

Abstract
Significance: Angiogenesis plays a critical role in wound healing. A defect in the formation of a neovasculature induces ulcer formation. One of the challenges faced by the clinician when devising strategies to promote healing of chronic wounds is the initiation of angiogenesis and the formation of a stable vasculature to support tissue regeneration. Understanding the molecular factors regulating angiogenesis during wound healing will lead to better therapies for healing chronic wounds.
Recent Advances: Classically, chronic wounds are treated with debridement to remove inhibitory molecules to reestablish angiogenesis and normal wound healing. The addition of platelet-derived growth factor (PDGF, becaplermin) has shown some promise as an adjunctive therapy, but better therapies are still needed. Current treatment strategies include investigating the outcome of augmenting cytokines locally to reduce the inflammatory response and promote angiogenesis.
Critical Issues: The failure of wounds to form a new vasculature results in the inability of the wound to fully heal, and thus may develop into a chronic ulcer if left untreated. Inhibition of neovascularization commonly results from an overactive inflammatory response that includes an excessive chemokine response. Therefore, understanding how the chemokine response regulates neoangiogenesis will enhance our ability to develop new treatment strategies to improve neovascularization and wound healing.
Future Directions: The ability to regulate the chemokine environment in chronic wounds may enhance the development of the neovasculature to reduce invasive treatments and enhance wound healing. Either inhibiting chemokines that promote a chronic inflammatory response or increasing the levels of proangiogenic chemokines may enhance angiogenesis in chronic wounds.

Richard J. Bodnar, PhD
Scope and Significance
Angiogenesis plays a major role in wound healing. The angiogenic response is needed to deliver immune cells, remove debris, and provide nutrients for tissue regeneration. A defect in the regulation of blood vessel growth can cause dehiscence and ulceration. This review will discuss the role of angiogenesis in wound healing and provide an overview of the chemokines that promote and inhibit angiogenesis. We will focus on the function of the CC and CXC chemokines on regulating angiogenesis and the signaling pathways of their receptors. In addition, the review will discuss some of the current research investigating the use of chemokines to enhance the angiogenic response as a therapeutic for the treatment of chronic wounds.
Translational Relevance
Improving our understanding of the role chemokines play in regulating both the inflammatory response and angiogenesis will have a significant impact on wound therapy. Connecting the relationship between chemokine signaling, inflammation, and angiogenesis may promote the development of novel therapies to reduce chronic inflammation and enhance angiogenesis in chronic wounds.
Clinical Relevance
It has been estimated that between 3 and 6 million people in the United States suffer from chronic ulcers, which is responsible for significant healthcare expenditures.1 The current clinical methods for the treatment of chronic wounds are typically labor intensive, costly, and do not always significantly enhance wound healing. Even if the treatment is successful, the recurrence rate for ulcers ranges from as low as 23% for pressure ulcers and as high as 70% for diabetic ulcers.1 There have been extensive investigations into the mechanisms responsible for chronic wound formation without the development of clinically effective therapies. A deficiency in vessel formation is one of the principal factors in the inability of a wound to heal and plays a major role in the development of chronic ulcers. Enhancing the angiogenic process has become an area of focus to enhance wound closure and treat chronic wounds. Ongoing research is investigating the use of angiogenic chemokines to enhance wound closure and healing. Thus, understanding the chemokines regulating angiogenesis may provide novel therapies to enhance the wound healing process to reduce the morbidity and mortality associated with chronic wounds.
Background
Angiogenesis in wound healing
The normal wound healing process occurs in four main phases; hemostasis, inflammation, granulation, and remodeling (Fig. 1). Angiogenesis is a critical factor in the ability of the tissue to repair itself. Formation of a new vasculature is essential for the removal of debris, providing nutrients and oxygen to the metabolic active wound bed. The hemostasis/coagulation phase is characterized by platelet activation and fibrin clot formation as a result of endothelium disruption. At this stage, activated platelets release a host of factors (e.g., CXCL4, angiostatin) that promotes initial inhibition of angiogenesis.2 The release of platelet factor 4 (PF4) sequesters various growth factors (vascular endothelial growth factor [VEGF], basic fibroblast growth factor [bFGF], platelet-derived growth factor [PDGF]), limiting their ability to promote angiogenesis.3,4 In addition, platelets release various cytokines (i.e., tumor necrosis factor alpha [TNF-α], transforming growth factor beta [TGF-β], CXCL8) to activate inflammatory cells to facilitate the initiation of the inflammatory phase.2 During the inflammatory phase of wound healing, there is an excessive amount of proangiogenic molecules (growth factors and cytokines) being secreted by macrophage, epithelial cells, and lymphocytes.5 The wound bed becomes an angiogenic sink, facilitating endothelial cell migration and proliferation, and promoting neovascularization of the wound bed. For example, endothelial progenitor cells (EPC) are mobilized to the inflamed wound site and undergo differentiation as they are incorporated into the newly forming vasculature (vasculogenesis) through the recruitment and induction of differentiation by various angiogenic factors, including stromal cell-derived factor 1 (SDF-1, CXCL12) and VEGF.5,6 Once in the wound bed, these cells release angiogenic factors to further support neovascularization. By day 4, (initiation of the granulation phase) angiogenesis is in full progression and neovascularization of the wound bed is occurring.
Figure 1.

Schematic of the wound healing phases. Wound healing occurs in four phases: hemostasis, inflammation, granulation, and resolving. This schematic depicts the stages of neovascularization during wound healing. Disruption of the vasculature causes platelet activation, releasing the contents of their dense α-granules to promote clot formation and activation and recruitment of monocytes and leukocytes. Activation of monocytes induces the secretion of angiogenic factors, enhancing the angiogenic response. During the granulation phase, there is an excess of vessels in the wound tissue. During the transition from the granulation to resolving phase, angiostatic chemokines (CXCL10 and CXCL11) are secreted inducing the dissociation of nonfunctional and nonessential vessels. bFGF, basic fibroblast growth factor; ENA-78, epithelial-derived neutrophil-activating peptide 78; GRO, growth-related oncogene; IP-10, interferon-γ–inducible factor-10 kDa; NAP-1, neutrophil-activating protein 1; PDGF, platelet-derived growth factor; PF4, platelet factor 4; TGF-β, transforming growth factor beta; TNF-α, tumor necrosis factor alpha; VEGF, vascular endothelial growth factor.
The formation of the granulation tissue relies on vascularization of the wound tissue. In the granulation phase, there is an excess (estimated to be five-fold) of vessels in the wound bed to meet the metabolic demands of the cells repairing the tissue (Fig. 1). Many of these vessels are not fully mature, leaky, or are not functional. Progression to the remodeling phase results in a significant decrease in the metabolic needs of the new tissue. At this phase, there is a decrease in angiogenic molecules and an increase in angiostatic molecules promoting vessel regression (Fig. 1).7 The exact mechanism for vessel regression is not well understood, but it is thought that an upregulation of CXCL10 (IP-10) and CXCL11 (IP-9, ITAC) secretion is one of the mechanisms promoting vessel regression.8,9 The neovasculature regresses back to a vessel density similar to prewounding. The reduction of the exuberant, immature, and leaky vasculature within the wound tissue is necessary to enhance the strength of the newly regenerated tissue and reduce scarring. Failure of the nonessential and nonfunctional vessels to regress causes the skin to be prone to dehiscence and ulceration.
Chemokines regulating angiogenesis
Chemokines are small (7–13 kDa) multifaceted signaling proteins that have been shown to regulate the immune response as well as promote homeostasis, chemotaxis, and angiogenesis (Table 1). They play an integral part in directing wound closure and healing. The chemokine family is divided into four subfamilies based on their structural properties. Chemokines are currently classified as C, CC, CXC, and CX3C (X indicates any amino acid) based on their folded protein structure.10 In wound healing, chemokines play a significant role in activating the immune response. Platelet activation and release of various chemokines (e.g., CXCL2, CXCL8) promote the activation of neutrophils and monocytes. Activation of these cells further induces the release of chemokines (e.g., CCL2, CCL3, CCL4, CCL5), promoting lymphocyte, keratinocyte, and endothelial cell activation.11
Table 1.
Chemokines
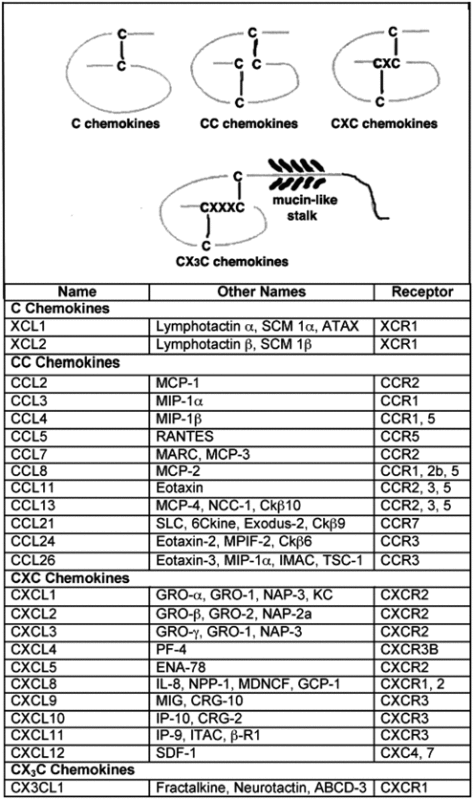 |
GRO, growth-related oncogene; IP-10, interferon-γ–inducible factor-10 kDa; MCP-1, monocyte chemotactic protein 1; MIP-1, macrophage inflammatory protein 1; PF4, platelet factor 4; RANTES, regulated on activation normal T-cell expressed and secreted.
The CC and CXC receptor families play a major role in regulating angiogenesis during the wound healing process. The CC and CXC subtype chemokine receptors are G-protein–coupled receptors, intracellular G-proteins function to mediate the outcome (e.g., cell survival or death) of ligand–receptor binding by inducing specific intracellular signaling pathways. For the most part, the CC (CCR1, CCR2, CCR3) and CXC (CXCR1, CXCR2, CXCR4) receptors directly promote angiogenesis with the exception of CXC receptor 3 (CXCR3) that has been shown to directly inhibit angiogenesis.3,8,12 Therefore, a defect in the levels of the angiogenic chemokines or in the levels of antiangiogenic chemokines will promote a dysfunction in angiogenesis and neovascular development (Table 2).
Table 2.
Chemokines directly regulating endothelial function
| Angiogenic Chemokines | Antiangiogenic Chemokines | ||
|---|---|---|---|
| Name | Other Name | Name | Other Name |
| CCL2 | MCP-1 | CXCL4 | PF4 |
| CCL5 | RANTES | CXCL9 | Mig |
| CXCL1 | GRO-α, GRO-1 | CXCL10 | IP-10 |
| CXCL2 | GRO-β, GRO-2 | CXCL11 | IP-9, ITAC |
| CXCL8 | IL-8 | ||
| CXCL12 | SDF-1 | ||
SDF-1, stromal cell-derived factor 1.
Chemokines can indirectly affect angiogenesis. The dysregulation of chemokines that activate neutrophils or monocytes/macrophages can negatively impact angiogenesis by limiting the secretion of proangiogenic factors. Similarly, dysfunction in epithelial cell secretion of chemokines can inhibit endothelial cell function. In addition, chemokines that enhance the activation of lymphocytes may lead to the release of antiangiogenic factor limiting angiogenesis. Thus, chemokines clearly regulate endothelial cell function and neovascularization through direct and indirect signaling mechanisms.
CXC chemokines and receptors
The predominant CXC ligands that are reported to be expressed in a wound and have been shown to regulate angiogenesis are: CXCL8 (IL-8), CXCL1 (GROα) and CXCL2 (GROβ), CXCL5 (ENA-78), CXCL7 (NAP-2), CXCL12 (SDF-1), CXCL11 (IP-9, ITAC), CXCL10 (IP-10), and CXCL9 (Mig). These chemokines are known to activate neutrophils, monocytes, lymphocytes, epithelial cells, and fibroblasts.11 These chemokines have been classified as angiogenic (CXCL1, CXCL2, CXCL5, CXCL7, CXCL8, and CXCL12) and angiostatic (CXCL9, CXCL10, and CXCL11). The ligands, CXCL1, CXCL7, CXCL8 bind to both CXCR1 and CXCR 2 receptors, whereas CXCL2 and CXCL5 have only been found to bind to CXCR1. The receptors activate two key pathways, phospholipase C (PLC)-β and PI3K. In turn, PLC-β activates protein kinase C (PKC) and phospholipase A2 (PLA2), and PI3K activates Ras/Raf/MAPK and Akt, leading to adhesion, migration, and proliferation13 (Fig. 2). CXCL8 and CXCL2 are initially released into the wound bed by platelets to promote angiogenesis. CXCL1, CXCL2, CXCL5, and CXCL12 are released by activated macrophages to further promote angiogenesis and enhance vascularization into the wound bed. CXCL8 and CXCL1 are predominantly expressed from days 1 to 4 then markedly decline.11 The exact role of the chemokine/receptor pair CXCL12/CXCR4 has in angiogenesis during wound healing is not well understood. It has been found to play a significant role in trafficking stem cell to the neovasculature, promoting vasculogenesis.14,15 A recent study by Xu et al., found that blockade of CXCR4 impaired bone marrow derived mesenchymal stem cell recruitment to the wound site decreased vessel formation and delayed wound healing.14 Other studies have shown that CXCL12 promotes the recruitment of circulating EPC and progenitor-mesenchymal stem cell–pericyte in the development and stabilization of nascent vessels.15–17 In addition, it has been shown to be involved in promoting endothelial migration18 and enhancing endothelial barrier function, which is necessary for vessel maturation.19 Taken together CXCL12 may enhance angiogenesis during wound healing by recruiting progenitor cells along with direct interaction with endothelial cells. Thus, these chemokines play a key role in promoting neovascularization during wound healing.
Figure 2.

Schematic of CXCR1 and 2 signaling pathways. These receptors are 7-transmembrane G-protein–coupled receptors that bind to Gαi, Gα14, and Gα16 and activate PKA, PKC, and MAPK pathways, to promote migration and proliferation. These receptors are highly expressed on macrophages and to a lesser extent, endothelial cells. These receptors promote angiogenesis. AKT, protein kinase B; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase; PKA, cAMP-dependent protein kinase; PLC, phospholipase C; PKC, protein kinase C.
The CXC ligands CXCL9, CXCL10, and CXCL11 acting through their receptor, CXCR3, promote the activation of lymphocytes, facilitating the immune response. These chemokines have also been identified to inhibit the function of fibroblasts and endothelial cells, promoting the transition to the remodeling phase.3,9,20 These chemokines are highly expressed during the granulation phase of wound healing by activated T-lymphocytes and are thought to contribute to vascular pruning of the wound bed.3,8 These ligands have been shown to inhibit endothelial migration, induce apoptosis, and promote vessel regression.3,8,21,22 CXCR3 activation inhibit endothelial migration through cAMP-dependent protein kinase (PKA)-mediated inhibition of m-calpain,3 promotes apoptosis through the activation of p38MAPK pathway,22 and cleavage of integrins inducing anoikis8 (Fig. 3). In addition, CXCR3-mediated signaling promotes the cleavage of integrins, αvβ3 facilitating vessel regression.8 Thus, the expression of CXCR3 ligands supports initiation of the inflammatory phase, and also plays an important role in transitioning into the remodeling phase by promoting regression of nonfunctioning and nonessential vasculature and reducing the migratory activity of fibroblasts.8,9 Platelet factor 4 (PF4, CXCL4) found in the α-granules of platelets and released upon platelet activation is also a CXCR3 ligand that inhibits endothelial function,3,21 but its role in regulating angiogenesis is not well understood. PF4 has been found to inhibit angiogenesis through sequestering VEGF and bFGF23,24 and activation of CXCR3.3,21 PF4 is sequestered to the fibrin clot due to its ability to bind to heprin.25,26 Thus, PF4 may promote hemostasis by initially sequestering VEGF and bFGF to inhibit premature initiation of angiogenesis and inhibiting endothelial immigration and subsequent vessel formation in the fibrin clot.
Figure 3.

Schematic of the CXCR3 inhibitory pathway. CXCR3 is expressed as two isoforms (CXCR3A and CXCR3B). Activation of CXCR3B, expressed on endothelial cells, promotes apoptosis through the activation of P38 and μ-calpain cleavage of integrins. CXCR3B also inhibits migration through the inhibition of m-calpain. Expression of CXCR3A is found on inflammatory cells and promotes migration and proliferation through the activation of MAPK and AKT pathways. IP3, inositol trisphosphate.
Additionally, endothelial cells express CXCR6, but to a significantly lower extent, and whether its expression is upregulated during wound healing is unclear, but it has been shown to be upregulated by IL-1β.27 Its ligand, CXCL16, is found in platelets and is also expressed by endothelial cells and leukocytes, but its role in wound healing has not been identified. CXCL16/CXCR6 ligand/receptor binding has been associated with various inflammatory diseases27,28 and may have some relevance in chronic wounds, but no correlation has been observed.
CC chemokines and receptors
The CC chemokines have less of a role in promoting angiogenesis compared with the CXC chemokines, but are thought to maintain vascular homeostasis during inflammation.29 CCL2 and CCL5 are expressed predominantly during the first week after wounding and contribute to monocytes, mast cell and leukocyte recruitment, and facilitate angiogenesis.6,11,30 Both chemokines are initially released from platelets and subsequently maintained by secretion from activated stromal cells.
Activation of keratinocytes, monocytes, and endothelial cells by IL-1β, TNF-α, growth factors, or oxidative stress induces CCL2 expression.31,32 CXCL2 is a well-known mediator of neovascularization and signals through CCR2 and CCR3 receptors to promote endothelial cell migration and cord formation in vitro.29 CCR2 baseline expression in senescent endothelial cells is upregulated under inflammatory conditions. The receptors activate three key pathways, mitogen-activated protein kinase (MAPK), PKC, and protein kinase B (AKT) (Fig. 4). Activation of MAPK and PKC pathway promotes endothelial migration and also promotes the expression of VEGF, bFGF, and MMP14 to further enhance angiogenesis.29,33 Activation of AKT supports cell survival and induces proliferation.
Figure 4.

Schematic of the CCR1, 2, and 5 signaling pathways. The CC receptor is a 7-transmembrane G-protein–coupled receptor that binds to Gαi, Gα14, and Gα16 and activates PKA, PKC, AKT, and MAPK pathways to promote migration, proliferation, and gene transcription. This receptor is expressed on inflammatory cells as well as endothelial cells.
CCL5 is initially secreted by platelet activation, but is also released from endothelial cells, macrophages, and T-cells and has been found to bind to CCR1, 3, and 5. The role of CCL5 in angiogenesis is not well understood. It may promote or inhibit angiogenesis depending on the CC receptor it activates. Binding to CCR1 and 2 has been shown to promote endothelial migration and cord formation.34 CCL5 activation of CCR5-mediated angiogenesis is controversial, as studies have shown its ability to both promote and inhibit endothelial cell function.35,36 Activation of the CCL5/CCR5 chemokine/receptor has been found to promote the recruitment of EPC to promote neovascularization during wound healing and shown to be important for sustaining the granulation phase.37
Chronic wounds
In nonhealing wounds, there is a defect in the ability of the wound to transition to the granulation stage. These wounds remain static in the inflammatory phase, promoting a hostile environment and leading to tissue necrosis. The exact mechanism for this disruption is not well understood and is thought to result, at least in part, from an increase in cytokine content and a decrease in vascularity of the wound tissue. Studies have shown that the delay in acute wound healing observed in skin wounds of type 2 diabetic (db/db) mice is due to a decrease in angiogenesis, persistent inflammation, delayed granulation tissue formation, and reduced collagen content.38 These wounds have also been found to have increased levels of inflammatory cytokines, which include TNFα, granulocyte colony-stimulating factor (G-CSF), growth-related oncogene (CXCL1, CXCL2), monocytes chemoattractant protein 1 (CCL2), interferon-induced protein of 10 kDa (CXCL10), interleukin-8 (CXCL8), and regulated on activation normal T-cell expressed and secreted (CCL5).38
A chronic ulcer is a wound that is persistent, lasting more than 4 weeks without significant healing, or persists as a recurrent wound. The cause of ulceration is usually multifactorial. However, ulcer development is believed to most commonly result from tissue ischemia due to an underlying disease such as hypertension atherosclerosis, diabetes, and any of a number of chronic vascular diseases. The majority of chronic ulcers develop on the lower extremity and without early intervention and effective treatment may lead to amputation. Current clinical approaches for the treatment of chronic ulcers include debridement along with the addition of skin substitutes or other commercially available advance wound care products, hyperbaric oxygen therapy, and negative pressure therapy. These treatments have varied success, likely due to the complexity of chronic wounds. Thus, novel therapies are needed to improve wound healing to reduce the incidence for the development of chronic wounds.
Discussion
The formation of new blood vessels, neovascularization, is a fundamental process that is necessary for proper wound healing. The disruption of angiogenesis or impaired neovascularization of the tissue is a central problem in the development of chronic ulcers. In chronic ulcers, it is hypothesized that disruption of neovascularization occurs as a result of multiple factors, which include sequestration of growth factors, reduced proangiogenic cytokines, and overproduction of antiangiogenic cytokines. Impediment of neovascularization along with inhibition of reepithelialization promotes ulcer formation and fosters the progression to a chronic ulcer. In addition, a hyperactive inflammatory response promotes extracellular matrix degradation and cell death and thus inhibits wound closure and further promotes ulceration.
Standard of care for chronic wounds starts with debridement of the necrotic tissue to remove the toxic factors to promote the healthy tissue and to reestablish the wound healing process. Additional therapies, including application of skin substitutes, hyperbaric oxygen therapy, and negative pressure, are used as added support to enhance wound healing. Since these therapies are invasive, time consuming, and often require repeated application, new methods are needed to improve the ability to heal chronic wounds.
A number of new and emerging therapies are being tested for their ability to accelerate wound healing; these include the addition of growth factors,39 cytokines,39 application of platelet gel,40 gene therapy,41 and stem cell therapy.42 Cytokines and chemokines are a novel target for the treatment of chronic ulcers since they are crucial to the wound healing process. Chemokines under investigation for the treatment of chronic wound are CCL2, CXCL8, and CXCL12.43–45
Because of the multifaceted function of CXCL12, that includes its ability to promote angiogenesis and reepithelialization, recruit EPC, and reduces tissue scarring, it is a potential target for the treatment of dermal wounds.43,46–48 It has been shown that direct application or lentiviral induction of CXCL12 enhances wound healing in diabetic mice.46,47 In a recent study by Rabbany et al., an alginate scaffold was used to deliver CXCL12 to accelerate wound closure.43 This study showed that alginate delivery of CXCL12 as a plasmid or protein had similar effects in their ability to accelerate healing and reduce scarring. In addition, the ability of CCL5 to directly control the homing of EPC to the wound tissue and contribute to the development of the neovasculature makes it a possible target for enhancing wound healing. Deletion of CCR5, the receptor for CCL5, not only showed a reduced sequestering of endothelial progenitor to the wounded tissue, but there was also a decrease in the angiogenic factors VEGF and TGF-β.37 Together, these studies suggest that targeting the recruitment of stem/progenitor cells to the wound tissue may be a possible therapeutic for enhancing angiogenesis and for use as a clinical treatment for chronic wound.
A recent study by Nishimura et al., investigated the ability of AMD3100, a CXCR4 antagonist, to promote diabetic wound healing.49 AMD3100 was first developed as an anti-HIV drug, but failed. It was subsequently found to promote stem cell mobilization and neovascularization after myocardial infarction through CXCR4-dependent and independent mechanisms.50 AMD3100 was also found to enhance chronic wound healing and promoted angiogenesis in diabetic mouse through direct enhancement of fibroblast and macrophage activation and through indirect stimulation of cytokine production and EPC mobilization.49 Thus, targeting CXCR4 may have clinical benefits in promoting the healing of chronic wounds.
In summary, targeting the chemokine signaling pathways has yet to reach the clinical level. Over the last decade, there have been a number of key advancements in understanding the signaling pathways initiated by chemokines under physiological and pathological conditions. However, there are still critical gaps in our knowledge of chemokine signaling and regulation of cell function. We are still trying to parse the effects a chemokine has on different cell types and how these redundancies affect angiogenesis, and the wound healing process. At present, chemokines have significant potential to make a substantive impact as a novel therapeutic in promoting angiogenesis and enhancing wound healing.
Summary
With significant advances in understanding the mechanisms and molecules involved in the wound healing process, few effective wound healing agents have been developed to promote a chronic nonhealing wound into an actively healing wound. Chemokines have been found to be crucial factors in physiological and pathological wound healing, therefore, chemokines may have significant potential to become targets for enhancing the wound healing response. In addition, the use of chemokine gene therapy may provide a novel tool for localized delivery of protein to the wound site. Although delivery of specific chemokines has significant potential for the treatment of chronic wounds, there are a number of hurdles that need to be overcome. The use of chemokines are challenging since the timing of delivery and concentration significantly affects the healing process, thus a multi-step approach may be necessary to enhance the repair response. The development of a method to deliver multiple factors that can promote angiogenesis along with modifying the inflammatory response will provide a greater success in promoting healing of chronic wounds and improve the lives of patients.
Take-Home Message.
• Angiogenesis is a major component of the wound healing process, therefore, disruption in the formation of vessels can cause the development of a nonhealing wound. Without the development of a sufficient vasculature, a wound is not able to completely heal.
• Chemokines and their receptors are critical mediators of neovascularization in both physiological and pathological conditions. The timing and pattern of chemokine release has a significant influence on neovascularization. Chemokines have been found to have the ability to facilitate the initiation of angiogenesis and promote the dissociation of nonessential vessels.
• Chemokines promote neovascularization through the stimulation of endothelial cell migration and proliferation (angiogenesis) and recruitment of EPC (e.g., vasculogenesis).
• The development of strategies to target angiogenic chemokines (CCl2, CXCL8, CXCL12) or their receptors (i.e., CCR2, CXCR4, CXCR2) individually or in combination may provide a novel therapy for the treatment of chronic wounds. Current strategies are focused on the CXCL12/CXCR4 ligand/receptor signaling pathway at the wound site. These studies are still in their infancy and more work is required to determine the efficacy of these treatments.
Abbreviations and Acronyms
- AKT
protein kinase B
- bFGF
basic fibroblast growth factor
- BMSC
bone marrow derived mesenchymal stem cell
- ENA-78
epithelial-derived neutrophil-activating peptide 78
- EPC
endothelial progenitor cell
- ERK
extracellular signal-regulated kinase
- G-CSF
granulocyte colony-stimulating factor
- GRO
growth-related oncogene
- IP-10
interferon-γ–inducible factor-10 kDa
- IP3
inositol trisphosphate
- MAPK
mitogen-activated protein kinase
- MCP-1
monocyte chemotactic protein 1
- MEK
mitogen-activated protein kinase kinase
- Mig
monokine induced by interferon-γ
- MIP-1
macrophage inflammatory protein 1
- MSC
mesenchymal stem cell
- NAP-2
neutrophil-activating protein 2
- PDGF
platelet-derived growth factor
- PF4
platelet factor 4
- PKA
cAMP-dependent protein kinase
- PKC
protein kinase C
- PLA2
phospholipase A2
- PLC
phospholipase C
- RANTES
regulated on activation, normal T-cell expressed and secreted
- TNF-α
tumor necrosis factor-alpha
- VEGF
vascular endothelial growth factor
Acknowledgments and Funding Sources
Support by grants from the National Institute of General Medical Sciences (NIGMS), the Department of Veterans Affairs, and the University of Pittsburgh, CMRF.
Author Disclosure and Ghostwriting
The author has no conflict of interest to disclose. The listed author is responsible for the writing of this article.
About the Author
Richard J. Bodnar, PhD, is a Research Assistant Professor in the Department of Pathology at the University of Pittsburgh. He also has a joint appointment as a Research Associate at the Veterans Affairs Healthcare System in Pittsburgh. In addition, he is a faculty member of the McGowan Institute for Regenerative Medicine and the Vascular Medicine Institute of Pittsburgh. Dr. Bodnar received his B.S. in Biochemistry from the University of Pittsburgh and his Ph.D. in Molecular Pharmacology from the University of Illinois at Chicago. The main focus of Dr. Bodnar's research is to understand the cellular and molecular mechanisms regulating angiogenesis during wound healing.
References
- 1.Werdin F, Tennenhaus M, Schaller HE, Rennekampff HO. Evidence-based management strategies for treatment of chronic wounds. Eplasty 2009;9:e19. [PMC free article] [PubMed] [Google Scholar]
- 2.Coppinger JA, Cagney G, Toomey S, Kislinger T, Belton O, McRedmond JP, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004;103:2096–2104 [DOI] [PubMed] [Google Scholar]
- 3.Bodnar RJ, Yates CC, Wells A. IP-10 blocks vascular endothelial growth factor-induced endothelial cell motility and tube formation via inhibition of calpain. Circ Res 2006;98:617–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gengrinovitch S, Greenberg SM, Cohen T, Gitay-Goren H, Rockwell P, Maione TE, et al. Platelet factor-4 inhibits the mitogenic activity of VEGF121 and VEGF165 using several concurrent mechanisms. J Biol Chem 1995;270:15059–15065 [DOI] [PubMed] [Google Scholar]
- 5.Bauer SM, Bauer RJ, Velazquez OC. Angiogenesis, vasculogenesis, and induction of healing in chronic wounds. Vasc Endovascular Surg 2005;39:293–306 [DOI] [PubMed] [Google Scholar]
- 6.Keeley EC, Mehrad B, Strieter RM. Chemokines as mediators of neovascularization. Arterioscler Thromb Vasc Biol 2008;28:1928–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wietecha MS, Cerny WL, DiPietro LA. Mechanisms of vessel regression: toward an understanding of the resolution of angiogenesis. Curr Top Microbiol Immunol 2013;367:3–32 [DOI] [PubMed] [Google Scholar]
- 8.Bodnar RJ, Yates CC, Rodgers ME, Du X, Wells A. IP-10 induces dissociation of newly formed blood vessels. J Cell Sci 2009;122(Pt 12):2064–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yates CC, Whaley D, Kulasekeran P, Hancock WW, Lu B, Bodnar R, et al. Delayed and deficient dermal maturation in mice lacking the CXCR3 ELR-negative CXC chemokine receptor. Am J Pathol 2007;171:484–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenkilde MM, Schwartz TW. The chemokine system—a major regulator of angiogenesis in health and disease. APMIS 2004;112:481–495 [DOI] [PubMed] [Google Scholar]
- 11.Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol 2001;69:513–521 [PubMed] [Google Scholar]
- 12.Romagnani P, Lasagni L, Annunziato F, Serio M, Romagnani S. CXC chemokines: the regulatory link between inflammation and angiogenesis. Trends Immunol 2004;25:201–209 [DOI] [PubMed] [Google Scholar]
- 13.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res 2008;14:6735–6741 [DOI] [PubMed] [Google Scholar]
- 14.Xu X, Zhu F, Zhang M, Zeng D, Luo D, Liu G, et al. Stromal cell-derived factor-1 enhances wound healing through recruiting bone marrow-derived mesenchymal stem cells to the wound area and promoting neovascularization. Cells Tissues Organs 2013;197:103–113 [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Xu Q. Stem/Progenitor cells in vascular regeneration. Arterioscler Thromb Vasc Biol 2014;34:1114–1119 [DOI] [PubMed] [Google Scholar]
- 16.Chen Y, Wong MM, Campagnolo P, Simpson R, Winkler B, Margariti A, et al. Adventitial stem cells in vein grafts display multilineage potential that contributes to neointimal formation. Arterioscler Thromb Vasc Biol 2013;33:1844–1851 [DOI] [PubMed] [Google Scholar]
- 17.Akhtar S, Gremse F, Kiessling F, Weber C, Schober A. CXCL12 promotes the stabilization of atherosclerotic lesions mediated by smooth muscle progenitor cells in Apoe-deficient mice. Arterioscler Thromb Vasc Biol 2013;33:679–686 [DOI] [PubMed] [Google Scholar]
- 18.Salcedo R, Wasserman K, Young HA, Grimm MC, Howard OM, Anver MR, et al. Vascular endothelial growth factor and basic fibroblast growth factor induce expression of CXCR4 on human endothelial cells: In vivo neovascularization induced by stromal-derived factor-1alpha. Am J Pathol 1999;154:1125–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi K, Sato K, Kida T, Omori K, Hori M, Ozaki H, et al. Stromal cell-derived factor-1alpha/C-X-C chemokine receptor type 4 axis promotes endothelial cell barrier integrity via phosphoinositide 3-kinase and Rac1 activation. Arterioscler Thromb Vasc Biol 2014;34:1716–1722 [DOI] [PubMed] [Google Scholar]
- 20.Luster AD, Cardiff RD, MacLean JA, Crowe K, Granstein RD. Delayed wound healing and disorganized neovascularization in transgenic mice expressing the IP-10 chemokine. Proc Assoc Am Physicians 1998;110:183–196 [PubMed] [Google Scholar]
- 21.Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med 2003;197:1537–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petrai I, Romboutsa K, Lasagni L, Annunziato F, Cosmi L, Romanelli RG, et al. Activation of p38MAPK mediates the angiostatic effect of the chemokine receptor CXCR3-B. Int J Biochem Cell Biol 2008;40:1764–1774 [DOI] [PubMed] [Google Scholar]
- 23.Jouan V, Canron X, Alemany M, Caen JP, Quentin G, Plouet J, et al. Inhibition of in vitro angiogenesis by platelet factor-4-derived peptides and mechanism of action. Blood 1999;94:984–993 [PubMed] [Google Scholar]
- 24.Sulpice E, Contreres JO, Lacour J, Bryckaert M, Tobelem G. Platelet factor 4 disrupts the intracellular signalling cascade induced by vascular endothelial growth factor by both KDR dependent and independent mechanisms. Eur J Biochem 2004;271:3310–3318 [DOI] [PubMed] [Google Scholar]
- 25.Sandset PM. CXCL4-platelet factor 4, heparin-induced thrombocytopenia and cancer. Thromb Res 2012;129 Suppl 1:S97–S100 [DOI] [PubMed] [Google Scholar]
- 26.Amelot AA, Tagzirt M, Ducouret G, Kuen RL, Le Bonniec BF. Platelet factor 4 (CXCL4) seals blood clots by altering the structure of fibrin. J Biol Chem 2007;282:710–720 [DOI] [PubMed] [Google Scholar]
- 27.Isozaki T, Arbab AS, Haas CS, Amin MA, Arendt MD, Koch AE, et al. Evidence that CXCL16 is a potent mediator of angiogenesis and is involved in endothelial progenitor cell chemotaxis: studies in mice with K/BxN serum-induced arthritis. Arthritis Rheum 2013;65:1736–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh ST, Schramme A, Tilgen W, Gutwein P, Reichrath J. Overexpression of CXCL16 in lesional psoriatic skin. Dermatoendocrinology 2009;1:114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weber KS, Nelson PJ, Grone HJ, Weber C. Expression of CCR2 by endothelial cells: implications for MCP-1 mediated wound injury repair and In vivo inflammatory activation of endothelium. Arterioscler Thromb Vasc Biol 1999;19:2085–2093 [DOI] [PubMed] [Google Scholar]
- 30.Engelhardt E, Toksoy A, Goebeler M, Debus S, Brocker EB, Gillitzer R. Chemokines IL-8, GROalpha, MCP-1, IP-10, and Mig are sequentially and differentially expressed during phase-specific infiltration of leukocyte subsets in human wound healing. Am J Pathol 1998;153:1849–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel JK, Clifford RL, Deacon K, Knox AJ. Ciclesonide inhibits TNFalpha- and IL-1beta-induced monocyte chemotactic protein-1 (MCP-1/CCL2) secretion from human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2012;302:L785–L792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 2009;29:313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jimenez-Sainz MC, Fast B, Mayor F, Jr., Aragay AM. Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Mol Pharmacol 2003;64:773–782 [DOI] [PubMed] [Google Scholar]
- 34.Suffee N, Hlawaty H, Meddahi-Pelle A, Maillard L, Louedec L, Haddad O, et al. RANTES/CCL5-induced pro-angiogenic effects depend on CCR1, CCR5 and glycosaminoglycans. Angiogenesis 2012;15:727–744 [DOI] [PubMed] [Google Scholar]
- 35.Barcelos LS, Coelho AM, Russo RC, Guabiraba R, Souza AL, Bruno-Lima G Jr., et al. , Role of the chemokines CCL3/MIP-1 alpha and CCL5/RANTES in sponge-induced inflammatory angiogenesis in mice. Microvasc Res 2009;78:148–154 [DOI] [PubMed] [Google Scholar]
- 36.Ambati BK, Anand A, Joussen AM, Kuziel WA, Adamis AP, Ambati J. Sustained inhibition of corneal neovascularization by genetic ablation of CCR5. Invest Ophthalmol Vis Sci 2003;44:590–593 [DOI] [PubMed] [Google Scholar]
- 37.Ishida Y, Kimura A, Kuninaka Y, Inui M, Matsushima K, Mukaida N, et al. Pivotal role of the CCL5/CCR5 interaction for recruitment of endothelial progenitor cells in mouse wound healing. J Clin Invest 2012;122:711–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galeano M, Torre V, Deodato B, Campo GM, Colonna M, Sturiale A, et al. Raxofelast, a hydrophilic vitamin E-like antioxidant, stimulates wound healing in genetically diabetic mice. Surgery 2001;129:467–477 [DOI] [PubMed] [Google Scholar]
- 39.Barrientos S, Brem H, Stojadinovic O, Tomic-Canic M. Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen 2014;22:569–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crovetti G, Martinelli G, Issi M, Barone M, Guizzardi M, Campanati B, et al. Platelet gel for healing cutaneous chronic wounds. Transfus Apher Sci 2004;30:145–151 [DOI] [PubMed] [Google Scholar]
- 41.Gruss CJ, Satyamoorthy K, Berking C, Lininger J, Nesbit M, Schaider H, et al. Stroma formation and angiogenesis by overexpression of growth factors, cytokines, and proteolytic enzymes in human skin grafted to SCID mice. J Invest Dermatol 2003;120:683–692 [DOI] [PubMed] [Google Scholar]
- 42.Navone SE, Pascucci L, Dossena M, Ferri A, Invernici G, Acerbi F, et al. Decellularized silk fibroin scaffold primed with adipose mesenchymal stromal cells improves wound healing in diabetic mice. Stem Cell Res Ther 2014;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rabbany SY, Pastore J, Yamamoto M, Miller T, Rafii S, Aras R, et al. Continuous delivery of stromal cell-derived factor-1 from alginate scaffolds accelerates wound healing. Cell Transplant 2010;19:399–408 [DOI] [PubMed] [Google Scholar]
- 44.Wood S, Jayaraman V, Huelsmann EJ, Bonish B, Burgad D, Sivaramakrishnan G, et al. Pro-inflammatory chemokine CCL2 (MCP-1) promotes healing in diabetic wounds by restoring the macrophage response. PLoS One 2014;9:e91574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang WG, Sanders AJ, Ruge F, Harding KG. Influence of interleukin-8 (IL-8) and IL-8 receptors on the migration of human keratinocytes, the role of PLC-gamma and potential clinical implications. Exp Ther Med 2012;3:231–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Badillo AT, Chung S, Zhang L, Zoltick P, Liechty KW. Lentiviral gene transfer of SDF-1alpha to wounds improves diabetic wound healing. J Surg Res 2007;143:35–42 [DOI] [PubMed] [Google Scholar]
- 47.Gallagher KA, Liu ZJ, Xiao M, Chen H, Goldstein LJ, Buerk DG, et al. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest 2007;117:1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sasaki T, Fukazawa R, Ogawa S, Kanno S, Nitta T, Ochi M, et al. Stromal cell-derived factor-1alpha improves infarcted heart function through angiogenesis in mice. Pediatr Int 2007;49:966–971 [DOI] [PubMed] [Google Scholar]
- 49.Nishimura Y, Ii M, Qin G, Hamada H, Asai J, Takenaka H, et al. CXCR4 antagonist AMD3100 accelerates impaired wound healing in diabetic mice. J Invest Dermatol 2012;132(3 Pt 1):711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jujo K, Hamada H, Iwakura A, Thorne T, Sekiguchi H, Clarke T, et al. CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc Natl Acad Sci U S A 2010;107:11008–11013 [DOI] [PMC free article] [PubMed] [Google Scholar]