

Abstract
Significance: Efficient recruitment of neutrophils to an injured skin lesion is an important innate immune response for wound repair. Defects in neutrophil recruitment lead to impaired wound healing.
Recent Advances: Chemokines and chemokine receptors are known to regulate neutrophil recruitment. Recent research advances reveal more mechanistic details about the regulation of chemokines and chemokine receptors on neutrophil egress from bone marrow, transmigration into the wound site, spatial navigation toward the necrotic skin tissue, and apoptosis-induced clearance by efferocytosis.
Critical Issues: Skin injury triggers local and systemic alterations in the expression of multiple chemotactic molecules and the magnitude of chemokine receptor-mediated signaling. The responses of a number of CXC and CX3C chemokines and their receptors closely associate with the temporal and spatial recruitment of neutrophils to wound sites during the inflammatory phase and promote the clearance of necrotic neutrophils during the transition into the proliferative phase. Functional aberrancy in these chemokines and chemokine receptor systems is recognized as one of the important mechanisms underlying the pathology of impaired wound healing.
Future Directions: Future research should aim to investigate the therapeutic modulation of neutrophil activity through the targeting of specific chemokines or chemokine receptors in the early inflammatory phase to improve clinical management of wound healing.

Ann Richmond, PhD
Scope and Significance
This review highlights the roles of chemoattractants and chemokine receptors in the regulated recruitment of neutrophils into acute skin wounds. It aims to provide readers with an overview of the multistep process of the trafficking and chemotaxis of neutrophils under the control of chemokines and chemokine receptors. It also reviews the recent advances in chemokine modulation of the resolution of neutrophilic inflammation during the progression from an inflammatory phase to proliferative phase during wound repair. Both temporal infiltration and clearance of wound neutrophils are critical for a normal wound healing.
Translational Relevance
The wound healing process can be divided into inflammatory, proliferative, and remodeling phases. Chemokines play important roles for the recruitment of leukocytes into the wound bed. During the inflammatory phase, neutrophils are the major immune cells recruited to the wounds to rebuild a provisional barrier against microbe invasion. Neutrophils produce reactive oxygen species (ROS) and proteases and also function to debride devitalized tissue. These functions are required in a timely manner. Chronic wounds usually associate with defect neutrophil function or with an extended period of wound inflammation.
Clinical Relevance
Defect of neutrophil infiltration makes wounds vulnerable to infection. An uncontrolled neutrophil activity with excessive production of proinflammatory mediators and proteases is often observed in chronic wound tissues. Chemokines and chemokine receptors are important regulators of the robust recruitment and activation of neutrophils in local skin lesions, as well as for promoting the egress of mature neutrophils from bone marrow into the peripheral blood pool. The wound chemokine profiles have been assessed to characterize the inflammatory status of wound tissues and evaluate the effectiveness of various treatments for chronic wounds.4–7
Overview
Damage to the skin immediately initiates host responses both topically and systemically. Depending on the severity of injury and the likely involvement of invasive pathogens, the skin tissues respond swiftly to wounding by mobilization of the host innate immune system, characterized by infiltration of abundant neutrophils, followed by infiltration of subsets of monocytic phagocytes and T lymphocytes. In addition, the major skin resident cells, such as fibroblasts, vascular endothelial cells, and keratinocytes, are activated to participate in the formation of granulation tissues and the rebuilding of the epidermal barrier. The final stage of wound healing is tissue remodeling characterized mainly by the clearance, deposition, and reorganization of the extracellular matrix. The above pathophysiology of wound healing is categorized into three succeeding and partially overlapping phases, namely, the inflammatory, proliferative, and remodeling phases. The initiation, transition, and duration of each phase is required to be precisely regulated by many biologically active molecules; otherwise, an impaired healing will result. Currently, these molecules are locally induced cytokines, chemokines, and growth factors capable of actively modulating the migration, metabolism, proliferation, and fate of cells in wound tissues.1 The biological activities of chemokines and chemokine receptors have been intensively studied in various cutaneous wound healing models and are therefore the main focus of this review.
Discussion of Findings and Relevant Literature
Neutrophil functions in wound inflammation
Acute inflammation is the first tissue response to wounding and is believed to be initiated by the immediate activation of platelets and monocytes entrapped in structurally destructed wound tissues through bleeding. Shortly, a cascade of activation or mobilization of more cell types leads to the swift formation of a proinflammatory microenvironment which, depending on the extent of tissue damage, normally lasts about 1 week, as shown in many experimental animal models. The influx of neutrophils is one of the notable events in acute wound inflammation. Neutrophils are the first immune cells recruited to the wound and they prevent microbe invasion through the processes of phagocytosis and generation of cellular oxidative species. Neutrophils also play roles in preparing the wound tissues for the following proliferative phase through release of vascular endothelial growth factor (VEGF) and proteolytic enzymes.2,3
Neutrophil recruitment to wound tissue
Chemokines are a family of small (8–11 kD) chemotactic peptides classified into CXC, CC, CX3C, or C chemokines based on the positioning of the conserved cysteine residues in the amino-terminus regions (Fig. 1). Forty-six chemokines have been discovered, and the expression of a number of chemokines has been observed in skin tissues,4 as summarized in Table 1.
Figure 1.

Classification of chemokines and chemokine receptors. Most chemokines interact with multiple receptors, and a single receptor can interact with multiple chemokines. This is the case for most CC and CXC chemokines. Decoy receptors can also bind multiple chemokines. By contrast, a minority of receptors has only one ligand (refer to Bachelerie et al.50).
Table 1.
Chemokines involved in neutrophil migration and in skin wound healing
| Chemokine name | Alternate name | Source | Function |
|---|---|---|---|
| CXCL4 | PF4 | Platelets | Neutrophil recruitment |
| CXCL8 | IL-8 | Keratinocytes, epithelial cells | Polymorphonuclear leukocyte attractant |
| CXCL12 | SDF-1 | Platelets | Proliferation and recruitment |
| CXCL10 | IP-10 | Multiple cell types | Recruitment of monocytes and T lymphocytes |
| CX3CL1 | Fractalkine | Lymphocytes, endothelial cells, macrophages, vascular smooth muscle cells | Monocyte/macrophage recruitment |
| CCL3 | MIP-1α | Macrophages, endothelial cells, fibroblasts, neutrophils | Polymorphonuclear and T-lymphocyte recruitment |
| CCL4 | MIP-1β | Neutrophils | Leukocyte recruitment |
| CCL5 | RANTES | Platelets, macrophages, CD4+ and CD8+ T lymphocytes | Recruitment of monocytes, memory T-helper cells, and eosinophils |
Neutrophil infiltration is one of the hallmarks of acute skin wounds (Fig. 2). Neutrophils constitute about 50% of all cell types in wounds at day 1 after injury.5 The abrupt infiltration of neutrophils is initiated by chemoattractants, which are abundantly produced and released locally in the wound microenvironment. The onset of the cascade of chemoattractant production lies on the immediate activation of platelets upon their adhesion and aggregation in wound blood clots. The activated platelets release a battery of biologically active molecules, including the platelet-derived growth factor (PDGF), transforming growth factor beta (TGFβ), VEGF, platelet factor 4 (PF4, CXCL4), and a number of other chemokines (Table 1).6 PF4 is a potent chemoattractant for neutrophils that likely along with cytokines from mast cells, triggers the cascade of persistent leukocyte recruitment. Stromal-derived growth factor 1 (SDF-1, CXCL12) is also released by activated platelets in skin lesions and its functions in skin lesions include the enhancement of proliferation of tissue-resident cells and recruitment of bone marrow-derived progenitor cells. Therefore, chemokines are the first-line local mediators responsible for initiating the inflammatory phase.
Figure 2.

Schematic illustration of the regulation of neutrophil recruitment into cutaneous wounds by both topical and systemic elements. Chemokine gradients are formed by secretions from platelets, monocytes, existing neutrophils, and resident cells in skin wound tissues shortly after injury. Circulating neutrophils transmigrate through venules and then follow a chemokine gradient to the site of the wound surface. Simultaneously, G-CSF (granulocyte colony-stimulating factor) and CXC-chemokines are released and delivered by blood circulation to bone marrow, leading to an imbalance between CXCR4- and CXCR2-mediated actions. Consequently, more matured bone marrow neutrophils egress into the peripheral pool and make their way to wound sites. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
As a large number of neutrophils are recruited to the wound site in such a short period of time postwounding, it is not surprising that many types of cells are involved in the intense secretion of chemoattractants in the early stage of acute inflammatory phase. Recent studies showed that elevations in IL-8 (CXCL8), IP-10 (CXCL10), MIP-1α (CCL3), and MIP-1β (CCL4) were detected in the wound fluid of human surgical wounds at the first postoperative day and the constituents of these chemokines favored a proinflammation profile.7 Moreover, the destruction of the integrity of the skin followed by invasion by microbes activates the Toll-like receptors (TLRs) through the multiple endogenous damage-associated molecular patterns (DAMPs) and the conserved pathogen-associated molecular patterns. TLRs are ubiquitously localized to the surface of many types of cells in skin tissues.8,9 Many of the receptors in the TLR family are capable of activating the signal transduction pathway of nuclear factor kappa-beta (NF-κB) in keratinocytes, vascular endothelial cells, fibroblasts, monocytes, and T lymphocytes in wounds leading to production of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6.9 Each of these proinflammatory cytokines is able to initiate production of cascades of more proinflammatory mediators, including chemokines in wound tissues (Table 1). Of note, the recruited neutrophils are an important source of chemokines, and the release of these chemokines plays a critical role for consistent recruitment of other neutrophils and leukocyte subtypes through extravasation.10,11 For example, the recruited neutrophils are responsible for increased production of KC (CXCL1), MIP-1α (CCL3), and RANTES (CCL5) in a skin air-pouch wounding model.10
Enforced action of chemokines and chemokine receptors on neutrophil recruitment
Persistent influx of neutrophils into wounding sites relies on extravasation of neutrophils from postcapillary venules in tissue lesions. The recruitment of neutrophils starts at the interface between hemopoietic cells and endothelial cells and encompasses several steps, in which neutrophils undergo slow rolling, adhesion strengthening, intraluminal crawling, paracellular and transcellular migration, and migration through the basement membrane.12,13 As more details in the molecular mechanisms of each step are unraveling, it is clear that the expression of chemokines on the abluminal surface of the endothelium is critical for the arrest of rolling neutrophils before transmigration pursues. To arrest the rolling of neutrophils, neutrophil surface integrin molecules need to bind to endothelium surface adhesion molecules with appropriate affinities. Therefore, integrins need to undergo conformational changes from low affinity to intermediate or high affinity to become active. The activation of integrins is completed in milliseconds and is highly dependent on the intracellular events triggered by the binding of chemoattractants to their corresponding receptors on the neutrophil surface. In a wounding induced acute inflammatory microenvironment, endothelial cells are activated to express adhesion molecules and chemokines. In addition, activated platelets and mast cells transfer microparticles or exocytosed intracellular granules containing chemokines, such as CXCL4, CXCL5, and RANTES (CCL5), to the nearby endothelial cells.14,15 CXCL8 is also transported to and immobilized on the luminal surface of endothelial cells through transcytosis after being secreted by activated cells in the inflamed tissues.16 Leukocytes expressing G-protein–coupled receptors (GPCRs), including chemokine receptors at the plasma membrane, roll along the surface of the endothelium and bind the ligands for these GPCRs presented on endothelial cells; this event swiftly initiates a cascade of intracellular signaling leading to integrin activation through an inside-out signaling process. As a result, the activated integrins are able to anchor the rolling leukocytes firmly on the endothelium allowing for initiation of their transmigration.17–19 The molecular mechanisms underlying chemokine receptor-dependent inside-out modulation of integrin activation are currently under vigorous investigation and have been known to be mediated by an early increase in intracellular Ca2+ followed by a late activation of the small GTPase Rap1.20,21 Furthermore, the paired immunoglobulin-like type 2 receptor (PILRα) has been found to negatively regulate the chemoattractant binding-induced integrin activation.22 The PILRα−/− neutrophils show robust transmigration activities in a number of acute inflammation models. These data reveal a pivotal role for neutrophil arrest in neutrophilic inflammation and render the related chemokines and their receptors as potential targets for modulation of wound inflammation (Table 1).
The simultaneous expression of multiple chemokines in inflamed wound tissues was previously considered to be redundant, ensuring a very robust infiltration of leukocytes. However, the chemoattractant expression and function are regulated in a restrictively temporal and spatial manner. For example, on one hand, the proinflammatory cytokine TNF-α −and IL-1–induced expression of CXCL1 is localized in keratinocytes at the stratum spinosum and granulosum, and in surface exudate of the wound during the early inflammatory phase.23 The CXCL8 expression also localizes at the superficial wound bed where a chemokine gradient is maintained. In addition, prominent expression of both CXCL1 and CXCL8 peaks at day 1 postwounding and persists for the first four postwounding days.23,24 On the other hand, the expression of chemokine receptors on leukocytes is dynamically and differentially modified in the wound microenvironment. For example, CXCL8 upregulates the expression of the neutrophil chemokine receptor CXCR1, while the expression of neutrophil CXCR2 is suppressed by the proinflammatory cytokine TNF-α in the inflamed tissues. Furthermore, experimental data show that CXCL8-induced respiratory burst in neutrophils depends on CXCR1-mediated signaling rather than that of CXCR2.24–26 Therefore, the timely and spatially expressed inflammatory mediators are setting the favorable tissue milieu not only for leukocytes to migrate precisely to the wounding sites but also to enforce their antimicrobial capability through the modulation of chemokine receptor expression.
Another interesting finding about persistent recruitment of neutrophils to the necrotic center of a tissue lesion is the ability of neutrophils to prioritize different receptor-mediated intracellular signaling. This occurs as neutrophils move through diverse gradient zones formed by chemotactic molecules, which are released either from necrotic neutrophils or activated resident cells. In a tissue injury model, distinct inner and outer chemoattractant zones surrounding the foci of the necrotic lesion are formed. The inner zone comprised the gradients of C5a, C3a, and N-formyl peptides. In the outer zone, the DAMPs and adenosine triphosphate molecules released by injured necrotic cells stimulate tissue-resident cells to secret IL-1β that upregulates intravascular intercellular adhesion molecule 1 (ICAM-1) expression. Meanwhile, intravascular gradients of CXCR2 ligands CXCL1 and CXCL2 are formed toward the necrotic site. When approaching from the outer zone into the inner zone, neutrophils at first prioritize the CXCR2-mediated phosphatidylinositol 3-kinase (PI3K) activation for intravascular chemotaxis, then prioritize the formyl peptide receptor 1 (FPR1)-mediated activation of both PI3K and P38 mitogen-activated protein kinase (MAPK) cascades for necrotaxis.27–30 Therefore, differential prioritization of receptors allows neutrophils to be recruited spatially and sequentially from peripheral regions toward the center of necrotic lesions (Fig. 3).
Figure 3.
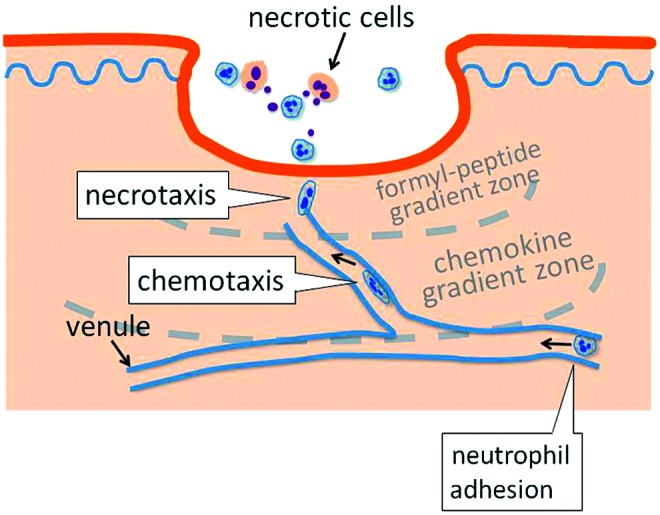
Multistep intravascular trafficking of neutrophils toward the site of injury. Necrotic cells induce spatially distinct chemoattractant gradient zones around the center of necrotic foci in the wound bed. When entering the wound adjacent area, neutrophils traffic at first by CXCR2-mediated chemotaxis along the endothelium lumen following intravascular chemokine gradients. Afterward, formyl-peptide receptor-dependent signaling dominates and neutrophils migrate into sites of necrosis by necrotaxis through a formyl-peptide gradient. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Among the chemokine receptors that play critical roles in wound repair, CXCR1 and CXCR2 have been shown as dominant receptors for leukocyte chemotaxis in human inflammatory tissues.31–35 In rodent models, CXCR2 is speculated as the major chemokine receptor for leukocyte recruitment into the wound sites. We previously observed that CXCR2−/− mice showed delayed healing in a skin excisional wounding model.32 Such deficiency in healing was also confirmed in CXCR2−/− mice with a chemical-induced skin injury model.36 Specifically, CXCR2 deficiency associates with a spectrum of dysfunctions of cell migration and proliferation in keratinocytes and endothelial cells leading to delayed reepithelialization, granulation tissue formation, and neovascularization. Notably, neutrophil infiltration was severely impaired in the inflammatory phase of wound healing that may likely negatively impact granulation tissue formation, neovascularization, and reepithelialization in the CXCR2−/− mice. In addition, a nonpeptide antagonist against CXCR2 abolished CXCR2-mediated signaling and delayed wound closure.36 In contrast, in mice with a β-arrestin-2 gene knockout that leads to constitutively upregulated CXCR2 signaling, neutrophil recruitment into both CXCL1 initiated skin inflammatory tissues and excisional skin wound tissues was significantly enhanced and the wound reepithelialization was expedited (Fig. 4).37
Figure 4.

Chemokine receptor CXCR2 plays important roles for cutaneous wound healing. Ligand binding-induced CXCR2 signaling is required for normal recruitment of neutrophils and macrophages and for neovascularization and wound reepithelialization (A). In CXCR2−/− mice (B), the lack of CXCR2-mediated signaling leads to delayed neutrophil and macrophage recruitment into wound tissues, compromised angiogenesis, and significantly delayed reepithelialization. β-arrestin2 (βarr2) binds to the G-protein–coupled receptor kinase phosphorylated C-terminus of CXCR2 and functions as a regulator for CXCR2 signaling. In comparison to normal CXCR2 signaling during wound healing (C), knockout of βarr2 impedes receptor internalization so that CXCR2 receptors are detained in the cell membrane, leading to enhanced CXCR2-mediated signaling. As a result, neutrophil recruitment, wound tissue vessel density, and reepithelialization are significantly improved (D). To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Mobilization of bone marrow leukocyte progenitor cells by chemokines and chemokine receptor systems
The infiltration of neutrophils into wounds requires the provision of a persistent supply from the peripheral vasculature of neutrophils that mature at and are released from the bone marrow. Abundant neutrophils are produced in the bone marrow and retained there for their whole lifespan unless released into the peripheral pool under certain pathological conditions. It has been shown that severe skin injury, infection, and other diseases are able to mobilize the production of bone marrow neutrophil precursor cells and promote the release of mature neutrophils into the peripheral circulation pool.38 Currently, a large body of data support the notion that the egress of neutrophils is antagonistically regulated by the CXCR2 and CXCR4 chemokine receptor systems (Fig. 5). In the bone marrow microenvironment, the CXCR2 ligands CXCL1 and CXCL2 are constitutively expressed mainly by endothelial cells, while the CXCR4 ligand CXCL12 is largely expressed by osteoblasts. Although bone marrow neutrophils express both CXCR2 and CXCR4 on their surface, the CXCL12 dominates to retain most neutrophils within bone marrow through augmented binding of α4 integrin very later antigen (VLA-4) on neutrophils to vascular cell adhesion molecule 1 (VCAM-1) on bone marrow endothelial and stromal cells.38,39 During maturation in bone marrow, the expression of neutrophil CXCR4 and VLA-4 are downregulated, resulting in a switch of signaling dominance from CXCR4 to CXCR2 that promotes the release of mature neutrophils into the peripheral pool. Under pathological conditions, however, skin injury and wound infection stimulate the production of CXCR2 ligands as well as granulocyte colony-stimulating factor (G-CSF). The G-CSF on one hand decreases the number of osteoblasts leading to the reduction of CXCL12 expression, while simultaneously upregulating the expression of CXCR2 ligands. On the other hand, G-CSF enforces or prioritizes the switch of hematopoiesis toward myeloid lineages through the G-CSF signal transducer and activator of transcription 3 (STAT3) axis.40,41 Consequently, more mature neutrophils are developed and released into peripheral circulation.
Figure 5.
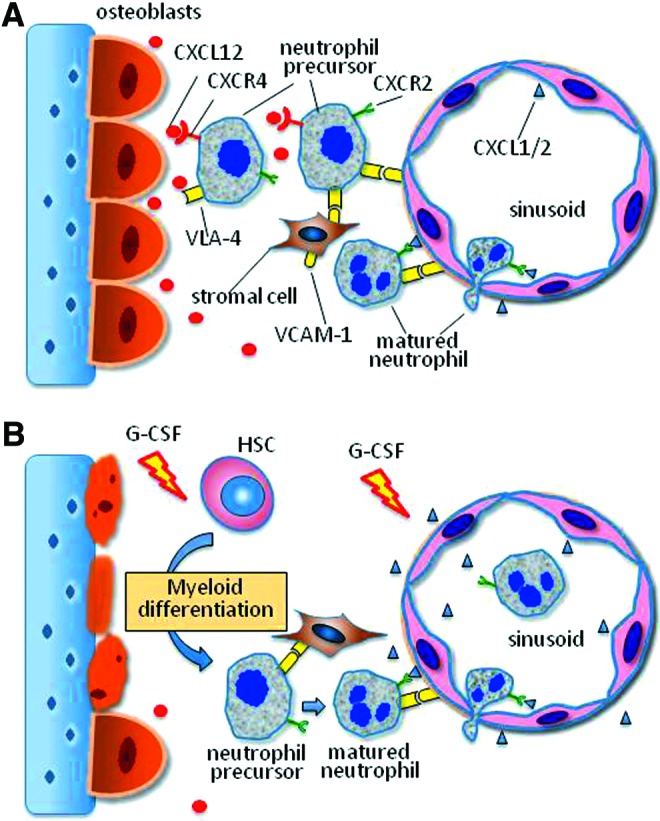
CXCR2/CXCR4 ligands antagonistically regulate neutrophil egress from bone marrow to peripheral pool. (A) Under physiological conditions, CXCR2 ligands CXCL1 and CXCL2 are constitutively expressed mainly by endothelial cells, while the CXCR4 ligand CXCL12 is largely expressed by osteoblasts in the bone marrow. Although bone marrow neutrophils express both CXCR2 and CXCR4, the CXCL12/CXCR4 interaction dominates, resulting in retention of most neutrophils within bone marrow through augmented binding of VLA-4 on neutrophils to VCAM-1 on bone marrow endothelial and stromal cells. During the maturation in bone marrow, the expression of neutrophil CXCR4 and VLA-4 are downregulated, resulting in a switch of dominant signaling from CXCR4 to CXCR2 that promotes egress of neutrophils into the peripheral pool. (B) Under pathological conditions, injury and infection stimulate production of CXCR2 ligands and G-CSF. G-CSF decreases the osteoblast numbers leading to reduction of CXCL12 expression, while simultaneously upregulating the expression of CXCR2 ligands. In addition, G-CSF prioritizes the switch of hematopoiesis toward myeloid lineages through the G-CSF-STAT3 axis. As a result, more mature neutrophils are developed and released into peripheral circulation. STAT3, signal transducer and activator of transcription 3; VCAM-1, vascular cell adhesion molecule 1; VLA-4, α4 integrin very later antigen. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Resolution of neutrophilic inflammation in wounds
Acute wound inflammation is regarded as an innate immune response to injury. The prominent neutrophil infiltration at the early inflammatory phase is critical for the restoration of tissue homeostasis. Neutrophils are capable of performing versatile functions upon their extravasation into wound tissues. After infiltrating into the wounds, the neutrophils normally function to debride the devitalized tissue, phagocytize microbe particles, synthesize and release ROS and proteases. Acute wounds require these functions of neutrophils during the early stage postwounding, so that the microenvironment of injured tissues will favor the progression of wound physiopathology from the inflammatory phase into the proliferative phase.42,43
Neutrophils along with macrophages are the major cell sources for protease and ROS production. Therefore, withdrawal of neutrophils and resolution of neutrophilic inflammation at the end of the inflammatory phase play important roles for appropriate wound healing. How neutrophil-associated inflammatory events are resolved is not fully understood. The cleavage of proinflammatory CXC chemokines by macrophage-specific matrix metalloproteinases (MMPs) and apoptotic neutrophil-induced clearance are pivotal steps for the resolution of wound inflammation. Following the infiltration of neutrophils, monocytes are recruited into wound tissues where they undergo differentiation into macrophages. Macrophage-specific MMP-12 was characterized in a skin air-pouch acute inflammation model and has been shown to effectively cleave several ELR+ CXC-chemokines at the ELR motif, thus rendering the chemokine incapable of activating its receptor. Several monocyte chemotactic proteins, namely, CCL2, 7, 8, and 13, are also modified by macrophage-specific MMP-12 and turned into antagonists.44 In mice, since the ELR+ CXC chemokines bind to the neutrophil chemotactic CXCR2 receptor, depletion of the functional CXC chemokines by cleavage of the ELR motif may markedly discontinue the neutrophil influx in the inflammatory wound tissues.45 Moreover, although the lifespan of neutrophils is usually prolonged after recruitment into inflamed tissues, local factors in the wound are capable of triggering neutrophils to undergo apoptosis, likely through the PI3K-mediated ROS production pathway.46,47 The apoptotic cells actively initiate their own clearance by macrophage efferocytosis, in which several find-me molecules establish unique signal cues for the attraction of macrophages to the site of apoptotic cells.48,49 CX3CL1 is one of the proposed find-me signaling molecules. As CX3CL1 and its receptor CX3CR1 are involved in macrophage recruitment and mediate skin wound healing,34 it is speculated that, similar to other inflammatory models, CX3CL1-mediated efferocytosis is an important mechanism for resolution of neutrophilic inflammation in wound tissues through clearance by recruited macrophages.
Take-Home Message.
• Infiltration of abundant neutrophils is critical for wound repair and prevention of microbe invasion.
• Efficient infiltration of wound neutrophils depends on multistep processes that include rolling, arrest, transmigration, and chemotaxis events. The infiltrated neutrophils are sourced from bone marrow where they differentiate and mature before egress into the peripheral blood pool.
• The injured skin tissues produce CXC and CCL chemokines and other chemoattractant molecules. These chemotactic substances establish gradients that guide neutrophils and other types of leukocytes to navigate into the wounding sites. Meanwhile, the injury triggers the production of G-CSF that converts the CXCR4 dominant signaling to that of CXCR2 in the bone marrow microenvironment, leading to the release of more mature neutrophils into the peripheral blood stream.
• The timely clearance of necrotic neutrophils from wound sites is critical for wound healing to progress from the early inflammatory phase to the next proliferative phase. The chemokine CX3CL1 and its receptor CX3CR1 plays important roles in the clearance of wound necrotic neutrophils.
Summary
Efficient recruitment of neutrophils to an injured skin lesion is the earliest crucial step for restoration of tissue homeostasis; moreover, neutrophilic infiltration is a prerequisite for the inflammatory-to-proliferative transition that is necessary for quality wound repair. Redundancy and overlapping functions among chemokines and chemokine receptors have complicated data interpretation of experiments designed to alter specific chemokine functions in wound tissues. However, insights gained from recent research into the spatial and temporal modulation of neutrophil chemotaxis and fate during wound repair underscore the differential actions of individual chemokines or chemokine receptors in the development of acute wound inflammation. Additional experimental data will be needed to provide the basis of therapeutic modulation of neutrophil activity through the targeting of specific chemokines or chemokine receptors in the early inflammatory phase to improve clinical management of wound healing.
Abbreviations and Acronyms
- C3
complement component 3
- C5
complement component 5
- CCL
chemokine (C-C motif) ligand
- CCR
chemokine (C-C motif) receptor
- CX3CL
chemokine (C-X3-C motif) ligand
- CXCL
chemokine (C-X-C motif) ligand
- CX3CR
chemokine (C-X3-C motif) receptor
- CXCR
chemokine (C-X-C motif) receptor
- DAMPs
damage-associated molecular patterns
- ELR
glutamate, leucine, arginine
- FPR1
formyl peptide receptor 1
- G-CSF
granulocyte colony-stimulating factor
- GPCRs
G-protein–coupled receptors
- ICAM-1
intercellular adhesion molecule 1
- IL
interleukin
- IP-10
interferon inducible protein-10
- KC
murine ortholog of CXCL1 or CXCL8
- MAPK
mitogen-activated protein kinase
- MMPs
matrix metalloproteinases
- MIP-1α
macrophage inflammatory protein 1-alpha
- MIP-1β
macrophage inflammatory protein 1-beta
- NF-κB
nuclear factor of kappa light polypeptide gene enhancer in B cells
- PDGF
platelet-derived growth factor
- PF4
platelet factor 4
- PI3K
phosphatidylinositol 3-kinases
- PILRα
paired immunoglobulin-like type 2 receptor
- RANTES
regulated upon activation normal T cell expressed and secreted factor
- ROS
reactive oxygen species
- SDF-1
stromal-derived growth factor 1
- STAT3
signal transducer and activator of transcription 3
- TGFβ
transforming growth factor beta
- TLRs
Toll-like receptors
- TNF-α
tumor necrosis factor alpha
- VEGF
vascular endothelial growth factor
- VLA-4
α4 integrin very later antigen
- VCAM-1
vascular cell adhesion molecule 1
Acknowledgments and Funding Sources
This work is partly supported by an award from Social Development Foundation of Shaanxi Province to Yingjun Su (No. 2012K16-01-05) and grants from the NCI (CA34590) and the Department of Veterans Affairs (Senior Research Career Development Award) to Ann Richmond.
Author Disclosure and Ghostwriting
The authors declare that no competing financial interests exit. The content of this article was expressly written by the authors listed. No ghostwriters were used to write this article.
About the Authors
Dr. Yingjun Su earned his MD and PhD in China and then did postdoctoral research in Yale and Vanderbilt Medical School, USA. He undertook positions as an instructor, then a research assistant professor in the Department of Cancer Biology, Vanderbilt School of Medicine. His research interests focus on wound healing and cancer biology of melanoma. He is currently an associate professor at the Institute of Plastic Surgery, Xijing Hospital, Xi'an, China. Professor Ann Richmond earned a PhD in Developmental Biology and did postdoctoral research in tumor biology at the Emory University School of Medicine. Then, she joined the faculty at Emory Medical School as assistant professor of medicine. She joined the Department of Cell Biology at Vanderbilt in 1989 as an associate professor and was promoted to full professor in 1995. In 2000, she became Ingram Professor and vice chairman of the Department of Cancer Biology. The primary focus of her research has been to characterize the role of inflammatory mediators and chemokines in tumor progression and metastasis. Dr. Richmond has authored over 164 scientific articles, reviews, and chapters and her work is highly cited. Her early research involved the purification of one of the first chemokines, CXCL1, from a melanoma culture medium, which she named MGSA.
References
- 1.Holzheimer RG, Steinmetz W. Local and systemic concentrations of pro- and anti-inflammatory cytokines in human wounds. Eur J Med Res 2000;5:347–355 [PubMed] [Google Scholar]
- 2.Gaudry M, Bregerie O, Andrieu V, El Benna J, Pocidalo MA, Hakim J. Intracellular pool of vascular endothelial growth factor in human neutrophils. Blood 1997;90:4153–4161 [PubMed] [Google Scholar]
- 3.Wysocki AB, Staiano-Coico L, Grinnell F. Wound fluid from chronic leg ulcers contains elevated levels of metalloproteinases MMP-2 and MMP-9. J Invest Dermatol 1993;101:64–68 [DOI] [PubMed] [Google Scholar]
- 4.Koelink PJ, Overbeek SA, Braber S, et al. Targeting chemokine receptors in chronic inflammatory diseases: an extensive review. Pharmacol Ther 2012;133:1–18 [DOI] [PubMed] [Google Scholar]
- 5.Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol 2001;69:513–521 [PubMed] [Google Scholar]
- 6.Anitua E, Andia I, Ardanza B, Nurden P, Nurden AT. Autologous platelets as a source of proteins for healing and tissue regeneration. Thromb Haemost 2004;91:4–15 [DOI] [PubMed] [Google Scholar]
- 7.Grimstad O, Sandanger O, Ryan L, et al. Cellular sources and inducers of cytokines present in acute wound fluid. Wound Repair Regen 2011;19:337–347 [DOI] [PubMed] [Google Scholar]
- 8.Lotze MT, Zeh HJ, Rubartelli A, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev 2007;220:60–81 [DOI] [PubMed] [Google Scholar]
- 9.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol 2005;17:1–14 [DOI] [PubMed] [Google Scholar]
- 10.Kasama T, Miwa Y, Isozaki T, Odai T, Adachi M, Kunkel SL. Neutrophil-derived cytokines: potential therapeutic targets in inflammation. Curr Drug Targets Inflamm Allergy 2005;4:273–279 [DOI] [PubMed] [Google Scholar]
- 11.Chou RC, Kim ND, Sadik CD, et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity 2010;33:266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev 2005;16:625–636 [DOI] [PubMed] [Google Scholar]
- 13.Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med 2006;203:2569–2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med 2003;9:61–67 [DOI] [PubMed] [Google Scholar]
- 15.von Hundelshausen P, Weber KS, Huo Y, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 2001;103:1772–1777 [DOI] [PubMed] [Google Scholar]
- 16.Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood 2002;100:3853–3860 [DOI] [PubMed] [Google Scholar]
- 17.Deevi RK, Koney-Dash M, Kissenpfennig A, et al. Vasodilator-stimulated phosphoprotein regulates inside-out signaling of beta2 integrins in neutrophils. J Immunol 2010;184:6575–6584 [DOI] [PubMed] [Google Scholar]
- 18.Hogg N, Patzak I, Willenbrock F. The insider's guide to leukocyte integrin signalling and function. Nat Rev Immunol 2011;11:416–426 [DOI] [PubMed] [Google Scholar]
- 19.Shamri R, Grabovsky V, Gauguet JM, et al. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat Immunol 2005;6:497–506 [DOI] [PubMed] [Google Scholar]
- 20.Bergmeier W, Goerge T, Wang HW, et al. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J Clin Invest 2007;117:1699–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyduk SJ, Chan JR, Duffy ST, et al. Phospholipase C, calcium, and calmodulin are critical for alpha4beta1 integrin affinity up-regulation and monocyte arrest triggered by chemoattractants. Blood 2007;109:176–184 [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Shiratori I, Uehori J, Ikawa M, Arase H. Neutrophil infiltration during inflammation is regulated by PILRalpha via modulation of integrin activation. Nat Immunol 2013;14:34–40 [DOI] [PubMed] [Google Scholar]
- 23.Nanney LB, Mueller SG, Bueno R, Peiper SC, Richmond A. Distributions of melanoma growth stimulatory activity of growth-regulated gene and the interleukin-8 receptor B in human wound repair. Am J Pathol 1995;147:1248–1260 [PMC free article] [PubMed] [Google Scholar]
- 24.Engelhardt E, Toksoy A, Goebeler M, Debus S, Brocker EB, Gillitzer R. Chemokines IL-8, GROalpha, MCP-1, IP-10, and Mig are sequentially and differentially expressed during phase-specific infiltration of leukocyte subsets in human wound healing. Am J Pathol 1998;153:1849–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones SA, Wolf M, Qin S, Mackay CR, Baggiolini M. Different functions for the interleukin 8 receptors (IL-8R) of human neutrophil leukocytes: NADPH oxidase and phospholipase D are activated through IL-8R1 but not IL-8R2. Proc Natl Acad Sci U S A 1996;93:6682–6686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moser B, Clark-Lewis I, Zwahlen R, Baggiolini M. Neutrophil-activating properties of the melanoma growth-stimulatory activity. J Exp Med 1990;171:1797–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foxman EF, Campbell JJ, Butcher EC. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J Cell Biol 1997;139:1349–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heit B, Robbins SM, Downey CM, et al. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat Immunol 2008;9:743–752 [DOI] [PubMed] [Google Scholar]
- 29.Khan AI, Heit B, Andonegui G, Colarusso P, Kubes P. Lipopolysaccharide: a p38 MAPK-dependent disrupter of neutrophil chemotaxis. Microcirculation 2005;12:421–432 [DOI] [PubMed] [Google Scholar]
- 30.McDonald B, Pittman K, Menezes GB, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010;330:362–366 [DOI] [PubMed] [Google Scholar]
- 31.Addison CL, Daniel TO, Burdick MD, et al. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol 2000;165:5269–5277 [DOI] [PubMed] [Google Scholar]
- 32.Devalaraja RM, Nanney LB, Du J, et al. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol 2000;115:234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heidemann J, Ogawa H, Dwinell MB, et al. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem 2003;278:8508–8515 [DOI] [PubMed] [Google Scholar]
- 34.Ishida Y, Gao JL, Murphy PM. Chemokine receptor CX3CR1 mediates skin wound healing by promoting macrophage and fibroblast accumulation and function. J Immunol 2008;180:569–579 [DOI] [PubMed] [Google Scholar]
- 35.Keeley EC, Mehrad B, Strieter RM. Chemokines as mediators of neovascularization. Arterioscler Thromb Vasc Biol 2008;28:1928–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milatovic S, Nanney LB, Yu Y, White JR, Richmond A. Impaired healing of nitrogen mustard wounds in CXCR2 null mice. Wound Repair Regen 2003;11:213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su Y, Raghuwanshi SK, Yu Y, Nanney LB, Richardson RM, Richmond A. Altered CXCR2 signaling in beta-arrestin-2-deficient mouse models. J Immunol 2005;175:5396–5402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosinski M, Yarmush ML, Berthiaume F. Quantitative dynamics of in vivo bone marrow neutrophil production and egress in response to injury and infection. Ann Biomed Eng 2004;32:1108–1119 [DOI] [PubMed] [Google Scholar]
- 39.Petty JM, Lenox CC, Weiss DJ, Poynter ME, Suratt BT. Crosstalk between CXCR4/stromal derived factor-1 and VLA-4/VCAM-1 pathways regulates neutrophil retention in the bone marrow. J Immunol 2009;182:604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gardner JC, Noel JG, Nikolaidis NM, et al. G-CSF drives a posttraumatic immune program that protects the host from infection. J Immunol 2014;192:2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wengner AM, Pitchford SC, Furze RC, Rankin SM. The coordinated action of G-CSF and ELR+CXC chemokines in neutrophil mobilization during acute inflammation. Blood 2008;111:42–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tecchio C, Cassatella MA. Neutrophil-derived cytokines involved in physiological and pathological angiogenesis. Chem Immunol Allergy 2014;99:123–137 [DOI] [PubMed] [Google Scholar]
- 43.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med 1989;320:365–376 [DOI] [PubMed] [Google Scholar]
- 44.Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE, Overall CM. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 2008;112:3455–3464 [DOI] [PubMed] [Google Scholar]
- 45.Clark-Lewis I, Schumacher C, Baggiolini M, Moser B. Structure-activity relationships of interleukin-8 determined using chemically synthesized analogs. Critical role of NH2-terminal residues and evidence for uncoupling of neutrophil chemotaxis, exocytosis, and receptor binding activities. J Biol Chem 1991;266:23128–23134 [PubMed] [Google Scholar]
- 46.Geering B, Gurzeler U, Federzoni E, Kaufmann T, Simon HU. A novel TNFR1-triggered apoptosis pathway mediated by class IA PI3Ks in neutrophils. Blood 2011;117:5953–5962 [DOI] [PubMed] [Google Scholar]
- 47.Geering B, Simon HU. A novel signaling pathway in TNFalpha-induced neutrophil apoptosis. Cell Cycle 2011;10:2821–2822 [DOI] [PubMed] [Google Scholar]
- 48.Elliott MR, Chekeni FB, Trampont PC, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009;461:282–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature 2002;417:182–187 [DOI] [PubMed] [Google Scholar]
- 50.Bachelerie F, Ben-Baruch A, Burkhardt AM, et al. International Union of Basic and Clinical Pharmocoloby. XXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev 2014; 16: 1–79 [DOI] [PMC free article] [PubMed] [Google Scholar]