

ABSTRACT
Different members of the Mycobacterium genus have evolved to cause tuberculosis in diverse human populations and in a variety of animal species. Our cumulative knowledge of mycobacterial genomes indicates that mutations in the PhoPR two-component virulence system were acquired not only during the natural evolution of mycobacterial species but also during in vitro subculture, which has given rise to the attenuated reference strain H37Ra or to different daughter strains of Mycobacterium bovis BCG. PhoPR is a well-known regulator of pathogenic phenotypes, including secretion of the virulence factor ESAT-6, biosynthesis of acyltrehalose-based lipids, and modulation of antigen export, in members of the Mycobacterium tuberculosis complex (MTBC). Evolutionarily conserved polymorphisms in PhoPR from Mycobacterium africanum, M. bovis, or M. tuberculosis H37Ra result in loss of functional phenotypes. Interestingly, some members of the MTBC have acquired compensatory mutations to counteract these polymorphisms and, probably, to maintain their pathogenic potential. Some of these compensatory mutations include the insertion of the IS6110 element upstream from phoPR in a particular M. bovis strain that is able to transmit between humans or polymorphisms in M. africanum and M. bovis that affect the regulatory region of the espACD operon, allowing PhoPR-independent ESAT-6 secretion. This review highlights the increasing knowledge of the significance of PhoPR in the evolution of the MTBC and its potential application in the construction of new attenuated vaccines based on phoPR inactivation. In this context, the live attenuated vaccine MTBVAC, based on a phoP fadD26 deletion mutant of M. tuberculosis, is the first vaccine of this kind to successfully enter into clinical development, representing a historic milestone in the field of human vaccinology.
MYCOBACTERIUM TUBERCULOSIS COMPLEX MEMBERS HAVE EVOLVED TO CAUSE TUBERCULOSIS IN HUMANS AND ANIMALS
Mycobacteria are widely distributed in the environment and are considered high-GC Gram-positive bacteria. In contrast to other Gram-positive bacteria, mycobacteria possess a multilayered cell envelope rich in uncommon lipids that are responsible for the acid-fast/Zielh-Neelsen staining of these organisms. The Mycobacterium genus is usually subdivided into fast- and slow-growing species, based on their ability to develop colonies in less or more than 7 days, respectively. Fast-growing species are in general opportunistic or nonpathogenic bacteria, whereas slow growers include human-pathogenic mycobacteria, such as M. tuberculosis, M. ulcerans, and M. leprae, causing tuberculosis, Buruli ulcer disease, and leprosy, respectively. In this review, we focus on the M. tuberculosis complex (MTBC), comprising a group of closely related tuberculosis-causing subspecies or ecotypes adapted to different animal hosts, including humans. According to phylogenetic distances, the MTBC species can be classified into eight major lineages (L1 to L8), which include the human-adapted ecotypes M. tuberculosis (L1 to L4 and L7), M. africanum (L5 and L6), and M. canettii and the animal-adapted ecotypes M. bovis, M. caprae, M. microti, M. pinnipedii, M. orygis, and M. mungi (1, 2), which are grouped into L8 (Fig. 1A). Similarly to L8 species, which have evolved to infect specific mammals, human-adapted strains have evolved to cause tuberculosis in human subpopulations. Accordingly, M. canettii was originally isolated in the Horn of Africa (3), M. africanum L5 and L6 strains are commonly found in West African countries (4), M. tuberculosis L7 is frequent in Ethiopia and L1 in the Indian Ocean rim, and M. tuberculosis L2, L3, and L4, which show wider distribution, predominantly infect people from East Asia, East India, and the Americas and Europe, respectively (2). It is important to consider that members of the MTBC are a highly clonal population, which is particularly well reflected by the evolutionary hyperconservation of human T-cell epitopes. Remarkably, 95% of 491 analyzed epitopes from across MTBC species showed no amino acid changes (5), a finding that might reflect a potential benefit for MTBC species of being recognized by the host immune system at some stage of the pathogenesis cycle.
FIG 1 .
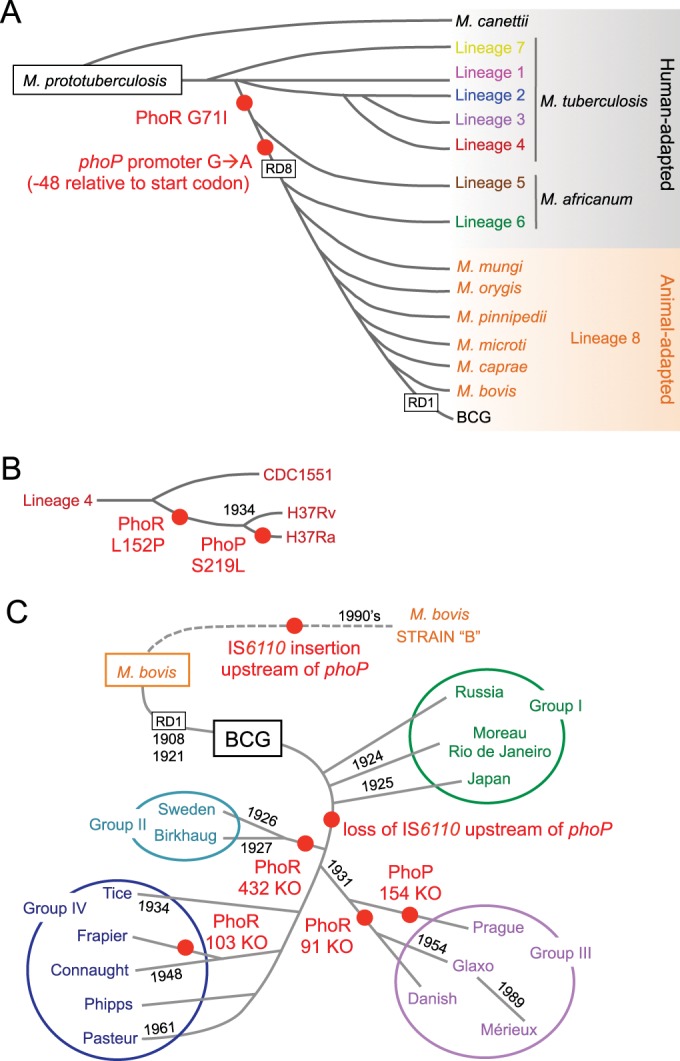
The phylogenies of the Mycobacterium tuberculosis complex and BCG include several polymorphisms in the PhoPR virulence system. (A) Evolutionary scenario of the MTBC showing the presence of the different members belonging to specific lineages. Adaptation of these species to humans and animals is also indicated. (B) Expanded view of lineage 4, showing representative strains. The Leu152Pro substitution in PhoR, exclusively in H37Rv and its attenuated form H37Ra, and a Ser219Leu substitution in PhoP from H37Ra are indicated. (C) Genealogy of M. bovis BCG daughter vaccine strains grouped by different phylogenetic markers. Deletion of RD1, responsible for BCG attenuation, and dates indicating genetic drifts are shown. Note the evolution of M. bovis strain B by acquisition of an IS6110 insertion upstream from the phoP gene. Other polymorphisms and RD deletions present in these phylogenies have been omitted for clarity; red-filled circles indicate phylogenetic locations of phoPR polymorphisms.
Aside from the natural evolution of the MTBC, the importance of the in vitro evolution during laboratory subcultivation is also worth noting. This process led to dissociation of the H37R strain into the H37Ra and H37Rv strains in 1934 (Fig. 1B) (6); both strains are widely used today as attenuated and virulent laboratory reference strains, respectively. Another example of in vitro attenuation comes from the generation of the parental Mycobacterium bovis BCG strain in 1921, which was originally distributed to different laboratories throughout the world and maintained by subculture passages under nonstandardized growth conditions, a process that led to several BCG daughter strains (Fig. 1C) (7).
This review will focus on the PhoPR virulence system and the polymorphisms acquired either during the natural evolution of the MTBC or during in vitro generation of H37Ra and BCG laboratory strains. As highlighted in this review, these polymorphisms could have an enormous impact on PhoPR-controlled virulence phenotypes. Taking into account the highly clonal population structure of the MTBC and the key role of PhoPR in MTBC virulence, we can hypothesize that mutations in PhoPR that apparently occurred randomly might rather have contributed to the evolution of the MTBC.
THE PhoPR TCS IN M. TUBERCULOSIS
Two-component systems (TCS) are highly conserved prokaryotic signal transduction pathways that consist of a histidine kinase (HK) as the sensor and a response regulator (RR) as the effector. The HK, often membrane associated, is responsible for detecting extracellular stimuli. In response to signal sensing, the HK autophosphorylates itself and then transfers its phosphate to the RR, which alters its conformation and modulates gene expression, usually through DNA binding. Overall, this mechanism enables bacterial adaptation to the initial stimulus (8). Intracellular pathogens usually use TCS to respond to host defenses and are often essential for virulence; the Salmonella PhoP is the most widely studied example (9).
Compared with environmental bacteria, the intracellular pathogen M. tuberculosis possesses few TCS; it has 11 operons (senX3-regX3, rv600c-rv601c-tcrA, phoPR, narL-rv0845, prrAB, mprAB, kdpDE, trcRS, dosRS-dosT, mtrAB, and tcrXY) and a paired HK and RR (pdtaR [rv1626]-pdtaS [rv3220c]), as well as 4 orphan RRs (rv0260, rv0818, rv2884, and rv3143) and an orphan HK (rv2027) (8). This relatively small number of TCS might very well reflect the adaptation of M. tuberculosis to the intracellular lifestyle, an assumption corroborated by the genetic decay of these transduction systems in the obligate intracellular pathogen M. leprae, with only 5 functional TCS. With the exception of rv600c-rv601c-tcrA, overexpression and mutant strains with modifications in these TCS have been constructed, revealing diverse roles in M. tuberculosis virulence and physiology (8). One of the most widely studied TCS in M. tuberculosis is the PhoPR (rv0757-rv0758) virulence system, originally annotated in accordance with its homology with phoPR from Bacillus subtilis, which responds to phosphate starvation (10). However, ever since the essential role of PhoPR in M. tuberculosis virulence was demonstrated (11–14), it has been assumed that M. tuberculosis phoPR acts as a virulence-controlling factor resembling phoPQ in the intracellular pathogen Salmonella spp.
PhoPR-DEPENDENT PHENOTYPES AND THEIR ROLE IN VIRULENCE
Pioneering studies demonstrated that a phoP mutant constructed in the M. tuberculosis strain MT103 clinical isolate was unable to replicate in bone marrow-derived macrophages and showed marked attenuation in intravenously inoculated BALB/c mice, as assessed by absence of replication in lungs, spleen, and liver (11). It was also observed that phoP mutants exhibited altered colony morphology, diminished cording formation, and smaller bacillary size than the wild type. These phenotypes, putatively related to the cell wall composition, led different laboratories to study the lipid content in phoP and phoPR mutants constructed in strains MT103 (L4), Beijing-GC1237 (L2), and H37Rv (L4), demonstrating that PhoP controls the biosynthesis of sulfolipid (SL) and di- and polyacyltrehaloses (DAT and PAT) (13, 15). These lipids are restricted to the pathogenic species of MTBC and are only absent in M. africanum L6 and M. bovis (14). Various roles have been proposed for SL, including inhibiting phagosome maturation (16), blocking monocyte priming (17), increasing superoxide production by neutrophils (18), increasing phagosome acidification (19), and restricting growth in human macrophages (20), among others. However, a pks2 mutant deficient in SL showed no effect on the persistence, replication, and pathogenicity of H37Rv in BALB/c mice and guinea pigs (21), whereas a mutant with a mutation in the SL transporter Mmpl8 constructed in the Erdman strain showed decreased virulence in C57BL/6 mice (22). Concerning DAT and PAT, the second group of PhoP-controlled lipids, their role in downmodulation of interleukin 2 (IL-2), IL-10, IL-12, and tumor necrosis factor alpha (TNF-α) responses in CD4+ and CD8+ human T cells has been demonstrated (23), and an H37Rv pks3/4 mutant deficient in DAT and PAT attached less efficiently to human phagocytic and nonphagocytic cells, although it did not display reduced virulence in mice (24). The coordinated regulation of SL and DAT/PAT by PhoP led us to think that the combined absence of these lipids might be required to attain attenuation in the mouse model. To test this hypothesis, a pks2-pks3/4 mutant deficient in SL and DAT/PAT was constructed in H37Rv and used for intravenous infection of BALB/c mice; however, unlike phoP mutants, the pks2-pks3/4 mutant exhibited very subtle changes in virulence as assessed by enumeration of viable bacteria per organ (25). This finding indicated that, in addition to the impaired synthesis of acyltrehalose-based lipids, other altered PhoPR-regulated phenotypes likely contribute to the attenuation of phoPR-deficient strains.
The explanation of this speculation arose from molecular characterization studies of the attenuated H37Ra strain, where it was demonstrated that PhoP controls the secretion of ESAT-6 by regulating the espACD gene cluster (26). This finding could explain the attenuation of phoP mutants, since ESAT-6 is considered one of the major virulence factors secreted by M. tuberculosis. In this context, it is important to mention that the deletion of region of difference 1 (RD1), which contains ESAT-6 and a major part of its secretion system (ESX-1), in the BCG vaccine strain (Fig. 1C) is considered the primary mechanism for the attenuation and safety of this strain (27, 28). Recent works have made major advances in elucidating the biological role of ESAT-6 in promoting phagosome escape (29, 30) and in mediating macrophage apoptosis (31, 32). The latter phenotype is involved in the cell-to-cell spread of pathogenic strains and, consequently, in the propagation of M. tuberculosis (33). These results are strengthened by those of in vivo infection studies using an RD1 mutant constructed in H37Rv, which displayed strong attenuation phenotypes both in vitro in human macrophages and in vivo in the C57BL/6 mouse model (34, 35). Aside from its role in virulence, RD1 was recently reported to be an important source of human T-cell epitopes, most of which are contained in ESAT-6 (36, 37). The key role of ESAT-6 secretion in virulence, together with the lack of its secretion observed in M. tuberculosis phoPR mutants, might represent a likely basis of attenuation for strains lacking a functional PhoPR system. Moreover, considering the immunogenic properties of RD1 and ESAT-6, attenuated M. tuberculosis phoPR mutants expressing genes contained in RD1 but not secreting the proteins they encode (including ESAT-6) could represent an attractive feature in the rational construction of new tuberculosis vaccines. Additionally, an M. tuberculosis phoP mutant displays increased immunogenicity and enhanced generation of memory T-cell subsets in mice compared to those of BCG (12, 38). In line with these data, a recent work has demonstrated an increased secretion of substrates (including immunodominant antigens) from the twin-arginine translocation (TAT) export pathway, a phenotype that depends on a PhoP-regulated noncoding RNA (39). Taken together, the adequate attenuation profile and higher immunogenicity exhibited by M. tuberculosis phoP mutants support the generation of potential phoP-based candidates as vaccines against human tuberculosis, as will be explained below.
THE PhoP VIRULENCE REGULATOR EXPLAINED AT THE MOLECULAR LEVEL
PhoP, encoded by the rv0757 gene of M. tuberculosis H37Rv, acts as the RR of the PhoPR TCS, and both genes are transcribed in a single operon and are autoregulated by PhoP, even though the phoR gene also has its own promoter (40). Similar to other RRs, PhoP binds its cognate DNA sequences as a dimer (41), as revealed by the structure of the protein forming a homodimer through its N-terminal receiver domain (42). As demonstrated by site-directed mutagenesis, the 27.5-kDa protein is phosphorylated at the Asp71 position in an Mg2+-dependent reaction (43). Phosphorylation enhances the binding affinity of PhoP for its target promoters, as demonstrated in in vitro experiments using the recombinant protein phosphorylated with the chemical phosphodonor acetyl phosphate (44, 45).
In an attempt to decipher the entire PhoP regulon, different laboratories compared the transcriptomes of wild-type and phoP mutant strains using microarrays, which led to the identification of PhoP-regulated genes in the M. tuberculosis H37Rv laboratory strain (13) and the MT103 clinical isolate (46). Both works generated comparable datasets and allowed the identification of the PhoP-controlled genes responsible for the absence of SL and DAT/PAT in phoP mutants. Among these genes, pks2, papA1, and mmpL8 are required for SL synthesis and export (22, 47, 48) and pks3, papA3, chp2 (rv1184c), and mmpl10 are involved in DAT/PAT biosynthesis (49). Furthermore, these microarray comparisons began the delineation of the molecular mechanism by which PhoP controls ESAT-6 secretion, as exemplified by differential expression of several ESX-1 genes coding for the ESAT-6 secretory apparatus (46). Although microarray analyses are useful to obtain a global landscape of gene expression, they frequently fail to answer some basic biological questions, such as whether a given gene is directly or indirectly regulated by transcription factors or whether there are transcripts in noncoding regions.
The systematic use of next-generation sequencing (NGS) techniques has solved these problems, and these techniques have provided an unprecedented level of detail in transcriptional regulation. A recent work applied two NGS techniques to study the PhoP regulatory network (39); chromatin immunoprecipitation sequencing (ChIP-seq) made it possible to detect all PhoP binding regions throughout the M. tuberculosis H37Rv chromosome and, consequently, to dissect those genes which are directly regulated by PhoP, and using high-resolution transcriptomic analysis (RNA-seq), it was possible to obtain a detailed transcriptome and to identify PhoP-dependent transcription across the entire genome, including intergenic or noncoding regions. Furthermore, by combining the PhoP ChIP-seq and RNA-seq data with ChIP-seq data for the RNA polymerase (50), it was possible to obtain a fine portrait of transcriptional regulation. Thus, PhoP is frequently positioned 100 bp upstream from its regulated promoters and 50 bp upstream from the RNA polymerase (Fig. 2A), which is consistent with the predominant role of PhoP as a transcriptional activator. However, it was observed that in those rare cases where PhoP is placed downstream from the RNA polymerase (e.g., PE8), it acts as a transcriptional repressor (39), possibly by interfering with RNA elongation. Since ChIP-seq captures PhoP-DNA complexes from whole bacteria, it covers virtually all PhoP binding sites, making it possible to obtain a reliable PhoP consensus sequence. Analysis of the PhoP bindome resulted in the direct-repeat consensus motif TCACAG(N5)TCACAG (Fig. 2A), which largely overlaps with the TCACAGC(N4)TCACAGC motif found in vitro by using iterative binding of recombinant PhoP to a random pool of nucleotides (51). Moreover, it was demonstrated that PhoP binds upstream from the pks2-papA1-mmpL8 and pks3-pks4-papA3-mmpL10 operons (Fig. 2B and C), which were previously reported as required for SL and DAT/PAT biosynthesis, respectively (22, 47–49). RNA-seq exploration confirmed previous findings that the aforementioned operons are downregulated in phoP mutants (39), thus explaining the absence of SL, DAT, and PAT in strains lacking a functional phoP.
FIG 2 .

Molecular characteristics of the PhoPR two-component system. (A) Positioning of the PhoP transcription factor relative to the RNA polymerase and its target genes measured as the most probable values from ChIP-seq data. The PhoP consensus motif [TCACAG(N5)TCACAG] is also indicated. (B) ChIP-seq reads from representative promoters of PhoP-regulated genes. Note the significant increase in ChIP-seq peaks in the wild-type strain relative to those in its phoP mutant, indicative of a specific interaction of PhoP with these regions. (C) PhoP binding motifs elucidated from ChIP-seq data from the analyses whose results are shown in panel B. Distances to the start codons of target genes are also indicated. ORF, open reading frame. (D) Genomic locations of relevant phoP polymorphisms discussed throughout the text. Insertions of IS6110 in the promoter region and their positions relative to the phoP start codon are shown. Amino acid positions of the Asp71 residue, which is involved in the phosphotransfer reaction, and the Ser219Leu substitution in H37Ra are also indicated. (E) Ribbon models of the DNA binding domain of PhoP superimposed over the structure of a PhoB-DNA complex. Solid spheres show the wild-type serine 219 in H37Rv (left) or the leucine residue that appears in H37Ra (right) (adapted from reference 40). The mutant leucine residue is expected to interfere with DNA binding and/or recognition. (F) Secondary structure of the PhoR sensor kinase indicating its membrane topology. Each domain has been colored individually, and α-helices (ovals) and β-strands (arrows) are indicated. Note the presence of PhoR polymorphisms in the sensor loop, which is located in the periplasmic space. The position of the histidine involved in the phosphotransfer reaction is also indicated.
Now that the molecular mechanism responsible for PhoP control of acyltrehalose-derived lipids has been characterized, the question of how PhoP regulates the secretion of TAT substrates and ESAT-6 remains to be answered. Among the PhoP ChIP-seq peaks, the most prominent interaction was detected within an intergenic region, and subsequent inspection of the adjacent RNA-seq profiles led to the proposal that a noncoding RNA named mcr7 is the most prominent PhoP-regulated region (Fig. 2B and C) (39). This regulation passed unnoticed in previous microarray experiments, probably because of the absence of probes within noncoding regions. An exhaustive characterization of mcr7 demonstrated that this noncoding RNA posttranscriptionally modulates the translation of the mRNA of tatC, an essential gene and a constituent of the TAT secretion system. As a consequence of the PhoP-mcr7 regulatory loop, the TAT secretion system is deregulated and M. tuberculosis phoP mutants secrete increased amounts of TAT substrates (including Ag85A and Ag85C) (39), which could contribute to the increased immunogenicity of these strains (38).
Regarding PhoP control of ESAT-6 secretion, in-depth exploration of NGS data has allowed the proposition that PhoP is a master regulator of different genetic networks controlling this phenotype, as shown in Fig. 3; PhoP binds to the promoter region of espR and regulates the expression of this gene (Fig. 2B and C) (39, 52), which has been proposed as a nucleoid-associated protein that otherwise transcriptionally regulates the espACD operon (53–55). Additionally, PhoP itself is also able to interact with the espA promoter, which possesses a consensus motif for PhoP [TCGCAG(N5)TTGCAG] (51). Taken together and considering the essential role of EspA in ESAT-6 secretion (26, 56, 57), PhoP is on the axis of the PhoP-EspR-EspA regulatory loop (Fig. 3). On the other hand, it was reported recently that PhoP regulates the expression of whiB6 by interacting with its promoter region (Fig. 2B and C) (58). WhiB6 is located adjacent to the ESX-1 region that codes for the ESAT-6 secretory apparatus, which points to WhiB6 as a putative regulator of this region. Indeed, the expression of whiB6 correlated with the expression of esxA (rv3875), the ESAT-6-coding gene, among other ESX-1 genes (58). ChIP-seq analysis using a WhiB6 overexpression system demonstrated that WhiB6 interacts with the promoter region of espA, as well as with the ESX-1 genes pe35 and espB (59), both required for ESAT-6 secretion (60, 61). Thus, a second regulatory network involving PhoP as the master regulator and WhiB6 as a secondary player is proposed to regulate ESAT-6 secretion, and consequently, whiB6 is included within the ESX-1 system (Fig. 3). The PhoP-WhiB6 regulatory network is of particular significance in M. tuberculosis research given that a recently reported single-nucleotide insertion in the whiB6 promoter of M. tuberculosis H37Rv has shifted the role of PhoP from transcriptional activator to transcriptional repressor. Consequently, ESAT-6 is expressed and secreted at lower levels in H37Rv than in other laboratory and clinical strains (58), a finding that highlights the importance of validating experimental data with different M. tuberculosis strains.
FIG 3 .

PhoP-dependent regulatory networks implicated in the control of ESAT-6 secretion. Different genes from the ESX-1 (whiB6 to mycP1) and the extended ESX-1 (espACD and espR) regions involved in ESAT-6 export are indicated. PhoP (blue ellipses) interacts with the espR, espA, and whiB6 promoters and controls the expression of these genes. EspR (pink ellipses) interacts with and activates the espACD locus. WhiB6 (green circles) also interacts with the promoter regions of espA, pe35, and espB genes. Overall, the PhoP-dependent EspR and WhiB6 regulatory circuits activate the espACD locus, which is required for ESAT-6 secretion. Locations of RD1, absent in BCG, and of RD8, absent in L6 and L8 lineages, as well as polymorphisms in espACD and whiB6 promoters (asterisks), are also indicated.
PhoR AS A SENSOR OF THE INTRACELLULAR NICHE IN M. TUBERCULOSIS
Similar to other HKs, the 52-kDa PhoR protein (encoded by rv0758 in H37Rv) is anchored to the inner phospholipid membrane by two transmembrane helices, which in turn are spanned by a 120-amino-acid segment (positions 36 to 155) that is responsible for signal sensing in the periplasmic space. The C terminus of the protein is located in the cytosol and contains domains required for signal transduction and phosphorylation. PhoR is autophosphorylated at the His259 position upon reception of the cognate signal (Fig. 2F). Several studies have attempted to identify the PhoPR-stimulating signal, and the first assumption was derived from comparison studies with PhoPQ of Salmonella spp., which is known to respond to Mg2+ (62). Interestingly, disruption of phoPR in M. tuberculosis prevents growth under Mg2+-limited conditions, although transcriptional profiling studies with the wild type and a phoPR mutant grown under high and low Mg2+ conditions showed no differences in genes involved in Mg2+ acquisition (13). Another assumption made using bioinformatics analyses is that the structure that is most similar to the putative PhoR external loop is the middle β-domain of the Escherichia coli YggB, a small membrane-spanning ion sensor that responds to mechanical stress; however, no experimental confirmation has been performed (63). A recent work has demonstrated that [Cl−] increases and pH decreases during phagosome maturation, linking PhoPR to sensing these intracellular cues via a pH- and chloride-dependent reporter system (64). In another study, the identification of the aprABC locus (for acid and phagosome regulated) as a novel PhoP-dependent gene led the authors to propose pH as an activating stimulus for the PhoPR TCS (65). In subsequent work, the authors identified two branches for pH adaptation in M. tuberculosis, a phoPR-dependent and a phoPR-independent branch, and found that the action of a certain branch depended on the carbon source present (66). Given the intracellular life cycle fitness of M. tuberculosis and the in vitro and in vivo attenuation showed by phoPR mutants (11, 12), it is plausible to link the PhoR sensor kinase with intracellular signals. Thus, one could speculate that, following phagocytosis of M. tuberculosis by resident macrophages, PhoR sensing of varying concentrations of Mg2+, Cl−, or H+ during phagosomal maturation would likely result in phosphorylation of PhoP. Upon phosphorylation, PhoP is expected to modulate the transcription of its regulon, which could result in positive regulation of SL and DAT/PAT synthesis and ESAT-6 secretion (Fig. 4) and in promoting immunomodulation of T cells and phagosome escape. However, further experiments are required to gain some insights into the molecular mechanism by which the sensor loop responds to these ions and to finely demonstrate PhoP phosphorylation in response to these stimuli. In addition, other as-yet-unexplored PhoR-stimulating signals that contribute to PhoPR activation, particularly considering possible nonconventional ligands, cannot be discarded.
FIG 4 .

Comparative illustration of PhoPR-regulated phenotypes in M. tuberculosis, M. bovis, M. africanum L6, and an M. tuberculosis phoP mutant. M. tuberculosis, carrying a functional PhoR, is able to sense its cognate stimulus and subsequently phosphorylate PhoP. Phosphorylated PhoP regulates three well-known phenotypes, including synthesis of SL and DAT/PAT (via pks2 and pks3 regulation), secretion of ESAT-6 (through espA regulation, as depicted in Fig. 3), and posttranscriptional regulation of tatC (mediated by the mcr7 noncoding RNA). M. bovis and M. africanum L6, carrying a defective PhoR G71I allele, are expected to have defects in PhoP phosphorylation, as a consequence of which these strains lack SL, DAT, and PAT. However, ESAT-6 secretion in these strains is restored by compensatory mutations in the espACD promoter region that include RD8 deletion and species-specific polymorphisms (asterisks). M. tuberculosis phoP mutants lack the aforementioned PhoP-regulated phenotypes and consequently do not synthesize SL, DAT, and PAT or secrete ESAT-6. These mutants also have a deregulated TAT system and, consequently, secrete larger amounts of TAT substrates, including the antigens Ag85A and Ag85C. Consequently, adequately attenuated M. tuberculosis phoP-based vaccine strains, such as MTBVAC, are expected to induce improved and longer-lasting immunogenicity compared to that of BCG in clinical trials.
PhoPR POLYMORPHISMS DURING MTBC EVOLUTION: SMALL CHANGES WITH HIGH IMPACT
Having reviewed the importance and molecular details of the PhoPR system, we will analyze diverse polymorphisms that have arisen either during the natural evolution process of MTBC or the in vitro development of M. bovis BCG or H37Rv and its attenuated form, H37Ra. As illustrated in Fig. 1, the MTBC and BCG phylogenies are spotted with several phoPR polymorphisms. Genomic sequence analysis of the attenuated H37Ra strain uncovered a Ser219Leu substitution (replacing Ser with Leu at position 219) in PhoP (Fig. 1B and 2E), which causes alterations in colony morphology and partially contributes to the avirulence of H37Ra (67). Parallel studies demonstrated that this mutation is responsible for the lack of SL, DAT, and PAT and the absence of ESAT-6 secretion in H37Ra (25, 26). Investigation of the position of this mutation within the PhoP structure revealed that it affects the C-terminal DNA binding domain (40), a finding that explains the absence of interaction of PhoP from H37Ra with its own promoter (25, 67). Aside from the PhoP mutation, other H37Ra polymorphisms, such as those responsible for the absence of phthiocerol dimycocerosates (PDIM), whose role in pathogenicity is well documented, could also contribute to the attenuated phenotype of H37Ra (25).
Comparisons of phoPR from representative MTBC strains revealed a Leu152Pro substitution in PhoR exclusively in H37Rv and H37Ra (Fig. 1B and 2F). Further analysis demonstrated that this mutation alters cell wall hydrophobicity in H37Rv, a finding that confirms the role of PhoPR in modeling the envelope composition in M. tuberculosis (68). It is important to recall that H37Rv and H37Ra are strains highly adapted to laboratory growth conditions, and thus, it is not surprising that some virulence-related phenotypes that are important for interactions with the host upon infection may be less expressed or even absent in these strains. An example is the recently identified single-nucleotide insertion in the PhoP-regulated gene whiB6 of H37Rv, which leads to decreased ESAT-6 expression and secretion in H37Rv compared to the levels in clinical strains of M. tuberculosis (58).
When the genetic comparison of phoPR was extended to the sequenced MTBC members, two additional polymorphisms were identified, one causing a Gly71Ile substitution in the sensor loop of PhoR (Fig. 1A and 2F) from M. africanum (L5 and L6) and from animal-adapted species (L8) and the second one located 48 bp upstream from the phoP start codon and affecting L8 species and M. africanum L6 (Fig. 1A and 2D). A series of genetic inactivation and complementation experiments demonstrated no effects for the phoP promoter mutation; however, a deleterious effect of the Gly71Ile mutation was observed in PhoPR from M. africanum L6 and from the animal-adapted species. These strains have a down-regulated PhoP regulon and, consequently, are unable to produce acyltrehalose-derived lipids; both phenotypes were efficiently restored upon complementation with the phoR allele from M. tuberculosis. Unexpectedly, these strains secreted ESAT-6 independently of the PhoR mutation (Fig. 4) (14), pointing to a compensatory mechanism, as detailed below.
PhoPR polymorphisms were also identified upon examination of the BCG phylogeny (Fig. 1C). Group I BCG strains differ from the others in having an IS6110 insertion upstream from phoPR that could affect PhoP autoregulation (Fig. 2D). Conversely, since the IS6110 transposon might function as a mobile promoter, this insertion might influence phoP expression; indeed, microarray studies showed increased expression of phoP in BCG Japan (group I) compared to its expression in BCG Pasteur (group IV). Other polymorphisms have been shown to result in deleterious proteins: BCG Sweden and BCG Birkhaug contain a deletion in phoR that truncates its C terminus; a frameshift in phoR from BCG Frappier abolishes its expression in this strain; and a frameshift in phoP from BCG Prague eliminates the C-terminal DNA binding domain, resulting in a natural phoP mutant. In addition, a 10-bp deletion in codon 91 of PhoR was found in some group III BCG strains (Glaxo, Merieux, and Danish). Notably, group III substrains are the most attenuated among all BCG daughter strains, but the precise role of the 91-bp knockout mutation in phoR remains to be elucidated (69, 70). Since BCG strains derive from an M. bovis isolate, all are affected by the PhoR Gly71Ile mutation described above, which abrogates SL, DAT, and PAT synthesis. Therefore, at present, it is unclear why additional mutations have been acquired in an already deleterious TCS, and this could reinforce the hypothesis that mutations in phoPR may not have occurred randomly and could instead represent evolutionary steps in the attenuation of MTBC species.
COMPENSATORY EVOLUTION: ARMS RACE AGAINST PhoPR POLYMORPHISMS
Previous observations regarding the ability of M. bovis and M. africanum strains to efficiently export ESAT-6 despite the deleterious Gly71Ile in their PhoR led to the hypothesis that compensatory mechanisms which can recover ESAT-6 secretion independently of PhoPR regulation might exist. A detailed inspection of the PhoP-EspR-EspA regulatory network revealed that several mutations upstream from the espACD locus have been acquired by M. bovis and M. africanum species, including the RD8 deletion, common to both species, and species-specific polymorphisms close to the PhoP and EspR binding sites (unpublished observations) (Fig. 3). Notably, introduction of the M. bovis espACD allele into an M. tuberculosis phoPR mutant restored ESAT-6 secretion, demonstrating that polymorphisms upstream from espA efficiently compensate the ESAT-6 secretion phenotype in M. bovis and M. africanum, which otherwise have a deleterious Gly71Ile substitution in their PhoR (Fig. 4) (14). The precise molecular reasons for this compensatory mechanism are unclear, although polymorphisms upstream from espA might increase the affinity of PhoP or EspR for this promoter region, resulting in espA expression in the absence of a fully functional PhoPR system. Due to the key role in the outcome of infection of ESAT-6 as a virulence factor (34, 35), species defective in ESAT-6 secretion or expression, such as BCG or M. tuberculosis phoPR mutants, could be condemned to an impaired person-to-person or animal-to-animal transmission capacity. Thus, compensatory evolution to restore ESAT-6 functionality in M. africanum and in animal-adapted species has presumably occurred to ensure the transmissibility of these species carrying a defective phoR allele.
An alternative example of compensatory evolution refers to a particular M. bovis isolate named strain B, which, contrary to other M. bovis strains, was able to transmit from human to human and caused high mortality among HIV-infected persons. Molecular characterization of strain B revealed the presence of an additional IS6110 insertion upstream from the phoP gene (Fig. 1C and 2D), and this insertion resulted in enhanced transcription of phoP (71). Subsequent studies with strain B demonstrated that this second IS6110 insertion restored the expression of the PhoP regulon and reestablished synthesis of PhoP-regulated lipids in M. bovis, resulting in increased virulence (14), which might explain the rare transmissibility of strain B between humans. Taken together, these findings exemplify the evolutionary arms race suffered by PhoP-dependent phenotypes in order to maintain the full pathogenic potential of virulent Mycobacterium species.
BIOLOGY LESSONS: INACTIVATE THE PhoP VIRULENCE REGULATOR TO CONSTRUCT NEW TUBERCULOSIS VACCINES
In conclusion, once the essential role of PhoPR in M. tuberculosis virulence and its implications in modeling the evolution of the MTBC have been reviewed, it is tempting to propose the potential use of M. tuberculosis phoPR mutants in constructing new safe and effective human tuberculosis vaccines. Unlike other infectious diseases, for which proper immunization relies on efficient humoral responses, protection against tuberculosis is thought to require an appropriate and long-lasting stimulation of the cellular arm of the immune system, although identification of biomarkers of protection against tuberculosis is urgently needed to confirm this hypothesis. Thus, our ideal conception for an effective tuberculosis vaccine would be a live attenuated M. tuberculosis strain that maintains the whole antigenic potential of the pathogen without causing disease, a classical approach to human vaccinology, as is the case for most currently licensed human whole-cell vaccines, which are safe and effective and are based on the human pathogen. In this context, live attenuated vaccine strains such as the current BCG are considered possible successful candidates. However, despite its long history of use and global coverage, BCG has failed to efficiently protect against tuberculosis, particularly in adult populations. This failure is probably due to the inability of BCG to mount an effective long-lasting immunity (72), considered to be due to a possible lack of an antigenic repertoire that is representative of the human pathogen. In this context, recent works have documented considerable epitope decay in BCG compared to the epitope repertoire of M. tuberculosis, and most of these epitopes have been shown to be located in RD1, RD2, and RD14, which are deleted in BCG strains (37). This handicap of BCG could be solved by using attenuated M. tuberculosis phoPR mutants to construct a new generation of live tuberculosis vaccines. In this context, an M. tuberculosis phoP mutant showed a safety profile comparable to that of BCG and improved protective efficacy in different animal models (mice, guinea pigs, and nonhuman primates) (12, 73). There are several potential advantages to employing live-attenuated M. tuberculosis phoPR-based vaccines: (i) unlike BCG, a derivative of cattle-evolved M. bovis, M. tuberculosis phoPR-based vaccines maintain the whole epitope repertoire of the human pathogen and are expected to stimulate a more natural immune response in the human host; (ii) M. tuberculosis phoPR mutants lack the cell wall lipids SL, DAT, and PAT, which are considered to interfere with the immune system upon host infection; (iii) impaired secretion and intact expression of ESAT-6 by these strains allows the loss of its virulence function in promoting phagosome escape and could possibly allow for greater immunogenicity due to the numerous specific human T-cell epitopes identified in ESAT-6; and (iv) as a consequence of the PhoP-mcr7-tatC regulatory circuit, phoP mutants have been shown to secrete larger amounts of TAT substrates, including antigens of the Ag85 complex, which are considered to result in increased immunogenicity (Fig. 4). In order to deal with the inherent safety risks associated with live attenuated vaccines based on M. tuberculosis, two stable deletion mutations were generated in the fadD26 gene (abrogating PDIM synthesis) and in phoP, removing antibiotic resistance markers. The final construct of the phoP fadD26 mutant was named MTBVAC and has been shown to have safety and biodistribution comparable to those of BCG and improved immunogenicity and efficacy against M. tuberculosis challenge in preclinical animal models (74). Thus, MTBVAC is the first live attenuated M. tuberculosis vaccine to fulfill the regulatory requirements to support successful entry into phase 1 clinical evaluation in healthy adults, and this first-in-human clinical trial began in 2013 (ClinicalTrials registration number NCT02013245) (74), markingg a milestone in the history of human vaccinology. The MTBVAC phase 1 trial ended successfully in November 2014, showing promising safety and immunogenicity that support advanced clinical development (F. Spertini et al., submitted for publication).
ACKNOWLEDGMENTS
We thank Dessislava Marinova for critical reading of the manuscript and her constructive comments.
This work was supported by projects TBVAC2020 (H2020-PHC-643381), PI12/01970, and BIO2014-52580P, funded by the European Commission Horizon 2020, Instituto de Salud Carlos III, and the Spanish Ministry of Science and Competitiveness, respectively. We also acknowledge the Gobierno de Aragón/Fondo Social Europeo. E.B. is the recipient of an FPI grant (reference BES-2012-052937).
Footnotes
Citation Broset E, Martín C, Gonzalo-Asensio J. 2015. Evolutionary landscape of the Mycobacterium tuberculosis complex from the viewpoint of PhoPR: implications for virulence regulation and application to vaccine development. mBio 6(5):e01289-15. doi:10.1128/mBio.01289-15.
REFERENCES
- 1.Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, Garnier T, Gutierrez C, Hewinson G, Kremer K, Parsons LM, Pym AS, Samper S, van Soolingen D, Cole ST. 2002. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A 99:3684–3689. doi: 10.1073/pnas.052548299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, Parkhill J, Malla B, Berg S, Thwaites G, Yeboah-Manu D, Bothamley G, Mei J, Wei L, Bentley S, Harris SR, Niemann S, Diel R, Aseffa A, Gao Q, Young D, Gagneux S. 2013. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet 45:1176–1182. doi: 10.1038/ng.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fabre M, Hauck Y, Soler C, Koeck J, van Ingen J, van Soolingen D, Vergnaud G, Pourcel C. 2010. Molecular characteristics of “Mycobacterium canettii” the smooth Mycobacterium tuberculosis bacilli. Infect Genet Evol 10:1165–1173. doi: 10.1016/j.meegid.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 4.de Jong BC, Antonio M, Gagneux S. 2010. Mycobacterium africanum—review of an important cause of human tuberculosis in West Africa. PLoS Negl Trop Dis 4:e744. doi: 10.1371/journal.pntd.0000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. 2010. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet 42:498–503. doi: 10.1038/ng.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steenken W, Oatway WH, Petroff SA. 1934. Biological studies of the tubercle bacillus: III. Dissociation and pathogenicity of the R and S variants of the human tubercle bacillus (H37). J Exp Med 60:515–540. doi: 10.1084/jem.60.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behr MA. 2002. BCG—different strains, different vaccines? Lancet Infect Dis 2:86–92. doi: 10.1016/S1473-3099(02)00182-2. [DOI] [PubMed] [Google Scholar]
- 8.Bretl DJ, Demetriadou C, Zahrt TC. 2011. Adaptation to environmental stimuli within the host: two-component signal transduction systems of Mycobacterium tuberculosis. Microbiol Mol Biol Rev 75:566–582. doi: 10.1128/MMBR.05004-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dalebroux ZD, Miller SI. 2014. Salmonellae PhoPQ regulation of the outer membrane to resist innate immunity. Curr Opin Microbiol 17:106–113. doi: 10.1016/j.mib.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salzberg LI, Botella E, Hokamp K, Antelmann H, Maass S, Becher D, Noone D, Devine KM. 2015. Genome-wide analysis of phosphorylated PhoP binding to chromosomal DNA reveals several novel features of the PhoPR-mediated phosphate limitation response in Bacillus subtilis. J Bacteriol 197:1492–1506. doi: 10.1128/JB.02570-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pérez E, Samper S, Bordas Y, Guilhot C, Gicquel B, Martín C. 2001. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol 41:179–187. doi: 10.1046/j.1365-2958.2001.02500.x. [DOI] [PubMed] [Google Scholar]
- 12.Martin C, Williams A, Hernandezpando R, Cardona P, Gormley E, Bordat Y, Soto C, Clark S, Hatch G, Aguilar D, Ausina V, Gicquel B. 2006. The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine 24:3408–3419. doi: 10.1016/j.vaccine.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 13.Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. 2006. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol 60:312–330. doi: 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalo-Asensio J, Malaga W, Pawlik A, Astarie-Dequeker C, Passemar C, Moreau F, Laval F, Daffe M, Martin C, Brosch R, Guilhot C. 2014. Evolutionary history of tuberculosis shaped by conserved mutations in the PhoPR virulence regulator. Proc Natl Acad Sci U S A 111:11491–11496. doi: 10.1073/pnas.1406693111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gonzalo Asensio J, Maia C, Ferrer NL, Barilone N, Laval F, Soto CY, Winter N, Daffe M, Gicquel B, Martin C, Jackson M. 2006. The virulence-associated two-component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J Biol Chem 281:1313–1316. doi: 10.1074/jbc.C500388200. [DOI] [PubMed] [Google Scholar]
- 16.Goren MB, D’Arcy Hart P, Young MR, Armstrong JA. 1976. Prevention of phagosome-lysosome fusion in cultured macrophages by sulfatides of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 73:2510–2514. doi: 10.1073/pnas.73.7.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pabst MJ, Gross JM, Brozna JP, Goren MB. 1988. Inhibition of macrophage priming by sulfatide from Mycobacterium tuberculosis. J Immunol 140:634–640. [PubMed] [Google Scholar]
- 18.Zhang L, Goren MB, Holzer TJ, Andersen BR. 1988. Effect of Mycobacterium tuberculosis-derived sulfolipid I on human phagocytic cells. Infect Immun 56:2876–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brodin P, Poquet Y, Levillain F, Peguillet I, Larrouy-Maumus G, Gilleron M, Ewann F, Christophe T, Fenistein D, Jang J, Jang M, Park S, Rauzier J, Carralot J, Shrimpton R, Genovesio A, Gonzalo-Asensio JA, Puzo G, Martin C, Brosch R, Stewart GR, Gicquel B, Neyrolles O. 2010. High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog 6:e1001100. doi: 10.1371/journal.ppat.1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilmore SA, Schelle MW, Holsclaw CM, Leigh CD, Jain M, Cox JS, Leary JA, Bertozzi CR. 2012. Sulfolipid-1 biosynthesis restricts Mycobacterium tuberculosis growth in human macrophages. ACS Chem Biol 7:863–870. doi: 10.1021/cb200311s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rousseau C, Turner OC, Rush E, Bordat Y, Sirakova TD, Kolattukudy PE, Ritter S, Orme IM, Gicquel B, Jackson M. 2003. Sulfolipid deficiency does not affect the virulence of Mycobacterium tuberculosis H37Rv in mice and guinea pigs. Infect Immun 71:4684–4690. doi: 10.1128/IAI.71.8.4684-4690.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Converse SE, Mougous JD, Leavell MD, Leary JA, Bertozzi CR, Cox JS. 2003. MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc Natl Acad Sci U S A 100:6121–6126. doi: 10.1073/pnas.1030024100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saavedra R, Segura E, Tenorio EP, López-Marín LM. 2006. Mycobacterial trehalose-containing glycolipid with immunomodulatory activity on human CD4+ and CD8+ T-cells. Microbes Infect 8:533–540. doi: 10.1016/j.micinf.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Rousseau C, Neyrolles O, Bordat Y, Giroux S, Sirakova TD, Prevost M, Kolattukudy PE, Gicquel B, Jackson M. 2003. Deficiency in mycolipenate- and mycosanoate-derived acyltrehaloses enhances early interactions of Mycobacterium tuberculosis with host cells. Cell Microbiol 5:405–415. doi: 10.1046/j.1462-5822.2003.00289.x. [DOI] [PubMed] [Google Scholar]
- 25.Chesne-Seck M-, Barilone N, Boudou F, Asensio JG, Kolattukudy PE, Martin C, Cole ST, Gicquel B, Gopaul DN, Jackson M. 2008. A point mutation in the two-component regulator PhoP-PhoR accounts for the absence of polyketide-derived acyltrehaloses but not that of phthiocerol dimycocerosates in Mycobacterium tuberculosis H37Ra. J Bacteriol 190:1329–1334. doi: 10.1128/JB.01465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frigui W, Bottai D, Majlessi L, Monot M, Josselin E, Brodin P, Garnier T, Gicquel B, Martin C, Leclerc C, Cole ST, Brosch R. 2008. Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS Pathog 4:e33. doi: 10.1371/journal.ppat.0040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. 2002. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 46:709–717. doi: 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- 28.Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR Jr. 2003. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A 100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Houben D, Demangel C, van Ingen J, Perez J, Baldeón L, Abdallah AM, Caleechurn L, Bottai D, van Zon M, de Punder K, van der Laan T, Kant A, Bossers-de Vries R, Willemsen P, Bitter W, van Soolingen D, Brosch R, van der Wel N, Peters PJ. 2012. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol 14:1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x. [DOI] [PubMed] [Google Scholar]
- 30.Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, Enninga J. 2012. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog 8:e1002507. doi: 10.1371/journal.ppat.1002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Derrick SC, Morris SL. 2007. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol 9:1547–1555. doi: 10.1111/j.1462-5822.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- 32.Aguiló N, Uranga S, Marinova D, Martín C, Pardo J. 2014. Bim is a crucial regulator of apoptosis induced by Mycobacterium tuberculosis. Cell Death Dis 5:e1343. doi: 10.1038/cddis.2014.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aguilo JI, Alonso H, Uranga S, Marinova D, Arbués A, de Martino A, Anel A, Monzon M, Badiola J, Pardo J, Brosch R, Martin C. 2013. ESX-1-induced apoptosis is involved in cell-to-cell spread of Mycobacterium tuberculosis. Cell Microbiol 15:1994–2005. doi: 10.1111/cmi.12169. [DOI] [PubMed] [Google Scholar]
- 34.Lewis K, Liao R, Guinn K, Hickey M, Smith S, Behr M, Sherman D. 2003. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J Infect Dis 187:117–123. doi: 10.1086/345862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherman D, Guinn K, Hickey M, Mathur S, Zakel K, Smith S. 2004. Mycobacterium tuberculosis H37Rv: delta RD1 is more virulent than M. bovis bacille Calmette-Guerin in long-term murine infection. J Infect Dis 190:123–126. doi: 10.1086/421472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang W, Zhang Y, Zheng H, Pan Y, Liu H, Du P, Wan L, Liu J, Zhu B, Zhao G, Chen C, Wan K. 2013. Genome sequencing and analysis of BCG vaccine strains. PLoS One 8:e71243. doi: 10.1371/journal.pone.0071243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Copin R, Coscollá M, Efstathiadis E, Gagneux S, Ernst JD. 2014. Impact of in vitro evolution on antigenic diversity of Mycobacterium bovis bacillus Calmette-Guerin (BCG). Vaccine 32:5998–6004. doi: 10.1016/j.vaccine.2014.07.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nambiar JK, Pinto R, Aguilo JI, Takatsu K, Martin C, Britton WJ, Triccas JA. 2012. Protective immunity afforded by attenuated, PhoP-deficient Mycobacterium tuberculosis is associated with sustained generation of CD4+ T-cell memory. Eur J Immunol 42:385–392. doi: 10.1002/eji.201141903. [DOI] [PubMed] [Google Scholar]
- 39.Solans L, Gonzalo-Asensio J, Sala C, Benjak A, Uplekar S, Rougemont J, Guilhot C, Malaga W, Martín C, Cole ST. 2014. The PhoP-dependent ncRNA Mcr7 modulates the TAT secretion system in Mycobacterium tuberculosis. PLoS Pathog 10:e1004183. doi: 10.1371/journal.ppat.1004183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalo-Asensio J, Soto CY, Arbues A, Sancho J, del Carmen Menendez M, Garcia MJ, Gicquel B, Martin C. 2008. The Mycobacterium tuberculosis phoPR operon is positively autoregulated in the virulent strain H37Rv. J Bacteriol 190:7068–7078. doi: 10.1128/JB.00712-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gupta S, Pathak A, Sinha A, Sarkar D. 2009. Mycobacterium tuberculosis PhoP recognizes two adjacent direct-repeat sequences to form head-to-head dimers. J Bacteriol 191:7466–7476. doi: 10.1128/JB.00669-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Menon S, Wang S. 2011. Structure of the response regulator PhoP from Mycobacterium tuberculosis reveals a dimer through the receiver domain. Biochemistry 50:5948–5957. doi: 10.1021/bi2005575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sinha A, Gupta S, Bhutani S, Pathak A, Sarkar D. 2008. PhoP-PhoP interaction at adjacent PhoP binding sites is influenced by protein phosphorylation. J Bacteriol 190:1317–1328. doi: 10.1128/JB.01074-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goyal R, Das AK, Singh R, Singh PK, Korpole S, Sarkar D. 2011. Phosphorylation of PhoP protein plays direct regulatory role in lipid biosynthesis of Mycobacterium tuberculosis. J Biol Chem 286:45197–45208. doi: 10.1074/jbc.M111.307447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cimino M, Thomas C, Namouchi A, Dubrac S, Gicquel B, Gopaul DN. 2012. Identification of DNA binding motifs of the Mycobacterium tuberculosis PhoP/PhoR two-component signal transduction system. PLoS One 7:e42876. doi: 10.1371/journal.pone.0042876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzalo-Asensio J, Mostowy S, Harders-Westerveen J, Huygen K, Hernández-Pando R, Thole J, Behr M, Gicquel B, Martín C. 2008. PhoP: a missing piece in the intricate puzzle of Mycobacterium tuberculosis virulence. PLoS One 3:e3496. doi: 10.1371/journal.pone.0003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sirakova TD, Thirumala AK, Dubey VS, Sprecher H, Kolattukudy PE. 2001. The Mycobacterium tuberculosis pks2 gene encodes the synthase for the hepta- and octamethyl-branched fatty acids required for sulfolipid synthesis. J Biol Chem 276:16833–16839. doi: 10.1074/jbc.M011468200. [DOI] [PubMed] [Google Scholar]
- 48.Kumar P, Schelle MW, Jain M, Lin FL, Petzold CJ, Leavell MD, Leary JA, Cox JS, Bertozzi CR. 2007. PapA1 and PapA2 are acyltransferases essential for the biosynthesis of the Mycobacterium tuberculosis virulence factor sulfolipid-1. Proc Natl Acad Sci U S A 104:11221–11226. doi: 10.1073/pnas.0611649104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Belardinelli JM, Larrouy-Maumus G, Jones V, Sorio de Carvalho LP, McNeil MR, Jackson M. 2014. Biosynthesis and translocation of unsulfated acyltrehaloses in Mycobacterium tuberculosis. J Biol Chem 289:27952–27965. doi: 10.1074/jbc.M114.581199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uplekar S, Rougemont J, Cole ST, Sala C. 2013. High-resolution transcriptome and genome-wide dynamics of RNA polymerase and NusA in Mycobacterium tuberculosis. Nucleic Acids Res 41:961–977. doi: 10.1093/nar/gks1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He X, Wang S. 2014. DNA consensus sequence motif for binding response regulator PhoP, a virulence regulator of Mycobacterium tuberculosis. Biochemistry 53:8008–8020. doi: 10.1021/bi501019u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao G, Howard ST, Zhang P, Wang X, Chen X-, Samten B, Pang X. 2015. EspR, a regulator of the ESX-1 secretion system in Mycobacterium tuberculosis, is directly regulated by the two-component systems MprAB and PhoPR. Microbiology 161:477–489. doi: 10.1099/mic.0.000023. [DOI] [PubMed] [Google Scholar]
- 53.Blasco B, Chen JM, Hartkoorn R, Sala C, Uplekar S, Rougemont J, Pojer F, Cole ST. 2012. Virulence regulator EspR of Mycobacterium tuberculosis is a nucleoid-associated protein. PLoS Pathog 8:e1002621. doi: 10.1371/journal.ppat.1002621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blasco B, Stenta M, Alonso-Sarduy L, Dietler G, Peraro MD, Cole ST, Pojer F. 2011. Atypical DNA recognition mechanism used by the EspR virulence regulator of Mycobacterium tuberculosis. Mol Microbiol 82:251–264. doi: 10.1111/j.1365-2958.2011.07813.x. [DOI] [PubMed] [Google Scholar]
- 55.Rosenberg OS, Dovey C, Tempesta M, Robbins RA, Finer-Moore JS, Stroud RM, Cox JS. 2011. EspR, a key regulator of Mycobacterium tuberculosis virulence, adopts a unique dimeric structure among helix-turn-helix proteins. Proc Natl Acad Sci U S A 108:13450–13455. doi: 10.1073/pnas.1110242108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, Bloom BR, Rubin EJ. 2005. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci U S A 102:10676–10681. doi: 10.1073/pnas.0504922102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garces A, Atmakuri K, Chase MR, Woodworth JS, Krastins B, Rothchild AC, Ramsdell TL, Lopez MF, Behar SM, Sarracino DA, Fortune SM. 2010. EspA acts as a critical mediator of ESX1-dependent virulence in Mycobacterium tuberculosis by affecting bacterial cell wall integrity. PLoS Pathog 6:e1000957. doi: 10.1371/journal.ppat.1000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Solans L, Aguilo N, Samper S, Pawlik A, Frigui W, Martin C, Brosch R, Gonzalo-Asensio J. 2014. A specific polymorphism in Mycobacterium tuberculosis H37Rv causes differential ESAT-6 expression and identifies WhiB6 as a novel ESX-1 component. Infect Immun 82:3446–3456. doi: 10.1128/IAI.01824-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Minch KJ, Rustad TR, Peterson EJ, Winkler J, Reiss DJ, Ma S, Hickey M, Brabant W, Morrison B, Turkarslan S, Mawhinney C, Galagan JE, Price ND, Baliga NS, Sherman DR. 2015. The DNA-binding network of Mycobacterium tuberculosis. Nat Commun 6:5829. doi: 10.1038/ncomms6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, Hinds J, Neyrolles O, Butcher PD, Leclerc C, Cole ST, Brosch R. 2006. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect Immun 74:88–98. doi: 10.1128/IAI.74.1.88-98.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu J, Laine O, Masciocchi M, Manoranjan J, Smith J, Du SJ, Edwards N, Zhu X, Fenselau C, Gao L. 2007. A unique mycobacterium ESX-1 protein co-secretes with CFP-10/ESAT-6 and is necessary for inhibiting phagosome maturation. Mol Microbiol 66:787–800. doi: 10.1111/j.1365-2958.2007.05959.x. [DOI] [PubMed] [Google Scholar]
- 62.Park S, Groisman EA. 2014. Signal-specific temporal response by the salmonella PhoP/PhoQ regulatory system. Mol Microbiol 91:135–144. doi: 10.1111/mmi.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ryndak M, Wang S, Smith I. 2008. PhoP, a key player in Mycobacterium tuberculosis virulence. Trends Microbiol 16:528–534. doi: 10.1016/j.tim.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 64.Tan S, Sukumar N, Abramovitch RB, Parish T, Russell DG. 2013. Mycobacterium tuberculosis responds to chloride and pH as synergistic cues to the immune status of its host cell. PLoS Pathog 9:e1003282. doi: 10.1371/journal.ppat.1003282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abramovitch RB, Rohde KH, Hsu F, Russell DG. 2011. aprABC: a Mycobacterium tuberculosis complex-specific locus that modulates pH-driven adaptation to the macrophage phagosome. Mol Microbiol 80:678–694. doi: 10.1111/j.1365-2958.2011.07601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baker JJ, Johnson BK, Abramovitch RB. 2014. Slow growth of Mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol Microbiol 94:56–69. doi: 10.1111/mmi.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JS, Krause R, Schreiber J, Mollenkopf H, Kowall J, Stein R, Jeon B, Kwak J, Song M, Patron JP, Jorg S, Roh K, Cho S, Kaufmann SHE. 2008. Mutation in the transcriptional regulator PhoP contributes to avirulence of Mycobacterium tuberculosis H37Ra strain. Cell Host Microbe 3:97–103. doi: 10.1016/j.chom.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 68.Schreuder LJ, Carroll P, Muwanguzi-Karugaba J, Kokoczka R, Brown AC, Parish T. 2015. Mycobacterium tuberculosis H37Rv has a single nucleotide polymorphism in PhoR which affects cell wall hydrophobicity and gene expression. Microbiology 161:765–773. doi: 10.1099/mic.0.000036. [DOI] [PubMed] [Google Scholar]
- 69.Brosch R, Gordon SV, Garnier T, Eiglmeier K, Frigui W, Valenti P, Dos Santos S, Duthoy S, Lacroix C, Garcia-Pelayo C, Inwald JK, Golby P, Garcia JN, Hewinson RG, Behr MA, Quail MA, Churcher C, Barrell BG, Parkhill J, Cole ST. 2007. Genome plasticity of BCG and impact on vaccine efficacy. Proc Natl Acad Sci U S A 104:5596–5601. doi: 10.1073/pnas.0700869104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu J, Tran V, Leung AS, Alexander DC, Zhu B. 2009. BCG vaccines: their mechanisms of attenuation and impact on safety and protective efficacy. Hum Vaccin 5:70–78. doi: 10.4161/hv.5.2.7210. [DOI] [PubMed] [Google Scholar]
- 71.Soto CY, Menendez MC, Perez E, Samper S, Gomez AB, Garcia MJ, Martin C. 2004. IS6110 mediates increased transcription of the phoP virulence gene in a multidrug-resistant clinical isolate responsible for tuberculosis outbreaks. J Clin Microbiol 42:212–219. doi: 10.1128/JCM.42.1.212-219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Orme IM. 2010. The Achilles heel of BCG. Tuberculosis (Edinb) 90:329–332. doi: 10.1016/j.tube.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 73.Verreck FAW, Vervenne RAW, Kondova I, van Kralingen KW, Remarque EJ, Braskamp G, van der Werff NM, Kersbergen A, Ottenhoff THM, Heidt PJ, Gilbert SC, Gicquel B, Hill AVS, Martin C, McShane H, Thomas AW. 2009. MVA.85A boosting of BCG and an attenuated, phoP deficient M. tuberculosis vaccine both show protective efficacy against tuberculosis in rhesus macaques. PLoS One 4:e5264. doi: 10.1371/journal.pone.0005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arbues A, Aguilo JI, Gonzalo-Asensio J, Marinova D, Uranga S, Puentes E, Fernandez C, Parra A, Cardona PJ, Vilaplana C, Ausina V, Williams A, Clark S, Malaga W, Guilhot C, Gicquel B, Martin C. 2013. Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine 31:4867–4873. doi: 10.1016/j.vaccine.2013.07.051. [DOI] [PubMed] [Google Scholar]