

ABSTRACT
BPLF1 of Epstein-Barr virus (EBV) is classified as a late lytic cycle protein but is also found in the viral tegument, suggesting its potential involvement at both initial and late stages of viral infection. BPLF1 possesses both deubiquitinating and deneddylating activity located in its N-terminal domain and is involved in processes that affect viral infectivity, viral DNA replication, DNA repair, and immune evasion. A recently constructed EBV BPLF1-knockout (KO) virus was used in conjunction with a humanized mouse model that can be infected with EBV, enabling the first characterization of BPLF1 function in vivo. Results demonstrate that the BPLF1-knockout virus is approximately 90% less infectious than wild-type (WT) virus. Transformation of human B cells, a hallmark of EBV infection, was delayed and reduced with BPLF1-knockout virus. Humanized mice infected with EBV BPLF1-knockout virus showed less weight loss and survived longer than mice infected with equivalent infectious units of WT virus. Additionally, splenic tumors formed in 100% of mice infected with WT EBV but in only 25% of mice infected with BPLF1-KO virus. Morphological features of spleens containing tumors were similar to those in EBV-induced posttransplant lymphoproliferative disease (PTLD) and were almost identical to cases seen in human diffuse large B-cell lymphoma. The presence of EBV genomes was detected in all mice that developed tumors. The results implicate BPLF1 in human B-cell transformation and tumor formation in humanized mice.
IMPORTANCE
Epstein-Barr virus infects approximately 90% of the world’s population and is the causative agent of infectious mononucleosis. EBV also causes aggressive lymphomas in individuals with acquired and innate immune disorders and is strongly associated with diffuse large B-cell lymphomas, classical Hodgkin lymphoma, Burkitt lymphoma, and nasopharyngeal carcinoma (NPC). Typically, EBV initially infects epithelial cells in the oropharynx, followed by a lifelong persistent latent infection in B-cells, which may develop into lymphomas in immunocompromised individuals. This work is the first of its kind in evaluating the effects of EBV’s BPLF1 in terms of pathogenesis and lymphomagenesis in humanized mice and implicates BPLF1 in B-cell transformation and tumor development. Currently, there is no efficacious treatment for EBV, and therapeutic targeting of BPLF1 may lead to a new path to treatment for immunocompromised individuals or transplant recipients infected with EBV.
INTRODUCTION
Epstein-Barr virus (EBV), a human tumor herpesvirus, can establish a lytic infection, in which the full complement of viral genes is expressed, or latent infection, in which only a small subset of viral genes is expressed. Most humans are infected with EBV by the second decade of life, typically asymptomatically in most healthy individuals, but EBV can cause infectious mononucleosis in adolescents and young adults. Additionally, EBV can cause lethal aggressive lymphomas in persons with acquired and innate immune disorders (1, 2) and is strongly linked to major malignancies, including Hodgkin lymphoma (3), high-grade non-Hodgkin lymphomas, and nasopharyngeal carcinoma (NPC) (4, 5). Patients who are immunosuppressed and have undergone bone marrow or solid organ transplant are at significant risk of B-cell lymphoproliferative disorders caused by EBV. These posttransplant lymphoproliferative diseases (PTLDs) may rapidly progress from polyclonal EBV infection to high-grade clonal neoplasms. Here, we make use of a humanized mouse model that can be infected with EBV, has fidelity to human infection in immunocompromised patients, and results in B-cell lymphomas that recapitulate posttransplant lymphoproliferative disorders seen in human patients (6, 7).
Typically, EBV-driven lymphomagenesis is thought to primarily involve latent gene products, but there is increasing evidence that lytic gene products and tegument proteins contribute to establishment of latency, lymphoproliferative disease, and tumor formation (8–11). Hong et al. demonstrated that knockout (KO) viruses of the early lytic gene products BZLF1 and BRLF1, which are required for activation of lytic replication and full lytic gene expression, result in drastic reduction of lymphoproliferative disease in SCID mice (8). Additionally, BZLF1 induces expression of interleukin-10 (IL-10) (12, 13), which promotes B-cell survival and proliferation (14), suggesting that BZLF1 expression may contribute to lymphoproliferative disease by modulating cellular signaling cascades (8). Two other EBV lytic genes, the BALF1 and BHRF1 genes, are expressed during infection of B cells and are necessary to establish latency (15, 16). BALF1 and BHRF1 have antiapoptotic properties; their deletion resulted in inhibition of B-cell transformation and establishment of latency (15, 17, 18).
The EBV tegument protein BNRF1 also contributes to the establishment of latency. Feederle et al. found that deletion of BNRF1 resulted in greatly reduced transformation of primary B cells compared with wild-type (WT) EBV (19). Tsai et al. found that BNRF1 binds to Daxx, modulates viral chromatin structure early during primary infection, and promotes latent viral gene expression and establishment of latency (20, 21). It is possible that expression of EBV immediate-early lytic proteins necessary for latency is regulated by tegument proteins, as observed for herpes simplex virus 1 (HSV-1) and human cytomegalovirus (HCMV). Transfected HSV-1 and HCMV DNA, along with constructs for tegument proteins pp71 and VP16, initiates lytic infection 10-fold more efficiently than transfected viral DNA alone (22, 23). Interestingly, EBV BPLF1 is reported to have homology to HSV-1 VP16 (24). Here, we report that infection with an EBV BPLF1-knockout (KO) virus results in decreased B-cell transformation and tumor formation in humanized mice.
BPLF1 is the large tegument protein of EBV (3,149 amino acids). It is classified as a late lytic cycle gene, but transcripts are detected as early as 6 to 8 h after infection (25, 26), and herpesvirus homologs to BPLF1 have both early and late functions during infection involving virus entry, transport, and assembly (27–30). Expression of short hairpin RNA (shRNA) against BPLF1 results in decreased EBV genome copy numbers (31).
BPLF1 contains deubiquitinating (DUB) activity within the first 205 amino acids of the N-terminal region. The DUB activity of BPLF1 and its catalytic triad (Cys-His-Asp), despite sequence diversity in this region, are strictly conserved across the Herpesviridae, and mutation of the active-site cysteine results in complete loss of enzymatic activity (32). BPLF1 DUB activity cleaves both K63 and K48 polyubiquitin chains (31). A functionally active BPLF1 fragment has recently been shown to block degradation of cytosolic and endoplasmic reticulum (ER) proteins by removal of ubiquitin from substrates targeted to the proteasome (33). BPLF1 interacts with EBV ribonucleotide reductase (RR), deubiquitinates its large subunit (RR1), and downregulates RR activity (31). BPLF1 also has deneddylating activity that utilizes the same active site as the deubiquitinating activity and interacts with and deneddylates cullin ring ligases, which potentially affects viral replication (25). In addition, the Kaposi’s sarcoma-associated herpesvirus (KSHV) DUB homolog was recently shown to decrease RIG-I ubiquitination and reduce RIG-I-mediated interferon (IFN) signaling, suggesting that the viral DUB activity antagonizes RIG-I signaling during KSHV infection (34). Several herpesviruses whose deubiquitinating activity has been deleted have been constructed, and all result in approximately a 10-fold decrease in viral infectivity (35–38). A knockout of the deubiquitinating activity of Marek’s disease virus, an alphaherpesvirus, reduced viral replication and severely impaired formation of T-cell lymphomas (39).
Recently, humanized mouse models have been developed that enabled creation of an in vivo system in which to study progression of infection and lymphomagenesis due to EBV (10, 40–45). Humanized mice were constructed by irradiating and injecting human hematopoietic stem cells into the liver of newborn Rag2−/− γC−/− double-knockout (DKO) mice (46, 47). Construction of a BPLF1-knockout virus by Saito et al. (48) allowed for characterization of BPLF1’s role in lymphomagenesis in humanized mice in which authentic B-cell tumors are generated. In this study, we have found that BPLF1-knockout virus results in decreased production of infectious virus, delayed ability to transform human B-cells, and retarded lymphoma formation in humanized mice. Mice infected with WT EBV develop tumors more quickly and frequently than mice infected with equivalent infectious units of BPLF1-knockout virus (here also called deltaBPLF1 or DUB KO). WT-infected mice lost weight and succumbed to infection more rapidly than did those infected with deltaBPLF1. Tumor incidence in DUB KO-infected mice was drastically reduced, and all mice with tumors were EBV positive. Histologically, tumors identified in WT-infected mice recapitulate large B-cell lymphomas seen in the posttransplant setting in human patients.
RESULTS
Loss of BPLF1 decreases viral infectivity.
Saito et al. (48) constructed a recombinant EBV BPLF1-knockout virus with the use of a previously described EBV bacmid as the template (49), in which the first 975 nucleotides of the BPLF1 open reading frame were replaced with neomycin resistance and streptomycin sensitivity genes, removing the start codon for BPLF1. They found that EBV deltaBPLF1 resulted in approximately a 3-fold decrease in intracellular viral DNA content, which could be partially restored by overexpression of the N-terminal region of WT BPLF1 but not with a C61A mutation that abolishes its deubiquitinating and deneddylating activity (31, 50). This result suggests that enzymatic activity of BPLF1 is at least partially responsible for the decrease in viral DNA replication. To investigate if EBV deltaBPLF1 affected viral infectivity, reactivation of the lytic cycle was induced by transfection of the EBV transactivator BZLF1, which resulted in production of infectious virus. The titers of infectious particles released into the medium were determined on Raji cells, and infectivity was monitored by detection of green fluorescent protein (GFP) encoded by the EBV bacmid construct (49, 51, 52) and measured by flow cytometry at 48 h and 72 h postinfection. Results in Fig. 1 indicate that BPLF1-knockout virus results in approximately a 70 to 90% decrease in infectious virus production (48-h titers for WT and BPLF1-KO virus were 4.6 × 104 and 9.5 × 103 infectious units/ml, respectively), which is in agreement with published findings for other herpesviral BPLF1 homologs (35–38). Thus, BPLF1 is an important determinant of viral infectivity.
FIG 1 .
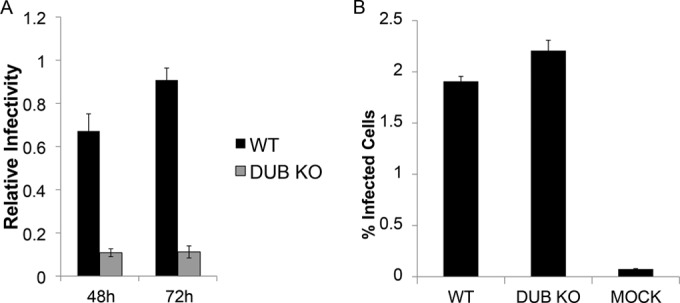
BPLF1-knockout virus is less infectious than WT EBV. 293 cells containing WT EBV or deltaBPLF1 (DUB KO) virus were transfected with the viral transactivator BZLF1 to induce lytic protein expression. (A) At 72 h postinduction, supernatant fluids containing viral particles with genomes encoding GFP were harvested and used to infect Raji cells. Infectivity in Raji cells was measured by detection of GFP 48 and 72 h postinfection. (B) Supernatants were concentrated to contain equivalent titers of infectious virus. Titers of concentrated WT and deltaBPLF1 virus were determined on freshly isolated B cells from human blood as detected by the presence of GFP.
For use in subsequent experiments, both WT and deltaBPLF1 viruses were concentrated to equivalent titers. Titers of WT and deltaBPLF1 virus were determined on primary human B cells isolated from blood. Figure 1B demonstrates that infection with equivalent titers of WT and deltaBPLF1, as determined by infection of Raji cells, results in equivalent titers on primary B-cells. Purified primary B-cells (3 × 105) were incubated with 3 × 104 infectious units (multiplicity of infection [MOI], 0.1) of WT and BPLF1-knockout virus. Titers were determined by detection of GFP by flow cytometry at 48 h postinfection. Approximately 2% of B-cells were infected with both WT and knockout virus. Titers detected in primary human B cells were approximately 4 × 103 /ml, a marked decrease from the 3 × 104 infectious units detected in Raji cells.
Absence of BPLF1 inhibits cellular transformation of human B-cells.
A long-established hallmark of EBV is its ability to transform human B-cells (53). Since BPLF1 is involved in viral DNA replication and interacts with several viral and cellular replication components (31, 48, 52, 54, 55), we examined if loss of BPLF1 could inhibit cellular transformation of human B-cells. Although BPLF1 is a late lytic gene, it is also present in the viral tegument (55, 56) and could affect early processes during infection that may influence B-cell transformation. Peripheral blood mononuclear cells (PBMCs) isolated from blood of three human donors were infected with either WT or deltaBPLF1 virus. Ten million cells were infected with 1.0 × 103 or 1.0 × 104 infectious particles of either WT or BPLF1-knockout virus. The percentage of B-cells in the total cell population was determined by flow cytometry after labeling with B-cell antibody (CD19) (Fig. 2). Initially, B-cells made up approximately 5 to 15% of total cells, but by 3 weeks, B-cell outgrowth was observed with WT virus for both donor A and donor B. A higher concentration of WT virus resulted in slightly faster B-cell outgrowth. B-cell outgrowth in cells infected with deltaBPLF1 virus either was always delayed or did not occur, suggesting that BPLF1 is necessary for efficient immortalization of B-cells. B-cell outgrowth of deltaBPLF1 was delayed approximately 2 weeks for donor A and 1 week for donor B compared to WT. Interestingly, for donor A, cells infected with 1.0 × 103 infectious particles of BPLF1-knockout virus resulted in B-cell outgrowth, whereas cells infected with 1.0 × 104 infectious particles of BPLF1-knockout virus did not. This result was unexpected and is likely a random event due to sample variation, as cell transformation by EBV is not always successful in cultured B-cells. A third donor (data not shown) resulted in B-cell outgrowth only for the WT 104-infectious particle sample. Overall, these results provide evidence that infection with BPLF1-knockout virus results in delayed and reduced B-cell outgrowth as detected by flow cytometry (Fig. 1 and 2) and suggest that lytic proteins, specifically BPLF1, may play roles in B-cell transformation and thus contribute to the tumorigenic properties of EBV.
FIG 2 .
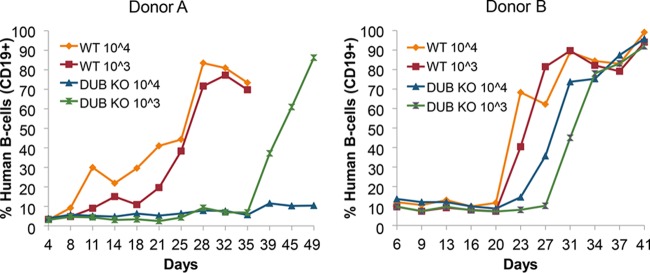
Human B-cell outgrowth is reduced and delayed in cells infected with BPLF1-knockout virus. Lymphocytes were isolated from blood of two different donors (A and B) by gradient centrifugation with Ficoll, and 3 × 106 lymphocytes were infected with 3 × 104 or 3 × 103 infectious units (MOIs of 0.01 and 0.001, respectively) of EBV WT or DUB KO virus. B-cells were counted with the use of PE-labeled antibody to the human B-cells marker CD19. Labeled B-cells were reported as percentages of total cells.
Detection of human leukocytes in humanized mice.
To create humanized mice, newborns are irradiated and injected with 2 × 105 human hematopoietic stem cells into the liver. Twelve weeks posttransplant, mice with stable human T/B/myeloid/natural killer cell reconstitution were identified by fluorescence-activated cell sorting (FACS) analysis of peripheral blood leukocytes with antibodies recognizing each cell type (46, 47). Figure 3 shows the percentage of human leukocytes (CD45+) and T cells (CD3+) for the 12 mice used in the study.
FIG 3 .
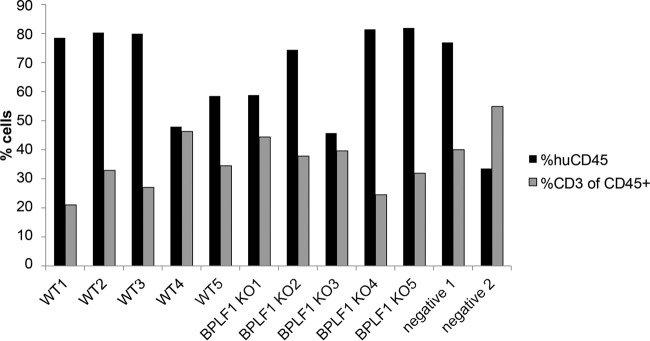
Detection of human leukocytes in mice. Twenty-four hours prior to infection, mice were bled and levels of human leukocytes were determined, indicating that mice contained sufficient levels for EBV infection. Flow cytometry was performed with cells isolated from blood for CD45 (human leukocytes) and the human T-cell marker CD3, and positive cells are graphed as percentages of total cells. %CD3 represents populations of CD45 cells that were CD3 positive.
Mice infected with BPLF1-KO virus lose weight more slowly and develop fewer tumors than do mice infected with equivalent titers of WT virus.
Mice were infected with 3 × 104 infectious particles of EBV WT or deltaBPLF1 (DUB KO) intraperitoneally (i.p.) and were weighed several times weekly until death or sacrifice due to 20% weight loss (Fig. 4A). Mice inoculated with WT virus (orange lines in Fig. 4A) were all terminated due to weight loss by day 30. Mice infected with deltaBPLF1 virus (red lines in Fig. 4A) survived significantly longer and in two cases until the end of the study (day 95) (Fig. 4B). One negative-control mouse survived until the end of the study, and the other died on day 64 due to seemingly unrelated causes. Three of the mice in the study died very early after infection (all by day 12); none contained detectable titers of EBV. It is likely that these animals did not die due to EBV infection (WT1, WT3, and DUB KO4).
FIG 4 .

Mice infected with BPLF1-KO (DUB KO) virus live longer and develop fewer tumors. Rag2−/− γC−/− double-knockout (DKO) mice were injected i.p. with 3 × 104 infectious particles of EBV WT or DUB KO virus in a final volume of 300 µl PBS. (A) Mice were weighed until death or sacrificed due to 20% weight loss. (B) All remaining mice were terminated on day 95. Mice receiving WT virus were all terminated due to weight loss by day 30. Mice receiving BPLF1-KO virus survived significantly longer, in two cases until the end of the study. (C) At time of death, spleens were removed and photographed. Representative photos are shown. (D) WT2, WT5, and KO3 all contained tumors and had enlarged spleens. Three/three mice infected with WT virus produced tumors, and 1/4 mice infected with DUB KO virus produced tumors. Neither of the 2 uninfected mice produced tumors (mice that died before day 12 due to unrelated causes were excluded).
At the time of death, spleens were removed and photographed (Fig. 4C). WT2, WT5, and DUB KO3 all contained visible tumors in the spleen and had enlarged spleens. Excluding the three mice that died early, 3/3 mice infected with WT produced tumors versus 1/4 mice infected with deltaBPLF1 (Fig. 4D). The 2 uninfected mice did not develop tumors. These results suggest that infection with BPLF1-knockout virus results in fewer tumors and delayed tumor formation in the spleen and longer survival times, supporting the conclusion that BPLF1 is involved in critical processes in EBV pathogenesis and oncogenesis. Tumor incidence for WT infections and survival times are in agreement with similar studies in humanized mice conducted by the Kenney laboratory (40).
EBV detected in splenic tumors of humanized mice.
At the time of harvest, spleen cells from infected mice were cultured to see if EBV could be detected by GFP. Approximately 1 week after plating the spleen cells, samples were examined by immunofluorescence microscopy to detect GFP-positive EBV genomes (Fig. 5A) (bright-field micrographs in upper panels; GFP fluorescence in lower panels). Samples that contained gross tumors were GFP positive, indicating the presence of EBV in the spleen. EBV was only detected in the spleens of animals with spleen tumors (WT2, WT4, WT5, and DUB KO3). Additionally, B-cell lines could only be established from spleens of infected mice that exhibited obvious tumors and presented with GFP-positive genomes. All other cell cultures did not survive.
FIG 5 .

EBV is detected in mice with tumors. (A) Portions of spleen tissue harvested from mice were disrupted and placed in culture medium. One week later, flasks were examined for GFP (EBV genomes) by immunofluorescence. Top panels are bright-field micrographs, and bottom panels are GFP fluorescence. (B) Detection of EBV genomes in blood of infected mice. Mice were bled weekly throughout the study. The last bleed prior to sacrifice was used to detect EBV genomes by quantitative PCR. Genomes were detected only in mice infected with WT EBV that developed tumors in the spleen. (C) EBV genomes detected in spleens of infected mice. DNA was extracted from splenic tissue at time of harvest, and EBV genome copies were detected with quantitative PCR. Viral genomes were present in all mice presenting with tumors but not in any other mice. EBV genomes were detected in WT2, WT4, WT5, and DUB KO3 (copy numbers 12.7, 99,712, 11.2, and 70.3, respectively) and varied greatly in number likely due to the region utilized for DNA extraction. Some regions of the spleen samples used appeared largely normal and likely resulted in lower genome copy numbers.
Real-time PCR was also performed on blood and spleen cells from infected mice toward the EBV BamHI repeat region to detect viral genome copies. One hundred microliters of blood was collected weekly, and DNA was harvested and screened for EBV genomes. EBV genomes were not detected in blood at early time points. In fact, EBV genomes were detected only just prior to sacrifice (within 1 week). Figure 5B shows detection of genome copies in the blood in the week prior to sacrifice for each animal. EBV genomes were detected in blood of WT-infected mice but not in any of the BPLF1-knockout virus-infected mice. Since EBV genomes were not detected early and were only detected in blood from animals with tumors, we speculate that genomes detected in the peripheral blood at late time points were likely circulating tumor cells. While DUB KO3 did have a splenic tumor and EBV genomes were detected (GFP) in the spleen cells, genome copies were not detected in the blood.
Additionally, DNA was extracted from spleen cells and EBV genomes were detected as done for blood samples (Fig. 5C). EBV genomes were only detected in spleens from WT2, WT4, WT5, and DUB KO3, findings which are in agreement with and support results with GFP detection in Fig. 5C. Interestingly, WT mice with tumors contained EBV genomes in both the spleen and blood, whereas the KO mouse had detectable levels of EBV genomes only in the spleen. These results show that EBV is detected only in mice in which splenic tumors developed, which suggests that it is the causative agent of the tumors, and infection with BPLF1-KO virus resulted in decreased detection of EBV and suppressed tumorigenesis. For high-grade B-cell lymphomas, leukemic involvement (i.e., peripheral blood involvement) is generally a late-stage finding, suggesting higher-stage disease. Although the sample size is small, failure to detect EBV in peripheral blood of mice infected with BPLF1-KO virus may further support the idea of less-aggressive disease in the absence of BPLF1. It is possible that EBV was cleared in mice that did not develop tumors, since it was not detected at the time of sacrifice; however, EBV genomes were not detected in blood initially after infection, leaving open the possibility that EBV may be present at undetectable levels.
Tumors from infected humanized mice closely resemble large B-cell lymphomas in humans.
Histopathology was performed on spleen tissue of infected mice. Spleens were sectioned and stained with hematoxylin and eosin (H&E) or subjected to immunohistochemistry (IHC) for human B- and T-cell markers (CD20 and CD3, respectively) or in situ hybridization for EBERs (EBV-encoded RNAs) (Fig. 6). Consistent with lack of infection or expansion of human lymphoid cells, mock-infected mice showed only scattered human T cells (CD3) and rare human B cells (CD20). EBER staining was negative, as expected. Although occasional granulomas were present within the spleen of these immunosuppressed animals, no abnormal lymphoid proliferation was identified.
FIG 6 .

B-cell lymphoma formation is decreased with BPLF1-knockout virus. Splenic sections from humanized mice were stained for B cells (CD20) and T cells (CD3). In situ hybridization was used for detection of EBV RNA (EBER), and hematoxylin and eosin staining was also performed. Negative control 2, B and T cells are seen in relatively normal abundance, and EBER staining is negative. WT4, detection of B-cell lymphoma (monomorphic posttransplant lymphoproliferative disease), strong EBER staining, and presence of dividing cells (H&E inset). WT5, detection of B-cell lymphoma, strong EBER staining, and presence of dividing cells (H&E inset). BPLF1 KO3, detection of B-cell lymphoma, strong EBER staining.
In contrast, tumors in WT-infected (WT2, WT4, and WT5) and BPLF1-mutant-infected (DUB KO3) mice consisted of collections of large, atypical lymphoid cells which variably exhibited nodular (DUB KO3, WT2, and WT5) to diffuse (WT4) pattern growth and were positive for EBER staining (Fig. 6; data not shown for WT2). Dividing cells, normally a rare occurrence, can be seen in the inset of H&E staining for WT4 and WT5 (Fig. 6). The morphological features are similar to those in EBV-induced PTLD, which is a continuum that progresses from polyclonal lymphoproliferative disease to lymphoma, most typically large B-cell lymphomas, similar to those seen in this study. These cases are on the monomorphic PTLD/lymphoma end of the spectrum, with case WT4 having morphological features that are nearly identical to what is seen in human diffuse large B-cell lymphoma. In case WT5, a contributing factor to the architectural appearance of multiple coalescing nodular masses of B-cells is the background of a substantial number of normal murine leukocytes, which may obscure/interrupt more-diffuse pattern growth.
All 3 WT-infected mice that survived past the early stages of the experiment presented with an EBV-positive large B-cell lymphoma. DUB KO-infected mice that did not develop tumors were EBV negative as determined by EBER staining, and relevant to the model system, no EBV-driven PTLD/lymphoma was identified. All mice with tumors were EBV positive and were representative of PTLD/large B-cell lymphoma in humans.
Tumors of DUB KO-infected mice proliferate at a rate similar to that of tumors of WT-infected mice.
To determine if there were differences in proliferation rates of lymphomas of mice infected with WT or deltaBPLF1 virus, sections of spleen were stained for the proliferation marker Ki67 (Fig. 7A). In contrast to the negative control, cellular proliferation in infected mice is greatly increased in WT4, WT5, and DUB KO3, as expected. Visually, it appeared that DUB KO3 had slightly less Ki67 staining, suggesting that the lymphoma produced by infection with DUB KO virus proliferates more slowly. To see if this were the case, Ki67-positive cells were counted over the entire tumor region. Examination determined that Ki67 staining was relatively constant over the entire tumor area, indicating that tumors formed with DUB KO virus (in this one instance) did not proliferate more slowly than those formed with WT EBV (Fig. 7B). A summary of data from infected mice is provided in Table 1.
FIG 7 .
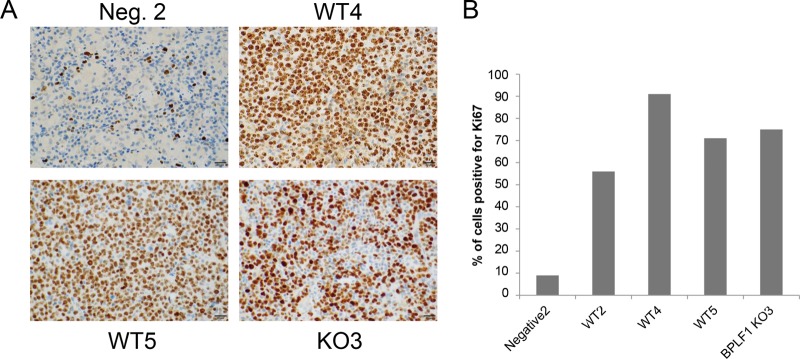
Increased proliferation in lymphoma region of spleens of infected mice with tumors. (A) Mice containing tumors showed increased rates of proliferation as determined by staining spleen sections with the proliferation marker Ki67. There were not significant differences between WT and DUB KO virus-infected tissue samples. (B) Ki67-positive cells from the tumor region were counted and graphed as percentages of total cells. The entire sample of negative control 2 was used for Ki67 staining.
TABLE 1 .
Summary of WT and BPLF1-KO virus infection results in humanized mice
| Mouse | Day of death | EBV detection in: |
Cell linea | Spleen tumor | Histopathology | |
|---|---|---|---|---|---|---|
| Blood | Spleen | |||||
| WT1 | 12 | No data | No data | No data | No data | Died early—no data |
| WT2 | 29 | Yes | Yes | Yes | Yes | Focal monomorphic PTLD/large B-cell lymphoma; EBER positive |
| WT3 | 4 | No data | No data | No data | No data | Died early—no data |
| WT4 | 26 | Yes | Yes | Yes | Yes | Monomorphic PTLD/diffuse large B-cell lymphoma; EBER positive |
| WT5 | 26 | Yes | Yes | Yes | Yes | Multifocal monomorphic PTLD/large B-cell lymphoma; EBER positive |
| DUB KO1 | 95 (end of study) | No | No | No | No | No lymphoma/PTLD identified; EBER negative |
| DUB KO2 | 95 (end of study) | No | No | No | No | No lymphoma/PTLD identified; EBER negative |
| DUB KO3 | 51 | No | Yes | Yes | Yes | Focal monomorphic PTLD/large B-cell lymphoma; EBER positive |
| DUB KO4 | 4 | No data | No data | No data | No data | Died early—no data |
| DUB KO5 | 26 | No | No | No | No | No lymphoma/PTLD identified; EBER negative |
| Negative 1 | 64 | No | No | No | No | No lymphoma/PTLD identified; EBER negative |
| Negative 2 | 95 (end of study) | No | No | No | No | No lymphoma/PTLD identified; EBER negative |
Ability to establish transformed cell line from spleen cells of infected mice.
DISCUSSION
The BPLF1 gene was found to encode deubiquitinating activity in 2005 (32) and deneddylating activity in 2010 (50). BPLF1 has become increasingly significant as more targets and functional effects are discovered. BPLF1 has been shown to be a key player in both cellular and viral processes, including immune evasion, DNA repair, viral replication and infectivity, and now cellular transformation and tumor formation (31, 48, 52, 54, 55, 57). Creation of the BPLF1-knockout virus in combination with a humanized mouse model has now made possible this first study of pathogenesis and in vivo effects of BPLF1. BPLF1 is positioned to affect both early and late stages of viral and host processes due to its presence in the viral tegument, which leads to its delivery into newly infected cells, and its expression as a late lytic cycle gene. These factors allow for BPLF1 to have effects on B-cell transformation, establishment of latency, and lymphomagenesis. This study indicates that BPLF1 helps drive B-cell immortalization and contributes to its lethal effects in humanized mice by increasing the incidence of lymphomagenesis.
Following construction of the deltaBPLF1 virus, Saito et al. reported that the recombinant virus resulted in an approximately 3-fold decrease in intracellular genome copy numbers (48). Infectivity studies disclosed almost a 90% decrease in viral titers (Fig. 1). Combined, these results suggest that not only does BPLF1-knockout virus produce fewer genomes but that the genomes produced by BPLF1-knockout virus result in less infectious virus than those produced by the WT (genomes produced by BPLF1-KO virus were approximately 60 to 70% less infectious than an equivalent number of genomes produced by WT EBV). Since BPLF1’s effects on viral genome replication alone cannot account for the loss of infectivity, our observations suggest that BPLF1 is likely involved in infectious virion assembly and on early stages of infection.
Human B-cells can routinely be transformed in cell culture by infection with EBV. A surprising finding was that infection of human B-cells with equivalent numbers of infectious units of BPLF1-knockout virus resulted in decreased B-cell outgrowth as a measure of cellular transformation. Establishment of latency in human B-cells depends on several factors, including circularization. The EBV genome is maintained in its linear form within the virion but as a circularized episome in latently infected B-cells (58). Additionally, transformation of B-cells has been shown to be dependent on expression of the latency genes for LMP1, EBNA2, and EBNA3A and EBNA3C (59, 60). EBNA1 is expressed in all types of latency and is important in genome replication and persistence (61, 62). EBNA3B and LMP2A were found to be dispensable for B-cell transformation (63–65). Perhaps, BPLF1 alters expression of latency proteins necessary for efficient B-cell transformation in a manner similar to that of its suggested homolog, HSV-1 VP16, which is capable of increasing HSV gene expression (23, 24).
When humanized mice were infected with 3 × 104 infectious units of WT or BPLF1-knockout virus, mice receiving BPLF1-knockout virus lost less weight due to infection (Fig. 4). Mice were terminated once body weight decreased by 20%, and all mice receiving WT EBV had to be terminated within the first month, whereas most of the deltaBPLF1 virus-infected mice maintained their weight through day 50 or more. These results suggest that pathogenesis produced by BPLF1-KO virus is reduced and slowed in comparison to that produced by the WT. Tumors were discovered in the spleen of 100% of WT-infected mice that survived beyond day 12 and in only 25% of mice infected with deltaBPLF1, indicating that lymphomagenesis progresses more slowly and with reduced frequency in the absence of BPLF1. How BPLF1 promotes lymphomagenesis and pathogenesis of EBV is unknown and will require further study. It has been suggested that early lytic protein expression contributes to EBV infection in primary B cells and that this event may be required for the establishment of latency (66). The presence of BPLF1 may possibly regulate early lytic gene expression in B-cells through its enzymatic activity by regulating transcription factors or by histone modifications. Additionally, immune evasion characteristics of BPLF1 may contribute to cellular transformation and increased pathogenesis (48, 55). Maintenance of latency in B cells may be altered in BPLF1-KO-infected cells as well.
EBV genomes were detected in blood and spleen of infected mice (Fig. 5). We demonstrate that, in all cases in which mice developed tumors, EBV was detectable. Spleen cells were cultured and analyzed for the presence of GFP-encoding genomes. EBV was detected only in the mice that developed tumors and not in the blood or spleen of other infected mice, leaving open the possibility that mice which did not develop tumors may have been able to clear the virus. EBV genomes were detected in the spleen of all the mice that developed tumors but, interestingly, were not detected in the blood of the DUB KO-infected mouse that did develop a tumor (DUB KO3). Virus detected in the blood may be due to metastasizing cells, since genomes were detected in the blood only immediately prior to termination. While only one KO mouse developed a tumor, the lack of EBV genomes in the blood suggests that BPLF1-KO virus infection may progress more slowly. It should also be noted that the amount of EBV genomes detected in the spleen varies greatly from mouse to mouse and is not an indication of total amounts of virus in the spleen but rather a reflection of the tissue section used for DNA extraction and genome detection (i.e., tissue directly from the lymphoma would have more genome copies than tissue farther away from the tumor). The objective was to show that spleen cells from animals with tumors did contain EBV genomes.
Histopathology was performed on spleen tissue of infected mice (Fig. 6). The uninfected sample shows fairly normal distribution of B and T cells within the spleen, whereas large B-cell lymphomas are clearly evident in DUB KO3, WT4, and WT5 samples. EBER staining is localized within regions of large B-cell lymphoma. H&E staining in these regions consisted of large atypical lymphoid cells that exhibited nodular to diffuse pattern growth and had high mitotic activity. These cases are consistent with monomorphic PTLD/lymphoma and map precisely with EBV detection, suggesting features nearly identical to those observed in immunocompromised individuals with large B-cell lymphomas due to EBV infection.
Last, we examined if tumors from WT-infected mice proliferated more rapidly than the tumor observed with BPLF1-KO virus. Staining for Ki67 shows highly elevated proliferation rates within the tumor region for all mice with lymphomas, but the proliferation rates appeared to be similar for WT and BPLF1-KO virus-infected mice. Based on this limited data set, it appears that once tumors are formed, there is not an apparent difference in proliferation rates between WT and BPLF1-KO virus. Interestingly, a study by Halder et al. showed a marked increase in Ki67 staining observed approximately 48 h postinfection and which increased through day 7 when primary human B cells were infected with EBV (66).
Together, these data demonstrate that BPLF1 is necessary for efficient infectious virus production and that BPLF1 plays an important role in EBV B-cell transformation, which likely accounts for the reduced and delayed tumor formation observed in humanized mouse studies. The mechanism by which BPLF1 inhibits these processes is still unknown and perhaps will be elucidated through follow-up studies examining episome formation, latency progression, and gene expression in cells infected with BPLF1-KO EBV. The current BPLF1-knockout construct removes the start codon and the first 975 bp, resulting in complete loss of both deubiquitinating and deneddylating activities. It remains to be determined if the enzymatic activity of BPLF1, perhaps by rescuing cellular and viral factors from degradation, is responsible for the observed effects or if other downstream regions of this very large protein contribute to its functional consequences.
MATERIALS AND METHODS
Cell lines, growth, and transfection.
HEK293T and 293EBV+ (49) and Raji cells were maintained as previously described (31, 52). EBV BPLF1-knockout virus (293 EBVdelta) uses 293EBV+ as a backbone, but the first 975 bp are deleted (48). Transfections for all cell lines were performed with Effectene (Qiagen) according to the manufacturer’s protocol.
Virus growth, concentration, and titration.
Infectious virus was produced by reactivation of the lytic cycle by transfection of BZLF1 and gp110 for both 293EBV+ and 293 BPLF1-knockout cell lines (10). Supernatant fluids were harvested 48 and 72 h posttransfection and cleared of cellular debris by centrifugation at 500 × g for 5 min. Virus was concentrated using an Amicon Ultra 15 10-kDa-molecular-mass-cutoff filter (Millipore) by centrifugation at 2,500 × g. For viral titration, 100 µl of cleared and concentrated supernatant fluids was placed on Raji cells and treated with 50 ng/ml phorbol-12-myristate-3-acetate and 3 mM sodium butyrate 24 h after infection (40). At 48 and 72 h postinfection, infectious titers were determined by detection of GFP-encoding EBV genomes by flow cytometry (52).
Viral infectivity was also determined on purified human primary B cells. B-cells were purified from freshly acquired buffy coat (Gulf Coast Labs) using the B-cell isolation kit II (Miltenyi Biotec) according to the manufacturer’s instructions. Purified B-cells (3.0 × 105) were infected with WT or BPLF1-knockout virus in culture medium (RPMI). Seventy-two hours postinfection, infectious titers were determined by detection of GFP by flow cytometry as described above.
Human B-cell outgrowth.
Whole blood from donors was diluted 1:2 with phosphate-buffered saline (PBS) and centrifuged through Ficoll (Ficoll-PaquePLUS from Pharmacia Biotech) for 30 min at 400 × g. Peripheral blood mononuclear cells were collected, washed twice with PBS, and counted. Approximately 3 million PBMCs in 3 ml RPMI supplemented with 20% fetal bovine serum (FBS) were used for each infection. Cells were infected with 3.0 × 103 and 3.0 × 104 infectious units of EBV WT or EBV BPLF1-knockout virus and placed in culture medium containing cyclosporine (RPMI 1640 plus 20% FBS plus 200 ng/ml cyclosporine). B-cells were counted twice weekly and were reported as a percentage of total cells. B-cells were detected by flow cytometry using CD19-phycoerythrin (PE) antibody (Miltenyi Biotec) according to the manufacturer’s instructions.
Rag2−/− γC−/− double-knockout (DKO) mouse construction and infection.
Newborn mice were irradiated and injected with 2 × 105 human hematopoietic stem cells into the liver. Twelve weeks after transplant, mice were screened for T/B/myeloid/natural killer cell reconstitution in peripheral blood by flow cytometry with antibodies recognizing each cell type (46, 47). Mice were injected intraperitoneally (i.p.) with 3.0 × 104 infectious units (as determined by GFP detection in Raji cells) of either WT EBV or BPLF1-KO virus diluted in a total volume of 120 µl PBS. Five mice received WT EBV, 5 mice received BPLF1-KO virus, and 2 mice, which received PBS, served as negative controls. Mice were treated with 5 µg antibody raised against CD3 (OKT3) (Imgenex) twice weekly for 14 days. Mice were bled once weekly for detection of EBV genomes. Mice were sacrificed at the end of the study (day 95) or when they lost approximately 20% body weight, and spleen and liver were harvested.
EBV genome detection in spleen and blood of infected mice.
DNA was extracted from weekly bleedings and from spleen at the time of harvest using the DNeasy Blood and Tissue kit (Qiagen). DNA from 100 µl blood or 350 ng of DNA from spleen tissue was used for real-time detection of EBV genomes. EBV genomes were detected with TaqMan probes targeting the BamHI repeat region as described previously (31). EBV genome copy numbers were determined using quantitated EBV genomes (Advanced Biotechnologies).
Culture of spleen cells from infected mice and detection of EBV genomes.
Portions of spleen tissue were ground and disrupted in 5 ml RPMI medium and centrifuged at 400 × g for 5 min. The cell pellet was resuspended in ACK lysing buffer (Lonza), incubated at room temperature for 5 min, and centrifuged to pellet (400 × g, 5 min). Pellets were resuspended in 2 ml RPMI medium, filtered through a cell strainer, plated in RPMI medium with 20% FBS, and placed at 37°C and 5% CO2. One week after plating, cells were examined by immunofluorescence microscopy to detect the presence of the GFP-tagged EBV genome.
H&E staining, IHC, and in situ hybridization.
Immunohistochemistry (IHC) and in situ hybridization were performed by the UNC Translational Pathology Laboratory and Animal Histopathology Core. Spleen tissue was formalin-fixed and paraffin-embedded. Consecutive sections were stained for hematoxylin and eosin, B-cell marker (CD20), T-cell marker (CD3), and the cell proliferation marker Ki67. Antibodies used were CD3 (Leica Biosystems; clone LN10; ready-to-use dilution [RTU]), CD20 (Leica Biosystems; clone MJ1/RTU), and Ki67 (Leica Biosystems; 1:300 dilution). Epstein-Barr virus-encoded RNAs (EBERs) were detected by in situ hybridization using reagents from Leica Microsystems. Histologic sections and associated stains were evaluated by a veterinary pathologist (S.A.M.) and a human pathologist (N.D.M.).
ACKNOWLEDGMENTS
This work was supported by NIH grants P01-CA19014-29 from NCI and 1R21A1095180 from NIAID. UNC Shared Resources are supported in part by an NCI Cancer Center Support Grant (2 P30 CA16086) to the UNC Lineberger Comprehensive Cancer Center.
We also acknowledge T. Tsurumi for the kind gift of BPLF1-knockout virus; UNC DLAM for mouse care and maintenance; the UNC Animal Histopathology Core and UNC Translational Pathology Laboratory for tissue sectioning, staining, and EBER in situ hybridization; and the UNC Lineberger Tissue Culture Facility for B-cell immortalization.
Footnotes
Citation Whitehurst CB, Li G, Montgomery SA, Montgomery ND, Su L, Pagano JS. 2015. Knockout of Epstein-Barr virus BPLF1 retards B-cell transformation and lymphoma formation in humanized mice. mBio 6(5):e01574-15. doi:10.1128/mBio.01574-15.
REFERENCES
- 1.MacMahon E, Glass JD, Hayward SD, Mann RB, Becker PS, Charache P, McArthur J, Ambinder RF. 1991. Epstein-Barr virus in AIDS-related primary central nervous system lymphoma. Lancet 338:969–973. doi: 10.1016/0140-6736(91)91837-K. [DOI] [PubMed] [Google Scholar]
- 2.Zhang L, Zhang J, Lambert Q, Der CJ, Del Valle L, Miklossy J, Khalili K, Zhou Y, Pagano JS. 2004. Interferon regulatory factor 7 is associated with Epstein-Barr virus-transformed central nervous system lymphoma and has oncogenic properties. J Virol 78:12987–12995. doi: 10.1128/JVI.78.23.12987-12995.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss LM, Movahed LA, Warnke RA, Sklar J. 1989. Detection of Epstein-Barr viral genomes in Reed-Sternberg cells of Hodgkin’s disease. N Engl J Med 320:502–506. doi: 10.1056/NEJM198902233200806. [DOI] [PubMed] [Google Scholar]
- 4.Raab-Traub N. 1992. Epstein-Barr virus and nasopharyngeal carcinoma. Semin Cancer Biol 3:297–307. [PubMed] [Google Scholar]
- 5.Zur Hausen HZ, Schulte-Holthausen H, Klein G, Henle W, Henle G, Clifford P, Santesson L. 1970. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 228:1056–1058. doi: 10.1038/2281056a0. [DOI] [PubMed] [Google Scholar]
- 6.Wang F-, Roy D, Gershburg E, Whitehurst CB, Dittmer DP, Pagano JS. 2009. Maribavir inhibits Epstein-Barr virus transcription in addition to viral DNA replication. J Virol 83:12108–12117. doi: 10.1128/JVI.01575-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitehurst CB, Sanders MK, Law M, Wang F-, Xiong J, Dittmer DP, Pagano JS. 2013. Maribavir inhibits Epstein-Barr virus transcription through the EBV protein kinase. J Virol 87:5311–5315. doi: 10.1128/JVI.03505-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong GK, Gulley ML, Feng W-, Delecluse H-, Holley-Guthrie E, Kenney SC. 2005. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J Virol 79:13993–14003. doi: 10.1128/JVI.79.22.13993-14003.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones RJ, Seaman WT, Feng W, Barlow E, Dickerson S, Delecluse H, Kenney SC. 2007. Roles of lytic viral infection and IL-6 in early versus late passage lymphoblastoid cell lines and EBV-associated lymphoproliferative disease. Int J Cancer 121:1274–1281. doi: 10.1002/ijc.22839. [DOI] [PubMed] [Google Scholar]
- 10.Ma S-, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, Tang W, Gumperz JE, Kenney SC. 2011. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J Virol 85:165–177. doi: 10.1128/JVI.01512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batchelor AH, O’Hare P. 1990. Regulation and cell-type-specific activity of a promoter located upstream of the latency-associated transcript of herpes simplex virus type 1. J Virol 64:3269–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitagawa N, Goto M, Kurozumi K, Maruo S, Fukayama M, Naoe T, Yasukawa M, Hino K, Suzuki T, Todo S, Takada K. 2000. Epstein-Barr virus-encoded poly(A) (−) RNA supports Burkitt’s lymphoma growth through interleukin-10 induction. EMBO J 19:6742–6750. doi: 10.1093/emboj/19.24.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahot S, Sergeant A, Drouet E, Gruffat H. 2003. A novel function for the Epstein-Barr virus transcription factor EB1/Zta: induction of transcription of the hIL-10 gene. J Gen Virol 84:965–974. doi: 10.1099/vir.0.18845-0. [DOI] [PubMed] [Google Scholar]
- 14.Masood R, Zhang Y, Bond MW, Scadden DT, Moudgil T, Law RE, Kaplan MH, Jung B, Espina BM, Lunardi-Iskandar Y. 1995. Interleukin-10 is an autocrine growth factor for acquired immunodeficiency syndrome-related B-cell lymphoma. Blood 85:3423–3430. [PubMed] [Google Scholar]
- 15.Penkert RR, Kalejta RF. 2011. Tegument protein control of latent herpesvirus establishment and animation. Herpesviridae 2:3. doi: 10.1186/2042-4280-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altmann M, Hammerschmidt W. 2005. Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol 3:e404. doi: 10.1371/journal.pbio.0030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bellows DS, Howell M, Pearson C, Hazlewood SA, Hardwick JM. 2002. Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J Virol 76:2469–2479. doi: 10.1128/jvi.76.5.2469-2479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oudejans JJ, van den Brule AJ, Jiwa NM, de Bruin PC, Ossenkoppele GJ, van der Valk P, Walboomers JM, Meijer CJ. 1995. BHRF1, the Epstein-Barr virus (EBV) homologue of the BCL-2 protooncogene, is transcribed in EBV-associated B-cell lymphomas and in reactive lymphocytes. Blood 86:1893–1902. [PubMed] [Google Scholar]
- 19.Feederle R, Neuhierl B, Baldwin G, Bannert H, Hub B, Mautner J, Behrends U, Delecluse HJ. 2006. Epstein-Barr virus BNRF1 protein allows efficient transfer from the endosomal compartment to the nucleus of primary B lymphocytes. J Virol 80:9435–9443. doi: 10.1128/JVI.00473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai K, Chan L, Gibeault R, Conn K, Dheekollu J, Domsic J, Marmorstein R, Schang LM, Lieberman PM. 2014. Viral reprogramming of the Daxx histone H3.3 chaperone during early Epstein-Barr virus infection. J Virol 88:14350–14363. doi: 10.1128/JVI.01895-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai K, Thikmyanova N, Wojcechowskyj JA, Delecluse H, Lieberman PM. 2011. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog 7:e1002376. doi: 10.1371/journal.ppat.1002376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldick CJ Jr, Marchini A, Patterson CE, Shenk T. 1997. Human cytomegalovirus tegument protein pp71 (ppUL82) enhances the infectivity of viral DNA and accelerates the infectious cycle. J Virol 71:4400–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Werstuck G, Bilan P, Capone JP. 1990. Enhanced infectivity of herpes simplex virus type 1 viral DNA in a cell line expressing the trans-inducing factor Vmw65. J Virol 64:984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmaus S, Wolf H, Schwarzmann F. 2004. The reading frame BPLF1 of Epstein-Barr virus: a homologue of herpes simplex virus protein VP16. Virus Genes 29:267–277. doi: 10.1023/B:VIRU.0000036387.39937.9b. [DOI] [PubMed] [Google Scholar]
- 25.Gastaldello S, Hildebrand S, Faridani O, Callegari S, Palmkvist M, Di Guglielmo C, Masucci MG. 2010. A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat Cell Biol 12:351–361. doi: 10.1038/ncb2035. [DOI] [PubMed] [Google Scholar]
- 26.Yuan J, Cahir-McFarland E, Zhao B, Kieff E. 2006. Virus and cell RNAs expressed during Epstein-Barr virus replication. J Virol 80:2548–2565. doi: 10.1128/JVI.80.5.2548-2565.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bottcher S, Granzow H, Maresch C, Mohl B, Klupp BG, Mettenleiter TC. 2007. Identification of functional domains within the essential large tegument protein pUL36 of pseudorabies virus. J Virol 81:13403–13411. doi: 10.1128/JVI.01643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol 74:11608–11618. doi: 10.1128/JVI.74.24.11608-11618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batterson W, Furlong D, Roizman B. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J Virol 45:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abaitua F, Daikoku T, Crump CM, Bolstad M, O’Hare P. 2011. A single mutation responsible for temperature-sensitive entry and assembly defects in the VP1-2 protein of herpes simplex virus. J Virol 85:2024–2036. doi: 10.1128/JVI.01895-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitehurst CB, Ning S, Bentz GL, Dufour F, Gershburg E, Shackelford J, Langelier Y, Pagano JS. 2009. The Epstein-Barr virus (EBV) deubiquitinating enzyme BPLF1 reduces EBV ribonucleotide reductase activity. J Virol 83:4345–4353. doi: 10.1128/JVI.02195-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, Ploegh HL. 2005. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol Cell 19:547–557. doi: 10.1016/j.molcel.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 33.Ernst R, Claessen JHL, Mueller B, Sanyal S, Spooner E, van der Veen AG, Kirak O, Schlieker CD, Weihofen WA, Ploegh HL. 2011. Enzymatic blockade of the ubiquitin-proteasome pathway. PLoS Biol 8:e1000605. doi: 10.1371/journal.pbio.1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inn K-, Lee S-, Rathbun JY, Wong L-, Toth Z, Machida K, Ou JJ, Jung JU. 2011. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J Virol 85:10899–10904. doi: 10.1128/JVI.00690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Loveland AN, Kattenhorn LM, Ploegh HL, Gibson W. 2006. High-molecular-weight protein (pUL48) of human cytomegalovirus is a competent deubiquitinating protease: mutant viruses altered in its active-site cysteine or histidine are viable. J Virol 80:6003–6012. doi: 10.1128/JVI.00401-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gredmark-Russ S, Isaacson MK, Kattenhorn L, Cheung EJ, Watson N, Ploegh HL. 2009. A gammaherpesvirus ubiquitin-specific protease is involved in the establishment of murine gammaherpesvirus 68 infection. J Virol 83:10644–10652. doi: 10.1128/JVI.01017-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bottcher S, Maresch C, Granzow H, Klupp BG, Teifke JP, Mettenleiter TC. 2008. Mutagenesis of the active-site cysteine in the ubiquitin-specific protease contained in large tegument protein pUL36 of pseudorabies virus impairs viral replication in vitro and neuroinvasion in vivo. J Virol 82:6009–6016. doi: 10.1128/JVI.00280-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim ET, Oh SE, Lee Y-O, Gibson W, Ahn J-H. 2009. Cleavage specificity of the UL48 deubiquitinating protease activity of human cytomegalovirus and the growth of an active-site mutant virus in cultured cells. J Virol 83:12046–12056. doi: 10.1128/JVI.00411-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jarosinski K, Kattenhorn L, Kaufer B, Ploegh H, Osterrieder N. 2007. A herpesvirus ubiquitin-specific protease is critical for efficient T cell lymphoma formation. Proc Natl Acad Sci U S A 104:20025–20030. doi: 10.1073/pnas.0706295104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma S-, Yu X, Mertz JE, Gumperz JE, Reinheim E, Zhou Y, Tang W, Burlingham WJ, Gulley ML, Kenney SC. 2012. An Epstein-Barr virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. J Virol 86:7976–7987. doi: 10.1128/JVI.00770-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wahl A, Linnstaedt SD, Esoda C, Krisko JF, Martinez-Torres F, Delecluse H-, Cullen BR, Garcia JV. 2013. A cluster of virus-encoded microRNAs accelerates acute systemic Epstein-Barr virus infection but does not significantly enhance virus-induced oncogenesis in vivo. J Virol 87:5437–5446. doi: 10.1128/JVI.00281-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujiwara S, Imadome K, Takei M. 2015. Modeling EBV infection and pathogenesis in new-generation humanized mice. Exp Mol Med 47:e135. doi: 10.1038/emm.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antsiferova O, Müller A, Rämer PC, Chijioke O, Chatterjee B, Raykova A, Planas R, Sospedra M, Shumilov A, Tsai M, Delecluse H, Münz C. 2014. Adoptive transfer of EBV specific CD8+ T cell clones can transiently control EBV infection in humanized mice. PLoS Pathog 10:e1004333. doi: 10.1371/journal.ppat.1004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chatterjee B, Leung CS, Münz C. 2014. Animal models of Epstein-Barr virus infection. J Immunol Methods 410:80–87. doi: 10.1016/j.jim.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 45.Fujiwara S. 2014. Reproduction of Epstein-Barr virus infection and pathogenesis in humanized mice. Immune Netw 14:1–6. doi: 10.4110/in.2014.14.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang Q, Zhang L, Wang R, Jeffrey J, Washburn ML, Brouwer D, Barbour S, Kovalev GI, Unutmaz D, Su L. 2008. FoxP3+CD4+ regulatory T cells play an important role in acute HIV-1 infection in humanized Rag2−/−gammaC−/− mice in vivo. Blood 112:2858–2868. doi: 10.1182/blood-2008-03-145946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L, Kovalev GI, Su L. 2007. HIV-1 infection and pathogenesis in a novel humanized mouse model. Blood 109:2978–2981. doi: 10.1182/blood-2006-07-033159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saito S, Murata T, Kanda T, Isomura H, Narita Y, Sugimoto A, Kawashima D, Tsurumi T. 2013. Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated NF-kappaB signaling during productive replication. J Virol 87:4060–4070. doi: 10.1128/JVI.02020-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delecluse H-, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc Natl Acad Sci U S A 95:8245–8250. doi: 10.1073/pnas.95.14.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gastaldello S, Hildebrand S, Faridani O, Callegari S, Palmkvist M, Di Guglielmo C, Masucci MG. 2010. A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat Cell Biol 12:351–361. doi: 10.1038/ncb2035. [DOI] [PubMed] [Google Scholar]
- 51.Hong GK, Delecluse H-, Gruffat H, Morrison TE, Feng W-, Sergeant A, Kenney SC. 2004. The BRRF1 early gene of Epstein-Barr virus encodes a transcription factor that enhances induction of lytic infection by BRLF1. J Virol 78:4983–4992. doi: 10.1128/JVI.78.10.4983-4992.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar R, Whitehurst CB, Pagano JS. 2014. The Rad6/18 ubiquitin complex interacts with the Epstein-Barr virus deubiquitinating enzyme, BPLF1, and contributes to virus infectivity. J Virol 88:6411–6422. doi: 10.1128/JVI.00536-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Henle W, Diehl V, Kohn G, Zur Hausen H, Henle G. 1967. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science 157:1064–1065. doi: 10.1126/science.157.3792.1064. [DOI] [PubMed] [Google Scholar]
- 54.Whitehurst CB, Vaziri C, Shackelford J, Pagano JS. 2012. Epstein-Barr virus BPLF1 deubiquitinates PCNA and attenuates polymerase eta recruitment to DNA damage sites. J Virol 86:8097–8106. doi: 10.1128/JVI.00588-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Gent M, Braem SGE, de Jong A, Delagic N, Peeters JGC, Boer IGJ, Moynagh PN, Kremmer E, Wiertz EJ, Ovaa H, Griffin BD, Ressing ME. 2014. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with Toll-like receptor signaling. PLoS Pathog 10:e1003960. doi: 10.1371/journal.ppat.1003960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johannsen E, Luftig M, Chase MR, Weicksel S, Cahir-McFarland E, Illanes D, Sarracino D, Kieff E. 2004. Proteins of purified Epstein-Barr virus. Proc Natl Acad Sci U S A 101:16286–16291. doi: 10.1073/pnas.0407320101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gastaldello S, Chen X, Callegari S, Masucci MG. 2013. Caspase-1 promotes Epstein-Barr virus replication by targeting the large tegument protein deneddylase to the nucleus of productively infected cells. PLoS Pathog 9:e1003664. doi: 10.1371/journal.ppat.1003664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hurley EA, Thorley-Lawson DA. 1988. B cell activation and the establishment of Epstein-Barr virus latency. J Exp Med 168:2059–2075. doi: 10.1084/jem.168.6.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tomkinson B, Robertson E, Kieff E. 1993. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J Virol 67:2014–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaye KM, Izumi KM, Kieff E. 1993. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci U S A 90:9150–9154. doi: 10.1073/pnas.90.19.9150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sugden B, Marsh K, Yates J. 1985. A vector that replicates as a plasmid and can be efficiently selected in B-lymphoblasts transformed by Epstein-Barr virus. Mol Cell Biol 5:410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yates J, Warren N, Reisman D, Sugden B. 1984. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci U S A 81:3806–3810. doi: 10.1073/pnas.81.12.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Longnecker R, Miller CL, Miao XQ, Tomkinson B, Kieff E. 1993. The last seven transmembrane and carboxy-terminal cytoplasmic domains of Epstein-Barr virus latent membrane protein 2 (LMP2) are dispensable for lymphocyte infection and growth transformation in vitro. J Virol 67:2006–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomkinson B, Kieff E. 1992. Use of second-site homologous recombination to demonstrate that Epstein-Barr virus nuclear protein 3B is not important for lymphocyte infection or growth transformation in vitro. J Virol 66:2893–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen A, Divisconte M, Jiang X, Quink C, Wang F. 2005. Epstein-Barr virus with the latent infection nuclear antigen 3B completely deleted is still competent for B-cell growth transformation in vitro. J Virol 79:4506–4509. doi: 10.1128/JVI.79.7.4506-4509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Halder S, Murakami M, Verma SC, Kumar P, Yi F, Robertson ES. 2009. Early events associated with infection of Epstein-Barr virus infection of primary B-cells. PLoS One 4:e7214. doi: 10.1371/journal.pone.0007214. [DOI] [PMC free article] [PubMed] [Google Scholar]