

Abstract
A challenge of X-ray radiation therapy is that high dose X-ray can damage normal cells and cause side effects. This paper describes a new nanoparticle-based method to reduce X-ray dose in radiation therapy by internalization of gold nanoparticles that are modified with cationic molecules into cancer cells. A cationic thiol molecule is synthesized and used to modify gold nanoparticles in a one-step reaction. The modified nanoparticles can penetrate cell membranes at high yield. By bring radio-sensitizing gold nanoparticles closer to nuclei where DNA is stored, the total X-ray dose needed to kill cancer cells has been reduced. The simulation of X-ray-gold nanoparticle interaction also indicates that Auger electrons contribute more than photoelectrons.
Graphical abstract
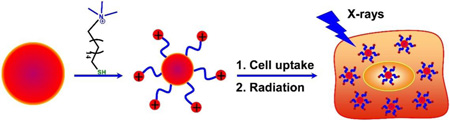
Gold nanoparticles with cationic surface modification can enhance X-ray radiation therapy by enhancing cellular uptake.
1. Introduction
A challenge of X-ray radiation therapy is that high dose radiation can damage normal cells and cause side effect due to low tumor selectivity.1 A variety of beam techniques have been developed to minimize dose on normal cells or maximize dose on cancer cells, but the methods are still limited by low precision of planning and positioning, low spatial resolution due to patient motion during treatment, and cannot treat hard-reaching tumors or tumors with undefined boundary.2–4 Radiosensitizers including oxygen, blood substitutes carrying oxygen, and radiosensitive drugs have been used to enhance efficacy of a given X-ray dose, but, damages to normal cells remain significant when X-ray dose is sufficient to kill tumors due to few reasons: inadequate delivery of radio-sensitive agents, finite targeting sites at tumor, large distance for free radicals to diffuse from sites of production (outside cell) to sites of action (inside cell), and early termination of free radical chain reactions.5–9 All these factors can cause ineffectiveness of radiation therapy, and therefore high dose X-ray is often required for cancer-killing.
In radiation therapy, X-ray photons generate photoelectrons and Auger electrons, which cause ionization of water and formation of reactive free radicals (mostly hydroxyl radicals). Free radicals diffuse through chain reactions into cells, and damage DNA in mitochondria and nuclei by extracting hydrogen atoms from ribose sugars, leading to cleavage of polynucleotide backbone.10–18 In normal condition, cells can repair damaged DNA. But, when the damage rate is higher than repair rate, damages are inherited and accumulated through cell division, causing cell to die or reproduce slowly.19–23 A typical diffusion length of hydroxyl free radical in an aqueous solution is ~200 nm (in the presence of scavenger), shorter than the distance from cell membrane to cell nucleus. If radiosensitizers can be placed in cancer cells or nuclei, the distance from site of production to nucleus will be reduced. The amount of free radicals available for DNA damage will be enhanced. The cell membrane penetrating ability of nanoparticle is dependent on the sizes, shapes and surface properties (charge and hydrophobicity).24–29 While neutral groups normally prevent nanoparticle adsorption, charged groups are primarily responsible for internalization in cells via endocytosis.30–32 A large amount of natural or synthetic nanoparticles with cationic surface charges can penetrate membrane, escape endosomes, and enter cytoplasm or nucleus. Nanoparticles modified with cell-penetrating peptides or antibodies can enter cells and chaperon cargoes in cytosol.33–35 But these methods require expensive reagents and multiple steps for modification.
Gold nanoparticles are considered bio-compatible and promising as radio-sensitizer. It is expected that the cationic modification of gold nanoparticles will enhance attachment of nanoparticles on cell membrane due to electrostatic attraction, which will lead to a higher chance of nanoparticle endocytosis. An issue for cationic modification of gold nanoparticles is that normal thiol chemistry leads to carboxyl terminated monolayers, and several additional operations will have to be taken sequentially to alter the surface charge polarity, where the multiple steps of washing, centrifuging and incubation tend to decrease yield of modification. This paper describes the synthesis and use of a thiol based cationic molecule that can be used to modify gold nanoparticles in a single step to form cationic nanoparticles that can be internalized in cancer cells at high yield. Upon irradiated with X-rays, cancer cells are killed at much lower dose.
2. Experiments
2.1 Materials and chemicals
Vybrant live/dead viability/cytotoxicity kit is from Invitrogen (Carlsbad, CA). RPMI 1640 media, penicillin, streptomycin, fetal bovine serum (FBS), and Dulbecco’s phosphate-buffered saline (D-PBS) and gold nanoparticles with the size of 10 nm at concentration of about 100 nM in 0.1 mM PBS are from Sigma–Aldrich (St. Louis, MO). Ultrapure water (18.2 MΩcm−1) from a Nanopure System (Barnstead, Kirkland, WA) is used throughout our experiments. The fluorescent images and dark field images are taken by a fluorescence microscope from Olympus (BX51M) in fluorescence mode and dark field mode, respectively. Synergy HT multimode microplate reader from Biotek (Winooski, VT) is used for absorbance and fluorescence measurements. In order to image nanoparticles, a suspension droplet of gold nanoparticles is dropped on carbon coated copper grid and allowed to dry at room temperature. A JEOL 1011 transmission electron microscope (TEM) operated at 100 kV is used to image nanoparticles. A Mini-X portable X-ray tube (Amptek, Bedford, MA) with a silver anode operating at 40 kV and 100 mA is used to generate primary X-rays and irradiate cells at a distance of 5 cm.
2.2 Synthesis of MTAB
16 mercapto-hexadecyl trimethylammonium bromide (MTAB) is synthesized according to Figure 1, which is following a literature method.36 3.93 g of triphenylphosphine (Ph3P) is added in 50 ml anhydrous tetrahydrofuran (THF); 2.67 g N-bromosuccinamide (NBS) is added into another 50 ml THF. Both solutions are mixed at 0 °C under vigorous stirring. Then, a solution of hexadecane-1,16-diol (1g) in 25 ml THF is slowly added to the mixture of NBS and Ph3P. The resulting solution is heated at 60 °C and stirred for 4 hours. After removing THF by rotary evaporation, the residue is re-crystallized from ethanol to obtain 1.1 g white powder (70% yield), which is tested by 1H NMR. 1H NMR (CDCl3, 400 MHz): δ 1.26–1.46 (m, 24 H), 1.85 (q, 4H), 3.41 (t, 4H). 1 g of 1,16-dibromohexadecane is dissolved in 40 ml methanol, and degassed in argon for 1 hour. 124 mg of sodium methoxide and 204 mg of thioacetic acid are dissolved in 12 ml anhydrous ice-cold methanol, and refluxed in argon. The content in the flask is slowly added to the solution over 4 hours duration. After reaction, the content of the flask is cooled to room temperature, and methanol is removed at reduced pressure. The yellow oil is purified by column chromatography (20% ethyl acetate in hexane) to obtain 480 mg of 16-bromo-1-hexadecane-thioacetate (50% yield). 4 ml of acetyl chloride is added drop-wise to a stirred solution of 16-bromo-1-hexa-decanethioacetate (400 mg) in 10 ml of methanol, followed by keeping at 50 °C for 4 hours. 200 ml of CH2Cl2 is added to the reaction mixture, and excess acetyl chloride and HCl are removed by extractions with deionized water. Methylene chloride is evaporated at reduced pressure to obtain 284 mg of 16-bromo-1-hexa-decanethiol as colorless oil (80% yield). 1H NMR (CDCl3, 400 MHz): δ 1.26–1.46 (m, 25H), 1.60 (m, 2H), 1.85 (q, 2H), 2.52 (q, 2H), 3.41 (t, 2H). 3 ml of 4.2 M ethanolic solution of trimethylamine is added to a solution of 16-bromo-1-hexadecanethiol (284 mg) in 5 ml of ethyl acetate. The mixture is vigorously stirred in argon for 4 days. The resulting white precipitate is filtered and washed with ethyl acetate to remove excess trimethylamine. The residue is dried in vacuum to obtain 270 mg of MTAB (80% yield). 1H NMR (CDCl3, 400 MHz): δ 1.26–1.46 (m, 25H), 1.60 (m, 2H), 1.85 (m, 2H), 2.52 (q, 2H), 3.5 (s, 9H), 3.55–3.7 (m, 2H). The final yield of MTAB is 22%.
Figure 1.

(A) Synthetic procedure of 16 mercapto-hexadecyl tri-methylammonium bromide (MTAB); (B) TEM image of MTAB-gold nanoparticles; (C) UV-Vis absorption spectra of gold nanoparticles before (black) and after (red) modification.
2.3 Cell culture and cell viability test
HeLa (CCL-2) cells are from American type culture collection (ATCC, Manassas, VA) and cultured in RMPI 1640 medium, supplemented with penicillin(100 U/ml), streptomycin (100 µg/ml), and 10% FBS, followed by a culture in a 5% CO2 incubator at 37 °C according to the protocol from ATCC. To determine cell cytotoxicity, 200 µl of suspension is seeded in each well of 96-well microplate at concentration of 1×105 cell/ml, followed by overnight culturing in 5% CO2 at 37 °C. Cells are exposed to different concentration (0.05, 0.1, 0.5, 1 nM) of citric acid (CA) or MTAB modified gold nanoparticles incubated for 24 hours. Cell viability after exposing to nanoparticles is determined by Calcein AM/EthD-1 assay, performed as follows. 100 µl of D-PBS is added in each well to wash cells and dilute serum-containing esterase, which can lead to false positive. A 100 µl of dual fluorescence calcein AM/EthD-1 assay reagents is added in each well and incubated for 30 min at room temperature prior to fluorescence measurement. The microplate is readout with Synergy HT multimode microplate reader from Biotek (Winooski, VT), where fluorescence signals are measured at 530 and 630nm, respectively. The backgrounds are subtracted before calculation by measuring a cell-free control. The percentages of live cells and dead cells are derived by dividing the fluorescence intensities of live or dead cells with the values obtained for controls. Each experimental condition is repeated for six times.
2.4 Quantifying number of internalized gold nanoparticles
The concentration of gold nanoparticles internalized into cells is determined with inductively coupled plasma mass spectroscopy (ICP-MS) (Complete Analysis Laboratories Inc., Parsippany, NJ). Briefly, HeLa cells are plated into a 6-well plate at 1×105 cells per each well. After co-incubation with CA- or MTAB-gold nanoparticles for 24 hours, the medium is removed, and cells are washed with 1× PBS for 3 times to remove nanoparticles that adhered to cell membrane. The washed cells are harvested from plate with trypsin-EDTA and then centrifuged to form pellet. The cell pellets are digested with 500 µl aqua regia for 20 min, and gold concentrations are measured by ICP-MS. Gold nanoparticles with known concentrations are used as standards.
About 105 cells are treated with 2 ml of medium containing 0.05, 0.1, 0.5 and 1 nM MTAB-nanoparticles. From ICP-MS, the total mass of gold nanoparticles taken by these cells is 0.7 µg/105 cells, or 7×10−12 g gold per cell at nanoparticle concentration of 1 nM. Given gold density of 19.30 g/cm3, the mass of a nanoparticle with 10 nm diameter will be 10−17 g. Thus, each cell contains approximately 6.9×105 gold nanoparticles.
3. Results and discussions
Fig. 1A shows the procedure of making the cationic molecule, 16 mercaptohexadecyl trimethylammonium bromide (MTAB). 1,16-hexadecanediol is converted into dibromide through standard bromination reaction, followed by conversion into monothioester. The resulting 16-bromo-1-hexadecanethiol is methylated to yield the final product. Purified MTAB is water-soluble, which allows ligand exchange with citric acid (CA)-coated gold nanoparticles (10nm) in aqueous medium. 5 mg of MTAB thiol is added into a 5 ml suspension of gold nanoparticles, followed by vigorous stirring in ambient condition for 24 hours. Gold nanoparticles is dialyzed against 0.1 mM PBS buffer solution with 3.5 kD cutoff dialysis membrane, and filtered with 0.2 µm membrane. After ligand exchange, the obtained MTAB-gold nanoparticles are stable in phosphate buffer saline (PBS). Fig. 1B shows a TEM image of MTAB-gold nanoparticles. The size of nanoparticles is ~10 nm, and no aggregation of gold nanoparticles is observed. Fig. 1C shows UV-Vis absorption spectra of gold nanoparticles before (black) and after (red) MTAB exchange, where the plasmonic peak shift is induced by MTAB monolayer formation on surfaces of gold nanoparticles. The zeta potential measurement is carried out to qualitatively describe the charge surrounding a nanoparticle. The zeta potential of the MTAB modified gold nanoparticles is found to be +53 mV, suggesting the cationic quaternary ammonium groups positioned around the gold nanoparticles.
HeLa cells are obtained from ATCC and grown in RPMI 1640 culture media that contain penicillin (100 Unit/ml), streptomycin (100 µg/ml), and 10% FBS in an incubator with 5% CO2 at 37 °C. After cell monolayer reaches 80% confluence, cells are incubated with CA- or MTAB-gold nanoparticles at concentrations of 0.05, 0.1, 0.5 and 1 nM, respectively. The growth media are removed after 24 hours, and cells are washed 3 times with 1× PBS to remove excess gold nanoparticles physically adsorbed on cell surface. Due to strong light scattering ability, nanoparticles can be observed with dark field optical microscopy. Fig. 2A–C are optical images of control cells, cells treated with CA-gold nanoparticles, and cells treated with MTAB-gold nanoparticles, respectively. Compared with CA modified ones, a large amount of MTAB-gold nanoparticles enter cells. Inductively coupled plasma-mass spectrometry (ICP-MS) that can detect metals at low concentration has been used to quantify the number of gold nanoparticles taken by cells. Fig. 2D shows that each cell uptakes an average of 690,000 gold nanoparticles after incubating with 1 nM suspension of MTAB-gold nanoparticles for 24 hours. The image shows that nanoparticles are clustered inside cells, suggesting that MTAB-gold nanoparticles enter cells via an endosomal pathway.36
Figure 2.

Dark field images of Hela cells (A), and Hela cells treated with 1 nM CA-gold nanoparticles (B) and 1 nM MTAB-gold nanoparticles (C); the numbers of MTAB-gold nanoparticles internalized into Hela cells (D).
Cells have been irradiated with X-ray (40 kVp, 100 µA) at a dose rate of 0.6 Gy/min. Cell viability is measured immediately after X-ray irradiation using Calcein AM/EthD-1 assay. No obvious cell death is observed when exposure time is less than 10 min. In order to minimize X-ray dose, the X-ray exposure time is set at 5 min. Fig. 3A–B shows the fluorescence images of cells treated with CA-gold nanoparticles and MTAB-gold nanoparticles, and irradiated with X-ray for 5min. The green and red colors show viable and dead cells, respectively. Most cells treated with CA-gold nanoparticles are alive; many cells treated with MTAB-gold nanoparticles are dead. Fig. 3C shows that cell viability depends on the concentration of MTAB-gold nanoparticles at irradiation time of 5 min, where cell viability decreases as the nanoparticle concentration increases. In contrast, MTAB-gold nanoparticles alone (black column), and X-ray alone (see supporting Fig. S1) do not cause much cell death. Thus, internalized gold nanoparticles cause the most cell death in the presence of X-ray.
Figure 3.

Fluorescent images of HeLa cells after exposed to 1 nM CA-gold nanoparticles (A), and Hela cells after exposed to MTAB-gold nanoparticles (B); MTAB-gold nanoparticles concentration dependent cell death in the presence and absence of X-ray irradiation (C); dose enhancement factors (DEF) of internalized gold nanoparticles, where Auger electrons contribute more than photoelectrons (D).
In order to understand effects of internalized gold nanoparticles in X-ray radiation based cell killing, an analytical approach is used to derive radio-sensitizing capabilities of MTAB-gold nanoparticles inside cells (dimension of 2×10×10 µm3).37 In this model, the radius of sphere with nanoparticle in center is equal to the range of emitted electrons (both photoelectrons and Auger electrons). The ratio of the dose delivered to cells with and without the nanoparticles is known as the dose enhancement factor (DEF). Fig. 3D shows the calculated dose enhancement factor (DEF) of electrons versus the number of internalized gold nanoparticles (100, 101, 102, 103, 104, 105 and 106) in each cell, respectively. Both of DEFs of Auger electrons and photoelectrons increases with the number of nanoparticles in cells. However, the DEFs of Auger electrons is considerably higher than those from photoelectrons at the same nanoparticle numbers. This is attributed to the short-range (< 1 µm) of Auger electrons, leading to deposition of more energy in vicinity of X-ray irradiated nanoparticles. As a result of near-particle energy deposition, dose enhancement within few hundred nanometers from nanoparticle is dominated by Auger electrons.
4. Conclusions
This paper describes a new surface chemistry method to enhance radiation killing of cancer cells by internalizing gold nanoparticles into cancer cells. The cationic monolayers around gold nanoparticles will allow a large number of nanoparticles interanlized into viable cells, which can lead to cell killing at much lower X-ray radiation dose. The adoption of this approach in radiation therapy will be of great importance, because 50% of cancer patients will have to use radiation therapy at certain stage of disease.
Supplementary Material
Acknowledgements
This project was supported by a NIH Director’s New Innovator Award (1DP2EB016572) to Ming Su.
Notes and references
- 1.Goldberg Z, Lehnert BE. Int. J. Oncol. 2002;21:337–349. [PubMed] [Google Scholar]
- 2.Barth RF, Soloway AH, F RG. Cancer Res. 1990;50:1061–1070. [PubMed] [Google Scholar]
- 3.Yu O. Averkov, Yakovenko VM. Plasma. Phys. Rep. 2002;28:453–410. [Google Scholar]
- 4.Adilakshmi T, Lease RA, Woodson SA. Nucleic Acids Res. 2006;34:e64. doi: 10.1093/nar/gkl291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosseinimehr SJ. Drug Discov Today. 2007;12:794–805. doi: 10.1016/j.drudis.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 6.Watts ME, Hodgkiss RJ, Jones NR, Fowler JF. Int. J. Radiat Biol. 1986;50:1009–1021. doi: 10.1080/09553008614551421. [DOI] [PubMed] [Google Scholar]
- 7.Ali H, van Lier JE. Chem. Rev. 1999;99:2379–2450. doi: 10.1021/cr980439y. [DOI] [PubMed] [Google Scholar]
- 8.Wardman P. Clin. Oncol. 2007;19:397–417. doi: 10.1016/j.clon.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 9.Werner ME, Copp JA, Karve S, Cummings ND, Sukumar R, Li C, Napier ME, Chen RC, Cox AD, Wang AZ. ACS Nano. 2011;5:8990–8998. doi: 10.1021/nn203165z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mark F, Becker U, Herak JN, Schulte-Frohlinde D. Radiat. Environ. Biophys. 1989;28:81–99. doi: 10.1007/BF01210293. [DOI] [PubMed] [Google Scholar]
- 11.Akar A, Gümüş H, Okumuşoğlu NT. Appl. Radiat. Isot. 2006;64:543–550. doi: 10.1016/j.apradiso.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 12.Borek C. J. Nutr. 2004;134:3207S–3209S. doi: 10.1093/jn/134.11.3207S. [DOI] [PubMed] [Google Scholar]
- 13.Chatgilialoglu C, O'Neill P. Exp. Gerontol. 2001;36:1459–1471. doi: 10.1016/s0531-5565(01)00132-2. [DOI] [PubMed] [Google Scholar]
- 14.Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free Radical Biol. Med. 2002;32:1102–1115. doi: 10.1016/s0891-5849(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 15.Wallace SS. Free Radical Biol. Med. 2002;33:1–14. doi: 10.1016/s0891-5849(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 16.Godley BF, Shamsi FA, Liang FQ, Jarrett SG, Davies S, Boulton M. J. Biol. Chem. 2005;280:21061–21066. doi: 10.1074/jbc.M502194200. [DOI] [PubMed] [Google Scholar]
- 17.Pogozelski WK, Tullius TD. Chem. Rev. 1998;98:1089–1108. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 18.Pratviel G, Bernadou J, Meunier B. Angew. Chem. Int. Ed. 1995;34:746–769. [Google Scholar]
- 19.Balasubramanian B, Pogozelski WK, Tullius TD. Proc. Natl. Acad. Sci. U. S. A. 1998;95:9738. doi: 10.1073/pnas.95.17.9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wood DK, Weingeist DM, Bhatia SN, Engelward BP. Proc. Natl. Acad. Sci. U. S. A. 2010;107:10008–10013. doi: 10.1073/pnas.1004056107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le XC, Xing JZ, Lee J, Leadon SA, Weinfeld M. Science. 1998;280:1066–1069. doi: 10.1126/science.280.5366.1066. [DOI] [PubMed] [Google Scholar]
- 22.Algan O, Stobbe CC, Helt AM, Hanks GE, Chapman JD. Radiat. Res. 1996;146:267–275. [PubMed] [Google Scholar]
- 23.Nevoie A, Pascariu M, Jitaru D, Ivanov I, Constantinescu D, Carasevici E, Luchian T. Dig. J. Nanomater. Bios. 2011;6:259–264. [Google Scholar]
- 24.Verma A, Stellacci F. Small. 2010;6:12–21. doi: 10.1002/smll.200901158. [DOI] [PubMed] [Google Scholar]
- 25.Juzenas P, Chen W, Sun Y-P, Coelho MAN, Generalov R, Generalova N, Christensen IL. Adv. Drug Del. Rev. 2008;60:1600–1614. doi: 10.1016/j.addr.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJAM, Geertsma RE. Biomaterials. 2008;29:1912–1919. doi: 10.1016/j.biomaterials.2007.12.037. [DOI] [PubMed] [Google Scholar]
- 27.Tkachenko AG, Xie H, Coleman D, Glomm W, Ryan J, Anderson MF, Franzen S, Feldheim DL. J. Am. Chem. Soc. 2003;125:4700–4701. doi: 10.1021/ja0296935. [DOI] [PubMed] [Google Scholar]
- 28.Chithrani BD, Ghazani AA, Chan WCW. Nano Lett. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 29.Gu Y-J, Cheng J, Lin C-C, Lam YW, Cheng SH, Wong W-T. Toxicol. Appl. Pharmacol. 2009;237:196–204. doi: 10.1016/j.taap.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Kaul G, Amiji M. Pharm. Res. 2002;19:1061–1067. doi: 10.1023/a:1016486910719. [DOI] [PubMed] [Google Scholar]
- 31.Kim SH, Jeong JH, Chun KW, Park TG. Langmuir. 2005;21:8852–8857. doi: 10.1021/la0502084. [DOI] [PubMed] [Google Scholar]
- 32.Terentyuk GS, Maslyakova GN, Suleymanova LV, Khlebtsov BN, Kogan BY, Akchurin GG, Shantrocha AV, Maksimova IL, Khlebtsov NG, Tuchin VV. J. Biophotonics. 2009;2:292–302. doi: 10.1002/jbio.200910005. [DOI] [PubMed] [Google Scholar]
- 33.Verma A, Uzun O, Hu Y, Hu Y, Han H-S, Watson N, Chen S, Irvine DJ, Stellacci F. Nat Mater. 2008;7:588–595. doi: 10.1038/nmat2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y, Lou D, Burkett J, Kohler H. J. Immunol. Methods. 2001;254:137–145. doi: 10.1016/s0022-1759(01)00410-0. [DOI] [PubMed] [Google Scholar]
- 35.Lindgren M, Hällbrink M, Prochiantz A, Langel Ü. Trends Pharmacol. Sci. 2000;21:99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 36.Vigderman L, Manna P, Zubarev ER. Angew. Chem. Int. Ed. 2012;51:636–641. doi: 10.1002/anie.201107304. [DOI] [PubMed] [Google Scholar]
- 37.Hossain M, Su M. J. Phys. Chem. C. 2012;116:23047–23052. doi: 10.1021/jp306543q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.