaminetransferase, (Ogt)[319,320].
Although earlier studies have suggested a role for TET1 and TET2 in ESC self-renewal[321], it was recently clarified that these enzymes are in fact required for proper differentiation of ESCs[322]. TET1 and TET2 are regulated by the Oct4-Sox2 complex in ESCs and TET1 knock down promotes the differentiation toward endoderm/mesoderm and trophoblast pathways[315]. A Tet1/2/3 triple knock-out mouse ESC line was unable to generate embryoid bodies, teratomas and could not give rise to healthy chimeras[322].
A current model proposes that the accumulation of epigenetic and or genetic changes in a normal adult stem cell generates eventually a heterogeneous tumor population that contains a subset of “CSCs” that are responsible for its long term maintenance[323]. Regardless of the hierarchical or stochastic nature of the CSCs, studying their epigenome is of paramount importance both for understanding the origin and evolution of cancer as well as applying novel epigenetic therapies. As a result, a growing number of studies has recently being directed in the elucidation of common or distinct epigenetic mechanisms that govern the self-renewal program of ESCs and CSCs.
Cancer epigenome exhibits global DNA hypomethylation and specific promoter hypermethylation[324,325]. DNA hypomethylation promotes cancer development by increasing genomic instability and activating growth-promoting genes such as R-Ras[326]. On the contrary site-specific hypermethylation favors oncogenesis by repressing tumor suppressor genes, other genes encoding transcription and DNA repair factors as well as PRC target genes[327,328]. The above studies provide compelling evidence for deregulated DNA methylation in cancer but do not address distinct cell subpopulations. In this line, Yasuda et al[329] studied 10 tumor suppressor genes (TSGs) in bulk and CSC enriched MCF7 breast cancer cells based on their ability to form tumor-spheres and found lower DNA methylation levels and H3K27m3 marks in the latter population. Ikegaki et al[330] proved that epigenetic modifiers can affect the expression of stemness genes and contribute to the establishment of CSCs. They showed that short-term treatment of Neuroblastoma cell lines with the DNA methylation inhibitor 5-aza-2′-deoxycytidine (5AdC) and/or the histone deacetylase inhibitor 4-phenylbutyrate, enhances their CSC phenotype[330].
The expression levels of DNMTs are elevated in several cancer types[331-333]. DNMT3b has been shown to play a crucial role in de novo hypermethylation of promoter CpG islands. In line with the role of DNMTs in cancer development, sustained overexpression of the murine dnmt1 gene in NIH3T3 cells results in cellular transformation[334]. Conversely, reduction of DNMT1 by an antisense DNMT-RNA reversed the transformed phenotype of the Y1 tumor cell line[335].
Morita et al[336] compared colorectal HCT116 WT and its dnmt1 knockout derivative and showed that the latter has reduced stem cell markers and contained less CSCs, as assessed by tumor formation following xenotransplantation. Again no difference in DNMT3a and DNMT3b was detected[336]. The importance of DNMT1 in CSC self-renewal was further confirmed by Trowbridge et al[337] using MLL-AF9 induced mouse leukemia. Additionally, a recent paper about the lyotropic reagent chloroquine reports that it can eliminate CSCs in a TNBC population through reduction of DNMT1 and Jak2 expression[338]. The above studies demonstrate the role of DNMT1 in the maintenance of the CSC properties and in vivo tumorigenicity.
Concerning the effects of DNA demethylase, 5hmC levels are decreased in a broad range of cancer cells[339]. TET1 has been found as a fusion partner of MLL in a subset of patients with acute myeloid leukemia[340,341]. TET2 has been reported as one of the most frequently mutated genes in hematopoietic cancer types, but many mutations appear in sites that don’t affect its enzymatic activity[342]. In agreement with this, it was recently found that enzymatically inactive TET1, acting as a transcriptional co-activator for Hif1a, is required for EMT[343]. Additionally, work by Song et al[344] has shown that TET2 is involved in breast cancer stemness and metastases due to the silencing of miR-200.
Investigating the methylation signature of CSCs permits identification of modifiers that can target their stemness properties, leading to increased tumor sensitivity to chemotherapy. Current DNMT inhibitors used in cancer therapy, such as Decitabine (5’-aza-2’deoxycitidine) act through incorporation into DNA therefore causing adverse side effects[345]. Less hazardous alternatives include use of small molecule inhibitors such as SGI-1027[346] and dietary phytochemicals[342].
Chromatin modifiers in ESCs and CSCs
It is the complex interplay of DNA methylation with the posttranslational modifications of the histone tails that determines the transcriptional activity of a particular locus[347]. The effect of histone modifications on diverse cellular functions, has caused intense interest in studying the chromatin modifying enzymes. These epigenetic modifiers include a variety of factors, such as histone methyltransferases (HMTs), demethylases (HDMs), acetyltransferases (HATs) and deacetylases (HDACs)[348,349]. All the above mentioned factors participate in the regulation of chromatin structure that in turn governs gene transcription.
ESCs are characterized by permissive chromatin structure and consequently higher transcriptional activity compared to differentiated cells. Generally, histone marks associated with active transcription are more abundant in ESCs and become reduced upon differentiation[350,351], whereas repressive marks appear in higher levels in differentiated cells. Another interesting feature most commonly found in ESCs are bivalent domains, which are defined by the presence of the active H3K4me3 mark alongside the repressive H3K27me3 mark and is believed to hold genes in a ‘‘transcription-ready’’ state[352,353]. However a recent study by Denissov et al[354] challenges the prevailing view by showing that not all bivalently marked genes in mESCs lose their differentiation responsiveness upon loss of H3K4me3. Thus, the role of bivalency and its association with pluripotency remains an open question.
Self-renewal in ESCs necessitates the action of chromatin repressive complexes in order to inhibit expression of differentiation-promoting genes. The best studied silencers of differentiation pathways in pluripotent cells are the PcG proteins, which are organized into two multiprotein complexes PRC1 and PRC2[355]. PRCs are highly expressed in ESCs and bind mainly to CpG-rich promoters of developmentally regulated genes[186,356].
The PRC2 complex has three core protein subunits: The enhancer of zeste homology (EZH2) component that catalyzes di- and trimethylation of H3k27, Embryonic ectoderm development (Eed) and suppressor of zeste (Suz12). PRC2 triggers gene silencing by recruiting PRC1, histone deacetylases and DNA methyltransferases[357]. The PRC1 complex composition is highly variable and the canonical complexes include CBX (polycomb), polycomb group factor (PCGF), human polyhomeotic homolog (HPH) and RING, the E3-ligase that catalyzes the monoubiquitination of histone H2A on lysine 119[358].
Depletion of PRC2 results in embryonic lethality in mice[359], while mESCs lacking Eed, Suz12 or Ezh2 show loss of H3K27me2/3, retention of self-renewal capacity and in vitro differentiation defects[360-363]. On the contrary, inactivation of PRC1 in mice leads to deficiencies concerning later developmental stages[364]. RING1b (subunit of PRC1 complex) deficient mESCs show a slight deregulation of some genes and loss of differentiation potential[365], whereas mESCs double mutated for Ring1a/Ring1b lose also the ability to self-renew[366]. In summary, PcG proteins seem to be required for proper ESC cell fate transition but not for their self-renewal. More information is required about their partners to fully understand their regulatory role in ESCs[367]. Moreover, it was shown that PRC1 and PRC2 can occupy distinct genomic sites and act independently[368].
The maintenance of the expression program that determines pluripotency requires the presence of both repressive and activatory chromatin modifiers. A complex that counteracts the repressing effect of Polycomb is the Trithorax/MLL[369]. Trithorax group (TrxG) complex contains a histone K4 tri-methyltransferase (Set1a/b, MLL1-4), a subunit that recognizes the H3k4me3 mark, tryptophan-aspartate repeat protein 5 (WDR5), absent-small-homeotic-2-like (Ash2L), retinoblastoma-binding protein 5 (RbBP5) and dumpy-30 (DPY-30)[369,370].
Both activating H3K4me3 and repressive H3K27me3 and H3K9me3 marks are removed by histone demethylases belonging to the Jumonji domain-containing protein family (Jmjd)[371]. Jmjd proteins connect ESC core transcriptional network with chromatin modulation. More specifically, Jmjd2c participates in stem cell maintenance by reversing H3K9me3 marks at the Nanog promoter, consequently protecting it from silencing, whereas Jmjd1a/2c gene expression is positively regulated by Oct4[372]. The diverse role of Jmjds is underlined by Pasini et al[362], who reported the involvement of JARID2 in the recruitment of PRC2 by differentiation-related genes in ES cells.
Another chromatin modifier called LSD1/KDM1 factor, is a histone demethylase that suppresses gene expression by removing methylation groups from H3K4. LSD1 has been found to colocalize with NuRD at the enhancer of pluripotency genes and down-regulates their expression upon differentiation[373]. Finally, Suv39H1, Suv39H2 and G9a methylases, generate H3K9me3 repressive mark important in ESCs[374].
Histone acetyltransferases (HATs) and the equivalent histone deacetylases (HDACs) constitute another critical category of chromatin modifiers acting as co-activators or co-repressors respectively[375]. Although deacetylation is associated with gene silencing, ChIP-sequencing studies show that HDACs also colocalize with acetyltransferases at transcriptionally active loci, probably to reset acetylation levels after gene activation[376].
HDAC1 and HDAC2, the most studied HDACs in ESCs, have been shown to be dispensable for mESCs self-renewal[377]. However, HDAC1 knockout- ESCs show differentiation defects. HDAC1 and HDAC2 are usually part of large complexes with repressive action like NuRD, CoREST and Sin3[378]. Alteration of the histone acetylation pattern interferes with stem cell pluripotency and differentiation[376] as well as reprogramming[379]. Most importantly inhibitors of HDACs are used as facilitators of reprogramming[380]. Noteworthy, NuRD complex, which couples chromatin remodeling capacity and histone deacetylation activity[381], has been shown to act as negative regulator of pluripotency associated genes byfine tuning their expression levels and sensitizing cells to differentiation stimuli[382]. The distinct repression targets of PRCs and NuRD may explain why reprogramming efficiency is increased by overexpression of PRCs components[383], but depletion of NuRD proteins[384].
Finally, small molecule drugs targeting histone demethylases or DNA demethylases are also valuable tools for reprogramming since they can substitute for transcription factors[385].
The information related to chromatin modifiers in ESCs has facilitated the elucidation of their role in tumorigenesis. In addition to DNA hypo-methylation, reduced histone acetylation but enhanced histone methylation is an epigenetic feature characteristic of cancer cells[386,387].
Polycomb HMT proteins are commonly upregulated in cancer[388]. EZH2, the best studied PcG protein has been found to promote tumor growth by inhibiting pro-differentiation pathways and enhancing cell cycle progression[389]. Furthermore HDMs, which reverse the action of HMTs, like LSD1, have also been implicated in oncogenesis[390]. Increased HDAC activity usually characterizes cancer cells[391]. Resetting normal acetylation levels through treatment with HDAC inhibitors (HDACi) has lowered tumorigenicity, suggesting HDACs as attractive targets for cancer epigenetic therapy[392].
The role of chromatin regulators in CSCs has recently started to be under study with Polycomb Group (PcGs) proteins to be in the spotlight. EZH2 proved to be essential for the maintenance of breast and pancreatic CSCs[393]. Intriguingly, EZH2 promotes NFκB signaling in ER-negative breast cancer cells[243], that in turn has been shown to contribute to the generation of CSCs through a positive feedback loop involving IL6[394]. In previous studies, silencing of EZH2 in glioblastoma CSCs significantly delayed intracranial tumor formation, demonstrating the necessity for EZH2 in CSC-driven tumorigenesis[395], whereas treatment of prostate cancer cells with the PRC2 inhibitor DZneP (3-Deazaneplanocin A) inhibited CSC spheroid formation and decreased CSC frequency[396]. Furthermore, EZH2 seems to contribute to the stemness phenotype of Ewing tumors by suppressing endothelial and neuroectodermal differentiation[397]. Gupta et al[398] developed a model of phenotypic transitions to study stochasticity in regulating cell-state equilibrium in cancer cells and EZH2 was used as one of the main phenotypic markers for the CSC population.
Increasing number of studies implicate BMI-1, another PcG member, in cancer stemness. Its expression has been found elevated in many CSC populations such as in salivary adenoid cystic carcinoma[399], prostate[400], esophageal[401], head and neck[402], cervical[403], colorectal[404], laryngeal[405] and ovarian[406] CSCs. In addition it has been reported that depletion of Suz12 - a component of PRC2 complex - results in the blockade of mammospheres formation[407] and increased apoptosis in colon CSCs[408]. These findings highlight Suz12, as an essential regulator of CSCs.
Downregulation of the histone methyltransferase MLL1 reduces CSC self-renewal and tumorigenicity suggesting a role in CSCs[409]. Additionally, HDMs like LSD1 are crucial for the biology of CSCs too. Wang et al[410] proved that inhibition of LSD1 inhibits the proliferation of pluripotent cancer cells but not that of normal somatic or non-pluripotent cancer cells. Except for histone methylation, histone acetylation is also essential for CSCs as a recent study showed that inhibition of HDACs limits the population of CSCs of head and neck cancer[411].
In conclusion, epigenetic regulators such as members of the Polycomb and Trithorax complexes, histone demethylases and histone deacetylases are essential for ESC pluripotency and at the same time can promote cancer stemness (Table 3). DNA methyltransferases are indispensable for both cell types and have been already reported as important targets for iPS generation and cancer chemotherapies (Table 3). Finally, DNA demethylases (TET 1, 2, 3) are critical targets for iPS generation, whereas their functions in CSCs await further clarification (Table 3).
MicroRNAs in ESCs and CSCs
The crucial role of microRNAs in mouse and human ESCs has been identified using Dicer and DGCR8 knockout mice. Dicer deletion resulted in embryonic lethality in mice[412], while DGCR8-deficient mouse ESCs retained self-renewal capacity[413].
Among the first families of microRNAs identified, miR-290-295 (miR-371 family, human homologous) and miR-302-367 clusters, which include the majority of miRNAs in mouse and human ESCs. Common characteristics of the two clusters is the promoter binding of the core pluripotency transcription factors (Oct4, Sox2 and Nanog)[414] and the decrease of their expression during differentiation[415]. Reintroduction of these miRNAs into Dicer1-knockout[416] and DGCR8-knockout[413] mice rescued proliferation and normal ESC self-renewal, respectively. It was found that these two clusters maintain the self-renewal by targeting retinoblastoma like 2 (Rbl2), a repressor of DNA methyltransferases (Dnmt3a and Dnmt3b). The latter methylates CpG islands and epigenetically silences Oct4[416,417]. In addition, these miRNAs were shown to regulate the G1/S transition of the ESC cell cycle, by repressing directly or indirectly the expression of the G1/S transition inhibitors (p21, Lats2, Rb1, Rbl2 and Rbl1)[418,419].
Other miRNAs acting on the hESC cell cycle were studied by Qi et al[420], showed that miR-195 and miR-372 promote the transition of G2/M and G1/S, by suppressing the G2/M checkpoint kinase WEE1 and CDKN1A respectively[420]. Furthermore, miR-92a and 92b were identified to target the CDKN1C gene and CDKN2B (known as p57), promoting in this way the G1/S transition[421].
Beside their function in maintaining pluripotency, miRNAs play important role in the differentiation of ESCs. Tay et al[422,423] demonstrated that miR-134, miR-296 and miR-470 bind to the coding regions of Oct4, Sox2 and Nanog and suppress their expression and thus the self-renewal state. Similarly, miR-200c, miR-203 and miR-183 target Sox2 and Klf4[267], while miR-145 was shown to repress human OCT4, Sox2 and KLF4 by binding to their 3′UTR, suggesting its role to regulate pluripotency[424]. In another study, induction of differentiation caused the increase of miR-21 expression levels revealing its crucial role in stem cell differentiation by targeting Nanog, Sox2 and Oct4[425]. Moreover, the expression of miR-22 was also detected in high levels during ESC differentiation[426]. Landgraf et al[427] demonstrated that miR-26a, miR-99b, miR-193, miR-199a-5p, and miR-218 are able to suppress the self-renewal of ESCs but the mechanism remains unclear. Another example of miRNAs that regulate the differentiation of ESCs is the miR-125 and miR-181 clusters[428]. A recent study showed that miR-125 and miR-181 families suppress Cbx7, the primary Polycomb ortholog of the PRC1 complex, in undifferentiated ESCs. Their overexpression leads to the differentiation of ESCs via regulation of Cbx7[428]. Furthermore, it was found that miR-34a, miR-100, and miR-137 are required for the differentiation of ESCs, and that they function in part by targeting Sirt1, Smarca5 and Jarid1b mRNAs[429].
Let-7 is another miRNA family which has been widely involved in the establishment of the differentiated cell fate. Melton et al[430] identified that silencing of pluripotency and self-renewal of ESCs can be caused by the introduction of let-7 into DGCR8-knockout ESCs. In addition, let-7 binds to 3’UTR, inhibits expression of several stemness factors (c-Myc, Sall4n, Lin28) and induces ESCs differentiation[430]. Interestingly, Lin28 forms a negative feedback loop with let-7, resulting from its blocking function during let-7 biogenesis at the Dicer processing step[431,432]. Let-7 can also target the G1/S transition activators (cdc25a, cdk6, cyclinD1 and cyclinD2) that increase susceptibility of G1 phase cells to pro-differentiation signals and promote differentiation of ESCs[431,432].
MiRNAs also play important roles in regulating CSC properties such as cell cycle exit and pluripotency, pro-survival and antistress mechanisms, EMT, migration and invasion, which contribute to tumor metastatic potential.
Because of the early discovery and better understanding of breast CSCs, miRNA studies are more advanced in this cancer type[433]. Let-7 regulates the properties of CSCs and its overexpression results in the reduction of mammosphere and tumor formation, metastasis and cell proliferation. Moreover, let-7 diminishes the expression of HMAG2, c-Myc, and RAS and controls CSC properties[434]. Another miRNA that is repressed in breast CSCs is miR-30. The inhibition of miR-30 induces metastasis and self-renewal of breast CSCs[435] and the transfection of both let-7 and miR-30, causes a more complete reduction of mammospheres formation and CSC abilities in breast CSCs[435].
Iliopoulos et al[436] showed the importance of an inflammatory positive feedback loop involving NF-κB, Lin28, let-7 and Il6 in the epigenetic maintenance of the cell transformation state following Src oncogenes activation. The same group using miRNA profiling defined a set of 22 miRNAs that are differentially expressed in normal vs CSCs that included the let-7 and the miR-200 families[407]. Interestingly, Shimono et al[437] also identified that miR-200 cluster is down-regulated in breast CSCs and miR-200c reduces the tumor formation. In addition, members of this family have the ability to regulate the breast CSC properties by repressing the BMI-1[437] and a subunit of a polycomb repressor complex, SUZ12[407]. Furthermore, several studies showed that miR-200 family and miR-205 modulate EMT, which is crucial for metastasis and tumor invasion. It was found that the expression of these miRNAs is decreased in cells undergoing TGF-beta induced EMT and their overexpression inhibits the EMT process. These miRNAs modulate the expression of EMT activators, ZEB1 and ZEB2, causing the inhibition of EMT program[438]. In contrast, ZEB1 and ZEB2 activate EMT by forming a double negative feedback loop with the miR-200 family, resulting from their binding to promoter regions of miR-200 family members[439]. As previously mentioned, miR-22 directly suppresses the expression of the TET family members (TET1-3), which are implicated in the demethylation of miR-200 promoter. In other words, miR-22 inhibits the activity of miR-200 cluster and promotes EMT and metastasis[344]. Polytarchou et al[440] identified that miR-15/16, miR-103/107, miR-145, miR-335, and miR-128b inhibit breast CSC function and growth by directly inhibiting the expression of Suz12, Bmi1, Zeb1, Zeb2, and Klf4.
MiR-9/9*, miR-17 and miR-106b are the mostly represented miRNAs in the CSC population in glioblastoma. It was demonstrated that knocking down of miR-9 and miR-17 causes a reduction of neurosphere formation[441]. Another study showed that miR-128 reduces glioma cell proliferation in vitro and glioma xenograft growth in vivo[442]. Furthermore, miR-128 overexpression modulates the properties of glioma[443] and prostate CSCs[400] by inhibiting BMI-1. Li et al[444] examined the role of miR-34a in brain tumor cells and human gliomas and they demonstrated that miR-34a causes cell cycle arrest, apoptosis or xenograft tumor repression, regulating the glioblastoma CSCs. In CD133+ CSCs of glioblastoma, miR-145 causes the inhibition of tumor formation as well as the reduction of CSC properties by targeting Sox2 and OCT4[445]. MiRNA expression profiles of glioblastoma stem cells and non-stem cells revealed that miR-451 is downregulated in the glioma CSCs. Transfection with the above miR leads to the inhibition of tumor growth of glioma CSCs and neurospheres formation[446]. Another miRNA which is important for CSCs of brain tumors is miR-199b-5p. It was shown that miR-199b-5p blocks Notch signaling and inhibits the self-renewal capacity of medulloblastoma cells by targeting HES-1, a transcription factor of the Notch signaling pathway[447].
Using prostate CSCs, Liu et al[448] performed miRNAs expression profiling and found that miR-34a, let-7b, miR-106a and miR-141 are downregulated whereas miR-301 and miR-452 are increased. They also showed that overexpression of miR-34a inhibits prostate CSC metastasis by targeting CD44, while knocking-down of miR-34a promotes tumor migration and development[448]. MiR-320 was also investigated for its role in prostate CSCs. Hsieh et al[449] illustrated that β-catenin is directly targeted by miR-320. Moreover, gene expression profile of miR-320-overexpressing prostate cancer cells revealed a diminished expression of both the genes involved in the Wnt/β-catenin pathway and the markers of CSCs[449].
As mentioned before, miR-34a does not play pivotal role only in glioma and prostate CSCs, but also in pancreatic and gastric CSCs[450,451]. Overexpression of miR-34a in these two cancer types reduces sphere formation and tumor regeneration[450,451]. Bu et al[452] reported that miR-34a inhibits colon CSC self-renewal and suppresses tumor formation. Particularly, miR-34a targets Notch-1 resulting in inhibition of Notch signaling which is frequently crucial in colorectal cancer[452].
LncRNAs in ESCs and CSCs
A number of studies investigated the role of lncRNAs in self-renewal and differentiation of mouse and human ESCs. By performing genome-wide screening, Sheik Mohamed et al[453] identified four lncRNAs residing proximally to the genomic binding sites of Oct4 and Nanog. Two of them, the AK028326 and the AK141205 have been shown to be the direct targets of Oct4 and Nanog, while their overexpression or inhibition resulted in dramatic changes in the mRNA expression levels of the two core transcription factors, revealing their involvement in the regulatory network[453]. Chakraborty et al[454] introduced a new technique combining knockdown and localization analysis of noncoding RNAs (c-KLAN) to study lncRNAs. By inhibiting Panct-1, a non-coding transcript, the amounts of Oct4 and Nanog mRNA were decreased, whereas the expression of differentiation markers increased (Gata6, Fgf5, and T-Brachyury)[454]. To further examine the role of lncRNAs in ESCs, Wang et al[418] demonstrated the lncRNA-RoR function as a critical regulator of self-renewal and differentiation in hESCs. LncRNA-RoR prevents the suppression of Oct4, Sox2 and Nanog by miRNAs (miR-145) and forms a feedback loop with the core transcription factors and microRNAs[418].
Concerning hESCs, the lncRNA_N1 lncRNA_N2 and lncRNA_N3 are important regulators of neuronal differentiation fate[455].
LncRNAs have also been investigated in cancer. Their role is not well understood in CSCs, but recent studies have correlated some lncRNAs with CSC activity, based on their ability to promote metastasis[103]. Gutschner et al[456] developed a metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) knockout model in human lung tumor cells, which confirmed the role of MALAT1 as a biomarker for lung cancer metastasis and revealed its ability to regulate genes associated with lung cancer metastasis[456]. Furthermore, MALAT1 was implicated in cervical cancer by regulating gene expression (caspase-3, -8, Bax, Bcl-2, BclxL), while in a more recent study it was shown that MALAT1 promotes cell migration and proliferation of cervical cancer cells[457,458]. HOTAIR is another lncRNA which has been widely studied for its role in cancer. Gupta et al[459] demonstrated that HOTAIR is a strong metastasis and tumor invasiveness biomarker in primary breast tumors. This function is due to the recruitment of PRC2 that silences metastasis suppressor genes[459]. Moreover, other studies suggest that HOTAIR is a potential biomarker for the existence of lymph node metastasis in hepatocellular carcinoma (HCCs)[460]. As discussed earlier, lncRNA-RoR is an important ESC self-renewal regulator. Recently, it has been shown that its expression was negatively regulated by the NRF2 transcription factor in mammary stem cells[461]. In particular, the inhibition of NRF2 led to an increase of both mammosphere formation in breast cancer cells and lncRNA-RoR levels supporting an involvement of lncRNA-RoR in tumorigenicity[461].
To summarize, the above studies demonstrate an essential role of ncRNAs in stemness of ESCs and CSCs (Table 3). A better understanding of ncRNA biology will ultimately provide further insights into the molecular mechanisms of tumorigenesis and lead to the development of new therapeutic strategies against cancer.
DISCUSSION
In recent years there is a tremendous expansion of research focused on the biology of stem cells. As understanding of the differences and similarities of stem cells of various normal or abnormal developmental origins increases, so is the appreciation of their great complexity. In this review we have presented an overall comparison between embryonic and CSCs in terms of biomarkers (Tables 1 and 2), signaling pathways (Figure 1, Table 3), transcriptional and epigenetic regulators (Table 3).
Figure 1.
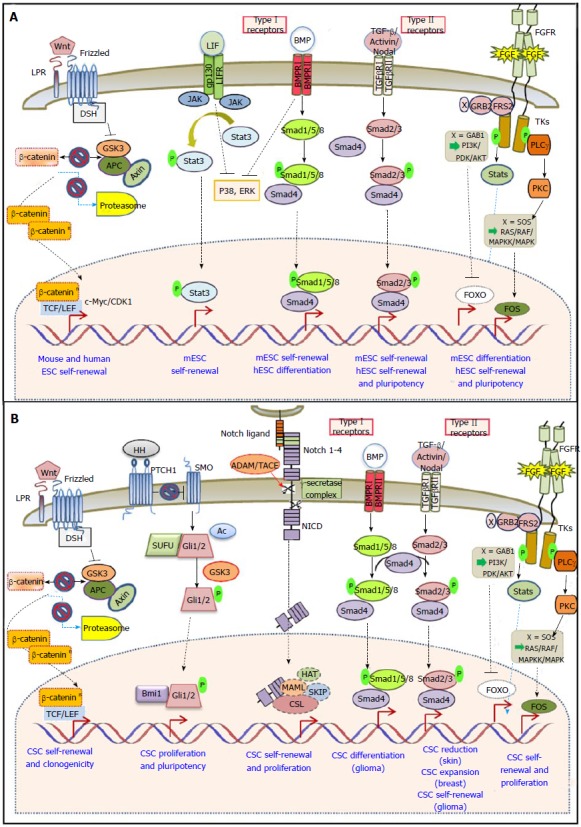
Core signaling pathways in embryonic stem cells and cancer stem cells. A: Mouse ESCs require concerted LIF/BMP signaling to sustain self-renewal and pluripotency, whereas hESC pluripotency depends on the activity of the TGF-β and the FGF pathway. Wnt signaling is important for pluripotency in both species; B: Wnt, Hedgehog, Notch and FGF signaling pathways are associated with CSC self-renewal, while BMP signaling is implicated in CSC differentiation. The TGF-β/Activin/Nodal pathway has a controversial role in CSCs depending on cancer type. ESCs: Embryonic stem cells; CSCs: Cancer stem cells; hESC: Human ESCs; APC: Adenomatous polyposis coli; Dsh: Dishevelled; FGF: Fibroblast growth factor; GSK3: Glycogen synthase kinase 3; JAK: Janus-family tyrosine kinase; LRP: Lipoprotein receptor-related protein; PKC: Protein kinase C; SUFU: Suppressor of fused homolog; TCF/LEF: Tcell factor/Lymphocyte enhancer binding factor.
Cell surface markers are very important for the characterization and separation of stem cells. Pluripotent (ES, iPS) cell markers are very useful for fast and effective enrichment when undifferentiated cells are required (positive selection). In addition, such markers are valuable in order to eliminate the pluripotent cells from their differentiated descendants (negative selection) before transplantation. Concerning cancer, a small proportion of cells that bears stem cell features - CSCs - are endowed with self-renewal and differentiation capacity and enhanced tumorigenicity. These cells are refractive to conventional therapies. In line with the above, promising therapies can be based on targeting CSC via their specific surface markers, as it has been shown in experimental models and clinical trials. An obvious disadvantage of such an approach is that while various markers are currently reported to mark or enrich for CSCs, no exclusive CSC markers are so far available to ensure high cell specificity of targeting. Table 2 shows combinations of markers that are used in order to identify CSCs from various cancer types. Among them, the major category are cell adhesion or communication molecules CD44, Esa/EpCAM, CD166, CD129 and receptor-associated proteins such as Lgr5, c-met, c-kit. The enzymatic activity of ALDH can be a useful tool for separating CSCs, however a detailed identification of the active isoforms that are expressed in various cancer types is required[462]. Most importantly, some ALDH isoforms can have functional roles in CSCs since they are directly involved in retinoic acid signaling and stemness. Another category of various CSC markers are the ABC transporters (Table 2) that actually render CSCs resistant to therapies. A recent promising method for CSC isolation is based on measurement of the intrinsic auto-fluorescence of epithelial CSCs due to the accumulation of riboflavin in cytoplasmic complexes containing ABCG2 transporters[463]. Some CSC markers are also common with those of ESCs such as CD133, SSEA1, EpCAM and the pluripotency transcription factors Oct4, Sox2 and Nanog. Brescia et al[464] have demonstrated that silencing CD133 expression in human GBM neurospheres disrupts the self-renewal and tumorigenic properties of the neurosphere cells, indicating that CD133 could potentially be used as a therapeutic target in these tumors. Transcription factors regulating pluripotency could be particularly useful as biomarkers since they are not expressed in tissue stem cells. However, pluripotency factors are intimately cross-regulated and difficult to assess unless convenient assays are used. The importance of these common markers in tumor aggression and metastasis awaits further investigation.
Cell differentiation during embryonic development and cell transformation that leads to oncogenesis share common signaling pathways, suggesting that deregulation of embryonic signaling pathways in adult tissues might result in tumor progression by transforming adult stem and progenitor cells. Many signaling pathways are involved in the stemness of both pluripotent (ES/iPS) and CSCs (Figure 1, Table 2). Wnt, TGF/BMP and FGF are important regulators of ESC self-renewal and differentiation and are able to enhance the CSC population. On the other hand, Hedgehog and Notch pathways have great importance for CSCs and only marginal role in ESCs. Chemical inhibitors of the Wnt, Notch and Hh pathways have been proven valuable in combating CSC populations from diverse solid tumors[79]. Agents that regulate Wnt signaling such as the CBP/β-catenin antagonist ICG-001, that forces β-catenin to bind p300 instead of CBP, are tested in pre-clinical trials for the eradication of CSC[464]. Gamma-secretase inhibitors are able to block the Notch signaling pathway resulting in inhibition of CSC proliferation and tumor regression[79]. Cyclopamine and synthetic small molecules that antagonize Hh pathway are used alone or in combination with conventional antitumor agents for the inhibition of pancreatic and glioblastoma CSCs[465].
Additional research on the regulation of normal stem cell response to external stimuli will offer alternative therapeutic means for cancer treatment.
Emerging evidence suggests that ESCs and CSCs may depend on common critical transcription factors (Table 3). In addition to the Myc oncogene that orchestrates a complex on its own (Myc-complex), CSCs also express members of the ESC core pluripotency complex such as Oct4, Sox2 and Nanog[466]. Although Oct4, Sox2 and Nanog expression positively correlates with the development of CSCs, the role of Klf4 is highly context-dependent[467]. While it promotes the self-renewal of various CSCs, it can inhibit EMT signaling pathway[260,265] and attenuate lung and liver metastases[468]. Pluripotency transcriptional factors may provide advantageous targets for the elimination of CSCs. For this purpose it would be important to characterize their regulatory circuits in CSCs to the same precision as it was previously attained in ESCs. This knowledge may provide novel approaches for combating cancer[247,469].
In addition to transcription factors, critical regulators of gene expression in embryonic and CSCs are non-coding RNAs (ncRNAs). Interestingly, ncRNAs, which are important for ESC pluripotency such as lncRNA-RoR, mir-145, miR-200, miR-34a and let-7, can affect the generation of CSCs (Table 3). Accumulating evidence indicates that individual miRNAs and lncRNAs behave as tumor suppressors or oncogenes. Among them, the miR-200 family in combination with the TGF-β signaling are major determinants of the EMT transition that correlates tightly with the appearance of CSCs[433,434]. Therefore, further investigation of their function in ESCs and CSCs may lead to clinical applications of non-coding RNAs in cancer diagnosis, treatment, and prognosis.
Epigenetic alterations are very important for the inheritance of the cellular phenotype properties. By governing chromatin state transitions and gene expression regulation, epigenetic processes have a crucial role in cell differentiation and tumorigenesis. Most importantly differentiated cancerous cells are driven into stemness via a profound epigenetic reprogramming that sustains the malignant phenotype[470,471]. Although there is already accumulating information regarding epigenetic alterations and therapeutic use of epigenetic modifiers in cancer, less is known about their role in CSCs. Muñoz et al[471] recently reviewed the role of epigenetic effectors (such as DNMT, EzH2, BMI1 and MLL1) in leukemia and solid tumors and proposed that epigenetic targeting of CSCs may contribute to therapy. The advent of somatic cell reprogramming to pluripotency (iPS) offered a new way to test the effect of extreme epigenetic changes in tumor cells. Can cancer cells be reprogrammed to pluripotency and then induced to differentiate? Although not abundant, recent results in this area suggest a cancer context dependent response to the iPS process[472]. In those cases, pluripotency was able to dominate over the cancerous phenotype leading to “normalization” although the neoplastic identity reappeared when cancer iPS cells were coaxed to differentiate to their lineage of origin[472]. Thus reprogramming could offer a new way to track the history of tumor progression.
In conclusion, studies in the fields of stem cells, iPS and CSCs provide strong evidence of cross-complementing benefits. Insight of the tumorigenic properties of stem cells and their differentiated descendants is required before their use in cell replacement therapies[473]. Understanding the biology and gene circuits shared by both normal stem cells and CSCs, is important for efficient cancer treatment. Cancer heterogeneity is contributed by both clonal and CSC components that are responsible for tumor aggressiveness, invasion and resistance to therapies. An emerging concept in cancer biology is that primary tumors may be composed of evolving and variable clones, each containing biologically distinct stem cells. As cell line models suffer from limited preservation of heterogeneity and newer models of patient derived xenografts are much more labor intensive and costly, alternative, ex vivo cancer models are important. As a result, use of the iPS approach may be useful. The hypothesis of normalizing cancer cells by epigenetic reprogramming has been recently under testing but its limits are not defined yet. Mixed initial results may reflect the heterogeneity of the experimental systems utilized, and various oncogenic and genome aberrations that may not be reversed by pluripotency. However, this is a new promising area that may generate diverse iPS-cancer clones from primary tumors. Thus even if the iPS process cannot “cure” cancer, it may provide, as with other human diseases, novel experimental models for studying cancer biology and drug discovery.
Footnotes
Supported by ESPA “Umbistem” 11ΣΥΝ_10_668.
Conflict-of-interest statement: The authors declare that they have no conflicting interests.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: February 5, 2015
First decision: April 15, 2015
Article in press: October 8, 2015
P- Reviewer: Andrisani OM, Barnes DW, Chen LY, Hsieh SY S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
References
- 1.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 2.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 3.Tesar PJ, Chenoweth JG, Brook FA, Davies TJ, Evans EP, Mack DL, Gardner RL, McKay RD. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- 4.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Theunissen TW, Jaenisch R. Molecular control of induced pluripotency. Cell Stem Cell. 2014;14:720–734. doi: 10.1016/j.stem.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cherry AB, Daley GQ. Reprogrammed cells for disease modeling and regenerative medicine. Annu Rev Med. 2013;64:277–290. doi: 10.1146/annurev-med-050311-163324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Solter D. From teratocarcinomas to embryonic stem cells and beyond: a history of embryonic stem cell research. Nat Rev Genet. 2006;7:319–327. doi: 10.1038/nrg1827. [DOI] [PubMed] [Google Scholar]
- 8.Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO Rep. 2014;15:244–253. doi: 10.1002/embr.201338254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goding CR, Pei D, Lu X. Cancer: pathological nuclear reprogramming? Nat Rev Cancer. 2014;14:568–573. doi: 10.1038/nrc3781. [DOI] [PubMed] [Google Scholar]
- 10.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–699. doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- 12.O’Connor ML, Xiang D, Shigdar S, Macdonald J, Li Y, Wang T, Pu C, Wang Z, Qiao L, Duan W. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014;344:180–187. doi: 10.1016/j.canlet.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13:497–512. doi: 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 15.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong DJ, Liu H, Ridky TW, Cassarino D, Segal E, Chang HY. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell. 2008;2:333–344. doi: 10.1016/j.stem.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao W, Ji X, Zhang F, Li L, Ma L. Embryonic stem cell markers. Molecules. 2012;17:6196–6236. doi: 10.3390/molecules17066196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knowles BB, Aden DP, Solter D. Monoclonal antibody detecting a stage-specific embryonic antigen (SSEA-1) on preimplantation mouse embryos and teratocarcinoma cells. Curr Top Microbiol Immunol. 1978;81:51–53. doi: 10.1007/978-3-642-67448-8_8. [DOI] [PubMed] [Google Scholar]
- 19.Solter D, Knowles BB. Monoclonal antibody defining a stage-specific mouse embryonic antigen (SSEA-1) Proc Natl Acad Sci USA. 1978;75:5565–5569. doi: 10.1073/pnas.75.11.5565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henderson JK, Draper JS, Baillie HS, Fishel S, Thomson JA, Moore H, Andrews PW. Preimplantation human embryos and embryonic stem cells show comparable expression of stage-specific embryonic antigens. Stem Cells. 2002;20:329–337. doi: 10.1634/stemcells.20-4-329. [DOI] [PubMed] [Google Scholar]
- 21.Kannagi R, Levery SB, Ishigami F, Hakomori S, Shevinsky LH, Knowles BB, Solter D. New globoseries glycosphingolipids in human teratocarcinoma reactive with the monoclonal antibody directed to a developmentally regulated antigen, stage-specific embryonic antigen 3. J Biol Chem. 1983;258:8934–8942. [PubMed] [Google Scholar]
- 22.Shevinsky LH, Knowles BB, Damjanov I, Solter D. Monoclonal antibody to murine embryos defines a stage-specific embryonic antigen expressed on mouse embryos and human teratocarcinoma cells. Cell. 1982;30:697–705. doi: 10.1016/0092-8674(82)90274-4. [DOI] [PubMed] [Google Scholar]
- 23.Giwercman A, Andrews PW, Jørgensen N, Müller J, Graem N, Skakkebaek NE. Immunohistochemical expression of embryonal marker TRA-1-60 in carcinoma in situ and germ cell tumors of the testis. Cancer. 1993;72:1308–1314. doi: 10.1002/1097-0142(19930815)72:4<1308::aid-cncr2820720426>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 24.Schopperle WM, DeWolf WC. The TRA-1-60 and TRA-1-81 human pluripotent stem cell markers are expressed on podocalyxin in embryonal carcinoma. Stem Cells. 2007;25:723–730. doi: 10.1634/stemcells.2005-0597. [DOI] [PubMed] [Google Scholar]
- 25.Tanabe K, Nakamura M, Narita M, Takahashi K, Yamanaka S. Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proc Natl Acad Sci USA. 2013;110:12172–12179. doi: 10.1073/pnas.1310291110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sundberg M, Jansson L, Ketolainen J, Pihlajamäki H, Suuronen R, Skottman H, Inzunza J, Hovatta O, Narkilahti S. CD marker expression profiles of human embryonic stem cells and their neural derivatives, determined using flow-cytometric analysis, reveal a novel CD marker for exclusion of pluripotent stem cells. Stem Cell Res. 2009;2:113–124. doi: 10.1016/j.scr.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 27.Evseenko D, Zhu Y, Schenke-Layland K, Kuo J, Latour B, Ge S, Scholes J, Dravid G, Li X, MacLellan WR, et al. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc Natl Acad Sci USA. 2010;107:13742–13747. doi: 10.1073/pnas.1002077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klauzinska M, Castro NP, Rangel MC, Spike BT, Gray PC, Bertolette D, Cuttitta F, Salomon D. The multifaceted role of the embryonic gene Cripto-1 in cancer, stem cells and epithelial-mesenchymal transition. Semin Cancer Biol. 2014;29:51–58. doi: 10.1016/j.semcancer.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parish CL, Parisi S, Persico MG, Arenas E, Minchiotti G. Cripto as a target for improving embryonic stem cell-based therapy in Parkinson’s disease. Stem Cells. 2005;23:471–476. doi: 10.1634/stemcells.2004-0294. [DOI] [PubMed] [Google Scholar]
- 30.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim MP, Fleming JB, Wang H, Abbruzzese JL, Choi W, Kopetz S, McConkey DJ, Evans DB, Gallick GE. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS One. 2011;6:e20636. doi: 10.1371/journal.pone.0020636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Afify A, Purnell P, Nguyen L. Role of CD44s and CD44v6 on human breast cancer cell adhesion, migration, and invasion. Exp Mol Pathol. 2009;86:95–100. doi: 10.1016/j.yexmp.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 33.Bourguignon LY, Spevak CC, Wong G, Xia W, Gilad E. Hyaluronan-CD44 interaction with protein kinase C(epsilon) promotes oncogenic signaling by the stem cell marker Nanog and the Production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance in breast tumor cells. J Biol Chem. 2009;284:26533–26546. doi: 10.1074/jbc.M109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schabath H, Runz S, Joumaa S, Altevogt P. CD24 affects CXCR4 function in pre-B lymphocytes and breast carcinoma cells. J Cell Sci. 2006;119:314–325. doi: 10.1242/jcs.02741. [DOI] [PubMed] [Google Scholar]
- 35.Allan AL. Cancer stem cells in solid tumors. New York: Humana Press; 2011. [Google Scholar]
- 36.Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, Fields JZ, Wicha MS, Boman BM. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69:3382–3389. doi: 10.1158/0008-5472.CAN-08-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreb JS. Aldehyde dehydrogenase as a marker for stem cells. Curr Stem Cell Res Ther. 2008;3:237–246. doi: 10.2174/157488808786734006. [DOI] [PubMed] [Google Scholar]
- 38.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 39.Irollo E, Pirozzi G. CD133: to be or not to be, is this the real question? Am J Transl Res. 2013;5:563–581. [PMC free article] [PubMed] [Google Scholar]
- 40.Salnikov AV, Gladkich J, Moldenhauer G, Volm M, Mattern J, Herr I. CD133 is indicative for a resistance phenotype but does not represent a prognostic marker for survival of non-small cell lung cancer patients. Int J Cancer. 2010;126:950–958. doi: 10.1002/ijc.24822. [DOI] [PubMed] [Google Scholar]
- 41.Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010–4015. doi: 10.1158/0008-5472.CAN-06-4180. [DOI] [PubMed] [Google Scholar]
- 42.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zinzi L, Capparelli E, Cantore M, Contino M, Leopoldo M, Colabufo NA. Small and Innovative Molecules as New Strategy to Revert MDR. Front Oncol. 2014;4:2. doi: 10.3389/fonc.2014.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi YH, Yu AM. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr Pharm Des. 2014;20:793–807. doi: 10.2174/138161282005140214165212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Read TA, Fogarty MP, Markant SL, McLendon RE, Wei Z, Ellison DW, Febbo PG, Wechsler-Reya RJ. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009;15:135–147. doi: 10.1016/j.ccr.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29:2672–2680. doi: 10.1038/onc.2010.35. [DOI] [PubMed] [Google Scholar]
- 47.Ishiwata T, Teduka K, Yamamoto T, Kawahara K, Matsuda Y, Naito Z. Neuroepithelial stem cell marker nestin regulates the migration, invasion and growth of human gliomas. Oncol Rep. 2011;26:91–99. doi: 10.3892/or.2011.1267. [DOI] [PubMed] [Google Scholar]
- 48.Nakai E, Park K, Yawata T, Chihara T, Kumazawa A, Nakabayashi H, Shimizu K. Enhanced MDR1 expression and chemoresistance of cancer stem cells derived from glioblastoma. Cancer Invest. 2009;27:901–908. doi: 10.3109/07357900801946679. [DOI] [PubMed] [Google Scholar]
- 49.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 50.Trzpis M, McLaughlin PM, de Leij LM, Harmsen MC. Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am J Pathol. 2007;171:386–395. doi: 10.2353/ajpath.2007.070152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weidle UH, Eggle D, Klostermann S, Swart GW. ALCAM/CD166: cancer-related issues. Cancer Genomics Proteomics. 2010;7:231–243. [PubMed] [Google Scholar]
- 53.Brizzi MF, Tarone G, Defilippi P. Extracellular matrix, integrins, and growth factors as tailors of the stem cell niche. Curr Opin Cell Biol. 2012;24:645–651. doi: 10.1016/j.ceb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 54.Wu XS, Xi HQ, Chen L. Lgr5 is a potential marker of colorectal carcinoma stem cells that correlates with patient survival. World J Surg Oncol. 2012;10:244. doi: 10.1186/1477-7819-10-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Lau WB, Snel B, Clevers HC. The R-spondin protein family. Genome Biol. 2012;13:242. doi: 10.1186/gb-2012-13-3-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi H, Ishii H, Nishida N, Takemasa I, Mizushima T, Ikeda M, Yokobori T, Mimori K, Yamamoto H, Sekimoto M, et al. Significance of Lgr5(+ve) cancer stem cells in the colon and rectum. Ann Surg Oncol. 2011;18:1166–1174. doi: 10.1245/s10434-010-1373-9. [DOI] [PubMed] [Google Scholar]
- 57.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 58.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 59.Li C, Wu JJ, Hynes M, Dosch J, Sarkar B, Welling TH, Pasca di Magliano M, Simeone DM. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology. 2011;141:2218–2227.e5. doi: 10.1053/j.gastro.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 60.Balic A, Dorado J, Alonso-Gómez M, Heeschen C. Stem cells as the root of pancreatic ductal adenocarcinoma. Exp Cell Res. 2012;318:691–704. doi: 10.1016/j.yexcr.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 61.Sharpe B, Beresford M, Bowen R, Mitchard J, Chalmers AD. Searching for prostate cancer stem cells: markers and methods. Stem Cell Rev. 2013;9:721–730. doi: 10.1007/s12015-013-9453-4. [DOI] [PubMed] [Google Scholar]
- 62.Tomao F, Papa A, Strudel M, Rossi L, Lo Russo G, Benedetti Panici P, Ciabatta FR, Tomao S. Investigating molecular profiles of ovarian cancer: an update on cancer stem cells. J Cancer. 2014;5:301–310. doi: 10.7150/jca.8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo L, Zeng J, Liang B, Zhao Z, Sun L, Cao D, Yang J, Shen K. Ovarian cancer cells with the CD117 phenotype are highly tumorigenic and are related to chemotherapy outcome. Exp Mol Pathol. 2011;91:596–602. doi: 10.1016/j.yexmp.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 64.La Porta CA, Zapperi S. Human breast and melanoma cancer stem cells biomarkers. Cancer Lett. 2013;338:69–73. doi: 10.1016/j.canlet.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 65.Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, Gritti A, Piccinini A, Porro D, Santinami M, et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007;43:935–946. doi: 10.1016/j.ejca.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 66.Redmer T, Welte Y, Behrens D, Fichtner I, Przybilla D, Wruck W, Yaspo ML, Lehrach H, Schäfer R, Regenbrecht CR. The nerve growth factor receptor CD271 is crucial to maintain tumorigenicity and stem-like properties of melanoma cells. PLoS One. 2014;9:e92596. doi: 10.1371/journal.pone.0092596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taghizadeh R, Noh M, Huh YH, Ciusani E, Sigalotti L, Maio M, Arosio B, Nicotra MR, Natali P, Sherley JL, et al. CXCR6, a newly defined biomarker of tissue-specific stem cell asymmetric self-renewal, identifies more aggressive human melanoma cancer stem cells. PLoS One. 2010;5:e15183. doi: 10.1371/journal.pone.0015183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kumar SM, Liu S, Lu H, Zhang H, Zhang PJ, Gimotty PA, Guerra M, Guo W, Xu X. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene. 2012;31:4898–4911. doi: 10.1038/onc.2011.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 70.Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, Wang CY, Hung SC, Chang YL, Tsai ML, Lee YY, et al. Oct4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS One. 2008;3:e2637. doi: 10.1371/journal.pone.0002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kimura M, Takenobu H, Akita N, Nakazawa A, Ochiai H, Shimozato O, Fujimura Y, Koseki H, Yoshino I, Kimura H, et al. Bmi1 regulates cell fate via tumor suppressor WWOX repression in small-cell lung cancer cells. Cancer Sci. 2011;102:983–990. doi: 10.1111/j.1349-7006.2011.01891.x. [DOI] [PubMed] [Google Scholar]
- 72.Yin S, Li J, Hu C, Chen X, Yao M, Yan M, Jiang G, Ge C, Xie H, Wan D, et al. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer. 2007;120:1444–1450. doi: 10.1002/ijc.22476. [DOI] [PubMed] [Google Scholar]
- 73.Mishra L, Banker T, Murray J, Byers S, Thenappan A, He AR, Shetty K, Johnson L, Reddy EP. Liver stem cells and hepatocellular carcinoma. Hepatology. 2009;49:318–329. doi: 10.1002/hep.22704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ji J, Wang XW. Clinical implications of cancer stem cell biology in hepatocellular carcinoma. Semin Oncol. 2012;39:461–472. doi: 10.1053/j.seminoncol.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Okamoto A, Chikamatsu K, Sakakura K, Hatsushika K, Takahashi G, Masuyama K. Expansion and characterization of cancer stem-like cells in squamous cell carcinoma of the head and neck. Oral Oncol. 2009;45:633–639. doi: 10.1016/j.oraloncology.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 77.Major AG, Pitty LP, Farah CS. Cancer stem cell markers in head and neck squamous cell carcinoma. Stem Cells Int. 2013;2013:319489. doi: 10.1155/2013/319489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dreesen O, Brivanlou AH. Signaling pathways in cancer and embryonic stem cells. Stem Cell Rev. 2007;3:7–17. doi: 10.1007/s12015-007-0004-8. [DOI] [PubMed] [Google Scholar]
- 79.Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8:97–106. doi: 10.1038/nrclinonc.2010.196. [DOI] [PubMed] [Google Scholar]
- 80.Karamboulas C, Ailles L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim Biophys Acta. 2013;1830:2481–2495. doi: 10.1016/j.bbagen.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 81.Niwa H, Burdon T, Chambers I, Smith A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998;12:2048–2060. doi: 10.1101/gad.12.13.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007;251:199–210. doi: 10.1016/j.canlet.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 83.Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C, Lin J. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res. 2011;71:7226–7237. doi: 10.1158/0008-5472.CAN-10-4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, Simms M, Bhasin D, Chettiar S, Li C, Li PK, et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013;73:5288–5298. doi: 10.1158/0008-5472.CAN-13-0874. [DOI] [PubMed] [Google Scholar]
- 85.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, Bloushtain-Qimron N, Kim JJ, Choudhury SA, Maruyama R, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44⁺CD24⁻ stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121:2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 87.Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T, et al. Wnt signaling through inhibition of β-catenin degradation in an intact Axin1 complex. Cell. 2012;149:1245–1256. doi: 10.1016/j.cell.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 88.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 89.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 90.ten Berge D, Kurek D, Blauwkamp T, Koole W, Maas A, Eroglu E, Siu RK, Nusse R. Embryonic stem cells require Wnt proteins to prevent differentiation to epiblast stem cells. Nat Cell Biol. 2011;13:1070–1075. doi: 10.1038/ncb2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10:55–63. doi: 10.1038/nm979. [DOI] [PubMed] [Google Scholar]
- 92.Wray J, Kalkan T, Gomez-Lopez S, Eckardt D, Cook A, Kemler R, Smith A. Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat Cell Biol. 2011;13:838–845. doi: 10.1038/ncb2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Valenta T, Gay M, Steiner S, Draganova K, Zemke M, Hoffmans R, Cinelli P, Aguet M, Sommer L, Basler K. Probing transcription-specific outputs of β-catenin in vivo. Genes Dev. 2011;25:2631–2643. doi: 10.1101/gad.181289.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Holland JD, Klaus A, Garratt AN, Birchmeier W. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol. 2013;25:254–264. doi: 10.1016/j.ceb.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 95.Niwa H. Wnt: what’s needed to maintain pluripotency? Nat Cell Biol. 2011;13:1024–1026. doi: 10.1038/ncb2333. [DOI] [PubMed] [Google Scholar]
- 96.Kelly KF, Ng DY, Jayakumaran G, Wood GA, Koide H, Doble BW. β-catenin enhances Oct4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell. 2011;8:214–227. doi: 10.1016/j.stem.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Davidson KC, Adams AM, Goodson JM, McDonald CE, Potter JC, Berndt JD, Biechele TL, Taylor RJ, Moon RT. Wnt/β-catenin signaling promotes differentiation, not self-renewal, of human embryonic stem cells and is repressed by Oct4. Proc Natl Acad Sci U S A. 2012;109:4485–4490. doi: 10.1073/pnas.1118777109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Blauwkamp TA, Nigam S, Ardehali R, Weissman IL, Nusse R. Endogenous Wnt signalling in human embryonic stem cells generates an equilibrium of distinct lineage-specified progenitors. Nat Commun. 2012;3:1070. doi: 10.1038/ncomms2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 100.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730–735. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 101.Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 102.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 103.Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481:85–89. doi: 10.1038/nature10694. [DOI] [PubMed] [Google Scholar]
- 104.Qu Q, Sun G, Li W, Yang S, Ye P, Zhao C, Yu RT, Gage FH, Evans RM, Shi Y. Orphan nuclear receptor TLX activates Wnt/beta-catenin signalling to stimulate neural stem cell proliferation and self-renewal. Nat Cell Biol. 2010;12:31–40; sup pp 1-9. doi: 10.1038/ncb2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mazumdar J, O’Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, Simon MC. O2 regulates stem cells through Wnt/β-catenin signalling. Nat Cell Biol. 2010;12:1007–1013. doi: 10.1038/ncb2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Santoyo-Ramos P, Likhatcheva M, García-Zepeda EA, Castañeda-Patlán MC, Robles-Flores M. Hypoxia-inducible factors modulate the stemness and malignancy of colon cancer cells by playing opposite roles in canonical Wnt signaling. PLoS One. 2014;9:e112580. doi: 10.1371/journal.pone.0112580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Barakat MT, Humke EW, Scott MP. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol Med. 2010;16:337–348. doi: 10.1016/j.molmed.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu SM, Choo AB, Yap MG, Chan KK. Role of Sonic hedgehog signaling and the expression of its components in human embryonic stem cells. Stem Cell Res. 2010;4:38–49. doi: 10.1016/j.scr.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 109.Heo JS, Lee MY, Han HJ. Sonic hedgehog stimulates mouse embryonic stem cell proliferation by cooperation of Ca2+/protein kinase C and epidermal growth factor receptor as well as Gli1 activation. Stem Cells. 2007;25:3069–3080. doi: 10.1634/stemcells.2007-0550. [DOI] [PubMed] [Google Scholar]
- 110.Scales SJ, de Sauvage FJ. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol Sci. 2009;30:303–312. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 111.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, Piccirillo S, Vescovi AL, DiMeco F, Olivi A, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Merchant AA, Matsui W. Targeting Hedgehog--a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–3140. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, Karikari C, Alvarez H, Iacobuzio-Donahue C, Jimeno A, et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67:2187–2196. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chiba S. Notch signaling in stem cell systems. Stem Cells. 2006;24:2437–2447. doi: 10.1634/stemcells.2005-0661. [DOI] [PubMed] [Google Scholar]
- 117.Lowell S, Benchoua A, Heavey B, Smith AG. Notch promotes neural lineage entry by pluripotent embryonic stem cells. PLoS Biol. 2006;4:e121. doi: 10.1371/journal.pbio.0040121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Noggle SA, Weiler D, Condie BG. Notch signaling is inactive but inducible in human embryonic stem cells. Stem Cells. 2006;24:1646–1653. doi: 10.1634/stemcells.2005-0314. [DOI] [PubMed] [Google Scholar]
- 119.Fox V, Gokhale PJ, Walsh JR, Matin M, Jones M, Andrews PW. Cell-cell signaling through NOTCH regulates human embryonic stem cell proliferation. Stem Cells. 2008;26:715–723. doi: 10.1634/stemcells.2007-0368. [DOI] [PubMed] [Google Scholar]
- 120.Yu X, Zou J, Ye Z, Hammond H, Chen G, Tokunaga A, Mali P, Li YM, Civin C, Gaiano N, et al. Notch signaling activation in human embryonic stem cells is required for embryonic, but not trophoblastic, lineage commitment. Cell Stem Cell. 2008;2:461–471. doi: 10.1016/j.stem.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu J, Sato C, Cerletti M, Wagers A. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr Top Dev Biol. 2010;92:367–409. doi: 10.1016/S0070-2153(10)92012-7. [DOI] [PubMed] [Google Scholar]
- 122.Koch U, Lehal R, Radtke F. Stem cells living with a Notch. Development. 2013;140:689–704. doi: 10.1242/dev.080614. [DOI] [PubMed] [Google Scholar]
- 123.Abel EV, Kim EJ, Wu J, Hynes M, Bednar F, Proctor E, Wang L, Dziubinski ML, Simeone DM. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One. 2014;9:e91983. doi: 10.1371/journal.pone.0091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, Fitch-Bruhns M, Lazetic S, Park IK, Sato A, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009;5:168–177. doi: 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 125.Sikandar SS, Pate KT, Anderson S, Dizon D, Edwards RA, Waterman ML, Lipkin SM. NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res. 2010;70:1469–1478. doi: 10.1158/0008-5472.CAN-09-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, Eberhart CG. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–7452. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 127.Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64:7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 128.Ayyanan A, Civenni G, Ciarloni L, Morel C, Mueller N, Lefort K, Mandinova A, Raffoul W, Fiche M, Dotto GP, et al. Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch-dependent mechanism. Proc Natl Acad Sci USA. 2006;103:3799–3804. doi: 10.1073/pnas.0600065103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Farnie G, Clarke RB, Spence K, Pinnock N, Brennan K, Anderson NG, Bundred NJ. Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J Natl Cancer Inst. 2007;99:616–627. doi: 10.1093/jnci/djk133. [DOI] [PubMed] [Google Scholar]
- 130.Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–718. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gallahan D, Callahan R. Mammary tumorigenesis in feral mice: identification of a new int locus in mouse mammary tumor virus (Czech II)-induced mammary tumors. J Virol. 1987;61:66–74. doi: 10.1128/jvi.61.1.66-74.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Derynck R. TGF-beta-receptor-mediated signaling. Trends Biochem Sci. 1994;19:548–553. doi: 10.1016/0968-0004(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 133.Oshimori N, Fuchs E. The harmonies played by TGF-β in stem cell biology. Cell Stem Cell. 2012;11:751–764. doi: 10.1016/j.stem.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.James D, Levine AJ, Besser D, Hemmati-Brivanlou A. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005;132:1273–1282. doi: 10.1242/dev.01706. [DOI] [PubMed] [Google Scholar]
- 135.Ogawa K, Saito A, Matsui H, Suzuki H, Ohtsuka S, Shimosato D, Morishita Y, Watabe T, Niwa H, Miyazono K. Activin-Nodal signaling is involved in propagation of mouse embryonic stem cells. J Cell Sci. 2007;120:55–65. doi: 10.1242/jcs.03296. [DOI] [PubMed] [Google Scholar]
- 136.Vallier L, Alexander M, Pedersen RA. Activin/Nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J Cell Sci. 2005;118:4495–4509. doi: 10.1242/jcs.02553. [DOI] [PubMed] [Google Scholar]
- 137.Qi X, Li TG, Hao J, Hu J, Wang J, Simmons H, Miura S, Mishina Y, Zhao GQ. BMP4 supports self-renewal of embryonic stem cells by inhibiting mitogen-activated protein kinase pathways. Proc Natl Acad Sci USA. 2004;101:6027–6032. doi: 10.1073/pnas.0401367101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ying QL, Nichols J, Chambers I, Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- 139.Xu RH, Chen X, Li DS, Li R, Addicks GC, Glennon C, Zwaka TP, Thomson JA. BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat Biotechnol. 2002;20:1261–1264. doi: 10.1038/nbt761. [DOI] [PubMed] [Google Scholar]
- 140.Xu RH, Peck RM, Li DS, Feng X, Ludwig T, Thomson JA. Basic FGF and suppression of BMP signaling sustain undifferentiated proliferation of human ES cells. Nat Methods. 2005;2:185–190. doi: 10.1038/nmeth744. [DOI] [PubMed] [Google Scholar]
- 141.Zhang P, Li J, Tan Z, Wang C, Liu T, Chen L, Yong J, Jiang W, Sun X, Du L, et al. Short-term BMP-4 treatment initiates mesoderm induction in human embryonic stem cells. Blood. 2008;111:1933–1941. doi: 10.1182/blood-2007-02-074120. [DOI] [PubMed] [Google Scholar]
- 142.Brons IG, Smithers LE, Trotter MW, Rugg-Gunn P, Sun B, Chuva de Sousa Lopes SM, Howlett SK, Clarkson A, Ahrlund-Richter L, Pedersen RA, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- 143.Smith AG. Embryo-derived stem cells: of mice and men. Annu Rev Cell Dev Biol. 2001;17:435–462. doi: 10.1146/annurev.cellbio.17.1.435. [DOI] [PubMed] [Google Scholar]
- 144.Beattie GM, Lopez AD, Bucay N, Hinton A, Firpo MT, King CC, Hayek A. Activin A maintains pluripotency of human embryonic stem cells in the absence of feeder layers. Stem Cells. 2005;23:489–495. doi: 10.1634/stemcells.2004-0279. [DOI] [PubMed] [Google Scholar]
- 145.Xiao L, Yuan X, Sharkis SJ. Activin A maintains self-renewal and regulates fibroblast growth factor, Wnt, and bone morphogenic protein pathways in human embryonic stem cells. Stem Cells. 2006;24:1476–1486. doi: 10.1634/stemcells.2005-0299. [DOI] [PubMed] [Google Scholar]
- 146.Vallier L, Reynolds D, Pedersen RA. Nodal inhibits differentiation of human embryonic stem cells along the neuroectodermal default pathway. Dev Biol. 2004;275:403–421. doi: 10.1016/j.ydbio.2004.08.031. [DOI] [PubMed] [Google Scholar]
- 147.Vallier L, Mendjan S, Brown S, Chng Z, Teo A, Smithers LE, Trotter MW, Cho CH, Martinez A, Rugg-Gunn P, et al. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development. 2009;136:1339–1349. doi: 10.1242/dev.033951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xu RH, Sampsell-Barron TL, Gu F, Root S, Peck RM, Pan G, Yu J, Antosiewicz-Bourget J, Tian S, Stewart R, et al. NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell. 2008;3:196–206. doi: 10.1016/j.stem.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mullen AC, Orlando DA, Newman JJ, Lovén J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, Young RA. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011;147:565–576. doi: 10.1016/j.cell.2011.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Besser D. Expression of nodal, lefty-a, and lefty-B in undifferentiated human embryonic stem cells requires activation of Smad2/3. J Biol Chem. 2004;279:45076–45084. doi: 10.1074/jbc.M404979200. [DOI] [PubMed] [Google Scholar]
- 151.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 153.Massagué J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007;12:313–327. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McLean GW, Komiyama NH, Serrels B, Asano H, Reynolds L, Conti F, Hodivala-Dilke K, Metzger D, Chambon P, Grant SG, et al. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004;18:2998–3003. doi: 10.1101/gad.316304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schober M, Fuchs E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-β and integrin/focal adhesion kinase (FAK) signaling. Proc Natl Acad Sci USA. 2011;108:10544–10549. doi: 10.1073/pnas.1107807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, Sanders M, Stanford J, Cook RS, Arteaga CL. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123:1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Peñuelas S, Anido J, Prieto-Sánchez RM, Folch G, Barba I, Cuartas I, García-Dorado D, Poca MA, Sahuquillo J, Baselga J, et al. TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009;15:315–327. doi: 10.1016/j.ccr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 159.Ikushima H, Todo T, Ino Y, Takahashi M, Miyazawa K, Miyazono K. Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell. 2009;5:504–514. doi: 10.1016/j.stem.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 160.Piccirillo SG, Reynolds BA, Zanetti N, Lamorte G, Binda E, Broggi G, Brem H, Olivi A, Dimeco F, Vescovi AL. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 161.Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, Kotliarova S, Kotliarov Y, Walling J, Ahn S, et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer Cell. 2008;13:69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lanner F, Rossant J. The role of FGF/Erk signaling in pluripotent cells. Development. 2010;137:3351–3360. doi: 10.1242/dev.050146. [DOI] [PubMed] [Google Scholar]
- 163.Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 164.Kunath T, Saba-El-Leil MK, Almousailleakh M, Wray J, Meloche S, Smith A. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development. 2007;134:2895–2902. doi: 10.1242/dev.02880. [DOI] [PubMed] [Google Scholar]
- 165.Stavridis MP, Lunn JS, Collins BJ, Storey KG. A discrete period of FGF-induced Erk1/2 signalling is required for vertebrate neural specification. Development. 2007;134:2889–2894. doi: 10.1242/dev.02858. [DOI] [PubMed] [Google Scholar]
- 166.Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Toyooka Y, Shimosato D, Murakami K, Takahashi K, Niwa H. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development. 2008;135:909–918. doi: 10.1242/dev.017400. [DOI] [PubMed] [Google Scholar]
- 168.Amit M, Carpenter MK, Inokuma MS, Chiu CP, Harris CP, Waknitz MA, Itskovitz-Eldor J, Thomson JA. Clonally derived human embryonic stem cell lines maintain pluripotency and proliferative potential for prolonged periods of culture. Dev Biol. 2000;227:271–278. doi: 10.1006/dbio.2000.9912. [DOI] [PubMed] [Google Scholar]
- 169.Xu C, Inokuma MS, Denham J, Golds K, Kundu P, Gold JD, Carpenter MK. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechnol. 2001;19:971–974. doi: 10.1038/nbt1001-971. [DOI] [PubMed] [Google Scholar]
- 170.Wang G, Zhang H, Zhao Y, Li J, Cai J, Wang P, Meng S, Feng J, Miao C, Ding M, et al. Noggin and bFGF cooperate to maintain the pluripotency of human embryonic stem cells in the absence of feeder layers. Biochem Biophys Res Commun. 2005;330:934–942. doi: 10.1016/j.bbrc.2005.03.058. [DOI] [PubMed] [Google Scholar]
- 171.Armstrong L, Hughes O, Yung S, Hyslop L, Stewart R, Wappler I, Peters H, Walter T, Stojkovic P, Evans J, et al. The role of PI3K/AKT, MAPK/ERK and NFkappabeta signalling in the maintenance of human embryonic stem cell pluripotency and viability highlighted by transcriptional profiling and functional analysis. Hum Mol Genet. 2006;15:1894–1913. doi: 10.1093/hmg/ddl112. [DOI] [PubMed] [Google Scholar]
- 172.Ding VM, Ling L, Natarajan S, Yap MG, Cool SM, Choo AB. FGF-2 modulates Wnt signaling in undifferentiated hESC and iPS cells through activated PI3-K/GSK3beta signaling. J Cell Physiol. 2010;225:417–428. doi: 10.1002/jcp.22214. [DOI] [PubMed] [Google Scholar]
- 173.Li J, Wang G, Wang C, Zhao Y, Zhang H, Tan Z, Song Z, Ding M, Deng H. MEK/ERK signaling contributes to the maintenance of human embryonic stem cell self-renewal. Differentiation. 2007;75:299–307. doi: 10.1111/j.1432-0436.2006.00143.x. [DOI] [PubMed] [Google Scholar]
- 174.Li W, Wei W, Zhu S, Zhu J, Shi Y, Lin T, Hao E, Hayek A, Deng H, Ding S. Generation of rat and human induced pluripotent stem cells by combining genetic reprogramming and chemical inhibitors. Cell Stem Cell. 2009;4:16–19. doi: 10.1016/j.stem.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 175.Xu Y, Zhu X, Hahm HS, Wei W, Hao E, Hayek A, Ding S. Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molecules. Proc Natl Acad Sci USA. 2010;107:8129–8134. doi: 10.1073/pnas.1002024107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Niwa H, Ogawa K, Shimosato D, Adachi K. A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature. 2009;460:118–122. doi: 10.1038/nature08113. [DOI] [PubMed] [Google Scholar]
- 177.Dirks PB. Brain tumor stem cells: bringing order to the chaos of brain cancer. J Clin Oncol. 2008;26:2916–2924. doi: 10.1200/JCO.2008.17.6792. [DOI] [PubMed] [Google Scholar]
- 178.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 179.Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, Kuperwasser C. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci USA. 2010;107:21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guthridge MA, Seldin M, Basilico C. Induction of expression of growth-related genes by FGF-4 in mouse fibroblasts. Oncogene. 1996;12:1267–1278. [PubMed] [Google Scholar]
- 181.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 182.McKeon F. p63 and the epithelial stem cell: more than status quo? Genes Dev. 2004;18:465–469. doi: 10.1101/gad.1190504. [DOI] [PubMed] [Google Scholar]
- 183.Kosaka N, Sakamoto H, Terada M, Ochiya T. Pleiotropic function of FGF-4: its role in development and stem cells. Dev Dyn. 2009;238:265–276. doi: 10.1002/dvdy.21699. [DOI] [PubMed] [Google Scholar]
- 184.Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 185.Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 187.Orkin SH, Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB, Orkin SH. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Orkin SH, Wang J, Kim J, Chu J, Rao S, Theunissen TW, Shen X, Levasseur DN. The transcriptional network controlling pluripotency in ES cells. Cold Spring Harb Symp Quant Biol. 2008;73:195–202. doi: 10.1101/sqb.2008.72.001. [DOI] [PubMed] [Google Scholar]
- 191.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 192.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 195.Rosner MH, Vigano MA, Ozato K, Timmons PM, Poirier F, Rigby PW, Staudt LM. A POU-domain transcription factor in early stem cells and germ cells of the mammalian embryo. Nature. 1990;345:686–692. doi: 10.1038/345686a0. [DOI] [PubMed] [Google Scholar]
- 196.Schöler HR, Dressler GR, Balling R, Rohdewohld H, Gruss P. Oct4: a germline-specific transcription factor mapping to the mouse t-complex. EMBO J. 1990;9:2185–2195. doi: 10.1002/j.1460-2075.1990.tb07388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Cauffman G, Van de Velde H, Liebaers I, Van Steirteghem A. Oct4 mRNA and protein expression during human preimplantation development. Mol Hum Reprod. 2005;11:173–181. doi: 10.1093/molehr/gah155. [DOI] [PubMed] [Google Scholar]
- 198.Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Schöler H, Smith A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 199.Nichols J, Evans EP, Smith AG. Establishment of germ-line-competent embryonic stem (ES) cells using differentiation inhibiting activity. Development. 1990;110:1341–1348. doi: 10.1242/dev.110.4.1341. [DOI] [PubMed] [Google Scholar]
- 200.Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- 201.Shimozaki K, Nakashima K, Niwa H, Taga T. Involvement of Oct3/4 in the enhancement of neuronal differentiation of ES cells in neurogenesis-inducing cultures. Development. 2003;130:2505–2512. doi: 10.1242/dev.00476. [DOI] [PubMed] [Google Scholar]
- 202.Aksoy I, Jauch R, Chen J, Dyla M, Divakar U, Bogu GK, Teo R, Leng Ng CK, Herath W, Lili S, et al. Oct4 switches partnering from Sox2 to Sox17 to reinterpret the enhancer code and specify endoderm. EMBO J. 2013;32:938–953. doi: 10.1038/emboj.2013.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Atlasi Y, Mowla SJ, Ziaee SA, Bahrami AR. Oct4, an embryonic stem cell marker, is highly expressed in bladder cancer. Int J Cancer. 2007;120:1598–1602. doi: 10.1002/ijc.22508. [DOI] [PubMed] [Google Scholar]
- 204.Ezeh UI, Turek PJ, Reijo RA, Clark AT. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer. 2005;104:2255–2265. doi: 10.1002/cncr.21432. [DOI] [PubMed] [Google Scholar]
- 205.Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007;67:4807–4815. doi: 10.1158/0008-5472.CAN-06-4608. [DOI] [PubMed] [Google Scholar]
- 206.Liu CG, Lu Y, Wang BB, Zhang YJ, Zhang RS, Lu Y, Chen B, Xu H, Jin F, Lu P. Clinical implications of stem cell gene Oct4 expression in breast cancer. Ann Surg. 2011;253:1165–1171. doi: 10.1097/SLA.0b013e318214c54e. [DOI] [PubMed] [Google Scholar]
- 207.Hu T, Liu S, Breiter DR, Wang F, Tang Y, Sun S. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer Res. 2008;68:6533–6540. doi: 10.1158/0008-5472.CAN-07-6642. [DOI] [PubMed] [Google Scholar]
- 208.Chiou SH, Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH, Chou SH, Chien CS, Ku HH, Lo JF. Positive correlations of Oct4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. 2008;14:4085–4095. doi: 10.1158/1078-0432.CCR-07-4404. [DOI] [PubMed] [Google Scholar]
- 209.Rentala S, Mangamoori LN. Oct4 expression maintained stem cell properties in prostate cancer-derived cd133 mdr1 cells. Trop J Pharm Res. 2009;8:3–9. [Google Scholar]
- 210.Beltran AS, Rivenbark AG, Richardson BT, Yuan X, Quian H, Hunt JP, Zimmerman E, Graves LM, Blancafort P. Generation of tumor-initiating cells by exogenous delivery of OCT4 transcription factor. Breast Cancer Res. 2011;13:R94. doi: 10.1186/bcr3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wang YD, Cai N, Wu XL, Cao HZ, Xie LL, Zheng PS. OCT4 promotes tumorigenesis and inhibits apoptosis of cervical cancer cells by miR-125b/BAK1 pathway. Cell Death Dis. 2013;4:e760. doi: 10.1038/cddis.2013.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Koo BS, Lee SH, Kim JM, Huang S, Kim SH, Rho YS, Bae WJ, Kang HJ, Kim YS, Moon JH, et al. Oct4 is a critical regulator of stemness in head and neck squamous carcinoma cells. Oncogene. 2015;34:2317–2324. doi: 10.1038/onc.2014.174. [DOI] [PubMed] [Google Scholar]
- 213.Kamachi Y, Uchikawa M, Kondoh H. Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet. 2000;16:182–187. doi: 10.1016/s0168-9525(99)01955-1. [DOI] [PubMed] [Google Scholar]
- 214.Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, Okochi H, Okuda A, Matoba R, Sharov AA, et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol. 2007;9:625–635. doi: 10.1038/ncb1589. [DOI] [PubMed] [Google Scholar]
- 216.Boer B, Kopp J, Mallanna S, Desler M, Chakravarthy H, Wilder PJ, Bernadt C, Rizzino A. Elevating the levels of Sox2 in embryonal carcinoma cells and embryonic stem cells inhibits the expression of Sox2: Oct-3/4 target genes. Nucleic Acids Res. 2007;35:1773–1786. doi: 10.1093/nar/gkm059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kopp JL, Ormsbee BD, Desler M, Rizzino A. Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells. 2008;26:903–911. doi: 10.1634/stemcells.2007-0951. [DOI] [PubMed] [Google Scholar]
- 218.Basu-Roy U, Seo E, Ramanathapuram L, Rapp TB, Perry JA, Orkin SH, Mansukhani A, Basilico C. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene. 2012;31:2270–2282. doi: 10.1038/onc.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lou X, Han X, Jin C, Tian W, Yu W, Ding D, Cheng L, Huang B, Jiang H, Lin B. SOX2 targets fibronectin 1 to promote cell migration and invasion in ovarian cancer: new molecular leads for therapeutic intervention. OMICS. 2013;17:510–518. doi: 10.1089/omi.2013.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tian T, Zhang Y, Wang S, Zhou J, Xu S. Sox2 enhances the tumorigenicity and chemoresistance of cancer stem-like cells derived from gastric cancer. J Biomed Res. 2012;26:336–345. doi: 10.7555/JBR.26.20120045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Herreros-Villanueva M, Zhang JS, Koenig A, Abel EV, Smyrk TC, Bamlet WR, de Narvajas AA, Gomez TS, Simeone DM, Bujanda L, et al. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis. 2013;2:e61. doi: 10.1038/oncsis.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Rybak AP, Tang D. SOX2 plays a critical role in EGFR-mediated self-renewal of human prostate cancer stem-like cells. Cell Signal. 2013;25:2734–2742. doi: 10.1016/j.cellsig.2013.08.041. [DOI] [PubMed] [Google Scholar]
- 223.Singh S, Trevino J, Bora-Singhal N, Coppola D, Haura E, Altiok S, Chellappan SP. EGFR/Src/Akt signaling modulates Sox2 expression and self-renewal of stem-like side-population cells in non-small cell lung cancer. Mol Cancer. 2012;11:73. doi: 10.1186/1476-4598-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Santini R, Pietrobono S, Pandolfi S, Montagnani V, D’Amico M, Penachioni JY, Vinci MC, Borgognoni L, Stecca B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene. 2014;33:4697–4708. doi: 10.1038/onc.2014.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Favaro R, Appolloni I, Pellegatta S, Sanga AB, Pagella P, Gambini E, Pisati F, Ottolenghi S, Foti M, Finocchiaro G, et al. Sox2 is required to maintain cancer stem cells in a mouse model of high-grade oligodendroglioma. Cancer Res. 2014;74:1833–1844. doi: 10.1158/0008-5472.CAN-13-1942. [DOI] [PubMed] [Google Scholar]
- 226.Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, Vrana J, Jones K, Grotewold L, Smith A. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–1234. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- 227.Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell. 2003;113:631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- 228.Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 229.Silva J, Nichols J, Theunissen TW, Guo G, van Oosten AL, Barrandon O, Wray J, Yamanaka S, Chambers I, Smith A. Nanog is the gateway to the pluripotent ground state. Cell. 2009;138:722–737. doi: 10.1016/j.cell.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hyslop L, Stojkovic M, Armstrong L, Walter T, Stojkovic P, Przyborski S, Herbert M, Murdoch A, Strachan T, Lako M. Downregulation of NANOG induces differentiation of human embryonic stem cells to extraembryonic lineages. Stem Cells. 2005;23:1035–1043. doi: 10.1634/stemcells.2005-0080. [DOI] [PubMed] [Google Scholar]
- 231.Kuroda T, Tada M, Kubota H, Kimura H, Hatano SY, Suemori H, Nakatsuji N, Tada T. Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol Cell Biol. 2005;25:2475–2485. doi: 10.1128/MCB.25.6.2475-2485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Amsterdam A, Raanan C, Schreiber L, Freyhan O, Schechtman L, Givol D. Localization of the stem cell markers LGR5 and Nanog in the normal and the cancerous human ovary and their inter-relationship. Acta Histochem. 2013;115:330–338. doi: 10.1016/j.acthis.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 233.Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008;22:3696–3705. doi: 10.1096/fj.08-102590. [DOI] [PubMed] [Google Scholar]
- 234.Chiou SH, Wang ML, Chou YT, Chen CJ, Hong CF, Hsieh WJ, Chang HT, Chen YS, Lin TW, Hsu HS, et al. Coexpression of Oct4 and Nanog enhances malignancy in lung adenocarcinoma by inducing cancer stem cell-like properties and epithelial-mesenchymal transdifferentiation. Cancer Res. 2010;70:10433–10444. doi: 10.1158/0008-5472.CAN-10-2638. [DOI] [PubMed] [Google Scholar]
- 235.Lu X, Mazur SJ, Lin T, Appella E, Xu Y. The pluripotency factor nanog promotes breast cancer tumorigenesis and metastasis. Oncogene. 2014;33:2655–2664. doi: 10.1038/onc.2013.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Guo Y, Liu S, Wang P, Zhao S, Wang F, Bing L, Zhang Y, Ling EA, Gao J, Hao A. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology. 2011;59:763–775. doi: 10.1111/j.1365-2559.2011.03993.x. [DOI] [PubMed] [Google Scholar]
- 237.Lin T, Ding YQ, Li JM. Overexpression of Nanog protein is associated with poor prognosis in gastric adenocarcinoma. Med Oncol. 2012;29:878–885. doi: 10.1007/s12032-011-9860-9. [DOI] [PubMed] [Google Scholar]
- 238.Lee M, Nam EJ, Kim SW, Kim S, Kim JH, Kim YT. Prognostic impact of the cancer stem cell-related marker NANOG in ovarian serous carcinoma. Int J Gynecol Cancer. 2012;22:1489–1496. doi: 10.1097/IGJ.0b013e3182738307. [DOI] [PubMed] [Google Scholar]
- 239.Meng HM, Zheng P, Wang XY, Liu C, Sui HM, Wu SJ, Zhou J, Ding YQ, Li J. Over-expression of Nanog predicts tumor progression and poor prognosis in colorectal cancer. Cancer Biol Ther. 2010;9:295–302. doi: 10.4161/cbt.9.4.10666. [DOI] [PubMed] [Google Scholar]
- 240.Nagata T, Shimada Y, Sekine S, Hori R, Matsui K, Okumura T, Sawada S, Fukuoka J, Tsukada K. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer. 2014;21:96–101. doi: 10.1007/s12282-012-0357-y. [DOI] [PubMed] [Google Scholar]
- 241.Ibrahim EE, Babaei-Jadidi R, Saadeddin A, Spencer-Dene B, Hossaini S, Abuzinadah M, Li N, Fadhil W, Ilyas M, Bonnet D, et al. Embryonic NANOG activity defines colorectal cancer stem cells and modulates through AP1- and TCF-dependent mechanisms. Stem Cells. 2012;30:2076–2087. doi: 10.1002/stem.1182. [DOI] [PubMed] [Google Scholar]
- 242.Jeter CR, Liu B, Liu X, Chen X, Liu C, Calhoun-Davis T, Repass J, Zaehres H, Shen JJ, Tang DG. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833–3845. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9:50–63. doi: 10.1016/j.stem.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 244.Niu CS, Li DX, Liu YH, Fu XM, Tang SF, Li J. Expression of NANOG in human gliomas and its relationship with undifferentiated glioma cells. Oncol Rep. 2011;26:593–601. doi: 10.3892/or.2011.1308. [DOI] [PubMed] [Google Scholar]
- 245.Zbinden M, Duquet A, Lorente-Trigos A, Ngwabyt SN, Borges I, Ruiz i Altaba A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010;29:2659–2674. doi: 10.1038/emboj.2010.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Zhou X, Zhou YP, Huang GR, Gong BL, Yang B, Zhang DX, Hu P, Xu SR. Expression of the stem cell marker, Nanog, in human endometrial adenocarcinoma. Int J Gynecol Pathol. 2011;30:262–270. doi: 10.1097/PGP.0b013e3182055a1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Wang ML, Chiou SH, Wu CW. Targeting cancer stem cells: emerging role of Nanog transcription factor. Onco Targets Ther. 2013;6:1207–1220. doi: 10.2147/OTT.S38114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.He A, Qi W, Huang Y, Feng T, Chen J, Sun Y, Shen Z, Yao Y. CD133 expression predicts lung metastasis and poor prognosis in osteosarcoma patients: A clinical and experimental study. Exp Ther Med. 2012;4:435–441. doi: 10.3892/etm.2012.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, Fink LM, Ma Y, Wong MP. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One. 2010;5:e14062. doi: 10.1371/journal.pone.0014062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Han J, Zhang F, Yu M, Zhao P, Ji W, Zhang H, Wu B, Wang Y, Niu R. RNA interference-mediated silencing of NANOG reduces cell proliferation and induces G0/G1 cell cycle arrest in breast cancer cells. Cancer Lett. 2012;321:80–88. doi: 10.1016/j.canlet.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 251.Siu MK, Wong ES, Kong DS, Chan HY, Jiang L, Wong OG, Lam EW, Chan KK, Ngan HY, Le XF, et al. Stem cell transcription factor NANOG controls cell migration and invasion via dysregulation of E-cadherin and FoxJ1 and contributes to adverse clinical outcome in ovarian cancers. Oncogene. 2013;32:3500–3509. doi: 10.1038/onc.2012.363. [DOI] [PubMed] [Google Scholar]
- 252.Bourillot PY, Aksoy I, Schreiber V, Wianny F, Schulz H, Hummel O, Hubner N, Savatier P. Novel STAT3 target genes exert distinct roles in the inhibition of mesoderm and endoderm differentiation in cooperation with Nanog. Stem Cells. 2009;27:1760–1771. doi: 10.1002/stem.110. [DOI] [PubMed] [Google Scholar]
- 253.Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM, Yang VW. Krüppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res. 2005;15:92–96. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Bourillot PY, Savatier P. Krüppel-like transcription factors and control of pluripotency. BMC Biol. 2010;8:125. doi: 10.1186/1741-7007-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, Schafer X, Lun Y, Lemischka IR. Dissecting self-renewal in stem cells with RNA interference. Nature. 2006;442:533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- 256.Li Y, McClintick J, Zhong L, Edenberg HJ, Yoder MC, Chan RJ. Murine embryonic stem cell differentiation is promoted by SOCS-3 and inhibited by the zinc finger transcription factor Klf4. Blood. 2005;105:635–637. doi: 10.1182/blood-2004-07-2681. [DOI] [PubMed] [Google Scholar]
- 257.Nakatake Y, Fukui N, Iwamatsu Y, Masui S, Takahashi K, Yagi R, Yagi K, Miyazaki J, Matoba R, Ko MS, et al. Klf4 cooperates with Oct3/4 and Sox2 to activate the Lefty1 core promoter in embryonic stem cells. Mol Cell Biol. 2006;26:7772–7782. doi: 10.1128/MCB.00468-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, Robson P, Zhong S, Ng HH. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol. 2008;10:353–360. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 259.Yasunaga M, Tada S, Torikai-Nishikawa S, Nakano Y, Okada M, Jakt LM, Nishikawa S, Chiba T, Era T, Nishikawa S. Induction and monitoring of definitive and visceral endoderm differentiation of mouse ES cells. Nat Biotechnol. 2005;23:1542–1550. doi: 10.1038/nbt1167. [DOI] [PubMed] [Google Scholar]
- 260.Aksoy I, Giudice V, Delahaye E, Wianny F, Aubry M, Mure M, Chen J, Jauch R, Bogu GK, Nolden T, et al. Klf4 and Klf5 differentially inhibit mesoderm and endoderm differentiation in embryonic stem cells. Nat Commun. 2014;5:3719. doi: 10.1038/ncomms4719. [DOI] [PubMed] [Google Scholar]
- 261.Wong CW, Hou PS, Tseng SF, Chien CL, Wu KJ, Chen HF, Ho HN, Kyo S, Teng SC. Krüppel-like transcription factor 4 contributes to maintenance of telomerase activity in stem cells. Stem Cells. 2010;28:1510–1517. doi: 10.1002/stem.477. [DOI] [PubMed] [Google Scholar]
- 262.Hoffmeyer K, Raggioli A, Rudloff S, Anton R, Hierholzer A, Del Valle I, Hein K, Vogt R, Kemler R. Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science. 2012;336:1549–1554. doi: 10.1126/science.1218370. [DOI] [PubMed] [Google Scholar]
- 263.Yu F, Li J, Chen H, Fu J, Ray S, Huang S, Zheng H, Ai W. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–2172. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Yu T, Chen X, Zhang W, Li J, Xu R, Wang TC, Ai W, Liu C. Krüppel-like factor 4 regulates intestinal epithelial cell morphology and polarity. PLoS One. 2012;7:e32492. doi: 10.1371/journal.pone.0032492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Yori JL, Johnson E, Zhou G, Jain MK, Keri RA. Kruppel-like factor 4 inhibits epithelial-to-mesenchymal transition through regulation of E-cadherin gene expression. J Biol Chem. 2010;285:16854–16863. doi: 10.1074/jbc.M110.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Leng Z, Tao K, Xia Q, Tan J, Yue Z, Chen J, Xi H, Li J, Zheng H. Krüppel-like factor 4 acts as an oncogene in colon cancer stem cell-enriched spheroid cells. PLoS One. 2013;8:e56082. doi: 10.1371/journal.pone.0056082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 268.Okuda H, Xing F, Pandey PR, Sharma S, Watabe M, Pai SK, Mo YY, Iiizumi-Gairani M, Hirota S, Liu Y, et al. miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res. 2013;73:1434–1444. doi: 10.1158/0008-5472.CAN-12-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Ma J, Yao Y, Wang P, Liu Y, Zhao L, Li Z, Li Z, Xue Y. MiR-152 functions as a tumor suppressor in glioblastoma stem cells by targeting Krüppel-like factor 4. Cancer Lett. 2014;355:85–95. doi: 10.1016/j.canlet.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 270.Cole MD, Henriksson M. 25 years of the c-Myc oncogene. Semin Cancer Biol. 2006;16:241. doi: 10.1016/j.semcancer.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 271.Laurenti E, Wilson A, Trumpp A. Myc’s other life: stem cells and beyond. Curr Opin Cell Biol. 2009;21:844–854. doi: 10.1016/j.ceb.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 272.Meyer N, Kim SS, Penn LZ. The Oscar-worthy role of Myc in apoptosis. Semin Cancer Biol. 2006;16:275–287. doi: 10.1016/j.semcancer.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 273.Cartwright P, McLean C, Sheppard A, Rivett D, Jones K, Dalton S. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development. 2005;132:885–896. doi: 10.1242/dev.01670. [DOI] [PubMed] [Google Scholar]
- 274.Smith KN, Singh AM, Dalton S. Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell Stem Cell. 2010;7:343–354. doi: 10.1016/j.stem.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Chappell J, Sun Y, Singh A, Dalton S. MYC/MAX control ERK signaling and pluripotency by regulation of dual-specificity phosphatases 2 and 7. Genes Dev. 2013;27:725–733. doi: 10.1101/gad.211300.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kidder BL, Yang J, Palmer S. Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS One. 2008;3:e3932. doi: 10.1371/journal.pone.0003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Sridharan R, Tchieu J, Mason MJ, Yachechko R, Kuoy E, Horvath S, Zhou Q, Plath K. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009;136:364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Yin XY, Grove L, Datta NS, Katula K, Long MW, Prochownik EV. Inverse regulation of cyclin B1 by c-Myc and p53 and induction of tetraploidy by cyclin B1 overexpression. Cancer Res. 2001;61:6487–6493. [PubMed] [Google Scholar]
- 280.Eilers M, Eisenman RN. Myc’s broad reach. Genes Dev. 2008;22:2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al. Pten and p53 converge on c-Myc to control differentiation, self-renewal, and transformation of normal and neoplastic stem cells in glioblastoma. Cold Spring Harb Symp Quant Biol. 2008;73:427–437. doi: 10.1101/sqb.2008.73.047. [DOI] [PubMed] [Google Scholar]
- 282.Ho C, Wang C, Mattu S, Destefanis G, Ladu S, Delogu S, Armbruster J, Fan L, Lee SA, Jiang L, et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology. 2012;55:833–845. doi: 10.1002/hep.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Wang J, Wang H, Li Z, Wu Q, Lathia JD, McLendon RE, Hjelmeland AB, Rich JN. c-Myc is required for maintenance of glioma cancer stem cells. PLoS One. 2008;3:e3769. doi: 10.1371/journal.pone.0003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Salcido CD, Larochelle A, Taylor BJ, Dunbar CE, Varticovski L. Molecular characterisation of side population cells with cancer stem cell-like characteristics in small-cell lung cancer. Br J Cancer. 2010;102:1636–1644. doi: 10.1038/sj.bjc.6605668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Walter D, Satheesha S, Albrecht P, Bornhauser BC, D’Alessandro V, Oesch SM, Rehrauer H, Leuschner I, Koscielniak E, Gengler C, et al. CD133 positive embryonal rhabdomyosarcoma stem-like cell population is enriched in rhabdospheres. PLoS One. 2011;6:e19506. doi: 10.1371/journal.pone.0019506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Civenni G, Malek A, Albino D, Garcia-Escudero R, Napoli S, Di Marco S, Pinton S, Sarti M, Carbone GM, Catapano CV. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer Res. 2013;73:6816–6827. doi: 10.1158/0008-5472.CAN-13-0615. [DOI] [PubMed] [Google Scholar]
- 287.Akita H, Marquardt JU, Durkin ME, Kitade M, Seo D, Conner EA, Andersen JB, Factor VM, Thorgeirsson SS. MYC activates stem-like cell potential in hepatocarcinoma by a p53-dependent mechanism. Cancer Res. 2014;74:5903–5913. doi: 10.1158/0008-5472.CAN-14-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Hitchler MJ, Rice JC. Genome-wide epigenetic analysis of human pluripotent stem cells by ChIP and ChIP-Seq. Methods Mol Biol. 2011;767:253–267. doi: 10.1007/978-1-61779-201-4_19. [DOI] [PubMed] [Google Scholar]
- 289.Revill K, Wang T, Lachenmayer A, Kojima K, Harrington A, Li J, Hoshida Y, Llovet JM, Powers S. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology. 2013;145:1424–35.e1-25. doi: 10.1053/j.gastro.2013.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12:206–222. doi: 10.1002/cbic.201000195. [DOI] [PubMed] [Google Scholar]
- 291.Fatemi M, Hermann A, Gowher H, Jeltsch A. Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur J Biochem. 2002;269:4981–4984. doi: 10.1046/j.1432-1033.2002.03198.x. [DOI] [PubMed] [Google Scholar]
- 292.Walton EL, Francastel C, Velasco G. Maintenance of DNA methylation: Dnmt3b joins the dance. Epigenetics. 2011;6:1373–1377. doi: 10.4161/epi.6.11.17978. [DOI] [PubMed] [Google Scholar]
- 293.Jackson M, Krassowska A, Gilbert N, Chevassut T, Forrester L, Ansell J, Ramsahoye B. Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol. 2004;24:8862–8871. doi: 10.1128/MCB.24.20.8862-8871.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Zvetkova I, Apedaile A, Ramsahoye B, Mermoud JE, Crompton LA, John R, Feil R, Brockdorff N. Global hypomethylation of the genome in XX embryonic stem cells. Nat Genet. 2005;37:1274–1279. doi: 10.1038/ng1663. [DOI] [PubMed] [Google Scholar]
- 295.Fouse SD, Shen Y, Pellegrini M, Cole S, Meissner A, Van Neste L, Jaenisch R, Fan G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell. 2008;2:160–169. doi: 10.1016/j.stem.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Bibikova M, Chudin E, Wu B, Zhou L, Garcia EW, Liu Y, Shin S, Plaia TW, Auerbach JM, Arking DE, et al. Human embryonic stem cells have a unique epigenetic signature. Genome Res. 2006;16:1075–1083. doi: 10.1101/gr.5319906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zhao L, Sun MA, Li Z, Bai X, Yu M, Wang M, Liang L, Shao X, Arnovitz S, Wang Q, et al. The dynamics of DNA methylation fidelity during mouse embryonic stem cell self-renewal and differentiation. Genome Res. 2014;24:1296–1307. doi: 10.1101/gr.163147.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Arand J, Spieler D, Karius T, Branco MR, Meilinger D, Meissner A, Jenuwein T, Xu G, Leonhardt H, Wolf V, et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012;8:e1002750. doi: 10.1371/journal.pgen.1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7:e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 303.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 304.Biniszkiewicz D, Gribnau J, Ramsahoye B, Gaudet F, Eggan K, Humpherys D, Mastrangelo MA, Jun Z, Walter J, Jaenisch R. Dnmt1 overexpression causes genomic hypermethylation, loss of imprinting, and embryonic lethality. Mol Cell Biol. 2002;22:2124–2135. doi: 10.1128/MCB.22.7.2124-2135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291–2298. doi: 10.1093/nar/27.11.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Pawlak M, Jaenisch R. De novo DNA methylation by Dnmt3a and Dnmt3b is dispensable for nuclear reprogramming of somatic cells to a pluripotent state. Genes Dev. 2011;25:1035–1040. doi: 10.1101/gad.2039011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Globisch D, Münzel M, Müller M, Michalakis S, Wagner M, Koch S, Brückl T, Biel M, Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA, Marques CJ, Andrews S, Reik W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 314.Robertson J, Robertson AB, Klungland A. The presence of 5-hydroxymethylcytosine at the gene promoter and not in the gene body negatively regulates gene expression. Biochem Biophys Res Commun. 2011;411:40–43. doi: 10.1016/j.bbrc.2011.06.077. [DOI] [PubMed] [Google Scholar]
- 315.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, Nardone J, Laiho A, Tahiliani M, Sommer CA, Mostoslavsky G, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Huang Y, Chavez L, Chang X, Wang X, Pastor WA, Kang J, Zepeda-Martínez JA, Pape UJ, Jacobsen SE, Peters B, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad Sci USA. 2014;111:1361–1366. doi: 10.1073/pnas.1322921111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Wu H, Zhang Y. Tet1 and 5-hydroxymethylation: a genome-wide view in mouse embryonic stem cells. Cell Cycle. 2011;10:2428–2436. doi: 10.4161/cc.10.15.16930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, Rappsilber J, Helin K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Vella P, Scelfo A, Jammula S, Chiacchiera F, Williams K, Cuomo A, Roberto A, Christensen J, Bonaldi T, Helin K, et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol Cell. 2013;49:645–656. doi: 10.1016/j.molcel.2012.12.019. [DOI] [PubMed] [Google Scholar]
- 320.Shi FT, Kim H, Lu W, He Q, Liu D, Goodell MA, Wan M, Songyang Z. Ten-eleven translocation 1 (Tet1) is regulated by O-linked N-acetylglucosamine transferase (Ogt) for target gene repression in mouse embryonic stem cells. J Biol Chem. 2013;288:20776–20784. doi: 10.1074/jbc.M113.460386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Freudenberg JM, Ghosh S, Lackford BL, Yellaboina S, Zheng X, Li R, Cuddapah S, Wade PA, Hu G, Jothi R. Acute depletion of Tet1-dependent 5-hydroxymethylcytosine levels impairs LIF/Stat3 signaling and results in loss of embryonic stem cell identity. Nucleic Acids Res. 2012;40:3364–3377. doi: 10.1093/nar/gkr1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Dawlaty MM, Breiling A, Le T, Barrasa MI, Raddatz G, Gao Q, Powell BE, Cheng AW, Faull KF, Lyko F, et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev Cell. 2014;29:102–111. doi: 10.1016/j.devcel.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Dumitrescu RG, Verma M. Cancer epigenetics: Methods and protocols. Totowa NJ: Humana. London: Springer [distributor]; 2012. [Google Scholar]
- 324.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 325.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 326.Nishigaki M, Aoyagi K, Danjoh I, Fukaya M, Yanagihara K, Sakamoto H, Yoshida T, Sasaki H. Discovery of aberrant expression of R-RAS by cancer-linked DNA hypomethylation in gastric cancer using microarrays. Cancer Res. 2005;65:2115–2124. doi: 10.1158/0008-5472.CAN-04-3340. [DOI] [PubMed] [Google Scholar]
- 327.Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H. Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest. 2001;81:77–82. doi: 10.1038/labinvest.3780213. [DOI] [PubMed] [Google Scholar]
- 328.Lahtz C, Pfeifer GP. Epigenetic changes of DNA repair genes in cancer. J Mol Cell Biol. 2011;3:51–58. doi: 10.1093/jmcb/mjq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Yasuda H, Soejima K, Watanabe H, Kawada I, Nakachi I, Yoda S, Nakayama S, Satomi R, Ikemura S, Terai H, et al. Distinct epigenetic regulation of tumor suppressor genes in putative cancer stem cells of solid tumors. Int J Oncol. 2010;37:1537–1546. doi: 10.3892/ijo_00000807. [DOI] [PubMed] [Google Scholar]
- 330.Ikegaki N, Shimada H, Fox AM, Regan PL, Jacobs JR, Hicks SL, Rappaport EF, Tang XX. Transient treatment with epigenetic modifiers yields stable neuroblastoma stem cells resembling aggressive large-cell neuroblastomas. Proc Natl Acad Sci USA. 2013;110:6097–6102. doi: 10.1073/pnas.1118262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Patra SK, Patra A, Zhao H, Dahiya R. DNA methyltransferase and demethylase in human prostate cancer. Mol Carcinog. 2002;33:163–171. doi: 10.1002/mc.10033. [DOI] [PubMed] [Google Scholar]
- 332.Oh BK, Kim H, Park HJ, Shim YH, Choi J, Park C, Park YN. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int J Mol Med. 2007;20:65–73. [PubMed] [Google Scholar]
- 333.Melki JR, Warnecke P, Vincent PC, Clark SJ. Increased DNA methyltransferase expression in leukaemia. Leukemia. 1998;12:311–316. doi: 10.1038/sj.leu.2400932. [DOI] [PubMed] [Google Scholar]
- 334.Wu J, Issa JP, Herman J, Bassett DE, Nelkin BD, Baylin SB. Expression of an exogenous eukaryotic DNA methyltransferase gene induces transformation of NIH 3T3 cells. Proc Natl Acad Sci USA. 1993;90:8891–8895. doi: 10.1073/pnas.90.19.8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.MacLeod AR, Szyf M. Expression of antisense to DNA methyltransferase mRNA induces DNA demethylation and inhibits tumorigenesis. J Biol Chem. 1995;270:8037–8043. doi: 10.1074/jbc.270.14.8037. [DOI] [PubMed] [Google Scholar]
- 336.Morita R, Hirohashi Y, Suzuki H, Takahashi A, Tamura Y, Kanaseki T, Asanuma H, Inoda S, Kondo T, Hashino S, et al. DNA methyltransferase 1 is essential for initiation of the colon cancers. Exp Mol Pathol. 2013;94:322–329. doi: 10.1016/j.yexmp.2012.10.004. [DOI] [PubMed] [Google Scholar]
- 337.Trowbridge JJ, Sinha AU, Zhu N, Li M, Armstrong SA, Orkin SH. Haploinsufficiency of Dnmt1 impairs leukemia stem cell function through derepression of bivalent chromatin domains. Genes Dev. 2012;26:344–349. doi: 10.1101/gad.184341.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Choi DS, Blanco E, Kim YS, Rodriguez AA, Zhao H, Huang TH, Chen CL, Jin G, Landis MD, Burey LA, et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells. 2014;32:2309–2323. doi: 10.1002/stem.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Mariani CJ, Madzo J, Moen EL, Yesilkanal A, Godley LA. Alterations of 5-hydroxymethylcytosine in human cancers. Cancers (Basel) 2013;5:786–814. doi: 10.3390/cancers5030786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Lorsbach RB, Moore J, Mathew S, Raimondi SC, Mukatira ST, Downing JR. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10; 11)(q22; q23) Leukemia. 2003;17:637–641. doi: 10.1038/sj.leu.2402834. [DOI] [PubMed] [Google Scholar]
- 341.Ono R, Taki T, Taketani T, Taniwaki M, Kobayashi H, Hayashi Y. LCX, leukemia-associated protein with a CXXC domain, is fused to MLL in acute myeloid leukemia with trilineage dysplasia having t(10; 11)(q22; q23) Cancer Res. 2002;62:4075–4080. [PubMed] [Google Scholar]
- 342.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara O, Bhat R, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Tsai YP, Chen HF, Chen SY, Cheng WC, Wang HW, Shen ZJ, Song C, Teng SC, He C, Wu KJ. TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 2014;15:513. doi: 10.1186/s13059-014-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, Beringer G, Brikbak NJ, Yuan X, Cantley LC, et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. 2013;154:311–324. doi: 10.1016/j.cell.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Plimack ER, Stewart DJ, Issa JP. Combining epigenetic and cytotoxic therapy in the treatment of solid tumors. J Clin Oncol. 2007;25:4519–4521. doi: 10.1200/JCO.2007.12.6029. [DOI] [PubMed] [Google Scholar]
- 346.Cortez CC, Jones PA. Chromatin, cancer and drug therapies. Mutat Res. 2008;647:44–51. doi: 10.1016/j.mrfmmm.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 348.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25:1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 350.Azuara V, Perry P, Sauer S, Spivakov M, Jørgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 351.Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol. 2006;7:540–546. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- 352.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 353.Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev. 2013;27:1318–1338. doi: 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Denissov S, Hofemeister H, Marks H, Kranz A, Ciotta G, Singh S, Anastassiadis K, Stunnenberg HG, Stewart AF. Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development. 2014;141:526–537. doi: 10.1242/dev.102681. [DOI] [PubMed] [Google Scholar]
- 355.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Young MD, Willson TA, Wakefield MJ, Trounson E, Hilton DJ, Blewitt ME, Oshlack A, Majewski IJ. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011;39:7415–7427. doi: 10.1093/nar/gkr416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Laugesen A, Helin K. Chromatin repressive complexes in stem cells, development, and cancer. Cell Stem Cell. 2014;14:735–751. doi: 10.1016/j.stem.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 358.Gil J, O’Loghlen A. PRC1 complex diversity: where is it taking us? Trends Cell Biol. 2014;24:632–641. doi: 10.1016/j.tcb.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 359.Faust C, Schumacher A, Holdener B, Magnuson T. The eed mutation disrupts anterior mesoderm production in mice. Development. 1995;121:273–285. doi: 10.1242/dev.121.2.273. [DOI] [PubMed] [Google Scholar]
- 360.Chamberlain SJ, Yee D, Magnuson T. Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells. 2008;26:1496–1505. doi: 10.1634/stemcells.2008-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Montgomery ND, Yee D, Chen A, Kalantry S, Chamberlain SJ, Otte AP, Magnuson T. The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr Biol. 2005;15:942–947. doi: 10.1016/j.cub.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 362.Pasini D, Bracken AP, Hansen JB, Capillo M, Helin K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol Cell Biol. 2007;27:3769–3779. doi: 10.1128/MCB.01432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, Yuan GC, Orkin SH. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell. 2008;32:491–502. doi: 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Akasaka T, van Lohuizen M, van der Lugt N, Mizutani-Koseki Y, Kanno M, Taniguchi M, Vidal M, Alkema M, Berns A, Koseki H. Mice doubly deficient for the Polycomb Group genes Mel18 and Bmi1 reveal synergy and requirement for maintenance but not initiation of Hox gene expression. Development. 2001;128:1587–1597. doi: 10.1242/dev.128.9.1587. [DOI] [PubMed] [Google Scholar]
- 365.Leeb M, Wutz A. Ring1B is crucial for the regulation of developmental control genes and PRC1 proteins but not X inactivation in embryonic cells. J Cell Biol. 2007;178:219–229. doi: 10.1083/jcb.200612127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Endoh M, Endo TA, Endoh T, Fujimura Y, Ohara O, Toyoda T, Otte AP, Okano M, Brockdorff N, Vidal M, et al. Polycomb group proteins Ring1A/B are functionally linked to the core transcriptional regulatory circuitry to maintain ES cell identity. Development. 2008;135:1513–1524. doi: 10.1242/dev.014340. [DOI] [PubMed] [Google Scholar]
- 367.Aloia L, Di Stefano B, Di Croce L. Polycomb complexes in stem cells and embryonic development. Development. 2013;140:2525–2534. doi: 10.1242/dev.091553. [DOI] [PubMed] [Google Scholar]
- 368.Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4:e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 370.Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol. 2011;12:799–814. doi: 10.1038/nrm3230. [DOI] [PubMed] [Google Scholar]
- 371.Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 372.Loh YH, Zhang W, Chen X, George J, Ng HH. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007;21:2545–2557. doi: 10.1101/gad.1588207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem. 2001;276:25309–25317. doi: 10.1074/jbc.M101914200. [DOI] [PubMed] [Google Scholar]
- 375.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 376.Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci USA. 2010;107:8242–8247. doi: 10.1073/pnas.1000478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Kelly RD, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41:741–749. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- 379.Kretsovali A, Hadjimichael C, Charmpilas N. Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells Int. 2012;2012:184154. doi: 10.1155/2012/184154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Hayakawa T, Nakayama J. Physiological roles of class I HDAC complex and histone demethylase. J Biomed Biotechnol. 2011;2011:129383. doi: 10.1155/2011/129383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Reynolds N, Latos P, Hynes-Allen A, Loos R, Leaford D, O’Shaughnessy A, Mosaku O, Signolet J, Brennecke P, Kalkan T, et al. NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell Stem Cell. 2012;10:583–594. doi: 10.1016/j.stem.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Luo M, Ling T, Xie W, Sun H, Zhou Y, Zhu Q, Shen M, Zong L, Lyu G, Zhao Y, et al. NuRD blocks reprogramming of mouse somatic cells into pluripotent stem cells. Stem Cells. 2013;31:1278–1286. doi: 10.1002/stem.1374. [DOI] [PubMed] [Google Scholar]
- 385.Yu C, Liu K, Tang S, Ding S. Chemical approaches to cell reprogramming. Curr Opin Genet Dev. 2014;28:50–56. doi: 10.1016/j.gde.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 388.Li G, Warden C, Zou Z, Neman J, Krueger JS, Jain A, Jandial R, Chen M. Altered expression of polycomb group genes in glioblastoma multiforme. PLoS One. 2013;8:e80970. doi: 10.1371/journal.pone.0080970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 390.Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg R, Ora I, Pajtler K, Klein-Hitpass L, Kuhfittig-Kulle S, et al. Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69:2065–2071. doi: 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- 391.Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189. doi: 10.1002/pros.20022. [DOI] [PubMed] [Google Scholar]
- 392.Carew JS, Giles FJ, Nawrocki ST. Histone deacetylase inhibitors: mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008;269:7–17. doi: 10.1016/j.canlet.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 393.van Vlerken LE, Kiefer CM, Morehouse C, Li Y, Groves C, Wilson SD, Yao Y, Hollingsworth RE, Hurt EM. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl Med. 2013;2:43–52. doi: 10.5966/sctm.2012-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. 2011;108:1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Suvà ML, Riggi N, Janiszewska M, Radovanovic I, Provero P, Stehle JC, Baumer K, Le Bitoux MA, Marino D, Cironi L, et al. EZH2 is essential for glioblastoma cancer stem cell maintenance. Cancer Res. 2009;69:9211–9218. doi: 10.1158/0008-5472.CAN-09-1622. [DOI] [PubMed] [Google Scholar]
- 396.Crea F, Duhagon MA, Farrar WL, Danesi R. Pharmacogenomics and cancer stem cells: a changing landscape? Trends Pharmacol Sci. 2011;32:487–494. doi: 10.1016/j.tips.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Richter GH, Plehm S, Fasan A, Rössler S, Unland R, Bennani-Baiti IM, Hotfilder M, Löwel D, von Luettichau I, Mossbrugger I, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci USA. 2009;106:5324–5329. doi: 10.1073/pnas.0810759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 399.Chang B, Li S, He Q, Liu Z, Zhao L, Zhao T, Wang A. Deregulation of Bmi-1 is associated with enhanced migration, invasion and poor prognosis in salivary adenoid cystic carcinoma. Biochim Biophys Acta. 2014;1840:3285–3291. doi: 10.1016/j.bbagen.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 400.Jin J, Lv X, Chen L, Zhang W, Li J, Wang Q, Wang R, Lu X, Miao D. Bmi-1 plays a critical role in protection from renal tubulointerstitial injury by maintaining redox balance. Aging Cell. 2014;13:797–809. doi: 10.1111/acel.12236. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 401.Yu X, Jiang X, Li H, Guo L, Jiang W, Lu SH. miR-203 inhibits the proliferation and self-renewal of esophageal cancer stem-like cells by suppressing stem renewal factor Bmi-1. Stem Cells Dev. 2014;23:576–585. doi: 10.1089/scd.2013.0308. [DOI] [PubMed] [Google Scholar]
- 402.Nör C, Zhang Z, Warner KA, Bernardi L, Visioli F, Helman JI, Roesler R, Nör JE. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia. 2014;16:137–146. doi: 10.1593/neo.131744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403.Liu XF, Yang WT, Xu R, Liu JT, Zheng PS. Cervical cancer cells with positive Sox2 expression exhibit the properties of cancer stem cells. PLoS One. 2014;9:e87092. doi: 10.1371/journal.pone.0087092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, Cao L, Baiazitov R, Du W, Sydorenko N, et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014;20:29–36. doi: 10.1038/nm.3418. [DOI] [PubMed] [Google Scholar]
- 405.Yu D, Liu Y, Yang J, Jin C, Zhao X, Cheng J, Liu X, Qi X. Clinical implications of BMI-1 in cancer stem cells of laryngeal carcinoma. Cell Biochem Biophys. 2015;71:261–269. doi: 10.1007/s12013-014-0194-z. [DOI] [PubMed] [Google Scholar]
- 406.Zhang S, Cui B, Lai H, Liu G, Ghia EM, Widhopf GF, Zhang Z, Wu CC, Chen L, Wu R, et al. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer-stem-cell therapy. Proc Natl Acad Sci USA. 2014;111:17266–17271. doi: 10.1073/pnas.1419599111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39:761–772. doi: 10.1016/j.molcel.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Benoit YD, Witherspoon MS, Laursen KB, Guezguez A, Beauséjour M, Beaulieu JF, Lipkin SM, Gudas LJ. Pharmacological inhibition of polycomb repressive complex-2 activity induces apoptosis in human colon cancer stem cells. Exp Cell Res. 2013;319:1463–1470. doi: 10.1016/j.yexcr.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Heddleston JM, Wu Q, Rivera M, Minhas S, Lathia JD, Sloan AE, Iliopoulos O, Hjelmeland AB, Rich JN. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012;19:428–439. doi: 10.1038/cdd.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, Liu Y, Ward D, Quan J, Ye T, et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res. 2011;71:7238–7249. doi: 10.1158/0008-5472.CAN-11-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Giudice FS, Pinto DS, Nör JE, Squarize CH, Castilho RM. Inhibition of histone deacetylase impacts cancer stem cells and induces epithelial-mesenchyme transition of head and neck cancer. PLoS One. 2013;8:e58672. doi: 10.1371/journal.pone.0058672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential for mouse development. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 413.Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, Guenther MG, Johnston WK, Wernig M, Newman J, et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Sinkkonen L, Hugenschmidt T, Berninger P, Gaidatzis D, Mohn F, Artus-Revel CG, Zavolan M, Svoboda P, Filipowicz W. MicroRNAs control de novo DNA methylation through regulation of transcriptional repressors in mouse embryonic stem cells. Nat Struct Mol Biol. 2008;15:259–267. doi: 10.1038/nsmb.1391. [DOI] [PubMed] [Google Scholar]
- 417.Benetti R, Gonzalo S, Jaco I, Muñoz P, Gonzalez S, Schoeftner S, Murchison E, Andl T, Chen T, Klatt P, et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15:268–279. doi: 10.1038/nsmb.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Wang Y, Xu Z, Jiang J, Xu C, Kang J, Xiao L, Wu M, Xiong J, Guo X, Liu H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25:69–80. doi: 10.1016/j.devcel.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 419.Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat Genet. 2008;40:1478–1483. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Qi J, Yu JY, Shcherbata HR, Mathieu J, Wang AJ, Seal S, Zhou W, Stadler BM, Bourgin D, Wang L, et al. microRNAs regulate human embryonic stem cell division. Cell Cycle. 2009;8:3729–3741. doi: 10.4161/cc.8.22.10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Sengupta S, Nie J, Wagner RJ, Yang C, Stewart R, Thomson JA. MicroRNA 92b controls the G1/S checkpoint gene p57 in human embryonic stem cells. Stem Cells. 2009;27:1524–1528. doi: 10.1002/stem.84. [DOI] [PubMed] [Google Scholar]
- 422.Tay YM, Tam WL, Ang YS, Gaughwin PM, Yang H, Wang W, Liu R, George J, Ng HH, Perera RJ, et al. MicroRNA-134 modulates the differentiation of mouse embryonic stem cells, where it causes post-transcriptional attenuation of Nanog and LRH1. Stem Cells. 2008;26:17–29. doi: 10.1634/stemcells.2007-0295. [DOI] [PubMed] [Google Scholar]
- 423.Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008;455:1124–1128. doi: 10.1038/nature07299. [DOI] [PubMed] [Google Scholar]
- 424.Xu N, Papagiannakopoulos T, Pan G, Thomson JA, Kosik KS. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell. 2009;137:647–658. doi: 10.1016/j.cell.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 425.Singh SK, Kagalwala MN, Parker-Thornburg J, Adams H, Majumder S. REST maintains self-renewal and pluripotency of embryonic stem cells. Nature. 2008;453:223–227. doi: 10.1038/nature06863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Gangaraju VK, Lin H. MicroRNAs: key regulators of stem cells. Nat Rev Mol Cell Biol. 2009;10:116–125. doi: 10.1038/nrm2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.O’Loghlen A, Muñoz-Cabello AM, Gaspar-Maia A, Wu HA, Banito A, Kunowska N, Racek T, Pemberton HN, Beolchi P, Lavial F, et al. MicroRNA regulation of Cbx7 mediates a switch of Polycomb orthologs during ESC differentiation. Cell Stem Cell. 2012;10:33–46. doi: 10.1016/j.stem.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Tarantino C, Paolella G, Cozzuto L, Minopoli G, Pastore L, Parisi S, Russo T. miRNA 34a, 100, and 137 modulate differentiation of mouse embryonic stem cells. FASEB J. 2010;24:3255–3263. doi: 10.1096/fj.09-152207. [DOI] [PubMed] [Google Scholar]
- 430.Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621–626. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Newman MA, Thomson JM, Hammond SM. Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA. 2008;14:1539–1549. doi: 10.1261/rna.1155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Huang TH, Esteller M. Chromatin remodeling in mammary gland differentiation and breast tumorigenesis. Cold Spring Harb Perspect Biol. 2010;2:a004515. doi: 10.1101/cshperspect.a004515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–1123. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 435.Yu F, Deng H, Yao H, Liu Q, Su F, Song E. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene. 2010;29:4194–4204. doi: 10.1038/onc.2010.167. [DOI] [PubMed] [Google Scholar]
- 436.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Gregory PA, Bracken CP, Bert AG, Goodall GJ. MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle. 2008;7:3112–3118. doi: 10.4161/cc.7.20.6851. [DOI] [PubMed] [Google Scholar]
- 439.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Polytarchou C, Iliopoulos D, Struhl K. An integrated transcriptional regulatory circuit that reinforces the breast cancer stem cell state. Proc Natl Acad Sci USA. 2012;109:14470–14475. doi: 10.1073/pnas.1212811109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Schraivogel D, Weinmann L, Beier D, Tabatabai G, Eichner A, Zhu JY, Anton M, Sixt M, Weller M, Beier CP, et al. CAMTA1 is a novel tumour suppressor regulated by miR-9/9* in glioblastoma stem cells. EMBO J. 2011;30:4309–4322. doi: 10.1038/emboj.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G, Raychaudhury A, Newton HB, Chiocca EA, Lawler S. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68:9125–9130. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 443.Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Li Y, Guessous F, Zhang Y, Dipierro C, Kefas B, Johnson E, Marcinkiewicz L, Jiang J, Yang Y, Schmittgen TD, et al. MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res. 2009;69:7569–7576. doi: 10.1158/0008-5472.CAN-09-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Yang YP, Chien Y, Chiou GY, Cherng JY, Wang ML, Lo WL, Chang YL, Huang PI, Chen YW, Shih YH, et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials. 2012;33:1462–1476. doi: 10.1016/j.biomaterials.2011.10.071. [DOI] [PubMed] [Google Scholar]
- 446.Gal H, Pandi G, Kanner AA, Ram Z, Lithwick-Yanai G, Amariglio N, Rechavi G, Givol D. MIR-451 and Imatinib mesylate inhibit tumor growth of Glioblastoma stem cells. Biochem Biophys Res Commun. 2008;376:86–90. doi: 10.1016/j.bbrc.2008.08.107. [DOI] [PubMed] [Google Scholar]
- 447.Garzia L, Andolfo I, Cusanelli E, Marino N, Petrosino G, De Martino D, Esposito V, Galeone A, Navas L, Esposito S, et al. MicroRNA-199b-5p impairs cancer stem cells through negative regulation of HES1 in medulloblastoma. PLoS One. 2009;4:e4998. doi: 10.1371/journal.pone.0004998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–215. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Hsieh IS, Chang KC, Tsai YT, Ke JY, Lu PJ, Lee KH, Yeh SD, Hong TM, Chen YL. MicroRNA-320 suppresses the stem cell-like characteristics of prostate cancer cells by downregulating the Wnt/beta-catenin signaling pathway. Carcinogenesis. 2013;34:530–538. doi: 10.1093/carcin/bgs371. [DOI] [PubMed] [Google Scholar]
- 450.Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, Xiang D, Desano JT, Bommer GT, Fan D, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009;4:e6816. doi: 10.1371/journal.pone.0006816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Ji Q, Hao X, Meng Y, Zhang M, Desano J, Fan D, Xu L. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer. 2008;8:266. doi: 10.1186/1471-2407-8-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Bu P, Chen KY, Chen JH, Wang L, Walters J, Shin YJ, Goerger JP, Sun J, Witherspoon M, Rakhilin N, et al. A microRNA miR-34a-regulated bimodal switch targets Notch in colon cancer stem cells. Cell Stem Cell. 2013;12:602–615. doi: 10.1016/j.stem.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Sheik Mohamed J, Gaughwin PM, Lim B, Robson P, Lipovich L. Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA. 2010;16:324–337. doi: 10.1261/rna.1441510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Chakraborty D, Kappei D, Theis M, Nitzsche A, Ding L, Paszkowski-Rogacz M, Surendranath V, Berger N, Schulz H, Saar K, et al. Combined RNAi and localization for functionally dissecting long noncoding RNAs. Nat Methods. 2012;9:360–362. doi: 10.1038/nmeth.1894. [DOI] [PubMed] [Google Scholar]
- 455.Ng SY, Johnson R, Stanton LW. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012;31:522–533. doi: 10.1038/emboj.2011.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Gutschner T, Hämmerle M, Eissmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Gross M, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–1189. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Guo F, Li Y, Liu Y, Wang J, Li Y, Li G. Inhibition of metastasis-associated lung adenocarcinoma transcript 1 in CaSki human cervical cancer cells suppresses cell proliferation and invasion. Acta Biochim Biophys Sin (Shanghai) 2010;42:224–229. doi: 10.1093/abbs/gmq008. [DOI] [PubMed] [Google Scholar]
- 458.Jiang Y, Li Y, Fang S, Jiang B, Qin C, Xie P, Zhou G, Li G. The role of MALAT1 correlates with HPV in cervical cancer. Oncol Lett. 2014;7:2135–2141. doi: 10.3892/ol.2014.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Geng YJ, Xie SL, Li Q, Ma J, Wang GY. Large intervening non-coding RNA HOTAIR is associated with hepatocellular carcinoma progression. J Int Med Res. 2011;39:2119–2128. doi: 10.1177/147323001103900608. [DOI] [PubMed] [Google Scholar]
- 461.Zhang Y, Xia J, Li Q, Yao Y, Eades G, Gernapudi R, Duru N, Kensler TW, Zhou Q. NRF2/long noncoding RNA ROR signaling regulates mammary stem cell expansion and protects against estrogen genotoxicity. J Biol Chem. 2014;289:31310–31318. doi: 10.1074/jbc.M114.604868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Marcato P, Dean CA, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle. 2011;10:1378–1384. doi: 10.4161/cc.10.9.15486. [DOI] [PubMed] [Google Scholar]
- 463.Miranda-Lorenzo I, Dorado J, Lonardo E, Alcala S, Serrano AG, Clausell-Tormos J, Cioffi M, Megias D, Zagorac S, Balic A, et al. Intracellular autofluorescence: a biomarker for epithelial cancer stem cells. Nat Methods. 2014;11:1161–1169. doi: 10.1038/nmeth.3112. [DOI] [PubMed] [Google Scholar]
- 464.Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–3162. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 465.Justilien V, Fields AP. Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin Cancer Res. 2015;21:505–513. doi: 10.1158/1078-0432.CCR-14-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Kim J, Orkin SH. Embryonic stem cell-specific signatures in cancer: insights into genomic regulatory networks and implications for medicine. Genome Med. 2011;3:75. doi: 10.1186/gm291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006;6:11–23. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- 468.Yori JL, Seachrist DD, Johnson E, Lozada KL, Abdul-Karim FW, Chodosh LA, Schiemann WP, Keri RA. Krüppel-like factor 4 inhibits tumorigenic progression and metastasis in a mouse model of breast cancer. Neoplasia. 2011;13:601–610. doi: 10.1593/neo.11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014;135:2741–2748. doi: 10.1002/ijc.28690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Suvà ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Muñoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012;6:620–636. doi: 10.1016/j.molonc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Kim J, Zaret KS. Reprogramming of human cancer cells to pluripotency for models of cancer progression. EMBO J. 2015;34:739–747. doi: 10.15252/embj.201490736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Knoepfler PS. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells. 2009;27:1050–1056. doi: 10.1002/stem.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Bianco C, Rangel MC, Castro NP, Nagaoka T, Rollman K, Gonzales M, Salomon DS. Role of Cripto-1 in stem cell maintenance and malignant progression. Am J Pathol. 2010;177:532–540. doi: 10.2353/ajpath.2010.100102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Pacheco-Pinedo EC, Durham AC, Stewart KM, Goss AM, Lu MM, Demayo FJ, Morrisey EE. Wnt/β-catenin signaling accelerates mouse lung tumorigenesis by imposing an embryonic distal progenitor phenotype on lung epithelium. J Clin Invest. 2011;121:1935–1945. doi: 10.1172/JCI44871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Lukacs RU, Memarzadeh S, Wu H, Witte ON. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell. 2010;7:682–693. doi: 10.1016/j.stem.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.di Martino E, Tomlinson DC, Knowles MA. A Decade of FGF Receptor Research in Bladder Cancer: Past, Present, and Future Challenges. Adv Urol. 2012;2012:429213. doi: 10.1155/2012/429213. [DOI] [PMC free article] [PubMed] [Google Scholar]