

Abstract
Fragile X is the most common inherited cause of mental retardation with a prevalence of 1 in 4000 for males and 1 in 5000 to 8000 for females. The American College of Medical Genetics and Genomics has recommended diagnostic testing for fragile X in symptomatic persons, women with ovarian dysfunction, and persons with tremor/ataxia syndrome. Although medical and scientific professionals do not currently recommend screening nonsymptomatic populations, improvements in current treatment approaches and ongoing clinical trials have generated growing interest in screening for fragile X. Here, we briefly review the relevant molecular basis of fragile X and fragile X testing and compare three different molecular technologies available for fragile X screening in both males and females. These technologic approaches include destabilizing the CGG-repeat region with betaine and using chimeric CGG-targeted PCR primers, using heat pulses to destabilize C-G bonds in the PCR extension step, and using melting curve analysis to differentiate expanded CGG repeats from normals. The first two-step method performed with high sensitivity and specificity. The second method provided agarose gel images that allow identification of males with expanded CGG repeats and females with expanded CGG-repeat bands which are sometimes faint. The third melting curve analysis method would require controls in each run to correct for shifting optimal cutoff values.
Reduction of the protein coded for by the fragile X mental retardation gene (FMR1) causes fragile X syndrome, a genetic condition that causes a range of developmental problems, including learning disabilities, cognitive impairment, and behavioral abnormalities.1 Expansion of CGG repeats in the 5′ untranslated region (UTR) of the FMR1 gene is associated with hypermethylation and inactivation of gene expression (Figure 1).
Figure 1.

Expansion of the CGG repeats in the 5′ untranslated region of the FMR1 gene on the X chromosome can result in decreased mRNA and FMRP production, causing fragile X. FMRP, FMR1 protein.
Function of the FMR1 Gene
Evidence suggests that the protein coded for by the FMR1 gene on the long arm of chromosome X (Xq27.3) binds mRNA and associates with polyribosomes in neurons.2 The protein likely shuttles mRNA from the nucleus through the cytoplasm and localizes dendritic mRNA where it represses synaptic protein synthesis. After the group 1 metabotropic glutamate receptor is stimulated, regulatory FMR1 protein (FMRP) production is believed to repress mRNA translation and protein synthesis and to control permanent physical changes that alter synaptic connections linked with the process of learning and memory.3–6
Mutations and Associated Conditions
Normal Range
Greater than 99% of fragile X cases are caused by expansions of CGG repeats in the FMR1 5′ UTR.7 The other 1% is caused by a variety of other mutations, primarily including gross deletions and duplications, regulatory mutations, and missense and nonsense mutations. The CGG-repeat numbers of individual genes have been categorized, and the borders of these categories are approximate. The normal range of CGG repeats is considered generally to be as high as 44 CGG repeats, and these repeats are interrupted typically every 9 or 10 repeats by an AGG triplet. These AGG triplets likely anchor the region and prevent slippage during DNA replication. The number and spacing of AGG triplets within CGG-repeat regions may help predict risk of expansion of <100 repeats.8
Gray Zone (Intermediate Range)
The range of 45 to 54 CGG repeats is referred to as the gray zone or intermediate range; for alleles of this size, neither disease associations nor the rate of expansion are fully understood. However, this range is not associated with fragile X syndrome, and gray zone alleles expanding to a full mutation in one generation have not been observed.7
Premutation Range
Alleles with approximately 55 to 199 CGG repeats are considered premutations. These alleles are transmitted unstably from parent to child, and expansions from this range to the full-mutation range typically occur during maternal transmission. Because mutations of this size possibly can have somatic mosaicism that includes a full mutation, careful examination of the range of allele sizes is warranted.7 The smallest FMR1 premutation that was reported to expand to a full mutation (to approximately 538 CGG repeats) in a single generation is 56 CGG repeats. In addition in this case, two AGG interruptions in the grandfather’s gray zone allele of 52 CGG repeats were absent when transmitted to his daughter.9
Expansion of an allele into the premutation range perturbs gene expression,7 and two conditions are associated with this range of expansions. Reductions in FMRP occur in this range and are associated with increased FMR1 mRNA. Premutation alleles may shift transcription of the FMR1 mRNA to an upstream site, and this use of an alternative start site may correlate with increased transcription levels. This RNA-mediated toxicity is associated with fragile X-associated tremor/ataxia syndrome,10,11 a late-onset, progressive development of intention tremor and ataxia frequently accompanied by cognitive and behavioral difficulties. Although most persons with pre-mutations do not show fragile X-related features, females with premutations generally >80 CGG repeats are at approximately 20% risk of fragile X-associated premature ovarian insufficiency.7 Older males and females with premutations are at risk of fragile X-associated tremor/ ataxia syndrome, with higher risk in males. The penetrance of fragile X-associated tremor/ataxia syndrome increases with age and CGG repeat length.
Full Mutations
Expansion of CGG repeats to >200 results in full mutations.7 Full mutations typically range from several hundred to several thousand CGG repeats. Hypermethylation is present typically on most or all DNA copies except DNA extracted from chorionic villus sampling; hypermethylation results typically in lower or no production of FMRP. Relatively small studies have indicated that, when a full mutation on the single X chromosome in males is completely methylated, the most severe form of fragile X results.12 Methylation of large CGG expansions can vary, however, leading to variable phenotypes. Females have two X chromosomes; during normal development, one is methylated randomly and inactivated in different tissues. Therefore, females with full mutations may experience a range of symptoms, depending on whether a normal-X allele or the full-mutation allele is active in a specific tissue.
For full mutations, size mosaics of CGG repeats and methylation mosaics are observed. Analysis of full mutations typically indicates that multiple sizes of CGG repeats are present. Tissue-specific differences can be seen, and persons with size and methylation mosaicism may be higher functioning than persons with completely methylated full mutations.7
Other Mutations in the FMR1 Gene
Currently, the Human Gene Mutation Database Professional 2014.3 Free version (http://www.hgmd.cf.ac.uk/ac/index.php, last accessed November 17, 2014)includes 65known mutations in the FMR1 gene, including the CGG repeat variations in the 5′ UTR. Almost all mutations are associated with fragile X and its related conditions, developmental delay, neurodevelopmental dysfunction, intellectual disability, or syndromic mental retardation. Mutations other than expanded CGG repeats are estimated to cause <1% of fragile X.7 More than one-half of these additional mutations are gross deletions (n = 33). These generally range from several hundred base pairs to millions of base pairs and frequently include the entire FMR1 gene. The next most common nonrepeat mutations are regulatory mutations (n =6). These are point mutations 83 to 332 bp upstream of the initiation codon (n =4) and 760 to 1174 bp downstream of thetermination codon (n =2). Next most common mutations are gross duplications (n =5). All but one include duplication of the entire FMR1 gene. Four point mutations include three missense mutations (p.Arg138Gln, c.413G>A; p.Ile304Asn, c.911T>A; and p.Leu578Phe, c.1732C>T) and one nonsense mutation (p.Ser27Term, c.80C>A). The remaining nonrepeat mutations include two small deletions, one splicing mutation, one small insertion/deletion, and one complex rearrangement.
Approaches to Detecting FMR1 Mutations
Detecting FMR1 Mutations Clinically
The American College of Medical Genetics and Genomics has recommended diagnostic testing for fragile X in symptomatic persons, women with ovarian dysfunction, and persons with tremor/ataxia syndrome, and the Committee on Genetics of the American Academy of Pediatrics has recommended testing for fragile X in all persons with intellectual disability for whom there is no strongly suspected diagnosis.13,14 Fragile X and CGG repeat abnormalities are also a heritable cause of approximately 0.46% to 2% of autism.15,16 Chromosomal microarray analysis, a method that detects submicroscopic genomic deletions and duplications, is used commonly to test for autism, but it does not detect fragile X. The methods for measuring and estimating CGG repeats clinically include Southern blot analysis for size and methylation estimation.7,17–20 In addition, PCR methods were developed to screen for and to clinically diagnose fragile X. Widely used commercial reagents allow the following: i) PCR amplification and quantification of normal and gray zone CGG repeats, ii) amplification and verification of premutations and full mutations by using CGG repeat-targeted primers, iii) determination of methylation with methylation-specific PCR, and iv) identification of AGG interruptions to help assess risk of expansion of < 100 CGG repeats.8,21–25
Sequencing FMR1
Although the coding portion of the FMR1 gene can be sequenced by conventional methods, the tri-nucleotide repeats in the 5′ UTR can approach 100% CG content, depending on the number of AGG interruptions. The tight binding of CG-rich regions causes difficulty in sequencing. The longer the CGG repeat, the more difficult it is to sequence, particularly in the full-mutation range. This means that expanded CGG repeats cannot be sequenced with conventional Sanger sequencing or commonly used next-generation sequencers. However, a new technology, single-molecule real-time sequencing, that may provide a means of better estimating the number of expanded CGG repeats in the future was reported recently.26
Detecting Deletions and Duplications
Gross deletions and insertions or duplications account for 38 of the 65 currently known mutations in FMR1 (Human Gene Mutation Database Professional 2014.3, available at http://www.hgmd.cf.ac.uk/ac/index.php, last accessed November 17, 2014). Methods that detect these types of mutations include Southern blot analyses, SALSA multiplex ligation-dependent probe amplification assay (MRC Holland MLPA, Amsterdam, the Netherlands), and comparative genomic hybridization array analysis.
Screening for Expanded CGG Repeats
The estimated prevalence of males with full mutations is approximately 1 in 4000; prevalence of females with full mutations is approximately 1 in 5000 to 8000.7 All major ethnic groups and races appear to be susceptible to expansion of CGG repeats in FMR1. Although varying prevalences in different ethnic groups occur, few ethnicities have a higher prevalence than the white population. Because of this relatively high prevalence in different ethnicities, improvements in current treatments, and several ongoing clinical trials that are evaluating agents to ameliorate or prevent the damage that occurs in fragile X (Clinical Trials available at http://www.nichd.nih.gov/health/topics/fragilex/clinicaltrials/Pages/default.aspx, last accessed May 2, 2014; FRAXA Research Foundation Clinical Trials available at: http://www.fraxa.org/toward-a-cure/clinical-trials, last accessed June 19, 2014), a growing interest exists in screening for expanded CGG repeats in FMR1.
In addition to methods that use Southern blot analysis, researchers have taken different technical approaches to screening for fragile X. Although commercial reagents are used for testing and screening in a clinical setting, in the newborn screening (NBS), public health setting NBS programs test every baby born in the United States and in other countries. The cost per test must be manageable for states with varying resources. Ideally, the supplies and reagents for a first-tier NBS test should cost ≤ $5 per test, and fragile X commercial reagent kits have higher costs. Various technical approaches to screening large populations target expanded CGG repeats, methylation of the promotor region, or FMRP itself.
Some techniques that detect fragile X in males are not able to detect full mutations reliably in females. These methods include an assessment of FMR1 methylation in DNA isolated from dried blood spots, developed by Warren and colleagues,27 and a recent method that targets FMRP detection by using an immunoassay with a novel standard.28 Although both of these techniques identify males with fragile X, they cannot reliably detect females with full mutations because of either the complexities of random X-methylation and inactivation or the overlap of normal FMRP levels in females with FMRP levels of females with full mutations on one X chromosome.
Several molecular techniques were published that attempt to detect both males and females with expanded CGG repeats. These methods use different technical approaches to facilitate the PCR amplification of the tightly bound repeat region in the 5′ UTR of FMR1. To evaluate the feasibility of using low-cost molecular techniques to screen for fragile X, we chose three approaches to this challenging problem that use novel molecular methods that had i) the potential to detect expanded CGG repeats in both males and females, ii) a target instrument and reagent cost of approximately $5 per test, and iii) procedures that were more amenable to high throughput than Southern blot analyses and methylation determinations.
Materials and Methods
Samples
We used commercially available DNA samples (including normal blood bank samples) to evaluate normal samples from 37 males and 54 females (n = 91), samples from one male and two females with gray zone mutations (n = 3), and samples from 26 males (4 with premutations and 22 with full mutations) and 12 females (7 with premutations and 5 with full mutations) (n = 38). Coriell Institute for Medical Research (Camden, NJ) provided the samples with expanded CGG repeats and samples from 10 male and 20 female subjects who were tested psychologically and were found to be normal (n = 30). All sample classifications were verified with the clinical AmplideX reagents from Asuragen (Austin, TX). Fragile X controls were provided by Asuragen, and the World Health Organization standards were run to validate all assays. For this study, we used DNA from commercial sources; thus, the work did not meet the definition of human subjects as specified in 45 CFR 46.102 (f).
Evaluation of Screening Methods
The first approach, developed by Tassone et al,29 uses betaine in the PCR master mix to amplify expanded FMR1 alleles in conjunction with a chimeric PCR primer that randomly targets the CGG region. This method uses a two-step screening strategy and can be used with DNA extracted from blood spots. In this procedure, a first round of PCR products are produced with sequence-based forward and reverse primers and are then analyzed by capillary electrophoresis (Figure 2A). Normal samples yield one or two normal chromatographic peaks for females and one normal peak for males. Samples that yield a single normal peak for females or no normal peak for males are tested again in a second round of PCR by using one sequenced-based primer and a chimeric primer that targets the CGG repeats (Figure 2B).
Figure 2.
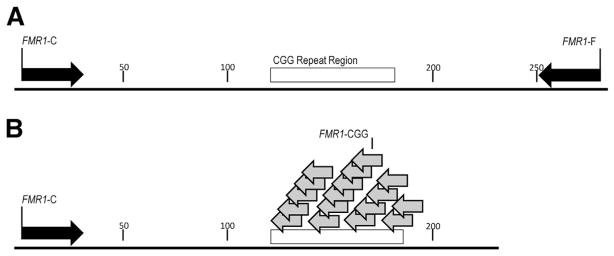
Tassone fragile X screening method29: A: The first step uses sequence-based forward and reverse PCR primers to amplify the CGG repeat region fragments which are analyzed on an ABI 3730 capillary electrophoresis DNA analyzer. B: If there are less than two peaks for females or no peak for males, the forward primer is run with a CGG-targeted reverse primer with capillary electrophoresis analysis of the resulting fragments.
The second approach, developed by Orpana and coworkers, uses heat pulses in the PCR extension step that destabilize the tight C-G bonds.30 In the PCR extension step gradual heating precedes multiple rapid heat pulses that destabilize the secondary structures caused by intramolecular folding of the DNA template and re-annealing of PCR products (Figure 3). Then the PCR products are analyzed with agarose gel electrophoresis, and length of the CGG repeat region is determined with standard molecular weight markers.
Figure 3.
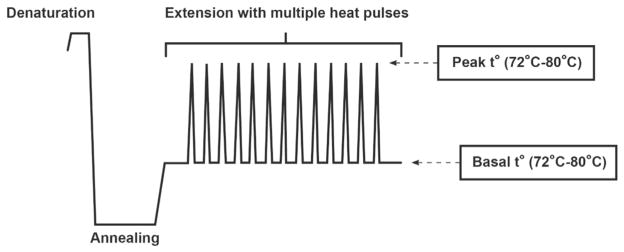
The Orpana fragile X screening method29 uses multiple heat pulses in the PCR extension step to destabilize secondary structures and to enhance the extension over the GC-rich sequence of the CGG repeat region. The method uses agarose gel electrophoresis to detect the resulting amplicons.
The third approach, developed by Teo et al,31 uses melting curve analysis on a real-time PCR instrument to detect FMR1 expansions in males and females. This approach consists of two complementary FMR1 triplet-primed PCR assays in the 3′ and 5′ directions (Figure 4). The 3′ and 5′ PCRs are performed separately under identical thermal cycling conditions in the LightCycler Real-Time PCR System (Roche Applied Science, Indianapolis, IN). The amplicons are melted after the thermocycling program is completed, and then the first derivative melting curves are analyzed. This method ascertains whether the temperature at which the first derivative melting curve returns to baseline is above or below a cutoff to distinguish premutations and full mutations from normal alleles. Gray zone mutations may fall in either category, and this method cannot identify them specifically.
Figure 4.
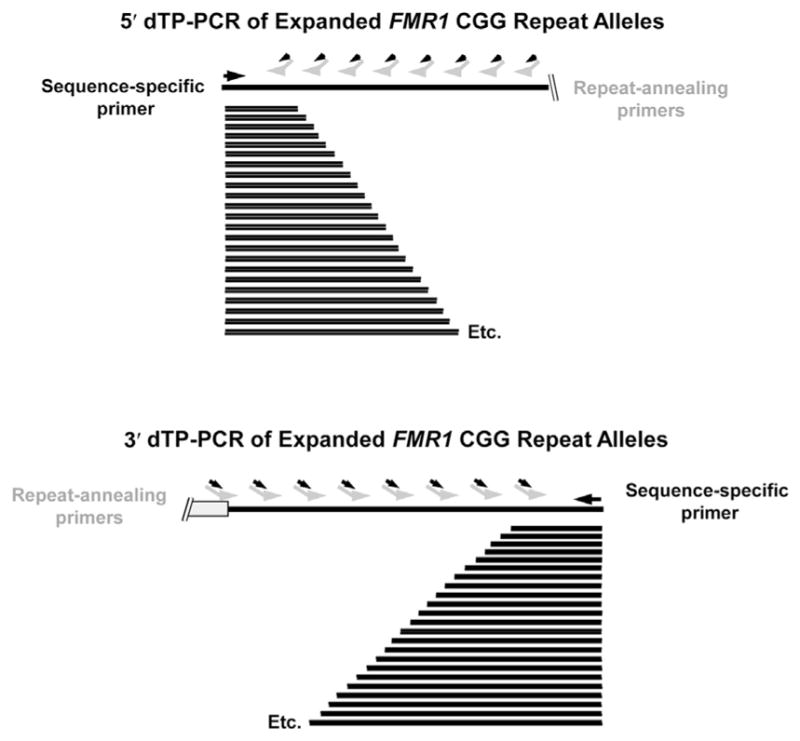
The Teo fragile X screening method31 uses a melting curve analysis of triplet-primed PCR products in the 5′ and 3′ directions. The repeat-annealing primers tail-CCGR and tail-CGGF anneal fully within the repeat sequence and are tailed at their 5′ ends with noncomplementary sequences to enhance the production of amplicons.
Statistical Analysis
The estimates of sensitivities, specificities, and 95% exact binomial CIs for those estimates were calculated with SAS version 9.3 (SAS Institute Inc., Cary, NC).
Results
Approximately 25% to 40% of the female population is homozygous for the same number of CGG repeats, so using the Tassone method a single normal peak for females analyzed with standard primers could indicate a homozygous female with two copies of the same normal repeat or a female with both a normal and a nonamplifying expanded CGG repeat. A male with no normal peak is likely to have an expanded CGG repeat. For expanded samples of males and females, the standard and chimeric primers produce a series of peaks of diminishing amplitude, referred to as a stutter, which extend beyond the normal range and confirm the expanded repeat. When normal samples are tested with standard primers and the resulting fragments are analyzed by capillary electrophoresis, the Tassone method yields one or two peaks in the normal range for heterozygous females and one peak in the normal range for males (Figure 5A). CGG repeats in the gray zone amplify and appear in the appropriate window (Figure 5B). Usually, larger pre-mutations and full mutations do not amplify with the first step of this method (Figure 5C). Samples that have only one peak for females and no peak for males require analysis with CGG-targeted primers (Figure 5D); this analysis does not distinguish between premutations and full mutations.
Figure 5.

Tassone method.29 A: A normal sample from a heterozygous female and a normal sample from a male analyzed with sequence-based forward and reverse primers. B: Samples from a female and male with a gray zone allele analyzed with sequence-based forward and reverse primers. C: A sample from a female with a normal 29 CGG-repeat allele and an expanded allele, and a sample from a male with an expanded CGG-repeat allele analyzed with sequence-based forward and reverse primers. D: Samples from a homozygous female with two normal 30 CGG-repeat alleles, a sample from a female with a normal and an expanded CGG allele, and a sample from a male with an expanded CGG allele run with a sequence-based primer and a chimeric CGG-targeted primer.
We analyzed 91 normal samples, 3 gray zone mutations, and 38 premutations and full mutations and correctly categorized all samples except one female sample with a normal and an expanded allele [100% specificity (95% CI, 96.0%–100%) and 97.6% sensitivity (95% CI, 87.1%–100.0%)] (Table 1). The incorrect categorization of the one female sample with an expanded allele was a random error; a repeat analysis correctly identified this sample. Identification of the expanded alleles depends on the stutter that results from the CGG-targeted PCR extending beyond the normal repeat range.
Table 1.
Sample Characterization
| Sample category | CGG repeat range | No. of samples tested | Sex | AmplideX method, range of CGG repeats
|
Tassone screening method, range of CGG repeats
|
Orpana screening method, range of bp*
|
Teo screening method†
|
|||
|---|---|---|---|---|---|---|---|---|---|---|
| Allele
|
Allele
|
Allele
|
Return to baseline temp, 3′ assay | |||||||
| 1 | 2 | 1 | 2 | 1 | 2 | |||||
| Normal | <45 | 37 | Male | 16–41 | NA | 16–41 | NA | 500–700 | NA | <91°C‡ |
| 54 | Female | 17–32 | 29–44 | 17–32 | 27–44 | 500–700 | 500–700 | <91°C‡ | ||
| Gray zone | 45–54 | 1 | Male | 54 | NA | 54 | NA | ND§ | NA | ND§ |
| 2 | Female | 29–30 | 52 | 29–30 | 52 | 500–700 | ND§ | ND§ | ||
| Premutation and full mutation | >55 | 26 | Male | 78–>200 | NA | 76–119 | NA | 700–1000 | NA | >91°C |
| 12 | Female | 20–34 | 79 to >200 | 17–33 | 77 to ≥193¶ | 500–700 | 700–1000|| | >91°C | ||
Approximate gel size range of the amplicon that contains the CGG repeats.
Results for 3′ Teo assay by using a cutoff of 91°C. Normals, n = 81 (M/F: 34/47); premutation and full mutations, n = 25 (M/F: 13/12).
For the Teo method, four false positives reached baseline above 91°C, three male samples and one female sample.
Gray zone alleles are not reliably distinguishable from normals, premutations, and full mutations.
For the Tassone method, there was one false negative female sample.
For the Orpana method, there were two false negative female samples.
F, female; M, male; NA, not applicable; ND, not distinguishable.
When normal alleles are present with the Orpana method, a distinct band is evident on the agarose gel (Figure 6). This method works well for males with premutations and full mutations because no normal band is present (Figure 6A) even if an expanded allele is not evident. Therefore, for males we correctly identified all normal samples and all samples with premutations and full mutations (Table 1). Quantitating gray zone mutations is difficult for males and females because of the resolution of the gels. In female samples the normal expected band can out-compete the expanded allele in the PCR (Figure 6B). Large expanded alleles in female samples may be faint, and the sensitivity and specificity will depend on the analyst’s ability to distinguish the faint expanded alleles from normal background in that region. We detected expanded bands for 10 of 12 female premutation and full mutation samples. Overall specificity and sensitivity estimates for the Orpana method, excluding gray zone CGG repeats, are 100% specificity (95% CI, 96.0%–100%) and 94.7% sensitivity (95% CI, 82.2%–99.4%).
Figure 6.
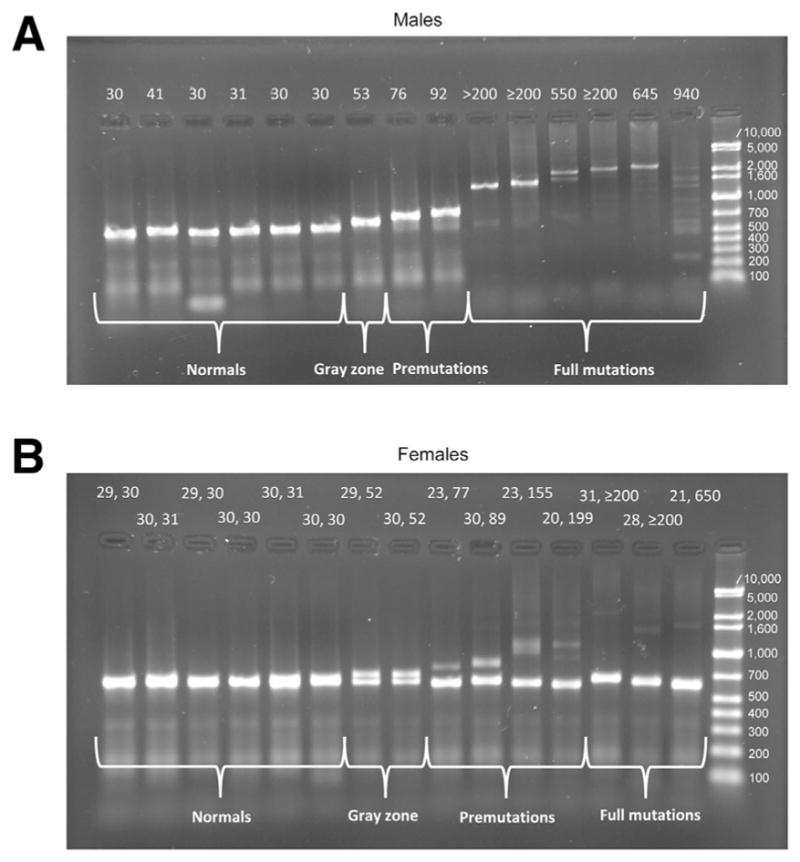
Orpana method.30 Agarose gels of amplicons generated by heat-pulse PCR from males (A) and females (B). The allele sizes are indicated above the tracks.
We obtained characteristic first-derivative melting curves for normal and expanded alleles with the Teo method (Figure 7), and the sensitivity and specificity of the allele characterization depended on the cutoffs used (Figure 8). Only 81 normal, 3 gray zone samples, and 25 premutation and full mutation samples could be run by this method because of the availability of the loaned instrument. All premutations and full mutations tested positive by using the more robust 3′ assay and a cutoff of 91°C (Table 1). Under these conditions, we called one gray zone negative, one positive, and we could not call the third because it returned to baseline too close to the cutoff line. The Teo method does not claim to distinguish gray zone samples from normal or premutations and full mutations. At this cutoff four of the normal samples tested positive, including three males and one female. Of these false positives, two males and one female had alleles with CGG repeats in the high normal range from 41 to 44, and the remaining normal male had 36 CGG repeats. With the use of a cutoff of 91.0°C for the more robust 3′ assay, the specificity and sensitivity estimates for the Teo method are 95.1% specificity (95% CI, 87.8%–98.6%) and 100.0% sensitivity (95% CI, 86.3%–100.0%) for premutations and full mutations.
Figure 7.
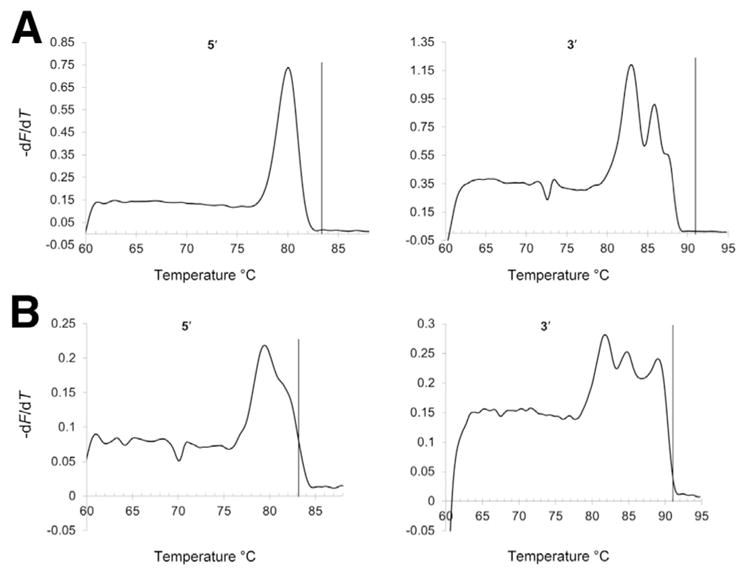
Teo method.31 Melting curves from a normal sample from a female with 18 and 29 CGG repeats (A) and a full mutation sample from a female with 21 and 650 CGG repeats (B). The vertical reference lines represent cutoffs of 83.3°C for the 5′ assay and 91°C for the 3′ assay.
Figure 8.

Teo method.31 The sensitivity and specificity of the 3′ and 5′ assay at different temperature cutoffs.
Discussion
Studies of population screening for fragile X conducted in a variety of settings have been systematically reviewed.32 Of the studies that met the inclusion criteria for the review, most prevalent were studies of women of reproductive age and of newborns. Studies were included when screening was offered to a general population and when psychosocial aspects of population screening of fragile X were addressed. Of the 11 studies that offered population screening, all but one screened women of reproductive age to determine whether they carried an expanded CGG repeat allele. The screening methods used relied on various combinations of PCR and Southern blot analysis, and one study quoted a cost of $100 per test. Southern blot analysis is too labor intensive and costly to be appropriate for NBS. Only one study offered NBS, and parents were only offered the option of testing their male newborns because the method used resulted in unreliable results in samples from females.
A recurrent theme across screening studies of women was the need for information and counseling because few participants had heard of fragile X before being offered screening and struggled to understand the clinical features of the disease.32 Although screening women of reproductive age was well received and early knowledge of the potential to develop fragile X-associated premature ovarian insufficiency was valued by women, screening of newborns was more contentious because the benefits of early intervention have not been established. Complex ethical and policy issues need to be resolved before mandated NBS is likely to be recommended. These issues include whether to screen only boys or both sexes, how to deal with incidental chromosomal findings, and whether to report only full mutations to parents or premutation expansions as well, which have adult-onset implications. The authors of the review by Hill et al32 recommend research to address these issues, in addition to further clinical trials to establish the benefit of early interventions.
Of the three molecular screening methods potentially appropriate for NBS that we evaluated in samples of males and females, the Tassone method achieved high sensitivity and specificity for all samples tested. The Orpana method works particularly well for male samples. Expanded alleles in females can be faint and more difficult to detect and are best evaluated by an analyst experienced in reading gels. Appropriate cutoff temperatures for the Teo method were not consistent from laboratory to laboratory or even from run to run in our laboratory. This circumstance would not allow establishing a universal cutoff and would necessitate using controls in each run to detect shifts in temperature cutoffs. Of the two Teo assays, we obtained the most robust performance with the 3′ assay.
In NBS one central laboratory usually performs screening for large populations. California has approximately 500,000 births per year and has several contract laboratories to perform this function. In other states one laboratory performs all of the analyses for one or more states, and the sample throughput demand can be quite high. The reagent, supply, and instrument cost per test is important with a goal of approximately ≤$5 per test to accommodate state NBS programs with varying financial resources. To determine whether these methods would be applicable to NBS laboratories, we estimated the cost of supplies and reagents for each screening method with amortization of major equipment over 5 years and the assumption of 55,602 samples per year, which was the median 2010 annual number of births by state in the United States. Assuming operation 52 weeks per year and 5 days per week, approximately 214 samples would be analyzed per day. This is a feasible number for each of these methods with the specified equipment. The estimated cost does not include labor. The Tassone method cost of $4.25 per sample and includes the forward and reverse primer assays and the CGG-targeted assay when needed. This method uses the ABI 3730 capillary electrophoresis DNA analyzer and the estimate assumes four thermal cyclers for this throughput. The Orpana method cost of $2.25 per sample includes the use of three gold block thermal cyclers. The Teo method cost of $5.71 per sample includes the use of the Roche LightCycler 480. In a high-throughput environment, the Tassone method has the advantage of potential automation of the PCR and capillary electrophoresis steps. Gel methods such as the Orpana method are less amenable to automation. In conclusion, in addition to available methods for clinical diagnosis of fragile X and its associated conditions in symptomatic persons,7,8,17–25 our study shows that there are appropriate methods to screen larger populations of males and females at low cost so that early interventions may help prevent or delay the disability of fragile X.
Acknowledgments
Supported by the Centers for Disease Control and Prevention, U.S. Department of Health and Human Services.
We thank Maya Sternberg, Research Math Statistician (National Center for Environmental Health at CDC) for performing the statistical analysis of the sensitivities and specificities for the three screening methods; the authors of the published methods for their input; and Drs. Don Bailey (Research Triangle Institute) and Catherine Riley (National Center on Birth Defects and Developmental Disabilities at CDC) for reviewing the manuscript and their valuable comments. We also thank Asuragen for donating clinical reagents, controls, and selected sample confirmations and Roche Applied Science for loaning instruments, supplies, and reagents.
J.I.L. analyzed the Orpana and Teo methods, drafted parts of the manuscript, suggested revisions, and approved the manuscript as submitted. G.R.K. conducted the analyses of the Tassone and Asuragen methods, including data analysis, drafted parts of the manuscript, suggested revisions, and approved the manuscript as submitted. P.W.M. conceptualized and designed the study; drafted parts of the manuscript; assembled, reviewed, and revised the manuscript; and approved the final manuscript as submitted.
Footnotes
Disclosures: Asuragen contributed clinical fragile X reagents, controls, and confirmatory assays. Roche Applied Science loaned instrumentation, supplies, and reagents.
Disclaimers: Reference to any specific commercial products, process, service, manufacturer, company, or trademark does not constitute its endorsement or recommendation by the US government, the Department of Health and Human Services, or the Centers for Disease Control and Prevention. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
References
- 1.Saul RA, Tarleton JC. FMR1-related disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong C-T, Smith RJH, Stephens K, editors. GeneReviews [Internet] Copyright University of Washington; Seattle: 1998. Available at http://www.ncbi.nlm.nih.gov/books/NBK1384, last revised April 26, 2012. [Google Scholar]
- 2.Antar LN, Dictenberg JB, Polciniak M, Afroz R, Bassess GJ. Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 2005;4:350–359. doi: 10.1111/j.1601-183X.2005.00128.x. [DOI] [PubMed] [Google Scholar]
- 3.Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown V, Small K, Lakkis L, Feng Y, Gunter C, Wilkinson KD, Warren ST. Purified recombinant Fmrp exhibits selective RNA binding as an intrinsic property of the fragile X mental retardation protein. J Biol Chem. 1998;273:15521–15527. doi: 10.1074/jbc.273.25.15521. [DOI] [PubMed] [Google Scholar]
- 5.Vanderklish PW, Edelman GM. Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2002;99:1639–1644. doi: 10.1073/pnas.032681099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monaghan KG, Lyon E, Spector EB American College of Medical Genetics and Genomics. ACMG Standards and Guidelines for fragile X testing: a revision to the disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics and Genomics. Genet Med. 2013;15:575–586. doi: 10.1038/gim.2013.61. [DOI] [PubMed] [Google Scholar]
- 8.Yrigollen CM, Durbin-Johnson B, Gane L, Nelson DL, Hagerman R, Hagerman PJ, Tassone F. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet Med. 2012;14:729–736. doi: 10.1038/gim.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Carvajal I, Lopez Posadas B, Pan R, Raske C, Hagerman PJ, Tassone F. Expansion of an FMR1 grey zone allele to a full mutation in two generations. J Mol Diagn. 2009;11:306–310. doi: 10.2353/jmoldx.2009.080174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belina A, Tassone F, Schwartz PH, Sahota P, Hagerman PJ. Redistribution of transcription start sites within the FMR1 promoter region with expansion of the downstream CGG-repeat element. Hum Mol Genet. 2004;13:543–549. doi: 10.1093/hmg/ddh053. [DOI] [PubMed] [Google Scholar]
- 11.Galloway JN, Nelson DL. Evidence for RNA-mediated toxicity in the fragile X-associated tremor/ataxia syndrome. Future Neurol. 2009;4:785–798. doi: 10.2217/fnl.09.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Vries BB, Janson CC, Duits AA, Verheij C, Willermsen R, van Hemel JO, van den Ouweland AM, Niermeijer MF, Oostra BA, Halley DJ. Variable FMR1 gene methylation of large expansions leads to variable phenotype in three males from one fragile X family. J Med Genet. 1996;33:1007–1010. doi: 10.1136/jmg.33.12.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moeschler JB, Shevell M Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014;134:e903–e918. doi: 10.1542/peds.2014-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherman S, Pletcher BA, Driscoll DA. Fragile X syndrome: diagnostic and carrier testing. Genet Med. 2005;7:584–587. doi: 10.1097/01.GIM.0000182468.22666.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, Miller KJ, Frazier JA, Silverstein I, Picker J, Weissman L, Raffalli P, Jeste S, Demmer LA, Peters HK, Brewster SJ, Kowalczyk SJ, Rosen-Sheidley B, McGowan C, Duda AW, 3rd, Lincoln SA, Lowe KR, Shonwald A, Robbins M, Hisama F, Wolff R, Becker R, Nasir R, Urion DK, Milunsky JM, Rappaport L, Gusella JF, Walsh CA, Wu BL, Miller DT Autism Consortium Clinical Genetics/DNA Diagnostics Collaboration. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–e735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reddy KS. Cytogenetic abnormalities and fragile-x syndrome in autism spectrum disorder. BMC Med Genet. 2005;6:3. doi: 10.1186/1471-2350-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rousseau F, Heitz D, Biancalana V, Blumenfeld S, Kretz C, Boue J, Tommerup N, Van Der Hagen C, DeLozier-Blanchet C, Croquette MF, Gilgenkrantz S, Jalbert P, Voelckel MA, Oberle I, Mandel JL. Direct diagnosis by DNA analysis of the fragile X syndrome of mental retardation. N Engl J Med. 1991;325:1673–1681. doi: 10.1056/NEJM199112123252401. [DOI] [PubMed] [Google Scholar]
- 18.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DPA, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang F, Eussen BE, van Ommen G-JB, Blonden LA, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostra BA, Warren ST. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 19.Nakahori Y, Knight SJ, Holland J, Schwartz C, Roche A, Tarleton J, Wong S, Flint TJ, Froster-Iskenius U, Bentley D. Molecular heterogeneity of the fragile X syndrome. Nucleic Acids Res. 1991;19:4355–4359. doi: 10.1093/nar/19.16.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu S, Pritchard M, Kremer E, Lynch M, Nancarrow J, Baker E, Holman K, Mulley JC, Warren ST, Schlessinger D, Sutherland GR, Richards RI. Fragile X genotype characterized by an unstable region of DNA. Science. 1991;252:1179–1181. doi: 10.1126/science.252.5009.1179. [DOI] [PubMed] [Google Scholar]
- 21.Filipovic-Sadic S, Sah S, Chen L, Krosting J, Sekinger E, Zhang W, Hagerman PJ, Stenzel TT, Hadd AG, Latham GJ, Tassone F. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem. 2010;56:399–408. doi: 10.1373/clinchem.2009.136101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seltzer MM, Baker MW, Hong J, Maenner M, Greenberg J, Mandel D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:589–597. doi: 10.1002/ajmg.b.32065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maenner MJ, Baker MW, Broman KW, Tian J, Barnes JK, Atkins A, McPherson E, Hong J, Brilliant MH, Mailick MR. FMR1 CGG expansions: prevalence and sex ratios. Am J Med Genet B Neuropsychiatr Genet. 2013;162B:466–473. doi: 10.1002/ajmg.b.32176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L, Hadd AG, Sah S, Houghton JF, Filipovic-Sadic S, Zhang W, Hagerman PJ, Tassone F, Latham GJ. High-resolution methylation polymerase chain reaction for fragile X analysis: evidence for novel FMR1 methylation patterns undetected in Southern blot analysis. Genet Med. 2011;13:528–538. doi: 10.1097/GIM.0b013e31820a780f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yrigollen CM, Tassone F, Durbin-Johnson B, Tassone F. The role of AGG interruptions in the transcription of FMR1 premutation alleles. PLoS One. 2011;6:e21728. doi: 10.1371/journal.pone.0021728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, McCalmon S, Hagerman RJ, Tassone F, Hagerman PJ. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res. 2013;23:121–128. doi: 10.1101/gr.141705.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coffee B, Keith K, Albizua I, Malone T, Mowrey J, Sherman SL, Warren ST. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85:503–514. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LaFauci G, Adayev T, Kascsak R, Kascsak R, Nolin S, Mehta P, Brown WT, Dobkin C. Fragile X screening by quantification of FMRP in dried blood spots by a Luminex Immunoassay. J Mol Diagn. 2013;15:508–517. doi: 10.1016/j.jmoldx.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 29.Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orpana A, Ho T, Stenman J. Multiple heat pulses during PCR extension enabling amplification of GC-rich sequences and reducing amplification bias. Anal Chem. 2012;84:2081–2087. doi: 10.1021/ac300040j. [DOI] [PubMed] [Google Scholar]
- 31.Teo CRL, Law HY, Lee CG, Chong SS. Screening for CGG repeat expansion in the FMR1 gene by melting curve analysis of combined 5′ and 3′ direct triplet-primed PCRs. Clin Chem. 2012;58:568–579. doi: 10.1373/clinchem.2011.174615. [DOI] [PubMed] [Google Scholar]
- 32.Hill MK, Archibald AD, Couns GDG, Cohen J, Metcalfe SA. A systematic review of population screening for fragile X syndrome. Genet Med. 2010;12:396–410. doi: 10.1097/GIM.0b013e3181e38fb6. [DOI] [PubMed] [Google Scholar]