

Abstract
Cystic Fibrosis is caused by mutations in the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) gene resulting in abnormal protein function. Recent advances of targeted molecular therapies and high throughput screening have resulted in multiple drug therapies that target many important mutations in the CFTR protein. In this review, we provide the latest results and current progress of CFTR modulators for the treatment of cystic fibrosis, focusing on potentiators of CFTR channel gating and Phe508del processing correctors for the Phe508del CFTR mutation. Special emphasis is placed on the molecular basis underlying these new therapies and emerging results from the latest clinical trials. The future directions for augmenting the rescue of Phe508del with CFTR modulators is also emphasized.
Keywords: Cystic fibrosis, CFTR modulators, lung disease, novel therapies, CFTR molecular defect
Introduction
The identification of the cystic fibrosis (CF) gene encoding the CF transmembrane conductance regulator (CFTR) protein in 19891,2 provided the CF research and clinical community with an unprecedented opportunity to begin understanding the molecular basis of this autosomal recessive disease and its clinical consequences. In the past decade, this knowledge has led to development of therapies that may be profoundly impacting the course of the disease in individuals with specific mutational classes. The progress that has ensued over the last 26 years has been remarkable and inspirational to patients and families living with CF as well as other rare disease communities. This review will focus on two important therapeutic approaches, termed “potentiators” and “correctors,” directed at modulating or repairing the function of the CFTR protein in those classes of disease causing mutations where some level of CFTR protein is produced.
CFTR is a protein kinase A (PKA) activated chloride and bicarbonate selective ion channel involved in salt and water transport across the apical membranes of epithelial cells located in multiple organs affected in individuals with CF3,4 including pancreas, liver, intestinal tract and the lung. Because pulmonary manifestations are the major cause of morbidity and mortality in CF, therapeutic development has been primarily focused on reversing the progressive obstructive lung disease. In the airway, there are several hypotheses linking loss of channel function to lung pathogenesis including airway surface dehydration5, and abnormal mucus viscoelastic properties6, and tethering to sub-mucosal glands7,8. These initial events likely lead to impaired mucus clearance and airway obstruction, making the airway more vulnerable to infection, inflammation and eventual structural damage9,10.
The structure of the dimeric CFTR channel at the apical surface is composed of multiple domains including two membrane spanning domains (MSD), two cytoplasmic nucleotide- binding domains (NBD), an intracellular regulatory (R) domain as well as several extra- and intracellular loops11,12. Thus, it is not surprising that the biogenesis of mature CFTR is multi-stepped and complex as the nascent, recently transcribed protein must fold properly and undergo glycosylation steps as it moves from the endoplasmic reticulum (ER) to the Golgi complex and is eventually transported to the cell surface. The cell's quality control mechanisms, ER-associated degradation (ERAD), rapidly remove and degrade any misfolded proteins. Even wild-type CFTR is a highly inefficient process where less than 20% of nascent protein successfully reaches the surface demonstrated in primary cell culture of human bronchial and intestinal epithelial cells culture systems13 with reduced expression in other heterologous cells systems. Once reaching the surface, the channel must be able to properly activate so that the channel is in an open state, allowing anion transport to occur. The proportion of time that the channel is actively transporting anions is termed its open probability (Po) and is a key measure of its function14.
Over 1900 CFTR mutations are known to cause CF15. Despite this large number, relatively few mutations account for the majority of CFTR alleles, and are particularly common in the northern European descent population. The most prevalent mutation is Phe508del (formerly F508del, or c.1521_1523delCTT), which causes a 3 base pair deletion resulting in omission of phenylalanine at position 508. This mutation accounts for ∼70% of all CFTR alleles, and as high as 86% in Northern European Caucasians15. Phe508del CFTR causes protein misfolding, resulting in efficient ERAD and minimal protein expression at the plasma membrane. Phe508del is the prototypical Class II mutation (mutations that result in premature degradation or incomplete maturation). Other mutations fall into several classes based on molecular mechanism10,16 (Figure 1). These classes include incomplete synthesis due to premature termination codons (PTCs, Class I); disordered regulation and gating, causing diminished ATP binding and hydrolysis (Class III), which includes the Gly551Asp (formerly known as G551D) mutation; defective chloride conductance (Class IV, which includes the Arg117His mutation formerly known as R117H); and a reduced number of CFTR transcripts due to a promoter or splicing abnormality (Class V). Class VI mutations are also defined as those that exhibit reduced cell surface half-life, a property shared by many Class II alleles.
Figure 1. Categories of CFTR mutations.
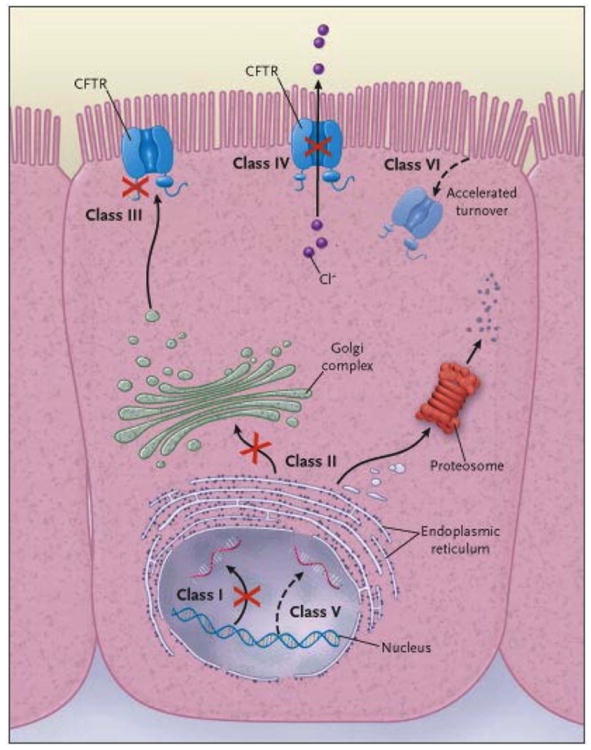
Classes of defects in the CFTR gene include the absence of synthesis (class I); defective protein maturation and premature degradation (class II); disordered regulation, such as diminished ATP binding and hydrolysis (class III); defective chloride conductance or channel gating (class IV); a reduced number of CFTR transcripts due to a promoter or splicing abnormality (class V); and accelerated turnover from the cell surface (class VI). From N Engl J Med, Rowe SM, Miller S, Sorscher EJ, Cystic Fibrosis 352:1992-2001. Copyright © 2005 Massachusetts Medical Society. Reprinted with permission.
A few caveats should be noted with regard to these categories. First, defects caused by a mutation may not be limited to one mechanistic category. For example, Phe508del has both reduced CFTR reaching the surface membrane but also abnormal channel activation (though less severe than Gly551Asp) and reduced residence time in the apical membrane. Second, phenotype studies have shown that mutational classes where little CFTR reaches the cell surface (Classes I, II, V) or channel activation is severely impaired (Class III) are associated with more severe disease including pancreatic insufficiency and more rapid lung progression. Third, to be successful, therapeutic approaches to correct dysfunctional CFTR must take into account these different mechanisms of CFTR dysfunction and it is likely that for more severe phenotypes (e.g., Phe508del) multiple approaches will be required. The complexity of repairing Phe508del is discussed in the correctors section.
Developing novel therapies
Since the discovery of the CFTR gene, there has been worldwide interest in pharmacologic approaches to stabilize the misfolded Phe508del protein to permit transport to the surface and to activate channel function. It is well established that low temperature could prevent Phe508del misfolding in vitro17 and that pharmacological chaperones would likely stabilize the misfolded protein18. Early drug development efforts utilized known pharmacologic compounds such as 4 phenyl-butyrate19, curcumin20 and 8-cyclopentyl-1,3-dipropylxanthine (CPX)21 with limited efficacy. These early experiences, however, emphasized the need for better predictive cell based model systems to prioritize therapeutic agents for future human trials. Fortunately, such model systems are now available.
In the past 15 years, drug development has been significantly accelerated by the introduction of cell based high-throughput screening (HTS) assays using fluorescence-based assays of membrane potential22 or halide efflux23, each markers of CFTR activity in vitro. Two major formats of HTS assays have been used to identify candidate CFTR modulators. First, chemical compounds have been screened without any pre-incubation period to identify “potentiators”, defined as pharmacologic agents that increase PKA regulated chloride channel gating of CFTR. A second assay has been designed to identify “correctors” defined as small molecules acting as chaperones that “rescue” misfolded CFTR and permit trafficking to the cell surface. In the corrector assays, cell lines used for HTS were preincubated with chemical compounds for ∼12-48 hours to allow time for CFTR de novo synthesis and trafficking to the cell surface. The compounds were then removed and a channel activator, such as genistein, was added for the fluorescence assay.
These HTS assays have permitted the screening of hundreds of thousands of chemical compounds with diverse structures14,24. The most promising compounds, called “hits”, have subsequently undergone medicinal chemical optimization to improve the potency and reduce potential toxicity of the compounds. This process has led to the successful identification of both potentiator and corrector drugs that have moved forward into human trials and even approval by U.S. and European regulatory agencies. This developmental process has been successfully implemented, using a variety of different cell types and reporter assays, by a number of pharmaceutical companies and academic drug-discovery units. The next sections of this review will describe the drug development process of first potentiators and then correctors for patients with CF.
Research efforts are being directed to other therapeutic approaches beyond correctors and potentiators. Examples include readthrough (or suppression) of premature termination codons (PTCs) to induce expression of full-length CFTR25,26, and other approaches such as gene replacement by viral and non-viral gene therapy are also under active development27,28, but are beyond the scope of this review. Since many patients (∼40%) are complex heterozygotes for more than one CFTR mutation29, a number of these approaches will form the basis of combination therapeutics. Treatment regimens of the future will be personalized to optimize the clinical benefit for the specific genetic mutations harbored by the individual patient30.
Therapies that Potentiate the CFTR Channel
Potentiators are compounds developed in hopes of restoring CFTR activity and function in patients with cystic fibrosis. These agents increase the time that activated CFTR channels at the epithelial cell surface remain open and functional. Potentiators improve gating and conductance defects by augmenting the open configuration of the CFTR channel. Class III and IV mutations are the categories most likely to respond to potentiator monotherapy, but any channel with some residual expression (like mild Class II defects or non-canonical Class V splicing mutations) have the potential to benefit31,32. While Class IV mutations are postulated to be responsive to potentiator therapy, it is unknown whether all Class IV mutations will have a clinically important improvement in CFTR function when treated with potentiator therapy like ivacaftor. Rather than a strict genotypic approach, a phenotypic approach may also be implemented, such as identifying potential candidates for therapy based on the presence of residual CFTR function, which implies that there may be some CFTR present to effectively potentiate. This concept is being tested in the recently completed “n-of-1” study of residual function mutations and the differential treatment effects of ivacaftor on these partial function CFTR mutations33.
Class III mutations are associated with a reduction in the gating mechanism or channel opening of the CFTR protein (see Figure 1)10,14. Class IV mutations allow limited chloride and bicarbonate ion transport, and are associated with reduced or defective anion conductance34. The most prevalent and well known Class III mutation, Gly551Asp, involves a substitution of glycine for aspartic acid at amino acid 551; it occurs in 4-5% of patients with CF29. This amino acid substitution occurs at a critical point in the NBD1, the interface with NBD235. Gly551Asp – CFTR reaches the plasma membrane of epithelial cells, but the protein contains a gating defect that abolishes ATP-dependent channel opening resulting in defective channel functioning with consequent little or no ion transport.
Among candidate compounds identified by high-throughput screening, ivacaftor (formerly VX-770) was felt to be the most promising potentiator suitable for drug development. It was found to increase the activity of both wild-type and defective cell surface CFTR protein in vitro14 and had favorable pharmacokinetic properties. The greatest effect was on cells with Gly551Asp-CFTR or similar gating mutations32. Addition of ivacaftor increased CFTR mediated chloride ion secretion in Gly551Asp/Phe508del human bronchial epithelium (HBE) cells from 0-5% to levels 35-50% of that measured in non-CF HBE. It additionally reduced sodium absorption. This pharmacologic effect has also been noted to be associated with an enhanced height in the apical airway surface liquid (ASL) allowing an increase in cilia beating to levels seen in the non-CF airway epithelium14,36.
The development of a CFTR-targeted drug that would potentially benefit CF patients was a remarkable breakthrough. The CFTR potentiator compound ivacaftor directly restored CFTR activity in vitro and so clinical trials in CF patients commenced. A Phase 2 clinical trial of ivacaftor evaluated its safety profile over 14-28 days of treatment37. Thirty-nine adults with CF with at least one Gly551Asp-CFTR allele participated in a randomized, placebo-controlled, double-blind, multicenter, multiple dose study. The frequency of adverse events was similar between study groups, and the safety profile provided support for further clinical evaluation. Significant within subject improvements were noted in two biomarkers of CFTR activity: respiratory (nasal potential difference) and non-respiratory (sweat chloride concentration). Additionally, improvement in lung function reflected by a significant change from baseline in FEV1 (forced expiratory volume in 1 second) was appreciated at several dose levels of ivacaftor. Those receiving a dosage of 150mg per day seemed most responsive, so that dose was selected for the Phase 3 studies.
Two Phase 3 clinical trials of ivacaftor, one in patients 12 years and older38 and a second in children 6-11 years39, evaluated improvement in lung function, risk of pulmonary exacerbations, patient-reported respiratory symptoms, weight, and concentration of sweat chloride, along with safety. One hundred sixty-one subjects with CF, 12 years of age or older (mean age 25.5 years) with at least one Gly551Asp-CFTR allele, participated in a randomized, double-blind, placebo-controlled trial, the STRIVE study38. Subjects received 150 mg of ivacaftor every 12 hours or placebo for 48 weeks. Adolescents and adults on ivacaftor participating in this study showed a 10.6% (absolute change) improvement in FEV1. Effects were noted by week 2 and sustained through week 48. Subjects receiving ivacaftor were 55% less likely to have a pulmonary exacerbation. Those receiving ivacaftor scored 8.6 points higher than controls on a standardized respiratory symptom questionnaire. At the end of 48 weeks, those receiving ivacaftor had gained 2.7 kg more weight than those on placebo. The change in sweat chloride concentration, a measure of CFTR activity, was also significant. The incidence of adverse events was similar between the two groups, and a lower percentage of serious adverse events were reported in those receiving ivacaftor.
A second Phase 3 trial was conducted in children ages 6-11 with CF with at least one Gly551Asp- CFTR allele39. This randomized, double-blind, placebo-controlled study, known as the ENVISION study, demonstrated a 10% improvement in FEV1, weight gain of 2.8 kg, and a marked decrease in sweat chloride concentration in those receiving ivacaftor. Ivacaftor was found to be both safe and effective in subjects 6 years of age and over.
Subjects participating in both the STRIVE and ENVISION studies were invited to enroll in the PERSIST study, an open label, roll-over continuation. The adults and adolescents who switched from placebo to ivacaftor showed improvement in FEV1, weight gain, and pulmonary exacerbation rate, similar to those treated with ivacaftor for the initial 48 weeks of the study40. Response was sustained, both at 96 weeks, and at 144 weeks for those who had received ivacaftor in the previous study.
A recent Phase 4 Gly551Asp observational study (GOAL) involved patients with at least one Gly551Asp-CFTR mutation and evaluated their clinical status before and after initiation of ivacaftor at the time of its approval by the US-FDA41. The GOAL study involved patients from 28 US CF care centers , including patients age 6 years and older who had no prior exposure to ivacaftor. Study assessments included spirometry, body weight, sweat chloride, and patient reported symptoms. Samples collected included blood, urine and sputum. The GOAL study also included additional substudies: evaluation of mucociliary clearance (MCC), gastrointestinal (GI) pH profiles, measure of sputum inflammation and microbiology. One hundred fifty-three participants were enrolled and 133 (88%) completed the six-month long study (there was no placebo control group). All participants had one copy of the Gly551Asp mutation and 72.2% had Phe508del as their second allele. Lung function as measured by FEV1 and FVC improved, except for the youngest age group where there were smaller changes noted, likely related to healthier lungs at baseline. Body weight and body mass index (BMI) both increased. Sweat chloride concentration declined significantly, to levels very similar to that observed in Phase 3 testing. Substantial improvements in quality of life measures and respiratory symptoms were seen. Remarkably, sputum cultures showed decreased endobronchial colonization with P. aeruginosa, providing evidence that CFTR modulation may alter CF microbiology, and suggesting that CFTR may play a role in host defense. A subsequent retrospective analysis confirmed these findings, and demonstrated patients with mild disease and more intermittent growth of P. aeruginosa were the most likely to clear their cultures, but also that patients with persistent mucoid Pseudomonas can also clear, albeit at lower rates42. An increased abundance of Prevotella, associated with higher lung function in CF43, was also observed in the GOAL study. Biomarkers of inflammation were not observed to change significantly. However, a large improvement in MCC was observed suggesting that CFTR has a central role in regulating MCC41.
With respect to ivacaftor's effect on body weight and body mass index (BMI), findings from a GOAL substudy suggest that CFTR may have a direct effect on gastrointestinal (GI) pH leading to improved absorption. The role of CFTR as a bicarbonate transporter involved in regulation of pancreatic and GI function was addressed in this study. Individuals with CF are known to have more acidic intestinal pH44. Before and after initiation of ivacaftor, a subgroup of patients swallowed capsules which measured pH throughout the intestinal tract. Ivacaftor significantly effected duodenal alkalization and throughout the distal intestine, suggesting that normalization of GI pH via CFTR potentiation may improve pancreatic enzyme function, nutrient digestion and absorption41 though studies testing this hypothesis directly have not been conducted to date.
The effect of ivacaftor on other Class III gating CFTR mutations has also been studied. A Phase 3 randomized, placebo-controlled, double-blind study of patients with genotype non-Gly551Asp gating mutation in at least 1 allele involved 39 patients ages 6 years and older45. This trial, known as the KONNECTION study, showed clinical benefit after 8 weeks of treatment with ivacaftor including improvement in FEV1, BMI, and sweat chloride concentration. Improvement in these parameters was noted to be similar to improvement seen in patients with Gly551Asp.
Class IV mutations, primarily located in the MSD, have defective conductance, allowing limited chloride and bicarbonate ion transport34. The efficacy of ivacaftor has been assessed in patients with Arg117His, traditionally characterized as a Class IV mutation that also exhibits reduced (but not complete abrogated) channel gating.
The KONDUCT study, a randomized, placebo-controlled, double-blind study of patients with genotype Arg117His in at least one allele, involved 69 patients ages 6 years and older. There was a 5% absolute improvement in FEV1 for all patients in the study, a trend that did not achieve statistical significance. There was, however, a 9% absolute improvement noted in patients greater than 18 years of age with more established lung disease as compared to the younger patients, which demonstrated no treatment benefit46. Recently the FDA approved ivacaftor for CF patients with the Arg117His mutation, and post-approval monitoring studies to help confirm clinical benefit and the appropriate treatment populations are planned.
Ivacaftor has been studied in patients with the most common CF genotype, Phe508del. Ivacaftor acts as a “modest potentiator”36 for the small amount of Phe508del-CFTR which is found at the cell surface. It increases the Phe508del channel open probability and chloride ion secretion in cultured HBE cells of some Phe508del homozygous CF patients in vitro14. Thus ivacaftor was studied in a Phase 2 clinical trial, known as the DISCOVER study involving 140 homozygous Phe508del patients47. This was a randomized, placebo-controlled, double-blind parallel study including patients greater than or equal to 12 years of age. The study was designed as a safety trial with a 4:1 randomization of ivacaftor to placebo and not powered to measure efficacy. The initial treatment duration was 16 weeks, with a follow-on open-label study for 96 weeks., Patients on this study receiving ivacaftor showed no statistically significant improvement in FEV1, rate of pulmonary exacerbation, or symptom scores possibly due to the lack of power.,. In the first part of the study, a small decrease in sweat chloride concentration occurred, but this was not substantiated in the open-label extension. While no efficacy was observed, the study helped establish the safety of the agent47.
Though the results of the studies described above have been truly extraordinary, potentiator montherapy benefits only a small number (<10%) of CF patients. Initial attempts at a phenotypic approach, involving patients with at least one mutation that produces some functional CFTR protein (termed residual activity), and case reports suggest ivacaftor may be beneficial in this group48. Since approximately 75% of CF patients carry at least one copy of the Phe508del mutation, successful therapy targeted to Phe508del mutation remains a great hope but a much greater challenge, as described in the next section.
The challenge of correcting Phe508del CFTR
The F508del mutation affects the protein's maturation at multiple steps including folding and activation making it much more challenging to restore49-51 (Figure 2). Deletion of Phe508 leads to pronounced loss of thermal stability due to NBD1 misfolding, resulting in retention in the ER and subsequent protein degradation. Thus, little mature protein reaches the plasma membrane50,52-54. Further impairment of Phe508del protein stability occurs because Phe508 facilitates binding between two important structural domains, NBD1 and MSD2. In addition, the NBD-MSD interface is key to opening the NBD-mediated anion channel so that the Phe508del mutation results in a significant gating abnormality55. Finally, through similar mechanisms as disordered protein folding, the small amounts of Phe508del that escape ERAD and reach the cell surface are also prematurely degraded and recycled, resulting in a substantially-shortened cell surface half-life56. These multiple distinct CFTR defects attributable to Phe508del point to the challenge of identifying a single agent capable of restoring mutant CFTR to therapeutically relevant levels, and helped predict the current era of multi-agent CFTR therapies (Figure 2).
Figure 2. Schematic of the molecular mechanism of Phe508del-CFTR.
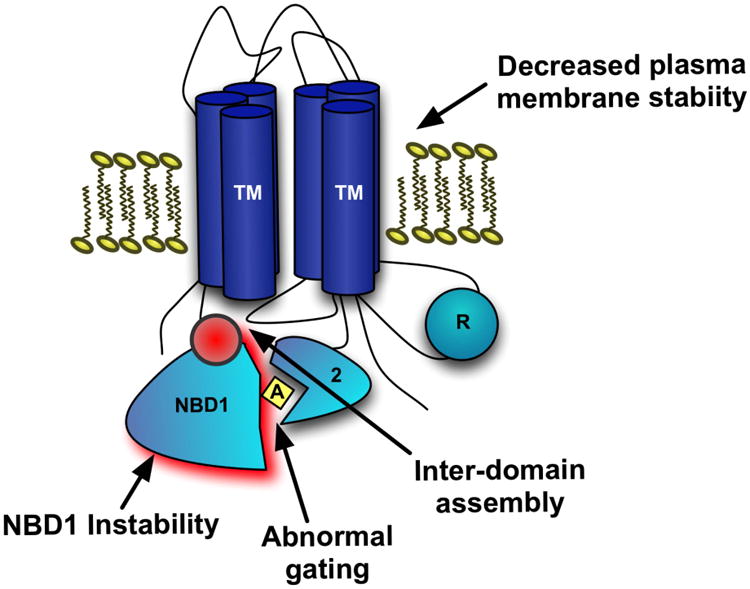
This schematic depicts the proposed mechanisms of abrogated F508del-CFTR function. The loss of the phenylalanine residue at the 508 position results in inherent instability of nucleotide binding domain-1 (NBD-1) and also promotes ineffective interactions of NBD-1 with the second membrane-spanning domain (MSD-2) via direct contact of the intracellular loop 4 (ICL-4) of MSD-2 with NBD-1. This is a potential molecular target for corrector therapy. This inter-domain assembly defect leads to abnormal channel gating and thermal and cell-surface instability resulting in increased cell-surface recycling to the ER.
Development of CFTR Modulators for Homozygous Phe508del Patients
Correctors and potentiators as monotherapies
As discussed in the introduction, more recent and successful CFTR modulator development efforts have resulted from high-throughput library screens for chloride channel function following incubation of test compounds with F508del expressing cells22,23,57. As postulated based on a successful screen22, Van Goor et al. demonstrated that the novel CFTR corrector lumacaftor (formerly VX-809) improved Phe508del-CFTR cellular processing24 and enhanced chloride secretion to ∼14% of non-CF in a human based cell model of F508del bronchial epithelial cells24, although the magnitude of this effect can vary depending on the cell model58. By contrast, ivacaftor monotherapy in vitro has only a modest effect on F508del HBE cells unless the cells have been pre-treated with a corrector14. The major effect of lumacaftor is likely stabilization of the NBD-MSD inter-domain assembly (shown in Figure 2) reducing cellular misprocessing49,50,59. Of note, the effect of lumacaftor on chloride transport in Phe508del cells is only a quarter to a third the effect observed with ivacaftor in G551D-CFTR cells. Fortunately, the effect of VX-809 on CFTR function can be enhanced 50-100% in Phe508del CFTR homozygous HBE cells following acute administration of ivacaftor24, providing in vitro evidence that a combination of a corrector and potentiator may be more efficacious than either therapy alone. Yet, even this combination in Phe508del homozygous cells remained inferior, compared to the effect of ivacaftor monotherapy on Gly551Asp heterozygous cells14.
An initial Phase 2a clinical trial testing lumacaftor monotherapy in patients homozygous for F508del-CFTR demonstrated the first steps towards effective rescue in the clinic60. Patients receiving lumacaftor monotherapy showed modest but detectable improvements in sweat chloride compared to placebo (8 mEq/L), but improved sweat chloride was not accompanied by significant changes in spirometry or other clinical measures60. While this established that some rescue of Phe508del CFTR in human subjects is achievable by a systemically delivered small molecule, these results indicated that the degree of CFTR rescue was insufficient to confer clinical improvement. Furthermore, because the highest dose tested (200 mg) demonstrated the maximum efficacy, it is possible that the most efficacious dose of lumacaftor was not reached. As noted previously, ivacaftor alone (DISCOVER trial) was safe but not efficacious in the homozygous Phe508del population 47.Thus, both in vitro and early clinical studies have demonstrated monotherapy with either correctors or potentiators have a low likelihood of clinical success in patients homozygous Phe508del emphasizing the need for combination therapeutic approaches.
Corrector and potentiators as combination therapy
Clinical development of combination corrector-potentiator therapy has been more successful, though challenges had to be managed. Lumacaftor and ivacaftor, the first combination therapy that progressed to human trials, have known drug-drug interactions. The early Phase 2 trial60 demonstrated that lumacaftor induced liver metabolism of ivacaftor in CF patients, thus reducing available concentrations of the potentiator. This same metabolic effect was observed in Phase 1 studies in normal healthy volunteers61. Based upon these pharmacokinetic data, the dose of ivacaftor was increased to 250 mg twice daily rather than the 150 mg twice daily monotherapy dose shown to be efficacious in Gly551Asp patients38,39. This higher ivacaftor dose, in combination with lumacaftor, demonstrated significant improvements in sweat chloride62 in a Phase 2 dose escalation study, and was therefore used for subsequent Phase 3 trials when used in combination with lumacaftor. This Phase 2 trial in patients either homozygous or heterozygous for Phe508del mutation also demonstrated that high dose co-administration of lumacaftor (600 mg qd or 400 mg twice daily) and ivacaftor (250 mg twice daily) resulted in significant improvements in FEV1 in the homozygous patients, but only when both agents were administered. Lumacaftor monotherapy for 28 days exhibited a slight but dose-dependent decrement in spirometry that is likely due to bronchospasm based on studies in normal volunteers (1-2% mean FEV1% decrement at the maximum doses)61. When ivacaftor was added for a subsequent 28 days, an FEV1% significant increase was observed (6% at maximum lumacaftor doses)62 in the homozygous Phe508del patients. The net effect was ∼3% FEV1% benefit within subject for lumacaftor- ivacaftor combination therapy that was significant as compared to placebo. This improvement in lung function was seen even though the sweat chloride change in Phe508del homozygous patients was quite modest (9.1mmol/L) and below expectations based upon in vitro findings24.
The same Phase 2 trial62 included a small cohort of Phe508del heterozygous individuals administered high dose lumacaftor-ivacaftor. In this group, no significant change in FEV1 was observed and so subsequent Phase 3 trials included only homozygous Phe508del individuals. The findings in the heterozygous cohort suggests that lumacaftor-ivacaftor is not efficacious enough to improve lung function in most patients with only one Phe508del allele unless the second allele is responsive to ivacaftor alone (e.g., Class III gating mutations or Arg117His)63.
Based on the encouraging lung function findings, lumacaftor-ivacaftor combination therapy was tested in two large multi-center 24 week, Phase 3, randomized control trials, termed TRAFFIC and TRANSPORT64. Each study tested the two highest doses of lumacaftor (600 mg once daily or 400 mg twice daily) in combination with high-dose ivacaftor (250 mg twice daily). TRAFFIC and TRANSPORT each enrolled ∼500 CF patients age 12 years and older who were homozygous for Phe508del-CFTR; development in younger CF patients is currently in progress65. The primary trial analysis confirmed a pooled mean absolute change in FEV1 of ∼2.5% in both treatment arms testing either lumacaftor 400 mg twice daily or 600 mg daily64. In addition, key secondary analyses demonstrated a reduction of CF pulmonary exacerbations (pooled reduction of ∼35%), increased BMI (+0.26kg/m2) and a statistically significant but modest improvements in a validated symptom score (CFQ-R)64. Both combination therapy doses were well tolerated, and most side effects were respiratory in nature. The most common significant adverse event in patients was elevation of liver function tests, requiring discontinuation of therapy in 7 patients on treatment. Currently, patients are being followed in a 52-week open-label extension study of both doses, which has exhibited a stable treatment effect upon long-term administration, and indicated a similar benefit among patients transitioned from placebo to active combination therapy66.
While the effects of lumacaftor-ivacaftor therapy appear beneficial among Phe508del homozygous patients, these effects are modest when compared with ivacaftor monotherapy in Class III mutations. Thus, there is strong motivation to develop more robust corrector molecules, especially for mutations that are difficult to fully restore, like Phe508del. As the community continues to look for effective combinations, the challenges of drug-drug interactions must also be considered. The impact of lumacaftor on ivacaftor metabolism in vivo60 was managed by increasing the ivacaftor dose. Recent in vitro studies utilizing primary cells and cell lines also suggest a deleterious effect of prolonged (> 48hours) ivacaftor administration on lumacaftor-corrected CFTR function by making lumacaftor corrected Phe508del CFTR67-69 less stable. For all these reasons, the search for the next generation of corrector/potentiator combinations must continue.
An alternative first generation corrector to lumacaftor is VX-661, which has a similar mechanism to lumacaftor but more advantageous pharmacologic properties. VX-661 has also demonstrated additive benefit when used in combination with ivacaftor in individuals homozygous for Phe508del CFTR. More robust clinical outcomes were seen in the optimized dosing (100 mg or 150 mg dosed daily both in combination with ivacaftor at 150 mg twice daily), including a 4.6% absolute improvement in FEV1 after 28 days of treatment in a Phase 2 clinical trial when compared to placebo70. Unlike lumacaftor, VX-661 was not associated with bronchospasm in normal volunteers. These promising findings have prompted the initiation of a series of registration-directed studies71. Because of the encouraging FEV1 response of VX-661, these Phase 2 and 3 studies include CF patients heterozygous for Phe508del CFTR as well as homozygous patients. The Phe508del heterozygous patients will be studied in several different cohorts based upon the class of mutation on the second allele. Some cohorts will have alleles that are nonresponsive to ivacaftor monotherapy in vitro (e.g., Class I, II) and others are responsive (e.g., Class III)32. A Phase 2 clinical trial was conducted in patients with Phe508del/Gly551asp genotype already receiving ivacaftor to determine if the addition of VX-661 (100 mg daily) for 28 days provided additional improvement in FEV1 and other clinical measures. FEV1 improved by 7.3% as compared with ivacaftor therapy alone. Sweat chloride and quality of life also improved in this cohort, further supporting efficacy data70. Aside from the development of VX-661, these data also indicate the potential benefit of maximizing CFTR activity in CF patients, even in the Gly551Asp population that has experienced strong and consistent effects of ivacaftor38,39
Future Therapeutic Approaches
Other mechanisms of corrector therapy are being pursued. Recently, the direct and indirect modulation of the nitric oxide (NO) pathway has been investigated as a possible corrector mechanism in F508del-CFTR. Riociguat is an oral soluble guanylate cyclase (sGC) stimulator in a NO-independent manner. It increases the sensitivity of sGC to NO, leading to increased production of cyclic guanosine monophosphate72. Recently, this drug was approved for the treatment of idiopathic pulmonary hypertension and chronic thromboembolic pulmonary hypertension (CTEPH)72. Riociguat is currently being studied in a Phase 2a trial in CF patients comparing riociguat to placebo for safety, tolerability and efficacy endpoints. Similarly, other pharmaceuticals programs have developed a novel class of molecules that indirectly increases epithelial and smooth muscle NO via inhibition of S-nitrosoglutathione reductase (GSNOR), the enzyme which degrades S-nitrosoglutathione (GSNO), a bioavailable storage molecule of NO73. GSNOR inhibitor compounds have been shown to increase cell surface localized Phe508del-CFTR and CFTR activity in human bronchial epithelial cells74 and in a murine model of CF in vivo75, possibly via reduction of chaperone-protein direction towards ERAD-mediated degradation of Phe508del CFTR protein. A recently completed Phase 1b dose escalation study of the intravenous GSNOR inhibitor in CF patients aged 18 years and older demonstrated safety and tolerability in a wide range of treatment doses; exploratory efficacy was assessed but demonstrated no significant improvements in CFTR activity compared to placebo76. Dose optimization of GSNOR inhibitors including a Phase 1b pharmacokinetics and safety study of the oral GSNOR inhibitor N91115 (SNO-3) in CF patients homozygous for Phe508del-CFTR has been completed. A Phase 2a, placebo controlled study of adults CF patients homozygous for F508del-CFTR has been initiated.
Phosphodiesterase inhibitors including sildenafil and other active analogues have been shown to improve surface localization of the mutant protein. The same agents augment chloride current in F508del CFTR expressing cell lines77, and enhance NPD in CF mice78,79, as did the related compound vardanafil80; these effects have yet to be confirmed in HBE cells and cell-free systems.
Since misfolded Phe508del CFTR exhibits two fundamentally distinct properties that alter is processing (namely NBD1 stability and interdomain assembly), it has been recognized that compounds that address these mechanisms independently can exhibit additive or synergistic affects49,50,59. Moreover, the global cellular response to misfolded protein may also represent a target. For example, treatment of CF cells with histone deacetylase (HDAC) inhibitors81 can modulate ER stress, and HDACs such as suberoylanilide hydroxamic acid (SAHA), as well as siRNA-silencing, increase levels of Phe508del CFTR in the cell membrane82. Additive or synergistic rescue of Phe508del CFTR using more than one such strategy may offer hope of achieving ion transport activity sufficient to confer a normal phenotype in CF respiratory epithelia57,83. Pharmacologic activity of such agents have also been reported to augment Phe508del CFTR half-life in the plasma membrane through altered surface recycling attributed to features of the cellular processing machinery84 or reduced endocytic trafficking85.
Conclusions
The past decade has yielded significant success in discovery and therapeutic development in the CF field. These new therapies are positively impacting the lives of some individuals with CF and the future for patients with the most common CFTR mutations has never been brighter. At the same time, the CF community has the unprecedented opportunity to understand the physiological changes created by the administration of CFTR modulators41. We have begun an iterative translational process where laboratory discovery leads to clinical application that yields additional laboratory and clinical knowledge. This translational wheel of progress is continuing at a record pace. Currently the Cystic Fibrosis Foundation and industry partners are supporting many discovery efforts to identify the future modulators of Phe508del CFTR. Whether this research will yield therapeutic approaches with a single or multiple drugs in combination is yet to be determined.
In this review, the focus has been on two classes of small molecule drugs that modulate dyfunctional CFTR. If these future drug combinations are sufficiently robust to correct CFTR function in individuals with only one copy of Phe508del, this modulator approach will ideally provide clinical benefit to over 90% of all patients with CF. The community cannot rest, however, until 100% of patients benefit. Thus, other approaches such as drugs that readthrough premature stop mutations25 and gene replacement or editing27,28 must continue.
Acknowledgments
Grant Funding: GS: CFF Solomon14YO; CFFT STOP-OB-13; Nivalis Therapeutics SNO-3
SR: NIH P30 DK072482; CLANCY09YO
BR: NIH P30DK089507; UL1TR000423
References
- 1.Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- 2.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 3.Berger HA, Anderson MP, Gregory RJ, Thompson S, Howard PW, Maurer RA, Mulligan R, Smith AE, Welsh MJ. Identification and regulation of the cystic fibrosis transmembrane conductance regulator-generated chloride channel. The Journal of clinical investigation. 1991;88(4):1422–1431. doi: 10.1172/JCI115450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440(7083):477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95(7):1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- 6.Birket SE, Chu KK, Liu L, Houser GH, Diephuis BJ, Wilsterman EJ, Dierksen G, Mazur M, Shastry S, Li Y, Watson JD, Smith AT, Schuster BS, Hanes J, Grizzle WE, Sorscher EJ, Tearney GJ, Rowe SM. A functional anatomic defect of the cystic fibrosis airway. American journal of respiratory and critical care medicine. 2014;190(4):421–432. doi: 10.1164/rccm.201404-0670OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, Awadalla MA, Moninger TO, Michalski AS, Hoffman EA, Zabner J, Stoltz DA, Welsh MJ. Cystic fibrosis. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science. 2014;345(6198):818–822. doi: 10.1126/science.1255825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. The New England journal of medicine. 2015;372(4):351–362. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. American journal of respiratory and critical care medicine. 2003;168(8):918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 10.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. The New England journal of medicine. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 11.Linsdell P. Functional architecture of the CFTR chloride channel. Molecular membrane biology. 2014;31(1):1–16. doi: 10.3109/09687688.2013.868055. [DOI] [PubMed] [Google Scholar]
- 12.Riordan JR. CFTR function and prospects for therapy. Annual review of biochemistry. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 13.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83(1):121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 14.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, Zhou J, McCartney J, Arumugam V, Decker C, Yang J, Young C, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu P. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(44):18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, Ramalho AS, Amaral MD, Dorfman R, Zielenski J, Masica DL, Karchin R, Millen L, Thomas PJ, Patrinos GP, Corey M, Lewis MH, Rommens JM, Castellani C, Penland CM, Cutting GR. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nature genetics. 2013;45(10):1160–1167. doi: 10.1038/ng.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okiyoneda T, Lukacs GL. Fixing cystic fibrosis by correcting CFTR domain assembly. The Journal of cell biology. 2012;199(2):199–204. doi: 10.1083/jcb.201208083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358(6389):761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 18.Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends in endocrinology and metabolism: TEM. 2004;15(5):222–228. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. The Journal of clinical investigation. 1997;100(10):2457–2465. doi: 10.1172/JCI119788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Egan ME, Pearson M, Weiner SA, Rajendran V, Rubin D, Glockner-Pagel J, Canny S, Du K, Lukacs GL, Caplan MJ. Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science. 2004;304(5670):600–602. doi: 10.1126/science.1093941. [DOI] [PubMed] [Google Scholar]
- 21.McCarty NA, Standaert TA, Teresi M, Tuthill C, Launspach J, Kelley TJ, Milgram LJ, Hilliard KA, Regelmann WE, Weatherly MR, Aitken ML, Konstan MW, Ahrens RC. A phase I randomized, multicenter trial of CPX in adult subjects with mild cystic fibrosis. Pediatric pulmonology. 2002;33(2):90–98. doi: 10.1002/ppul.10041. [DOI] [PubMed] [Google Scholar]
- 22.Van Goor F, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, Joubran J, Knapp T, Makings LR, Miller M, Neuberger T, Olson E, Panchenko V, Rader J, Singh A, Stack JH, Tung R, Grootenhuis PD, Negulescu P. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. American journal of physiology Lung cellular and molecular physiology. 2006;290(6):L1117–1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 23.Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, Verkman AS. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. The Journal of clinical investigation. 2005;115(9):2564–2571. doi: 10.1172/JCI24898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(46):18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Aviram M, Elfring GL, Northcutt VJ, Miller LL, Kerem B, Wilschanski M. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372(9640):719–727. doi: 10.1016/S0140-6736(08)61168-X. [DOI] [PubMed] [Google Scholar]
- 26.Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L, Hirawat S, Miller NL, Constantine S, Reha A, Ajayi T, Elfring GL, Miller LL. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. American journal of respiratory and critical care medicine. 2010;182(10):1262–1272. doi: 10.1164/rccm.201001-0137OC. [DOI] [PubMed] [Google Scholar]
- 27.Griesenbach U, Geddes DM, Alton EW. Update on gene therapy for cystic fibrosis. Current opinion in molecular therapeutics. 2003;5(5):489–494. [PubMed] [Google Scholar]
- 28.Prickett M, Jain M. Gene therapy in cystic fibrosis. Translational research : the journal of laboratory and clinical medicine. 2013;161(4):255–264. doi: 10.1016/j.trsl.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 29.Cystic Fibrosis Foundation Patient Registry. 2013 http://wwwcfforg/UploadedFiles/research/ClinicalResearch/PatientRegistryReport/2013_CFF_Patient_Registry_Annual_Data_Reportpdf.
- 30.Clancy JP, Jain M. Personalized Medicine in Cystic Fibrosis: Dawning of a New Era. American journal of respiratory and critical care medicine. 2012 doi: 10.1164/rccm.201204-0785PP. [DOI] [PubMed] [Google Scholar]
- 31.Sloane PA, Rowe SM. Cystic fibrosis transmembrane conductance regulator protein repair as a therapeutic strategy in cystic fibrosis. Curr Opin Pulm Med. 2010;16(6):591–597. doi: 10.1097/MCP.0b013e32833f1d00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. Journal of cystic fibrosis : official journal of the European Cystic Fibrosis Society. 2014;13(1):29–36. doi: 10.1016/j.jcf.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 33.Vertex Press Release: Vertex Announces Upcoming Presentations of Data at North American Cystic Fibrosis Conference - October 9 to 11, 2014. 2014 http://investorsvrtxcom/releasedetailcfm?ReleaseID=869555.
- 34.Sheppard DN, Rich DP, Ostedgaard LS, Gregory RJ, Smith AE, Welsh MJ. Mutations in CFTR associated with mild-disease-form Cl- channels with altered pore properties. Nature. 1993;362(6416):160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- 35.Bompadre SG, Sohma Y, Li M, Hwang TC. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. The Journal of general physiology. 2007;129(4):285–298. doi: 10.1085/jgp.200609667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sermet-Gaudelus I, de Blic J, LeBourgeois M, Pranke I, Edelman A, Ramsey BW. Potentiating and correcting mutant CFTR in patients with cystic fibrosis. In: Mall MA, Elborn JS, editors. Cystic Fibrosis. Vol. 64. Wakefield, UK: European Respiratory Society; 2014. pp. 129–149. [Google Scholar]
- 37.Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Sagel SD, Hornick DB, Konstan MW, Donaldson SH, Moss RB, Pilewski JM, Rubenstein RC, Uluer AZ, Aitken ML, Freedman SD, Rose LM, Mayer-Hamblett N, Dong Q, Zha J, Stone AJ, Olson ER, Ordonez CL, Campbell PW, Ashlock MA, Ramsey BW. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. The New England journal of medicine. 2010;363(21):1991–2003. doi: 10.1056/NEJMoa0909825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, Griese M, McKone EF, Wainwright CE, Konstan MW, Moss R, Ratjen F, Sermet-Gaudelus I, Rowe SM, Dong Q, Rodriguez S, Yen K, Ordonez C, Elborn JS, Group VXS. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. The New England journal of medicine. 2011;365(18):1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, Mainz JG, Rodriguez S, Li H, Yen K, Ordonez CL, Ahrens R, Group VXS. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. American journal of respiratory and critical care medicine. 2013;187(11):1219–1225. doi: 10.1164/rccm.201301-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKone EF, Borowitz D, Drevinek P, Griese M, Konstan MW, Wainwright CE, Ratjen F, Sermet-Gaudelus I, Plant B, Jiang Y, Gilmartin G, Davies JC. Long-term safety and efficacy of ivacaftor in patients with cystic fibrosis who have the G551D-CFTR mutation: response through 144 weeks of treatment (96 weeks of PERSIST) Pediatric Pulmonology Supplement. 2013;48(S36):287. [Google Scholar]
- 41.Rowe SM, Heltshe SL, Gonska T, Donaldson SH, Borowitz D, Gelfond D, Sagel SD, Khan U, Mayer-Hamblett N, Van Dalfsen JM, Joseloff E, Ramsey BW Network GIotCFFTD. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. American journal of respiratory and critical care medicine. 2014;190(2):175–184. doi: 10.1164/rccm.201404-0703OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heltshe SL, Mayer-Hamblett N, Burns JL, Khan U, Baines A, Ramsey BW, Rowe SM Network GIotCFFTD. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2015;60(5):703–712. doi: 10.1093/cid/ciu944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zemanick ET, Harris JK, Wagner BD, Robertson CE, Sagel SD, Stevens MJ, Accurso FJ, Laguna TA. Inflammation and airway microbiota during cystic fibrosis pulmonary exacerbations. PloS one. 2013;8(4):e62917. doi: 10.1371/journal.pone.0062917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gelfond D, Ma C, Semler J, Borowitz D. Intestinal pH and Gastrointestinal Transit Profiles in Cystic Fibrosis Patients Measured by Wireless Motility Capsule. Digestive diseases and sciences. 2013;58(8):2275–2281. doi: 10.1007/s10620-012-2209-1. [DOI] [PubMed] [Google Scholar]
- 45.De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, Higgins M. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. Journal of Cystic Fibrosis. 2014;13(6) doi: 10.1016/j.jcf.2014.09.005. published online September 26, 2014. [DOI] [PubMed] [Google Scholar]
- 46.Vertex Press Release: Vertex Submits Supplemental New Drug Application (sNDA) to U.S. Food and Drug Administration for Use of KALYDECO® (ivacaftor) in People 18 and Older with Cystic Fibrosis who have the R117H Mutation. 2014 http://investorsvrtxcom/releasedetailcfm?ReleaseID=857163.
- 47.Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordonez CL, Geller DE, Group VXS. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest. 2012;142(3):718–724. doi: 10.1378/chest.11-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yousef S, Solomon GM, Brody A, Rowe SM, Colin AA. Improved Clinical and Radiographic Outcomes After Treatment With Ivacaftor in a Young Adult With Cystic Fibrosis With the P67L CFTR Mutation. Chest. 2015;147(3):e79–82. doi: 10.1378/chest.14-1198. [DOI] [PubMed] [Google Scholar]
- 49.Mendoza JL, Schmidt A, Li Q, Nuvaga E, Barrett T, Bridges RJ, Feranchak AP, Brautigam CA, Thomas PJ. Requirements for efficient correction of DeltaF508 CFTR revealed by analyses of evolved sequences. Cell. 2012;148(1-2):164–174. doi: 10.1016/j.cell.2011.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, Roldan A, Lukacs GL. Correction of both NBD1 energetics and domain interface is required to restore DeltaF508 CFTR folding and function. Cell. 2012;148(1-2):150–163. doi: 10.1016/j.cell.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang C, Protasevich I, Yang Z, Seehausen D, Skalak T, Zhao X, Atwell S, Spencer Emtage J, Wetmore DR, Brouillette CG, Hunt JF. Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein science : a publication of the Protein Society. 2010;19(10):1932–1947. doi: 10.1002/pro.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aleksandrov AA, Kota P, Cui L, Jensen T, Alekseev AE, Reyes S, He L, Gentzsch M, Aleksandrov LA, Dokholyan NV, Riordan JR. Allosteric modulation balances thermodynamic stability and restores function of DeltaF508 CFTR. Journal of molecular biology. 2012;419(1-2):41–60. doi: 10.1016/j.jmb.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lazrak A, Fu L, Bali V, Bartoszewski R, Rab A, Havasi V, Keiles S, Kappes J, Kumar R, Lefkowitz E, Sorscher EJ, Matalon S, Collawn JF, Bebok Z. The silent codon change I507-ATC->ATT contributes to the severity of the DeltaF508 CFTR channel dysfunction. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2013;27(11):4630–4645. doi: 10.1096/fj.13-227330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bartoszewski RA, Jablonsky M, Bartoszewska S, Stevenson L, Dai Q, Kappes J, Collawn JF, Bebok Z. A synonymous single nucleotide polymorphism in DeltaF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. The Journal of biological chemistry. 2010;285(37):28741–28748. doi: 10.1074/jbc.M110.154575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(9):3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jurkuvenaite A, Varga K, Nowotarski K, Kirk KL, Sorscher EJ, Li Y, Clancy JP, Bebok Z, Collawn JF. Mutations in the amino terminus of the cystic fibrosis transmembrane conductance regulator enhance endocytosis. The Journal of biological chemistry. 2006;281(6):3329–3334. doi: 10.1074/jbc.M508131200. [DOI] [PubMed] [Google Scholar]
- 57.Pedemonte N, Tomati V, Sondo E, Galietta LJ. Influence of cell background on pharmacological rescue of mutant CFTR. American Journal of Physiology Cell Physiology. 298(4):C866–874. doi: 10.1152/ajpcell.00404.2009. [DOI] [PubMed] [Google Scholar]
- 58.Rowe SM, Pyle LC, Jurkevante A, Varga K, Collawn J, Sloane PA, Woodworth B, Mazur M, Fulton J, Fan L, Li Y, Fortenberry J, Sorscher EJ, Clancy JP. DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulmonary Pharmacology and Therapeutics. 2010;23(4):268–278. doi: 10.1016/j.pupt.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, Roldan A, Verkman AS, Kurth M, Simon A, Hegedus T, Beekman JM, Lukacs GL. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nature chemical biology. 2013;9(7):444–454. doi: 10.1038/nchembio.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clancy JP, Rowe SM, Accurso FJ, Aitken ML, Amin RS, Ashlock MA, Ballmann M, Boyle MP, Bronsveld I, Campbell PW, De Boeck K, Donaldson SH, Dorkin HL, Dunitz JM, Durie PR, Jain M, Leonard A, McCoy KS, Moss RB, Pilewski JM, Rosenbluth DB, Rubenstein RC, Schechter MS, Botfield M, Ordonez CL, Spencer-Green GT, Vernillet L, Wisseh S, Yen K, Konstan MW. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax. 2012;67(1):12–18. doi: 10.1136/thoraxjnl-2011-200393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marigowda G, Liu F, Waltz D. Effect of Bronchodilators in Health Individiuals Receiving Lumacaftor in Combination with Ivacaftor Peds Pulmonary. 2014;(Supplment 38) [Google Scholar]
- 62.Boyle MP, Bell SC, Konstan MW, McColley SA, Rowe SM, Rietschel E, Huang X, Waltz D, Patel NR, Rodman D on behalf of the VXSG. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: a phase 2 randomised controlled trial. The lancet Respiratory medicine. 2014;2(7):527–538. doi: 10.1016/S2213-2600(14)70132-8. [DOI] [PubMed] [Google Scholar]
- 63.Rowe SM, McColley SA, Rietschel E, Li X, Bell SC, Konstan MW, Marigowda G, Waltz D, Boyle MP. Effect of 8 Weeks of Lumacaftor in Combination with Ivacaftor in Patients with CF and Heterozygous for the F508del CFTR Mutation. Peds Pulmonary. 2014;(Supplement 38) [Google Scholar]
- 64.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP Traffic, Groups TS. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015 doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosenfeld M, Marigowda G, Liu F, Waltz D. Effect of Lumacaftor in Combination with Ivacaftor on FEV1 and Safety Measures in Patients Aged 6-11 years with CF Who are Homozygous for F508del-CFTR. Peds Pulmonary. 2014;(Supplement 38) [Google Scholar]
- 66.Vertex Press Release: Two 24-Week Phase 3 Studies of Lumacaftor in Combination with Ivacaftor Met Primary Endpoint with Statistically Significant Improvements in Lung Function (FEV1) in People with Cystic Fibrosis who have Two Copies of the F508del Mutation. 2014 http://investorsvrtxcom/releasedetailcfm?ReleaseID=856185.
- 67.Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, Borot F, Szollosi D, Wu YS, Finkbeiner WE, Hegedus T, Verkman AS, Lukacs GL. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Science translational medicine. 2014;6(246):246ra297. doi: 10.1126/scitranslmed.3008889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cholon DM, Quinney NL, Fulcher ML, Esther CR, Jr, Das J, Dokholyan NV, Randell SH, Boucher RC, Gentzsch M. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Science translational medicine. 2014;6(246):246ra296. doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu X, Dawson DC. Cystic fibrosis transmembrane conductance regulator (CFTR) potentiators protect G551D but not DeltaF508 CFTR from thermal instability. Biochemistry. 2014;53(35):5613–5618. doi: 10.1021/bi501007v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Donaldson SH, Pilewski JM, Cooke J, Himes-Lekstrom J. Addition of VX-661, An Investigational CFTR Corrector, to Ivacaftor, a CFTR Potentiator, in Patients with CF and Heterozygous for F508del/G551D-CFTR. Peds Pulmonary. 2014;(Supplement 38) [Google Scholar]
- 71.Vertex Press Release: Treatment with VX-661 and Ivacaftor in a Phase 2 Study Resulted in Statistically Significant Improvements in Lung Function in People with Cystic Fibrosis Who Have Two Copies of the F508del Mutation. 2013 http://investorsvrtxcom/releasedetailcfm?ReleaseID=757597.
- 72.DeSouza SA, Preston IR. The safety and effectiveness of riociguat to treat chronic thromboembolic pulmonary hypertension. Expert review of cardiovascular therapy. 2015:1–10. doi: 10.1586/14779072.2015.1027193. [DOI] [PubMed] [Google Scholar]
- 73.Green LS, Chun LE, Patton AK, Sun X, Rosenthal GJ, Richards JP. Mechanism of inhibition for N6022, a first-in-class drug targeting S-nitrosoglutathione reductase. Biochemistry. 2012;51(10):2157–2168. doi: 10.1021/bi201785u. [DOI] [PubMed] [Google Scholar]
- 74.Blonder JP, Look K, Sun X, Sui J, Qiu J, Scoggin C, Gabriel SE. A Novel GSNOR Inhibitor with Potent Bronchodilator Effects and CFTR Potentiation Activity. Peds Pulmonary. 2013;(Supplment 36) [Google Scholar]
- 75.Blonder JP, Quinney NL, Looker D, Sun X, Gentzsch M, Scoggin C, Gabriel SE. Intestinal Current Measurement to Assess Modulation of F508del-CFTR Function by GSNOR inhibitor Treatment in Vivo. Peds Pulmonary. 2013;(Supplement 36) [Google Scholar]
- 76.Donaldson SH, Taylor-Cousar JL, Rosenbluth DB, Zeitlin P, Chmiel J, Jain M, Mckoy KS, Zemanick ET. Safety, Tolerability, and Pharmacokinetics of the Intravenous S-Nitrosoglutathione Reductase Inhibitor N6022: An Ascending Dose Study in Subjects Homozygous for the F508Del-CFTR Mutation. Peds Pulmonary. 2014;(Supplement 38) [Google Scholar]
- 77.Robert R, Carlile GW, Pavel C, Liu N, Anjos SM, Liao J, Luo Y, Zhang D, Thomas DY, Hanrahan JW. Structural analog of sildenafil identified as a novel corrector of the F508del-CFTR trafficking defect. Molecular Pharmacology. 2008;73(2):478–489. doi: 10.1124/mol.107.040725. [DOI] [PubMed] [Google Scholar]
- 78.Lubamba B, Lecourt H, Lebacq J, Lebecque P, De Jonge H, Wallemacq P, Leal T. Preclinical evidence that sildenafil and vardenafil activate chloride transport in cystic fibrosis. American journal of respiratory and critical care medicine. 2008;177(5):506–515. doi: 10.1164/rccm.200703-344OC. [DOI] [PubMed] [Google Scholar]
- 79.Lubamba B, Lebacq J, Reychler G, Marbaix E, Wallemacq P, Lebecque P, Leal T. Inhaled PDE5 inhibitors restore chloride transport in cystic fibrosis mice. European Respiratory Journal. doi: 10.1183/09031936.00013510. [DOI] [PubMed] [Google Scholar]
- 80.Dhooghe B, Noel S, Bouzin C, Behets-Wydemans G, Leal T. Correction of Chloride Transport and Mislocalization of CFTR Protein by Vardenafil in the Gastrointestinal Tract of Cystic Fibrosis Mice. PloS one. 2013;8(10):e77314. doi: 10.1371/journal.pone.0077314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim Chiaw P, Wellhauser L, Huan LJ, Ramjeesingh M, Bear C. A chemical corrector modifies the channel function of F508del-CFTR. Molecular Pharmacology. 78(3):411–418. doi: 10.1124/mol.110.065862. [DOI] [PubMed] [Google Scholar]
- 82.Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, Thomas PJ, Matsumura Y, Skach WR, Gentzsch M, Riordan JR, Sorscher EJ, Okiyoneda T, Yates JR, 3rd, Lukacs GL, Frizzell RA, Manning G, Gottesfeld JM, Balch WE. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nature chemical biology. 2010;6(1):25–33. doi: 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR, 3rd, Segatori L, Kelly JW. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134(5):769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Varga K, Goldstein RF, Jurkuvenaite A, Chen L, Matalon S, Sorscher EJ, Bebok Z, Collawn JF. Enhanced cell-surface stability of rescued DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. Biochemical Journal. 2008;410(3):555–564. doi: 10.1042/BJ20071420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Young A, Gentzsch M, Abban CY, Jia Y, Meneses PI, Bridges RJ, Bradbury NA. Dynasore inhibits removal of wild-type and DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) from the plasma membrane. Biochemical Journal. 2009;421(3):377–385. doi: 10.1042/BJ20090389. [DOI] [PubMed] [Google Scholar]