

Abstract
Background
Streptococcus thermophilus is a major dairy starter used for manufacturing of dairy products. In the present study, we developed a multilocus sequence typing (MLST) scheme for this important food bacterium. Sequences of 10 housekeeping genes (carB, clpX, dnaA, murC, murE, pepN, pepX, pyrG, recA, and rpoB) were obtained for 239 S. thermophilus strains, which were isolated from home-made fermented dairy foods in 18 different regions of Mongolia and China.
Methods
All 10 genes of S. thermophilus were sequenced, aligned, and defined sequence types (STs) using the BioNumerics Software. The nucleotide diversity was calculated by START v2.0. The population structure, phylogenetic relationships and the role of recombination were inferred using ClonalFrame v1.2, SplitsTree 4.0 and Structure v2.3.
Results
The 239 S. thermophilus isolates and 18 reference strains could be assigned into 119 different STs, which could be further separated into 16 clonal complexes (CCs) and 38 singletons. Among the 10 loci, a total of 132 polymorphic sites were detected. The standardized index of association (IAS = 0.0916), split-decomposition and ρ/θ (relative frequency of occurrence of recombination and mutation) and r/m value (relative impact of recombination and mutation in the diversification) confirms that recombination may have occurred, but it occurred at a low frequency in these 10 loci. Phylogenetic trees indicated that there were five lineages in the S. thermophilus isolates used in our study. MSTree and ClonalFrame tree analyses suggest that the evolution of S. thermophilus isolates have little relationship with geographic locality, but revealed no association with the types of fermented dairy product. Phylogenetic analysis of 36 whole genome strains (18 S. thermophilus, 2 S. vestibularis and 16 S. salivarius strains) indicated that our MLST scheme could clearly separate three closely related species within the salivarius group and is suitable for analyzing the population structure of the other two species in the salivarius group.
Conclusions
Our newly developed MLST scheme improved the understanding on the genetic diversity and population structure of the S. thermophilus, as well as provided useful information for further studies on the genotyping and evolutionary research for S. thermophilus strains with global diversity.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-015-0551-0) contains supplementary material, which is available to authorized users.
Keywords: Home-made fermented dairy foods, Streptococcus thermophilus, Multilocus sequence typing, Population genetics, Food microbiology
Background
Streptococcus thermophilus is a lactic acid bacteria species that is essential for the manufacturing of many types of fermented dairy products, occurring naturally as well as in commercial starter cultures [1]. Because S. thermophilus can produce large amounts of lactic acid, exopolysaccharides, and flavor compounds in fermentation processes, it is considered the second most important industrial dairy starter after Lactococcus lactis [2, 3]. Therefore, analyses of the genetic diversity, population structure, and phylogenetic relationships of S. thermophilus are important for understanding the evolution of ecological or biological properties of bacterial strains and for optimizing their industrial exploitation. Especially, S. thermophilus is genetically similar to S. vestibularis and S. salivarius in S. salivarius group. Several restriction- or PCR-based typing methods have been applied to the typing of S. thermophilus [4–8]. More recently, a powerful method for population genetic studies is multilocus sequence typing (MLST), which is based on sequencing of conserved housekeeping genes. The method provides faster and less expensive nucleotide sequence determination, and has become the gold standard for studying the evolution and population genetics of pathogenic microbes [9].
MLST involves the sequencing of a small number (at least seven) of housekeeping genes for each strain. The sequences of each fragment are assigned allele numbers for each locus. All allele numbers are combined to define the allelic profile of the strain and each different allelic profile is assigned as a sequence type (ST), which is used to describe the strain [10]. Because the typing is based on nucleotide sequence, MLST is primarily used for the exchange of electronic data between laboratories via the internet and provides accurate information on strain evolution [11, 12]. MLST has been used successfully in molecular epidemiology studies [13–15], and for identifying and typing lactic acid bacteria of various species of Lactobacillus [16–18], Pediococcus [19], Enterococcus [20], Oenococcus [21], and Lactococcus [22, 23]. The MLST scheme also reported for S. thermophilus analyzed the nucleotide variability at eight genetic loci of 27 S. thermophilus and the phylogenic relationship of two oral streptococci S. salivarius and S. vestibularis in the salivarius group [24].
In this study, 239 S. thermophilus isolated from different ecological sources and geographical areas and 36 whole genome strains (18 S. thermophilus, 2 S. vestibularis and 16 S. salivarius strains) were used to: (i) developed an effective MLST scheme for characterization of S. thermophilus and distinguish three closely related species in S. salivarius group; (ii) apply MLST to assess phylogenetic relationship and evolutionary characteristics of these strains; (iii) compare the genetic diversity of S. thermophilus from different type of dairy foods and geographic origin.
Methods
Bacterial isolates and DNA extraction
A total of 239 isolates of S. thermophilus were selected from Lactic Acid Bacteria Collection Centre (LABCC) of Inner Mongolia Agriculture University in China. Those included strains isolated from home-made fermented dairy foods (cow milk, yak milk, goat milk, mare milk and Qula-a kind of traditional cheese) in 6 provinces of China and 11 provinces and 1 city of Mongolia from 2005 to 2009. All these isolates were identified based on 16S rRNA gene sequencing, and some representative isolate were tested for gas production from glucose, salt tolerance (3.0 and 6.5 %), growth at different temperature (5, 10, 45, and 50 °C) and pH (3.0, 3.5, 4.0, and 7.0) in MRS broth [25–29]. The information of 239 S. thermophilus strains is listed in Additional file 1. Thirty six completed genome strains (18 S. thermophilus, 2 S. vestibularis and 16 S. salivarius strains) were chosen as reference strains for evaluate the novel MLST scheme (Table 1). Sequences of those completed genome strains were obtained directly from the NCBI genome database.
Table 1.
36 whole genome strains belonging to the salivarius group used in this study
| Organism | Strains | GenBank Accession no. | Source | Location | Year |
|---|---|---|---|---|---|
| Streptococcus salivarius | HSISS1 | GCA_000448645 | Ileostomy effluent | Netherlands | 2010 |
| HSISS4 | GCF_000448685 | Ileostomy effluent | Netherlands | 2010 | |
| JIM 8777 | FR873482 | Oral cavity | France | 2004 | |
| NCTC8618 | CP009913 | / | / | / | |
| UC3162 | JYOY00000000 | / | / | / | |
| CCHSS3 | FR873481 | Human blood | France | 2002 | |
| KB0005 | JYOX00000000 | / | / | / | |
| 57.I | CP002888 | Dental plaque | / | / | |
| SK126 | ACLO00000000 | Human skin | / | / | |
| M18 | AGBV00000000 | Oral swab from healthy volunteer | New Zealand | / | |
| K12 | ALIF00000000 | Saliva from healthy child | / | / | |
| NU10 | JJMT00000000 | / | / | / | |
| YU10 | JJMS00000000 | / | / | / | |
| C150 | GCA_000187445 | Human airway | / | ||
| PS4 | AJFW00000000 | Milk from a healthy woman | spain | 2010 | |
| HSISS3 | GCF_000448605 | Ileostomy effluent | Netherlands | 2010 | |
| Streptococcus vestibularis | F0396 | GCA_000180075 | Oral cavity | England | 1988 |
| ATCC 49124 | GCA_000188295 | Oral cavity | England | 1988 | |
| Streptococcus thermophilus | M17PTZA496 | CM002372 | Fontina cheese | Italy | 1996 |
| MTCC 5461 | ALIL01000000 | Fermented milk product (curd) | India | 1984 | |
| MTCC 5460 | ALIK00000000 | Fermented milk product (curd) | India | 1984 | |
| 1F8CT | CM003138 | Curd of “Grana Padano” cheese | Italy | 2012 | |
| TH1477 | CM003135 | Cow milk | Italy | 2012 | |
| TH1436 | CM002370 | Artisanal goat cheese from raw milk | Italy | 2011 | |
| CNRZ1066 | CP000024 | Commercial yogurt | France | 1986 | |
| LMG 18311 | CP000023 | Commercial yogurt | United Kingdom | 1974 | |
| MTH17CL396 | CM002371 | Fontina cheese | Italy | 1996 | |
| JIM 8232 | FR875178 | Raw milk | France | 2002 | |
| ND03 | CP002340 | Naturally fermented yak milk | China | 2005 | |
| TH985 | CM003139 | Buffalo mozzarella whey | Italy | 2003 | |
| TH982 | CM003136 | Buffalo moarella curd | Italy | 2003 | |
| LMD-9 | CP000419 | Starter for yogurt and mozzarella cheese | USA | / | |
| TH1435 | CM002369 | Artisanal goat cheese from raw milk | Italy | 2011 | |
| MN-ZLW-002 | CP003499 | Yogurt block | China | / | |
| DGCC 7710 | AWVZ00000000 | Commercial starter culture | / | / | |
| ASCC 1275 | CP006819 | / | / | / |
All S. thermophilus strains were grown in 5.0 mL M17-broth (OXOID, CM0817B, Germany) supplemented with 5.0 g/L of lactose at 30 °C for 18–22 h, and then strains were harvested by centrifugation and cell pellets were used for DNA extraction. Total genomic DNA was extracted from cultures by using a previously reported method [30]. Purified DNA was diluted to a final concentration of 100 ng/μL for application.
MLST loci selection
Ten housekeeping loci (carB, clpX, dnaA, murC, murE, pepN, pepX, pyrG, recA and rpoB gene) were selected for MLST analysis of S. thermophilus isolates based on the chromosome locations (preferably evenly separated across the entire genome), functions of the encoded proteins (preferably conserved and well characterized) and presence in all the strains as a single copy [21, 31]. The primers of ten genes were designed by Primer Premier 5.0 program (Premier Biosoft International) on the basis of known genome in S. thermophilus ND03 [32], and the information of primers is listed in Table 2.
Table 2.
Genes and primers used for Multilocus sequence typing
| Gene | Amplicon size (bp) | Application size (bp) | Gene function | PCR primer | Sequences (5’-3’) | Temperature |
|---|---|---|---|---|---|---|
| carB | 652 | 499 | carBamoyl phosphate synthetase | carB_primerF | AAGGCTACAGTGTTGTTCT | 51 °C |
| carB_primerR | GATAAGGTTTGCGTTGG | |||||
| clpX | 768 | 630 | ATP-dependent Clp protease subunit X | clpX_primerF | GCAGGTTATGTGGGTGA | 52 °C |
| clpX_primerR | TACTGGAGCAGCTTTCC | |||||
| dnaA | 669 | 522 | chromosomal replication initiator protein dnaA | dnaA_primerF | CACCAGGAGCAACTTAT | 49 °C |
| dnaA_primerR | CTTCAATCGGAATGAGA | |||||
| murC | 592 | 451 | UDP-N-acetyl muramate-alanine ligase | murC_primerF | TTTGTTTACGGTGAGGA | 51 °C |
| murC_primerR | CCACTTTGGCAGGTTTA | |||||
| murE | 721 | 592 | UDP-N-acetylmuramyl tripeptide synthase | murE_primerF | AACACCCTCAAGACAAA | 48 °C |
| murE_primerR | ACGAATACCTTAGCACC | |||||
| pepN | 737 | 615 | lysyl-aminopeptidase, aminopeptidase N | pepN_primerF | CGCTGAAGAGGGCGATAC | 58 °C |
| pepN_primerR | CTAACCAACGGCGGAGC | |||||
| pepX | 664 | 524 | X-prolyl dipeptidyl aminopeptidase | pepX_primerF | ACATCCCTGTTAGTCCTG | 52 °C |
| pepX_primerR | TCTCCCTCCATCTTGTG | |||||
| pyrG | 736 | 624 | CTP synthase | pyrG_primerF | CACTGAAGTTGGTGGGA | 52 °C |
| pyrG_primerR | CATACCGAGGCAGACAC | |||||
| recA | 768 | 617 | recombinase A | recA_primerF | AAAGAAGGTGGCATCGC | 58 °C |
| recA_primerR | ATCGTCCTCATCTAGCTCAAC | |||||
| rpoB | 786 | 661 | RNA polymerase beta subunit | rpoB_primerF | CATTACACGCACTACGG | 47 °C |
| rpoB_primerR | GATAACAGCATCCTCGA |
PCR amplification and DNA sequencing
For each strain, the genomic DNA was used as a template for PCR amplification of MLST loci on the automatic thermal cycler (PTC-200, MJ Research, Waltham, MA). Thermal cycling conditions for PCR were: 94 °C for 5 min; 30 cycles of 94 °C for 1 min, corresponding temperature of each locus for 1 min, and 72 °C for 2 min; a final elongation step of 72 °C for 10 min. For each target, PCR mixture (50 μL) containing 150 ng of genomic DNA, 10 mM of each dNTP, 10 pmol of each primer, 2.5 U Taq polymerase in 1x PCR buffer (with 2.5 mmol/L Mg2+). PCR products were electrophoresed in a 1.2 % agarose gel. Sequencing of the PCR products was performed in Shanghai Majorbio Bio-pharm Technology Corporation. The same primers were used for PCR and sequencing on both DNA strands.
MLST data analysis
For MLST analysis, forward and reverse sequences were trimmed, aligned, and analyzed using MEGA 6.0 software package (version 6.0, www.megasoftware.net) [33]. Definition of alleles using a nonredundant dataset and minimum spanning tree analysis were preformed using the BioNumerics Software (version 6.6, Applied-Maths, Sint Maartens-Latem, Belgium). Different allelic sequences (with at least one nucleotide difference) were assigned arbitrary numbers. For each of the ten MLST loci, a unique nucleotide sequence defined an allele. Unique allelic profiles, consisting of the allele numbers at each of the ten MLST loci, defined STs. The same ST was used for several strains when they shared the same allelic profiles. Groups of isolates with closely related allelic profiles have been called clonal complexes (CCs). STs were grouped into CC with the eBURST program [34] and located in the BioNumerics program.
START version 2.0 program [35] was used for calculating the number of polymorphic sites, mol% G + C content, dN/dS, and IA and IAS value for linkage analysis of the population as well as recombination testing. The nucleotide diversity (π) per site was calculated using Dnasp version 5.0 [36]. The split decomposition method was used to assess the degree of tree-like structure for alleles of each locus and all STs using SplitsTree 4.0 [37]. The phi test for recombination based on individual loci of the whole strain collection was also done with SplitsTree. The phylogenetic trees from the concatenated sequences (5718 bp) were constructed by the neighbor-joining method with a Kimura two-parameter distance model using MEGA 6.0. Bootstrap analysis with 1,000 replicates was performed.
The software Structure v2. 3 with linkage model [38] was used to identify the ancestral subpopulations and assign ancestry proportions for each isolate. The K value that generated the highest posterior probability was used as the probable number of ancestral populations. Three independent runs were performed for each value of the number of populations K ranging from 3 to 15. Each run consisted of 100,000 Markov Chain Monte Carlo (MCMC) iterations, of which the first 20,000 iterations were discarded as burn-in. The K value that generated the highest median posterior probability was used as the probable number of ancestral populations.
The evolutionary relationships amongst of S. thermophilus strains were inferred using ClonalFrame v1.2 [39]. Three independent runs of ClonalFrame were performed each consisting of 200,000 MCMC iterations, and a posterior sampling of 300,000 iterations. The prior iterations were discarded and model parameters were sampled in the second period of the run every 100th iteration thereafter, resulting in 3,000 samples from the posterior. The genealogy of the population was summarized and the robustness of the tree topology was evaluated by concatenating the posterior samples of the 3 runs to built-up a 50 % majority rule consensus tree using the ClonalFrame GUI. The ρ/θ and r/m was also accounted by ClonalFrame.
Nucleotide sequence accession numbers
All MLST data of S. thermophilus in this study are available at http://pubmlst.org/sthermophilus/ [40]. Allele sequences of the ten MLST loci have been deposited in the GenBank database under accession numbers KF794203 to KF796592.
Results
Allelic profiles and sequence types
The 10 genes were successfully amplified for all strains and sequenced by bidirectional sequencing technology, and forward and reverse sequences were trimmed, aligned, and analyzed. Nucleotide positions of the sequences showing ambiguities were excluded from the analysis. Ultimately, the sequences of 10 housekeeping genes, ranging in size from 451 to 630 bp (Table 2), were used for MLST analysis.
Additional file 2 summarizes the allelic profiles of the 239 isolates and 18 S. thermophilus reference strains. A total of 119 different STs (ST1–ST119) were obtained; 83 STs corresponded to single isolates, 24 STs included 2–4 isolates, 8 STs included 5–9 isolates, and 4 STs included 11–15 isolates. The most represented STs were ST5 composed of 15 isolates (5.8 % of all isolates), ST2 (14 isolates, 5.4 %), ST79 (13 isolates, 5.1 %), and ST39 (11 isolates, 4.3 %). The breakdown by region was as follows: 140 strains from 12 different regions of Mongolia were identified as 67 STs, and 99 strains from 6 different provinces of China were assigned to 43 STs. Among the 119 STs identified in our collection, 14 STs were attributed to strains isolated in more than two provinces. The widest type was ST5, which was identified in strains isolated in as many as five different provinces across China and Mongolia. A similar distribution was observed for ST65, ST66, and ST91, which were identified in strains isolated from two provinces of China and Mongolia, respectively; the remaining ST strains were isolated in a specific province. Moreover, almost all genome strains were identified as a single ST, the exceptions being ND03, MN-ZLW-002, ASCC 1275, DGCC 7710, and LMD-9. Strain ND03 belonged to ST2, MN-ZLW-002 belonged to ST65, ASCC 1275 and DGCC 7710 belonged to ST66, and strain LMD-9 belonged to ST91.
Nucleotide sequence variation at each MLST locus
We calculated the nucleotide diversity at each locus of all strains (Table 3). All loci were polymorphic and the number of polymorphic nucleotide sites varied between 8 (recA and murC) and 20 (clpX) suggesting a different rate of evolution. Between 7 (recA) and 18 (clpX) alleles were found for each locus. A total of 5718 nucleotides comprised 132 polymorphic sites. The mol% G + C content observed for different gene fragments varied from 36.89 % (dnaA) to 44.14 % (pepN) and was 39.1 % for the full S. thermophilus ND03 genome [32]. The nucleotide diversity per site (π) among the 10 genes varied from 0.0040 in rpoB to 0.0056 in pepN. The dN/dS ratio of 10 loci ranged from 0.0311 (pyrG) to 0.2239 (pepX) and all were far less than 1 (Table 3), suggesting strong purifying selective pressure (negative selection) in these genes.
Table 3.
Descriptive analysis of MLST data genetic variability at Streptococcus thermophilus loci
| Gene | No. of | πa/site | G + C content (mol %) | d N/d S b | |
|---|---|---|---|---|---|
| alleles | polymorphic sites | ||||
| carB | 10 | 10 | 0.0050 | 43.49 | 0.1439 |
| clpX | 18 | 20 | 0.0055 | 41.62 | 0.0789 |
| dnaA | 9 | 12 | 0.0054 | 36.89 | 0.0819 |
| murC | 9 | 8 | 0.0047 | 40.45 | 0.0360 |
| murE | 14 | 19 | 0.0048 | 41.64 | 0.0910 |
| pepN | 17 | 14 | 0.0056 | 44.14 | 0.1734 |
| pepX | 15 | 16 | 0.0053 | 41.97 | 0.2239 |
| pyrG | 12 | 12 | 0.0055 | 42.07 | 0.0311 |
| recA | 7 | 8 | 0.0043 | 39.08 | 0.0807 |
| rpoB | 13 | 13 | 0.0040 | 43.41 | 0.1046 |
a: Mean pairwise nucleotide difference per site
b: d N/d S represents the ratio of nonsynonymous to synonymous substitutions
Evidence for recombination in S. thermophilus
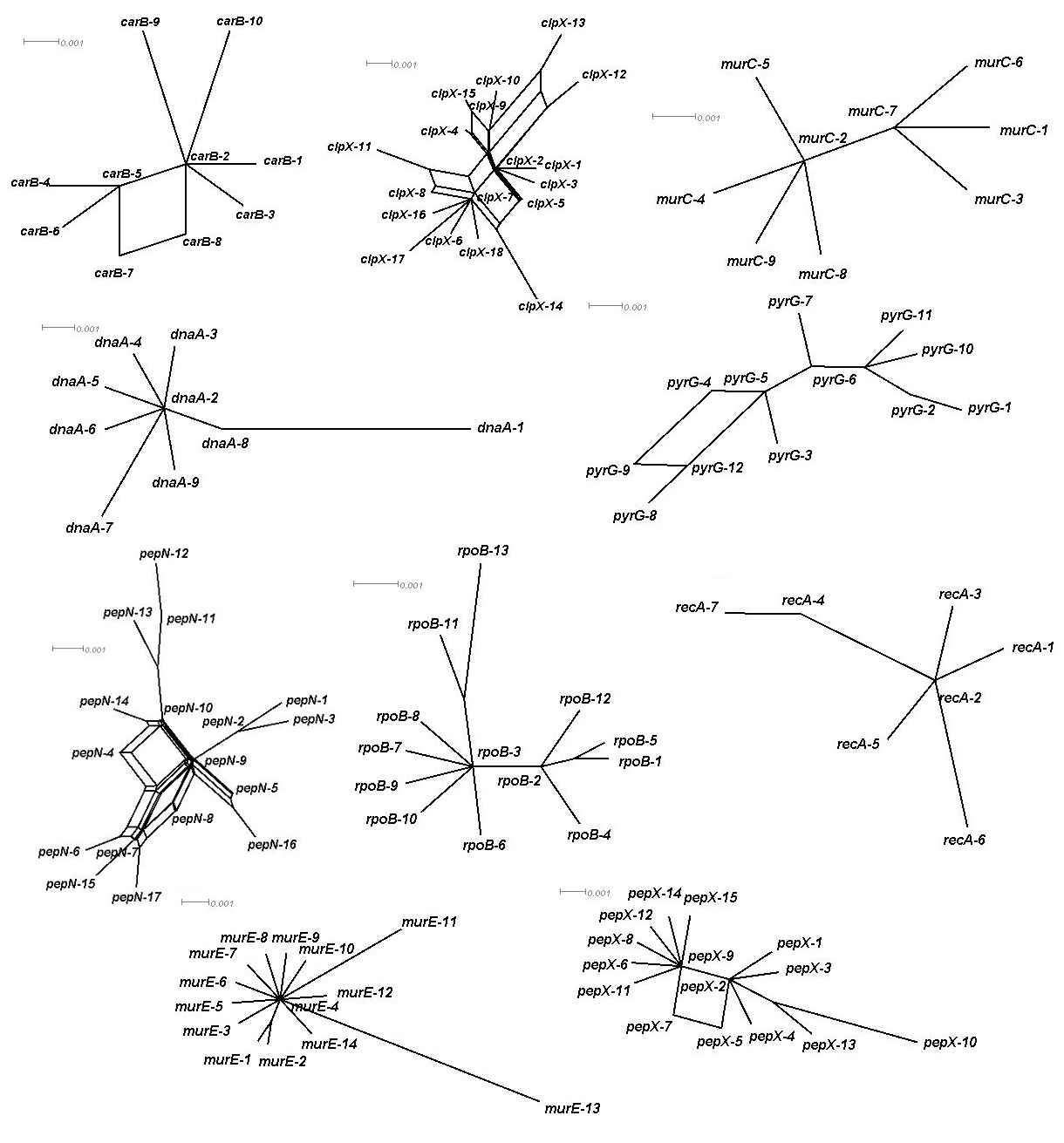
The multilocus linkage disequilibrium between MLST loci was measured using the index of association (IA) [41] and standardized index of association (IAS) [42]. To minimize linkage disequilibrium introduced by sampling bias or recent expansion of adaptive genotypes, only one strain from each ST was analyzed [22]. An IA of 0.8242 (P = 0.000) and an IAS of 0.0916 (P =0.000) for the 10 loci were obtained, which were greater than the value of 0 expected for a population at linkage equilibrium. This also shows the formation of an underlying clonal population structure of all the studied isolates. However, the low IAS value confirms that recombination played a role in the evolution of the analyzed genes, and the split graphs of all ten loci and the concatenated sequences corroborated this. From the split graphs (Additional files 3 and 4), we can see that some parallelogram structures were formed for the clpX, pepN, pepX, pyrG, and carB genes indicating that these genes have undergone a little intergenic recombination during evolution (Additional file 3). In addition, the concatenated sequences of the 10 loci displayed a complex network-like structure (Additional file 4), suggesting that there have been several recombination events for these 10 loci based on the phi test (P = 2.108E-9). It is deduced that the selection of 5 genes with a more recombination history maybe contribute to the recombination events.
The linkage disequilibrium analysis and split graphs proved that recombination are existent in the evolution of 257 S. thermophilus strains. Then ClonalFrame was used to further assess the recombination or mutation play a leading role in the evolution of S. thermophilus. The r/m value (relative impact of recombination and mutation in diversification) and ρ/θ value (relative frequency of occurrence of recombination and mutation) were 0.0036 (95 % Credibility Interval 0.001 - 0.034) and 0.0034 (95 % Credibility Interval 0.001 - 0.031), respectively. These low values indicate that recombination occurred less frequently than mutation at these loci in the evolution of 257 S. thermophilus.
Clonal complexes and minimum-spanning tree analysis
Assignment of STs to CCs by eBURST analysis divided 119 STs into 16 CCs (CC1–CC16) and 38 singletons (Additional file 2). The CC strains represented 73.2 % of all strains. The colored zones between some circle groups in Fig. 1 indicate profiles belonging to the same CC. The major CCs, CC1 (yellow zone), included 14 STs (corresponding to 25 strains) with ST49 identified as the ancestor genotype. CC1 comprised 24 strains isolated from seven different provinces in Mongolia, and only one strain isolated from Qinghai province in China (ST92). CC2 contained 36 isolates representing 10 STs (pink zone), with most of the strains isolated from Mongolia and 4 strains isolated from China. ST5 was identified as the ancestor ST of CC2. CC3 included 10 STs with ST67 identified as the ancestor genotype. 6 Mongolian isolates, 5 Chinese isolates, and reference strains JIM 8232, LMD-9, TH1435, TH1436, ASCC 1275, and DGCC 7710 comprise CC3 (red zone). CC4 consisted of 19 strains (light blue zone) and CC5 consisted of 15 strains (purple zone), all isolated from Xinjiang and Tibet, and Qinghai in China, respectively. All other CCs comprised less than 6 STs with a limited number of strains, and the strains in one CC were almost entirely from the same geographical location.
Fig. 1.

Minimum-spanning tree analysis of 239 Streptococcus thermophilus isolates and 18 reference strains based on the allelic profiles of 10 genes. Each circle corresponds to a sequence type (ST), and the circle size denotes the number of strains sharing the same ST. Colored zones between some groups of circles indicate that these profiles belong to the same clonal complex (CC). The strength of the link (bold, plain, or discontinuous) is related to the genetic similarity (number of common alleles) between profiles. Black line was drawn as boundaries of each branch
To analyze the relationship between the clusters, geographic origin, and the source dairy product among the 257 S. thermophilus isolates, allelic profile-based phylogenetic analysis was performed using a Minimum-Spanning Tree (MSTree) analysis with the BioNumerics v5.10 software. To facilitate analysis, Gansu and Sichuan province of China were grouped into Gannan region, and 11 provinces and one city in Mongolia were grouped into three larger regions (central, northwestern, and northeastern Mongolia) based on geographic position and environment. As shown in Fig. 1, strains of the same allelic profile were in the same circle, the size of which was proportional to the number of strains of that particular profile, and the different colors of the circles indicate different strain regions. The MSTree revealed four major branches and three minor branches. Branch 1 contained 39 strains from CC1, CC7 and 5 single STs. All strains were isolated from Mongolia except ST 92. Branch 2 contained three CCs (CC5, CC12, and CC16) and strain F8CT. The isolates in these branches were from Qinghai in China (19 strains) and central Mongolia (9 strains). Branch 3 contained three CCs (CC3, CC8, and CC15), with isolates from Xinjiang (6 strains), Gannan (13 strains), Qinghai (11 strains), Inner Mongolia (11 strains), central Mongolia (9 strains), and 13 reference strains. Regarding CC2 as the center, Branch 4 was divided into three minor branches (4a-c). Branch 4a included 38 strains of CC2, mainly from Mongolia (32 strains). Branch 4b, containing CC11 and CC13, contained 28 strains isolated from Mongolia. Branch 4c contained 55 strains of CC4, CC6, CC9, CC10, and CC14, most isolated from Xinjiang (20 strains), Tibet (3 strains), Gannan (9 strains), and central Mongolia (15 strains). Moreover, ST11, ST70, ST102, and ST122 do not belong to these four branches and are very different from ST 49. It is suggested that the phylogenetic relationships are more distant between these isolates compared to the others.
Population structure and phylogenetic relationships
We attempted to statistically estimate the number of ancestral subpopulations (K) within the genetic population of the 119 S. thermophilus STs (257 isolates) using STRUCTURE with the linkage model. A maximal posterior probability of K = 6 was found in our sample, meaning that the genetic diversity of S. thermophilus can be divided into six ancestral subpopulations. For each ST, the average proportion of genetic material derived from each ancestral population is shown in Fig. 2b. The 119 STs fell into six distinct populations based on the major ancestral source of genetic diversity. Subpopulation 1 (green) contained 25 STs, 10 from Mongolia and 15 from Tibet, Xinjinag province, and Gannan in China. Subpopulation 2 (yellow) comprised 13 STs from Mongolia (23/25 strains). Subpopulation 3 (pink) consisted of 37 STs, mainly from Mongolia (72/79 strains), and subpopulation 4 (blue) consisted of 20 STs derived from central Mongolia strains and 13 reference strains. Subpopulation 5 (red) comprised 17 STs represented by isolates from central Mongolia (17/36) and Qinhai province in China (19/36). Subpopulation 6 (orange) contained 5 STs from five reference strains and one Mongolian strain. There was little admixture of ancestral sources among these six subpopulations, suggesting high homogeneity for the STs of each subpopulation. Moreover, ST98 and ST101 contained a high degree of admixture, which seemed to have acted frequently both as donors and as recipients of recombination exchanges.
Fig. 2.
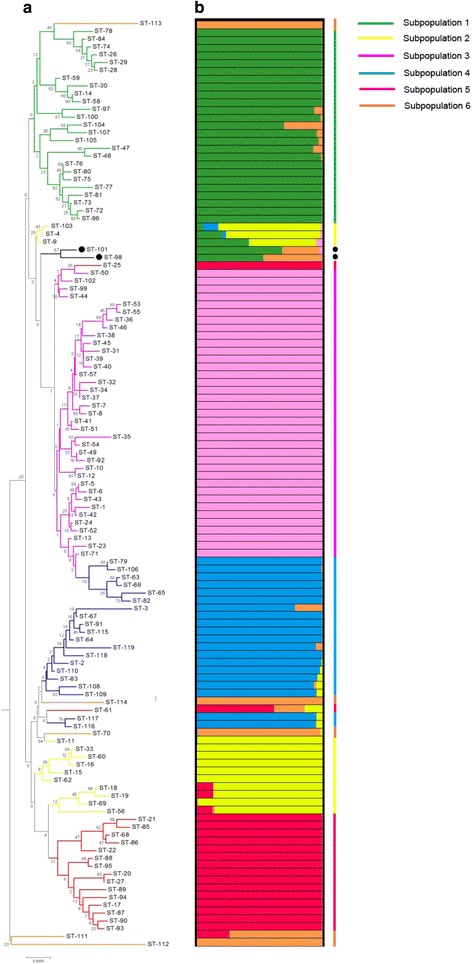
NJ tree and Ancestry of 119 Streptococcus thermophilus STs. a Neighbour-joining tree constructed from concatenated sequences of the 10 MLST loci. Bootstrap values are indicated for all branches. STs are colored according to their affiliation to one of the six ancestral subpopulations; admixed STs are in black. b Sources of ancestry of each unique ST from six ancestral subpopulations by Structure (assuming K = 6 populations and applying the linkage model). Six ancestral subpopulations are colored in Green (subpopulations 1), yellow (subpopulations 2), pink (subpopulations 3), blue (subpopulations 4), red (subpopulations 5) and orange (subpopulations 6). Each ST is represented by a single line with the ST designation at the top consisting of colored stacked bars that indicate the proportion of ancestry from each of six subpopulations
The phylogeny of the 119 STs was analyzed by constructing a NJ tree from the concatenated sequences of 10 loci. As shown in Fig. 2a, the NJ tree revealed four major branches. The colored branches of the NJ tree are in accordance with the color of the groups identified in STRUCTURE. As shown in Fig. 2, we found that the STs of subpopulations 2 (yellow) and 4 (blue), 6 (orange) were distributed in differenced branches of the NJ tree. To examine any possible influences of recombination on tree topology, we inferred a clonal genealogy from our data using ClonalFrame, which takes recombination into account during tree building. The ClonalFrame tree (Additional file 5) showed all isolates fell into five lineages, A-E. Five subpopulations (1–5) identified in STRUCTURE (Fig. 2a) corresponded to the subpopulations shown in Additional file 5 and are represented by corresponding colors. However, subpopulation 6, found in STRUCTURE, did not constitute a lineage in the NJ analysis and ST114 (MTH17CL396) fell into lineage C. The other STs formed small clades at this level of analysis based on the ClonalFrame results. Based on the combined evidence from the STRUCTURE and ClonalFrame analyses, the 257 S. thermophilus strains formed five lineages during evolution.
Phylogenetic analysis of S. thermophilus within the salivarius group
S. thermophilus belongs to the salivarius group, together with the two oral streptococci S. salivarius and S. vestibularis, and these three species are genetically very similar. A MLST scheme was created to analyze the phylogenetic relationships among these three species using five housekeeping genes (ddlA, thrS, pyrE, sodA, and dnaE) [43, 44]. S. thermophilus is the only nonpathogenic streptococcus originating from dairy products. Comparative genomic analysis found that the origin of genes acquired by S. thermophilus is suggested to be other LAB living in recurrent association with S. thermophilus in cheese or yogurt manufacture [45]. Therefore, we developed a new MLST scheme for S. thermophilus based on fragments of 10 housekeeping genes, which were used in a previous analysis of LAB. A phylogenetic tree was constructed using 36 whole genome strains, including 18 S. thermophilus, 16 S. salivarius, and 2 S. vestibularis, and the concatenated sequences of 10 loci (Fig. 3). It revealed four clear branches and the species S. thermophilus, S. salivarius, and S. vestibularis were supported by significant bootstrap values. The NJ tree also shows the existence of a fourth branch consisting of the three strains. In addition, 16 S. salivarius and 2 S. vestibularis were separated from each other, indicating that this MLST scheme is suitable for analyzing the population structure of all species in the salivarius group.
Fig. 3.

Phylogenetic tree of 18 Streptococcus thermophilus, 2 S. vestibularis and 16 S. salivarius strains, based on the concatenated sequences of 10 housekeeping genes
Discussion
S. thermophilus is a key species involved in the acidification of milk and the development of texture in various fermented dairy products. However, few studies have characterized the diversity of the genetic population and the evolutionary scheme of this food bacterium from different ecological origins. To contribute to the characterization of the population structure of S. thermophilus, we utilized the MLST method to examine 239 strains isolated from different ecological sources and geographical areas.
MLST was first used for pathogenic microbes [10]. This technique is useful for elucidating the phylogenetic relationships and evolution of isolates. Traditional MLST schemes generally include only five to seven gene loci, the lengths of which range from 400 to 500 bp. This small number of gene loci and short length may lead to a decrease in information and unreliable results. Based on previous MLST studies of LAB and the S. thermophilus ND03 genome, 12 gene targets (carB, clpX, dnaA, murC, murE, pepN, pepX, pyrG, recA, groEL, uvrC, and rpoB gene) were first selected for MLST analysis in this study. Two loci (groEL and uvrC) could not be amplified from all examined strains and were removed. From these 10 gene loci, 239 S. thermophilus isolates and 18 genome strains were identified as 119 STs. The number of alleles per locus ranged from 7 to 18. When any gene locus with the lowest number of alleles was removed from the analysis, the number of STs decreased. Finally, an optimization of the MLST scheme based on these 10 housekeeping genes was chosen for analyzing S. thermophilus. Compared to previous MLST schemes [24], an average of 12 distinguishable alleles per locus among the 119 STs were identified, which is more than the average of 6–7 alleles observed from 27 S. thermophilus isolates in a previous study. Phylogenetic analysis of whole genome strains within the salivarius group showed that our method could clearly separate three closely related species and also confirmed the existence of an additional cluster. Our MLST scheme has strong typing discrimination for S. thermophilus and could be applied to the other two species in the salivarius group.
Analysis of synonymous and non-synonymous changes in the allele sequences of a locus can be used to determine whether it is subject to selection pressure. In our analysis, all loci displayed a dN/dS ratio lower than 1, indicating a strong purifying selection preventing amino acid changes, which is a typical phenomenon for housekeeping genes and is desired in MLST schemes. The π of the 10 genes varied from 0.0040 in rpoB to 0.0056 in pepN. Previous reports on other lactic acid bacteria have ranged from 0.0004 to 0.0072 for L. plantarum [46], 0.0051 to 0.0096 for L. delbruecki [18], and 0.0038 to 0.0119 for Leuconostoc mesenteroides [47]. These results suggest that the examined S. thermophilus housekeeping genes are relatively conserved. Loss of gene function, recombination, and horizontal gene transfer (HGT) were proposed to contribute to the plasticity of the S. thermophilus genome [48, 49]. The low IAS value (0.0916) and network structure of the split-graph in this study confirms that recombination may have occurred, while the low ρ/θ and r/m value indicate that recombination has not played a major role in the evolution of these loci. We infer that the discrepancy between our data and the literature may be related to the selected loci and S. thermophilus isolates.
The population structure within the sequences was analyzed using the linkage model in Structure. It is suggested that the six ancestral subpopulations in our isolates and some STs contain a high degree of admixture (Fig. 2). To examine any possible influences of recombination on tree topology, we inferred a phylogenetic tree using ClonalFrame. The ClonalFrame tree (Additional file 5) revealed that subpopulation 6 identified in STRUCTURE did not represent a true lineage, because this subpopulation is, in fact, a random mixture of strains that did not fall into one of the five true lineages and does not represent a real evolutionary lineage. However, the NJ tree contains four major branches, but they are not well supported because of low bootstrap values. The low bootstrap values are representative of the inherently incongruent phylogenetic signals in the NJ topology. It is suggested that the admixture of STs possibly contributed to the overall poor support values for the NJ topology [50]. Compared with the analytical tools in ClonalFrame and STRUCTURE, our data demonstrate that NJ analysis is not necessarily a suitable method for analyzing phylogenetic relationships between bacteria using MLST data.
One interesting application of the MLST technique was the identification of an association between strains and their origin. Previous studies on 40 Lactobacillus casei isolates using MLST demonstrated specificity to particular ecological niches [31]. However, the MSTree (Fig. 1) and ClonalFrame tree (Additional file 5) analyses in this study suggest that the evolution of S. thermophilus isolates have little relationship with geographic locality. Often, most strains from the same location clustered together, while the rest were dispersed across other clusters. The isolates from the same region are likely to have been exposed to similar environment selective pressures. For instance, Tibet and Xinjiang are located in western China, and have similar climate characteristics of drought and extensive sunshine, thus most isolates from those regions belong to the same lineage. Reference strain ND03 was isolated from Qinghai province in China, so it belongs to ST2 along with the 13 Qinghai isolates. In addition, it is interesting that the central Mongolian strains are distributed in several lineages. Central Mongolia includes the city of UlaanBaatar, the capital of Mongolia. Because of the traffic network in UlaanBaatar, trade contacts were more frequent with the other provinces of Mongolia and China. We deduced that fermented food exchange was a factor that may have contributed to the distribution of the central Mongolian strains, but the reference strains, especially the 12 European strains, have no strong relationship to the lineages and origins of the examined strains that could be identified, based either on the isolation of dairy sources or the sampling location. Furthermore, no significant associations between lineages and the type of dairy products were found in our collection of S. thermophilus (data not shown), which was probably due to an unequal number of isolates from different fermented products. A total of 182 S. thermophilus were isolated from fermented cow milk, whereas only 57 strains were from other fermented dairy products (3 isolates from fermented mare milk, 11 isolates from fermented goat milk, 5 isolates from Qula, and 38 isolates from fermented yak milk). As more isolates are collected from different countries and types of fermented products, further evidence may become available for the grouping of S. thermophilus compared to worldwide isolates, which would provide a strong indication of the factors that have affected its evolutionary history.
Conclusions
MLST was used to study the genetic polymorphisms and evolutionary relationships of 239 isolates of S. thermophilus from different origins. Analysis of a geographically diverse and representative collection of isolates using MLST can provide a better understanding of S. thermophilus genome evolution, and provide information for future studies on the structure and genetic evolution of S. thermophilus globally.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Grant No. 31471711), National Key Technology R&D Program (2013BAD18B01), Program for International S&T Cooperation Projects of China (ISTCP, 2014DFR31150), the Hi-Tech Research and Development Program of China (863 Planning, Grant No. 2011AA100902), the China Agriculture Research System (Grant No. CARS-37), the Natural Science Foundation of Inner Mongolia (No. 2013MS1205) and the Open Projects of Inner Mongolia Natural Science Foundation (No. 20102010). This publication made use of the Streptococcus thermophilus MLST website (http://pubmlst.org/sthermophilus/) sited at the University of Oxford, U.K. We would like to thank Professor Jolley KA and Maiden MCJ for kind help with MLST data submission.
Additional files
Bacterial strains used in this study. (XLS 19 kb)
Allelic profiles of analyzed Streptococcus thermophilus strains. (XLS 30 kb)
{kind=link}
Split-decomposition of alleles for individual MLST loci of Streptococcus thermophilus strains. (JPEG 250 kb)
{kind=link}
Combined split-decomposition of alleles for the 10 MLST loci of Streptococcus thermophilus strains. (JPEG 191 kb)
Clonal genealogy inferred by ClonalFrame from the 239 Streptococcus thermophilus isolates and 18 reference strains. Six subpopulations identified in Fig. 2b by Structure corresponded to lineages of the ClonalFrame clonal genealogy and have therefore been colored with the same colors as in Fig. 2. (PDF 1528 kb)
Footnotes
Jie Yu and Zhihong Sun contributed equally to this work.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
JY, ZHS and STS designed the experiments. JY, XXX, YQS, HYX, QHB and QL performed the experiments. JY, ZHS, WJL, MHBLG and STS drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Jie Yu, Email: yujie8301@sina.com.
Zhihong Sun, Email: sunzhihong78@163.com.
Wenjun Liu, Email: wjliu168@163.com.
Xiaoxia Xi, Email: xixiaoxia22@163.com.
Yuqin Song, Email: songyqnd@163.com.
Haiyan Xu, Email: weiliangxhy@163.com.
Qiang Lv, Email: 1109701051@qq.com.
Qiuhua Bao, Email: nmgbqh@126.com.
Bilige Menghe, Email: mhblg@163.com.
Tiansong Sun, Email: sts9940@sina.com.
References
- 1.Coppola S, Parente E, Dumontet S, La Peccerella A. The microflora of natural whey cultures utilized as starters in the manufacture of Mozzarella cheese from water-buffalo milk. Lait. 1988;68(3):295–309. doi: 10.1051/lait:1988319. [DOI] [Google Scholar]
- 2.Hols P, Hancy F, Fontaine L, Grossiord B, Prozzi D, Leblond-Bourget N, Decaris B, Bolotin A, Delorme C, Dusko Ehrlich S, Guedon E, Monnet V, Renault P, Kleerebezem M. New insights in the molecular biology and physiology of Streptococcus thermophilus revealed by comparative genomics. FEMS Microbiol Rev. 2005;29(3):435–63. doi: 10.1016/j.femsre.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Iyer R, Tomar SK, Uma Maheswari T, Singh R. Streptococcus thermophilus strains: multifunctional lactic acid bacteria. Int Dairy J. 2010;20(3):133–41. doi: 10.1016/j.idairyj.2009.10.005. [DOI] [Google Scholar]
- 4.Lazzi C, Bove CG, Sgarbi E, Monica G, La Gioia F, Sandra T, Neviani E. Application of AFLP fingerprint analysis for studying the biodiversity of Streptococcus thermophilus. J Microbiol Methods. 2009;79(1):48–54. doi: 10.1016/j.mimet.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 5.Moschetti G, Blaiotta G, Aponte M, Catzeddu P, Villani F, Deiana P, Coppola S. Random amplified polymorphic DNA and amplified ribosomal DNA spacer polymorphism: powerful methods to differentiate Streptococcus thermophilus strains. J Appl Microbiol. 1998;85(1):25–36. doi: 10.1046/j.1365-2672.1998.00461.x. [DOI] [PubMed] [Google Scholar]
- 6.O'Sullivan TF, Fitzgerald GF. Comparison of Streptococcus thermophilus strains by pulse field gel electrophoresis of genomic DNA. FEMS Microbiol Lett. 1998;168(2):213–9. doi: 10.1111/j.1574-6968.1998.tb13276.x. [DOI] [PubMed] [Google Scholar]
- 7.Horvath P, Romero DA, Coûté-Monvoisin A-C, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol. 2008;190(4):1401–12. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El‐Sharoud W, Delorme C, Darwish M, Renault P. Genotyping of Streptococcus thermophilus strains isolated from traditional Egyptian dairy products by sequence analysis of the phosphoserine phosphatase (serB) gene with phenotypic characterizations of the strains. J Appl Bacteriol. 2012;112(2):329–37. doi: 10.1111/j.1365-2672.2011.05212.x. [DOI] [PubMed] [Google Scholar]
- 9.Maiden MC. Multilocus sequence typing of bacteria. Annu Rev Microbiol. 2006;60:561–88. doi: 10.1146/annurev.micro.59.030804.121325. [DOI] [PubMed] [Google Scholar]
- 10.Maiden MCJ, Bygraves JA, Edward F, Giovanna M, Joanne ER, Rachel U, Zhang Q, Zhou J. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci. 1998;95:3140–5. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miragaia M, Thomas JC, Couto I, Enright MC, de Lencastre H. Inferring a population structure for Staphylococcus epidermidis from multilocus sequence typing data. J Bacteriol. 2007;189(6):2540–52. doi: 10.1128/JB.01484-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Solyman SM, Black CC, Duim B, Perreten V, van Duijkeren E, Wagenaar JA, Eberlein LC, Sadeghi LN, Videla R, Bemis DA, Kania SA. Multilocus sequence typing for characterization of Staphylococcus pseudintermedius. J Clin Microbiol. 2013;51(1):306–10. doi: 10.1128/JCM.02421-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Octavia S, Salim A, Kurniawan J, Lam C, Leung Q, Ahsan S, Reeves PR, Nair GB, Lan R. Population structure and evolution of non-O1/non-O139 Vibrio cholerae by multilocus sequence typing. PLoS ONE. 2013;8(6):e65342. doi: 10.1371/journal.pone.0065342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui Y, Yu C, Yan Y, Li D, Li Y, Jombart T, Weinert LA, Wang Z, Guo Z, Xu L. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis. Proc Natl Acad Sci U S A. 2013;110(2):577–82. doi: 10.1073/pnas.1205750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogler AJ, Chan F, Wagner DM, Roumagnac P, Lee J, Nera R, Eppinger M, Ravel J, Rahalison L, Rasoamanana BW. Phylogeography and molecular epidemiology of Yersinia pestis in Madagascar. PLoS Neglect Trop D. 2011;5(9) doi: 10.1371/journal.pntd.0001319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diancourt L, Passet V, Chervaux C, Garault P, Smokvina T, Brisse S. Multilocus sequence typing of Lactobacillus casei reveals a clonal population structure with low levels of homologous recombination. Appl Environ Microbiol. 2007;73(20):6601–11. doi: 10.1128/AEM.01095-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Picozzi C, Bonacina G, Vigentini I, Foschino R. Genetic diversity in Italian Lactobacillus sanfranciscensis strains assessed by multilocus sequence typing and pulsed-field gel electrophoresis analyses. Microbiology. 2010;156(Pt 7):2035–45. doi: 10.1099/mic.0.037341-0. [DOI] [PubMed] [Google Scholar]
- 18.Tanigawa K, Watanabe K. Multilocus sequence typing reveals a novel subspeciation of Lactobacillus delbrueckii. Microbiology. 2011;157(Pt 3):727–38. doi: 10.1099/mic.0.043240-0. [DOI] [PubMed] [Google Scholar]
- 19.Calmin G, Lefort F, Belbahri L. Multi-loci sequence typing (MLST) for two lacto-acid bacteria (LAB) species: Pediococcus parvulus and P. damnosus. Mol Biotechnol. 2008;40(2):170–9. doi: 10.1007/s12033-008-9073-4. [DOI] [PubMed] [Google Scholar]
- 20.Burgos MJ, Lopez RL, Abriouel H, Omar NB, Galvez A. Multilocus sequence typing of Enterococcus faecalis from vegetable foods reveals two new sequence types. Foodborne Pathog Dis. 2009;6(3):321–7. doi: 10.1089/fpd.2008.0169. [DOI] [PubMed] [Google Scholar]
- 21.Bilhère E, Lucas PM, Claisse O, Lonvaud-Funel A. Multilocus sequence typing of Oenococcus oeni: detection of two subpopulations shaped by intergenic recombination. Appl Environ Microbiol. 2009;75(5):1291–300. doi: 10.1128/AEM.02563-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Passerini D, Beltramo C, Coddeville M, Quentin Y, Ritzenthaler P, Daveran-Mingot M-L, Le Bourgeois P. Genes but not genomes reveal bacterial domestication of Lactococcus lactis. PLoS ONE. 2010;5(12) doi: 10.1371/journal.pone.0015306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernández E, Alegría Á, Delgado S, Martín MC, Mayo B. Comparative phenotypic and molecular genetic profiling of wild Lactococcus lactis subsp. lactis strains of the L. lactis subsp. lactis and L. lactis subsp. cremoris genotypes, isolated from Starter-Free Cheeses made of raw milk. Appl Environ Microbiol. 2011;77(15):5324–35. doi: 10.1128/AEM.02991-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delorme C, Bartholini C, Bolotine A, Ehrlich SD, Renault P. Emergence of a cell wall protease in the Streptococcus thermophilus population. Appl Environ Microbiol. 2010;76(2):451–60. doi: 10.1128/AEM.01018-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu J, Wang WH, Menghe BL, Jiri MT, Wang HM, Liu WJ, Bao QH, Lu Q, Zhang JC, Wang F, Xu HY, Sun TS, Zhang HP. Diversity of lactic acid bacteria associated with traditional fermented dairy products in Mongolia. J Dairy Sci. 2011;94(7):3229–41. doi: 10.3168/jds.2010-3727. [DOI] [PubMed] [Google Scholar]
- 26.Sun ZH, Liu WJ, Zhang JC, Yu J, Gao W, Jiri M, Menghe B, Sun TS, Zhang HP. Identification and characterization of the dominant lactic acid bacteria isolated from traditional fermented milk in Mongolia. Folia Microbiol (Praha) 2010;55(3):270–6. doi: 10.1007/s12223-010-0040-7. [DOI] [PubMed] [Google Scholar]
- 27.Liu W, Bao Q, Jirimutu, Qing M, Siriguleng, Chen X, et al. Isolation and identification of lactic acid bacteria from Tarag in Eastern Inner Mongolia of China by 16S rRNA sequences and DGGE analysis. Microbiol Res. 2012, 167(2):110–5. [DOI] [PubMed]
- 28.Bao Q, Yu J, Liu W, Qing M, Wang W, Chen X, Wang F, Li M, Wang H, Lv Q, Zhang H. Predominant lactic acid bacteria in traditional fermented yak milk products in the Sichuan province of China. Dairy Sci Technol. 2012;92(3):309–19. doi: 10.1007/s13594-012-0061-x. [DOI] [Google Scholar]
- 29.Bao Q, Liu W, Yu J, Wang W, Qing M, Chen X, Wang F, Zhang J, Zhang W, Qiao J, Sun T, Zhang H. Isolation and identification of cultivable lactic acid bacteria in traditional yak milk products of Gansu province in China. J Gen Appl Microbiol. 2012;58(2):95–105. doi: 10.2323/jgam.58.95. [DOI] [PubMed] [Google Scholar]
- 30.Yu J, Sun Z, Liu W, Bao Q, Zhang J, Zhang H. Phylogenetic study of Lactobacillus acidophilus group, L. casei group and L. plantarum group based on partial hsp60, pheS and tuf gene sequences. Eur Food Res Technol. 2012;234(6):927–34. doi: 10.1007/s00217-012-1712-0. [DOI] [Google Scholar]
- 31.Cai H, Rodriguez BT, Zhang W, Broadbent JR, Steele JL. Genotypic and phenotypic characterization of Lactobacillus casei strains isolated from different ecological niches suggests frequent recombination and niche specificity. Microbiology. 2007;153(Pt 8):2655–65. doi: 10.1099/mic.0.2007/006452-0. [DOI] [PubMed] [Google Scholar]
- 32.Sun Z, Chen X, Wang J, Zhao W, Shao Y, Wu L, Zhou Z, Sun T, Wang L, Meng H, Zhang H, Chen W. Complete genome sequence of Streptococcus thermophilus strain ND03. J Bacteriol. 2011;193(3):793–4. doi: 10.1128/JB.01374-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feil EJ, Li BC, Aanensen DM, Hanage WP, Spratt BG. eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J Bacteriol. 2004;186(5):1518–30. doi: 10.1128/JB.186.5.1518-1530.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jolley KA, Feil EJ, Chan MS, Maiden MC. Sequence type analysis and recombinational tests (START) Bioinformatics. 2001;17(12):1230–1. doi: 10.1093/bioinformatics/17.12.1230. [DOI] [PubMed] [Google Scholar]
- 36.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 37.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23(2):254–67. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 38.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes. 2007;7(4):574–8. doi: 10.1111/j.1471-8286.2007.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007;175(3):1251–66. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jolley KA, Maiden MC. BIGSdb: Scalable analysis of bacterial genome variation at the population level. Bmc Bioinformatics. 2010;11(23):595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith JM, Smith NH, O'Rourke M, Spratt BG. How clonal are bacteria? Proc Natl Acad Sci. 1993;90(10):4384–8. doi: 10.1073/pnas.90.10.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haubold B, Hudson RR. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Linkage Analysis. Bioinformatics. 2000;16(9):847–8. doi: 10.1093/bioinformatics/16.9.847. [DOI] [PubMed] [Google Scholar]
- 43.Delorme C, Poyart C, Ehrlich SD, Renault P. Extent of horizontal gene transfer in evolution of Streptococci of the salivarius group. J Bacteriol. 2007;189(4):1330–41. doi: 10.1128/JB.01058-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delorme C, Abraham AL, Renault P, Guedon E. Genomics of Streptococcus salivarius, a major human commensal. Infect Genet Evol. 2014;33:381–92. doi: 10.1016/j.meegid.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 45.Bolotin A, Quinquis B, Renault P, Sorokin A, Ehrlich SD, Kulakauskas S, Lapidus A, Goltsman E, Mazur M, Pusch GD, Fonstein M, Overbeek R, Kyprides N, Purnelle B, Prozzi D, Ngui K, Masuy D, Hancy F, Burteau S, Boutry M, Delcour J, Goffeau A, Hols P. Complete sequence and comparative genome analysis of the dairy bacterium Streptococcus thermophilus. Nat Biotech. 2004;22(12):1554–8. doi: 10.1038/nbt1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Las RB, Marcobal A, Munoz R. Development of a multilocus sequence typing method for analysis of Lactobacillus plantarum strains. Microbiology. 2006;152(Pt 1):85–93. doi: 10.1099/mic.0.28482-0. [DOI] [PubMed] [Google Scholar]
- 47.Zhang W, Liu W, Song Y, Xu H, Menghe B, Zhang H, Sun Z. Multilocus sequence typing of a dairy-associated Leuconostoc mesenteroides population reveals clonal structure with intragenic homologous recombination. J Dairy Sci. 2015;98(4):2284–93. doi: 10.3168/jds.2014-9227. [DOI] [PubMed] [Google Scholar]
- 48.Lefébure T, Stanhope MJ. Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 2007;8(5):R71–R. doi: 10.1186/gb-2007-8-5-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marri PR, Hao W, Golding GB. Gene gain and gene loss in streptococcus: is it driven by habitat? Mol Biol Evol. 2006;23(12):2379–91. doi: 10.1093/molbev/msl115. [DOI] [PubMed] [Google Scholar]
- 50.Chaillou S, Lucquin I, Najjari A, Zagorec M, Champomier-Vergès M-C. Population genetics of Lactobacillus sakei reveals three lineages with distinct evolutionary histories. PLoS ONE. 2013;8(9):e73253. doi: 10.1371/journal.pone.0073253. [DOI] [PMC free article] [PubMed] [Google Scholar]