

Significance
Cerebral malaria (CM) is a deadly complication of Plasmodium falciparum infection in African children despite effective antimalarial treatment. Once signs of neurologic disease have commenced, there is no adjunctive treatment for CM, and overall mortality remains high. Thus, a treatment that arrests disease and promotes healing in the late stages is urgently needed. Here we report, in an animal model of CM, that the glutamine analog 6-diazo-5-oxo-L-norleucine (DON) is an effective therapy even when treatment is initiated after infected animals show neurological signs of disease. Within hours of DON treatment blood–brain barrier integrity was restored, and brain swelling was reduced. These results suggest DON as a strong candidate for an effective adjunctive therapy for CM in African children.
Keywords: cerebral malaria, adjunctive therapy, CD8+ T cells, glutamine metabolism, DON
Abstract
The most deadly complication of Plasmodium falciparum infection is cerebral malaria (CM) with a case fatality rate of 15–25% in African children despite effective antimalarial chemotherapy. There are no adjunctive treatments for CM, so there is an urgent need to identify new targets for therapy. Here we show that the glutamine analog 6-diazo-5-oxo-l-norleucine (DON) rescues mice from CM when administered late in the infection a time at which mice already are suffering blood–brain barrier dysfunction, brain swelling, and hemorrhaging accompanied by accumulation of parasite-specific CD8+ effector T cells and infected red blood cells in the brain. Remarkably, within hours of DON treatment mice showed blood–brain barrier integrity, reduced brain swelling, decreased function of activated effector CD8+ T cells in the brain, and levels of brain metabolites that resembled those in uninfected mice. These results suggest DON as a strong candidate for an effective adjunctive therapy for CM in African children.
The World Health Organization estimates that there are nearly 200 million clinical cases of Plasmodium falciparum malaria annually (1). For most individuals living in endemic areas, malaria is uncomplicated and resolves with time. However, in about 1% of cases, almost exclusively among young children, malaria becomes severe and life threatening, resulting in 525,000 deaths each year in Africa alone. One of the most deadly complications of P. falciparum infection in humans is cerebral malaria (HCM) characterized by the onset of severe neurological signs such as altered consciousness, seizures, and coma (2). Autopsy and MRI analyses of brains of children with HCM indicate sequestration of infected red blood cells (iRBCs), microhemorrhaging, breakdown of the blood–brain barrier (BBB) (3), and a fatal increase in intracranial pressure resulting from edema (4, 5). At present, despite effective antimalarial drug treatment, mortality for children presenting with HCM remains high, at 15–25%. HCM takes a second toll on African children, leaving survivors at risk for debilitating neurological defects (6). Thus, there is an urgent need for the development of effective adjunctive therapies that can be used in conjunction with antimalarials to treat children with HCM.
Experimental cerebral malaria (ECM) in mice is a widely used model of HCM and provides a valuable tool for elucidating the mechanisms involved in CM pathogenesis and identifying cellular and molecular targets for adjunctive therapy (7). In ECM, 6–7 d after infection with Plasmodium berghei ANKA (PbA), mice of susceptible strains, such as C57BL/6, develop ataxia, paralysis, seizures, and coma and ultimately die (8). ECM displays key features of HCM, including BBB breakdown, focal hemorrhaging, and brain swelling (9–11). ECM’s pathology also requires sequestration of iRBCs in the brain vasculature (12), a hallmark of HCM (3). Histological analysis of the brains of children who died of HCM showed leukocytes, primarily monocytes with phagocytized hemozoin and platelets but also intravasculature leukocytes, including CD8+ T cells, sequestered in the brain vessels (13, 14). In ECM monocytes and both CD4+ and CD8+ T cells have been shown to accumulate in the brain by both flow cytometry and by intravital imaging (15). Current evidence indicates that CD8+ T cells are the major mediators of death in ECM (16) and that antigen-specific CD8+ T cells engage parasite antigens cross-presented on MHC class I molecules on brain endothelium, resulting in endothelial cell dysfunction by a perforin-dependent mechanism (17).
A critical role for metabolic reprogramming in regulating immune responses is becoming increasingly appreciated. Upon activation, T cells undergo metabolic reprogramming to meet the increased energetic and biosynthetic demands of growth and effector T-cell functions (18–20). Reprogramming involves a shift to aerobic glycolysis and increased glutaminolysis. Activated T cells import large quantities of Gln and increase their expression of glutaminase (21–23). Because the pathology leading to death in CM is believed to be in part immune mediated, we hypothesized that blocking T-cell metabolism might effectively mitigate the pathology leading to death in HCM. To this end, in the present study we focus on targeting Gln metabolism for an adjunctive therapy for CM using the Gln analog 6-diazo-5-oxo-l-norleucine (DON). DON broadly inhibits Gln metabolism, in part by blocking Gln transport and inhibiting all three isoforms of glutaminase as well as other Gln-using enzymes such as the amidotransferases and glutamine synthetase (24). Consequently, DON has been shown to be a potent inhibitor of T-cell proliferation (22).
Here we show that DON treatment rescues PbA-infected mice from ECM at late stages in the disease, at a time when the animals show clinical signs of neurological damage and physical loss of BBB integrity, brain swelling, and hemorrhaging. The ability of DON to arrest disease is concomitant with a decrease in the effector function of parasite-specific CD8+ T cells in the brains of treated mice. However, the striking ability of DON treatment to arrest pathology and promote survival so late in the disease suggests a fundamental and potentially direct role for Gln metabolism in promoting neuropathology. Overall, these results suggest DON as a candidate for an adjunctive therapy for HCM in African children.
Results
DON Treatment Promotes Survival Even in Late-Stage ECM.
To determine if blocking Gln metabolism would inhibit death caused by immunopathology in a mouse model of ECM, C57BL/6 mice were infected with PbA on day 0 and were injected i.p. with DON (1.3 mg/kg) or saline, beginning at 7:00 AM on the morning of day 5 p.i. (day 5a p.i.), at 11:00 PM on day 5 p.i. (day 5p p.i.), or at 7:00 AM on day 6 p.i. (day 6a p.i.). DON treatments were repeated either every day or every other day (Fig. S1). The majority of untreated PbA-infected mice died during day 6 p.i., and all died by day 7 p.i. (Fig. 1A). In contrast, all the mice treated with DON beginning on day 5a p.i. and 80% of the mice treated on day 5p p.i. survived as followed until day 12 p.i. Remarkably, nearly 50% of mice treated as late as day 6a p.i. survived.
Fig. S1.
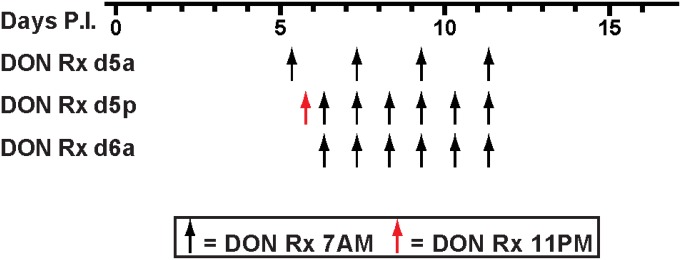
DON treatment schedule. DON treatment (1.3 mg/kg) was initiated on day 5 p.i. at 7:00 AM (d5a), day 5 p.i. at 11:00 PM (d5p), or day 6 p.i. at 7:00 AM (d6a) and was continued every day or every other day as shown.
Fig. 1.

DON treatment reduced the mortality associated with ECM. C57BL/6 mice were infected with PbA on day 0 and were injected i.p. with saline (NoRx) (n = 49) or with DON (1.3 mg/kg) beginning on day 5 p.i. at 7:00 AM (DON Rx d5a) (n = 28), on day 5 p.i. at 11:00 PM (DON Rx d5p) (n = 28), or on day 6 p.i. at 7:00 AM (DON Rx d6a) (n = 28). DON treatment was continued every day or every other day as shown in Fig. S1. (A) Kaplan–Meier survival plots. (B) Clinical scores from 0 (no symptoms) to 10 (moribund) of mice in A. (C) Peripheral blood parasitemia for mice in A. Data for DON Rx d5a were combined from three independent experiments, data for DON Rx d5p were combined from four independent experiments, and data for DON Rx d6a were combined from two independent experiments. Data in B and C are shown as mean and SEM. (D) Fold changes in PbA 18s RNA in brains of DON-treated and untreated PbA-infected mice on day 6a p.i. compared with PbA 18s RNA in brains of PbA-infected untreated mice on day 5p p.i. Each dot represents a mouse with the mean and SD given. The results shown are combined from three independent experiments, each having three or four mice per group. A Mann–Whitney test showed no significant difference.
Mice were evaluated for the development of neurological signs associated with ECM and were given clinical scores between 0 (no signs) to 10 (moribund) using previously described criteria (25). Nearly all the untreated mice that were infected with PbA developed neurological signs by day 5p p.i. that in most cases were severe (clinical score >6) by day 6a p.i. (Fig. 1B). Treatment with DON beginning on day 5a p.i. prevented the development of neurological symptoms in all PbA-infected mice (Fig. 1B). Treatment of mice with DON beginning on day 5p p.i., a point at which most mice had clinical scores of 2, not only prevented the worsening of clinical signs but promoted the rapid resolution of symptoms (Fig. 1B). Remarkably, treatment of mice on day 6a p.i., when many mice already had developed clinical scores of 5, blocked the progression of the disease and rapidly resolved the symptoms in half of the mice (Fig. 1B). Thus, DON was able to arrest disease and promote healing even when the mice already were displaying signs of neurologic damage. A scatter plot showing the clinical scores of individual mice on the day of treatment and the resulting outcome of treatment is given in Fig. S2. For mice treated with DON on d5p p.i., the clinical scores on the day of DON treatment (which ranged from 0 to 6) did not distinguish the mice that survived from those that died, even though the average clinical score was significantly higher for mice that died following treatment. For mice treated with DON on day 6a p.i., clinical scores below 8 did not distinguish the mice that survived from those that did not. However, all mice with clinical scores of 9 or 10 died following DON treatment, indicating that there is a tipping point clinically at which mice no longer can be rescued by DON treatment.
Fig. S2.

Clinical scores for mice that survived or died with or without DON treatment. (A) Clinical scores for mice that were not treated starting at day 5 p.i. at 11:00 PM (d5p) and then at day 6 p.i. at 7:00 AM (d6a). (B) Clinical scores measured on d5p and on d6a for mice that either survived or died after treatment with DON on d5p. The average clinical score at d5p is significantly higher for the mice that died than for mice that survived (P = 0.0045). (C) Clinical scores measured on d6a and on d7 at 7:00 AM (d7a) for mice treated on d6a with DON that survived or died. Each dot represents a mouse, and dots are staggered around time points. The average clinical score at d6a is significantly higher for the mice that died than for mice that survived (P = 0.017). A plus sign indicates a mouse that died after the first time point.
In both DON-treated and untreated PbA-infected mice parasitemia increased similarly with time, reaching peak levels of ∼8–12% between d5 and d6 p.i. (Fig. 1C). Nearly all untreated mice died by day 7 p.i., with peripheral parasitemia of 10–12%. In the DON-treated mice that survived beyond day 6 p.i., the peripheral parasitemia decreased beginning on day 7 p.i., reaching low levels by day 8 p.i. However, during the critical period of day 6 p.i., during which untreated mice die and DON treated mice survive, the parasite loads were indistinguishable in the brains of treated and untreated mice (Fig. 1D), suggesting that inhibition of parasite growth by DON is not the primary mechanism promoting survival. Consistent with the ability of DON to inhibit parasite replication, continued DON treatment suppressed parasitemia to under 5% as followed out to day 17 p.i., and when DON treatment was stopped, parasitemia increased to a mean of 40% by day 21 p.i. (Fig. S3).
Fig. S3.

Late cessation of DON Rx leads to increased parasitemia. C57BL/6 mice were infected with PbA on day 0 and given saline (No Rx) or DON (1.3 mg/kg) every other day beginning at 7:00 AM on day 1 p.i. (DON Rx d1a). The peripheral blood parasitemia was determined by flow cytometry on the days indicated.
DON Treatment Promotes Recovery of BBB Integrity and Reduction in Brain Swelling but Does Not Have an Immediate Effect on Brain Hemorrhaging.
Compromise of the BBB, brain swelling, and hemorrhaging are major components of the neuropathology observed in ECM. Thus, we assessed the effect of DON treatment on the integrity of the BBB by measuring the leakage of the dye Evans blue (EB) into the brains (26, 27). Mice were injected retroorbitally with EB and were killed 3 h later; then their brains removed, and EB was quantified. Both visual inspection (Fig. 2A) and EB quantification (Fig. 2B) showed significant leakage of EB into the brains of PbA-infected, untreated mice on day 5p p.i., and the leakage increased further on day 6a p.i. (Fig. 2 A and B). Remarkably, on day 6a p.i. EB leakage was significantly less in the brains of PbA-infected DON-treated mice than in the brains of untreated mice, and the EB leakage in the brains of PbA-infected DON-treated mice decreased significantly further by day 7a p.i. (Fig. 2 A and B).
Fig. 2.

DON treatment promoted BBB function and reduced brain swelling but did not acutely resolve brain hemorrhages in PbA-infected mice. All mice were infected with PbA and treated with saline or DON (1.3 mg/kg) on day 5p p.i., and the brains were removed and analyzed on the days and times indicated. (A) Representative images of the brains of mice injected with EB. (B) EB levels in the brains were quantified and expressed relative to the EB levels in the brains of PbA-infected, untreated mice on d6a p.i. Each symbol represents one mouse. The data are combined from three independent experiments and are shown as mean and SD. (C) Brain water content expressed as the weight of each brain after desiccation divided by the weight before desiccation × 100 is given. Data are combined from two independent experiments and are shown as mean and SD. (D) Representative images of brain sections; hemorrhages are indicated by white arrows. (E) Quantification of brain hemorrhages. Each symbol represents one mouse. Data are combined from three independent experiments. Mann–Whitney tests were used for comparison of groups (**P < 0.005, ***P < 0.0005).
We also determined the effect of DON treatment on the water content of the brain as a measure of cerebral edema by weighing the brains before and after desiccation. The brains of PbA-infected mice had significantly greater water content than the brains of uninfected mice on both day 5p p.i. and day 6a p.i. (Fig. 2C). Treating mice with DON on day 5p p.i. significantly reduced the water content of the brains measured on day 6a p.i. Thus, DON not only arrested the disease process; it also promoted resolution even when treatment was initiated at a time when significant BBB dysfunction and brain swelling already were manifest.
To quantify brain hemorrhages, brain sections were stained with H&E. PbA-infected, untreated mice developed petechial hemorrhages throughout the brain by day 5p p.i. that increased in number by day 6a p.i. (Fig. 2 D and E). Treatment of mice with DON beginning on day 5p p.i. had no significant effect on the number of hemorrhages that developed. However, by day 15 p.i. the hemorrhages were no longer evident (Fig. 2 D and E). Thus, DON-treated mice survived local hemorrhaging in the brain when the overall integrity of the BBB was restored and brain swelling was reduced.
DON Treatment Results in a Reduction in the Number of CD8+ Effector T Cells That Degranulate in the Brains of Infected Mice.
CD8+ T cells have been shown to play a major role in promoting death in ECM (16), and Gln metabolism is critical for the differentiation, proliferation, and function of effector T cells. Thus, we determined the effect of DON treatment on T cells in the brains of infected mice. DON was administered to PbA-infected mice on day 5p p.i., and on day 6a p.i. mice were terminally anesthetized and transcardially perfused with cold PBS. The brains were collected, and single-cell suspensions were prepared and analyzed by flow cytometry. First we examined immune cell infiltration of both the treated and untreated mice. Compared with uninfected mice, the brains of infected mice showed large increases in all immune cell types analyzed, including CD8+ and CD4+ T cells, neutrophils, macrophages, and natural killer (NK) cells. Interestingly, despite its ability to reverse disease, DON treatment had no effect on the numbers of immune cells in the brains of infected mice (Fig. 3 A–E).
Fig. 3.

DON treatment reduced CD8+ T-cell degranulation but not the accumulation of immune cells in the brains of PbA-infected mice. Uninfected mice and mice infected with PbA were treated with DON (1.3 mg/kg) or saline on day 5p p.i. (A–E) On day 6a p.i. mice were perfused with cold PBS, the brains and/or spleens were removed, and single-cell suspensions were prepared. Cells were analyzed by flow cytometry using the gating strategy shown in Fig. S4. Shown are the number of cells per brain for macrophage/dendritic cells (DC) (A), neutrophils (B), NK cells (C), CD 4+ T cells (D), and CD8+ T cells (E). Data were combined from three independent experiments. (F) Representative flow cytometry plots of GAP50 MHC class I Db tetramer-binding CD8+ T cells in unenriched spleen/lymph node cell populations (Left and Right) and in GAP50-tetramer–binding cells enriched by GAP50-tetramer-bound magnetic beads (Center). (G) The number of GAP50-tetramer–binding CD8+ T cells in spleen and brains. Data were combined from three independent experiments. (H) Representative flow cytometry plots of CD8+ T cells in the spleens of mice that received fluorescently labeled CD107-specific Abs i.v. 1 h before being killed to allow labeling of CD107-expressing cells in vivo. (I and J)The percent of total CD8+ T cells that expressed CD107 (I) and the percent of GAP50-tetramer–binding CD8+ T cells that expressed CD107 (J) are shown. Data were combined from three independent experiments. Mann–Whitney tests were used for statistical analysis. ns, not significant; *P < 0.05, **P < 0.005, ***P <0.0005.
Fig. S4.

Gating strategy to identify immune cells in the brain and spleens. Single-cell suspensions were gated, excluding very small cells or debris, and cell doublets were excluded by side-scatter width. The LIVE/DEAD aqua dye was used to label dead cells, and only living cells were gated. The pan leukocyte marker CD45.2 was used to gate on leukocytes. Cells were gated on CD3+ cells that include CD4+ and CD8+ T cells, NK T cells, and γδ T cells. CD3+ cells were gated further on CD8+ and CD4+ cells. The CD3− gate was used to subset cells further into neutrophils (Ly6G+, Ly6C+), macrophages/dendritic cells (DC cells) (Ly6G−, Ly6C+), and NK cells (NK1.1+). Leukocytes per spleen and brain preparation were counted on a hemocytometer, and the numbers of each cell type were calculated.
Next, we examined the effect of DON on the expansion and function of parasite-specific CD8+ T cells. The accumulation of PbA-specific CD8+ T cells in the brains and spleens was quantified using a peptide-MHC class I tetramer composed of the PbA glideosome-associated protein 50 (GAP50) peptide, SQLLNAKYL, bound to MHC class I Db (referred to as a GAP50-tetramer) that identifies ∼5% of splenic CD8+ T cells in PbA-infected mice (28). We verified that in the spleens of uninfected mice the frequency of GAP50-tetramer–binding CD8+ T cells was less than 0.1% (Fig. 3F, Left). Enrichment for GAP50-tetramer–binding cells in the spleen and lymph nodes of uninfected mice by GAP50-tetramer–bound magnetic bead purification showed ∼2,700 GAP50-tetramer–binding CD8+ T cells, the majority of which were resting CD44− cells (Fig. 3F, Center). In contrast, in PbA-infected mice the frequency of GAP50-tetramer–binding T cells in the spleen was ∼3%, and these were CD44+ effector T cells (Fig. 3F Right). DON treatment of PbA-infected mice resulted in a small decrease in the number of GAP50-tetramer–binding CD8+ T cells in the spleens but had little effect on the number of GAP50-tetramer–binding T cells in the brains (Fig. 3G).
Having observed that DON did not inhibit expansion of parasite-specific CD8+ T cells, we determined the effect of DON on CD8+ T-cell effector function. The development of ECM has been shown to be dependent on degranulation and perforin release of CD8+ T cells that accumulate in the brains of PbA-infected mice (16). Degranulation of CD8+ T cells results in the expression of CD107 on their plasma membranes. To assess the ability of DON to inhibit CD8+ T-cell degranulation in the brains of infected mice, fluorescently labeled CD107-specific mAbs were administered i.v. to mice 1 h before tissues were removed for analysis to allow in vivo labeling of CD107+ cells. Shown are representative flow cytometry plots showing the percent of CD44+, CD8+ T cells that express CD107 in the spleens of uninfected mice, PbA-infected mice, and PbA-infected mice treated with DON (Fig. 3H). DON treatment of PbA-infected mice resulted in significant decreases in the percent of CD8+ T cells that were CD107+ in both the spleens and brains (Fig. 3I). The percent of GAP50-tetramer–binding CD8+ T cells that were CD107+ also decreased significantly in the brains of PbA-infected mice (Fig. 3J). Thus, although the late-stage treatment of DON does not block the expansion of the PbA-specific CD8+ T cells, it appears to block their effector function as measured by degranulation.
The Metabolic Profiles of Brains from DON-Treated PbA-Infected Mice Resemble Those of Uninfected Mice.
The ability of DON to arrest ECM rapidly at such a late stage of disease and to promote recovery led us to wonder whether, in addition to inhibiting immune-mediated pathology, DON also might affect pathology induced by altered brain metabolism. To investigate this possibility, we profiled metabolites in the brain, liver, and serum from five groups of mice that differed in their infection, treatment, and/or clinical status (Table S1). Principal component analysis of all detected metabolites revealed distinct clustering of samples according to infection status in all tissues (Fig. 4 A–C). Notably, we observed that infected, DON-treated mice with low clinical scores (<6) clustered with uninfected mice for metabolites in the brain (Fig. 4A). In contrast, when we examine the effect of DON on systemic metabolism, as determined by serum metabolites, or the effect of DON on liver metabolism, this correlation did not hold (Fig. 4 B and C), suggesting that DON treatment in ECM results in specific effects on brain metabolites.
Table S1.
Infection and treatment groups for metabolomics studies
| Group description | Number of samples | ||
| Brain | Liver | Serum | |
| Uninfected, untreated | 5 | 5 | 5 |
| Uninfected, DON-treated | 5 | 5 | 5 |
| PbA-infected, DON-treated, low clinical scores (<6) on day 6 p.i. | 10 | 10 | 10 |
| PbA-infected, DON-treated, high clinical scores (≥6) on day 6 p.i. | 6 | 6 | 6 |
| PbA-infected, untreated | 8 | 7 | 7 |
Fig. 4.

DON significantly alters metabolism in the brain during ECM. (A–C) Principle components analysis for each of three tissue types collected from mice at day 6a p.i. for the five experimental groups defined by infection, treatment, and clinical outcome (Table S1). Principal components were determined from all detectable metabolites for the brain (438 metabolites) (A), liver (544 metabolites) (B), and serum (563 metabolites) (C). Each sphere represents one tissue sample from one mouse. (D) Venn diagram showing the number of differentially abundant brain metabolites for the two main comparisons: infected, untreated mice vs. uninfected, untreated mice and infected, DON-treated mice with low clinical scores vs. infected, untreated mice. Differential abundance thresholds were an absolute fold-change in abundance of ≥1.2 and a false-discovery rate of <5% (Welch’s t test).
Because DON blocks the initial step in cellular glutaminolysis by inhibiting glutaminase (24) (Fig. S5A), we first examined the metabolites of glutaminolysis and observed that PbA infection alone affects these metabolites differently in the brain than in the liver and serum. However, we did not observe a clear pattern predicted for treatment with a glutaminase inhibitor (Fig. S5B). Of the 144 brain metabolites that changed significantly in infected versus uninfected mice, 81 overlapped with the 89 significantly affected metabolites identified by comparing infected, DON-treated mice with low clinical scores and infected, untreated mice (Fig. 4D). Pathways significantly affected by PbA infection involved citrulline metabolism, the urea cycle, or nitric oxide biosynthesis (aspartate, citrulline, ornithine, and urea) and were similar to pathways affected by DON treatment of PbA-infected mice (Fig. S6). Strikingly, all 81 overlapping metabolites were metabolites that were reversed by DON treatment (Table S2). Unsupervised hierarchal clustering analysis on these 81 brain metabolites accurately grouped the samples by infection, treatment, and clinical status (Fig. 5), with 9 of 10 PbA-infected, DON-treated mice with low clinical scores (<6) clustering together and demonstrating remarkable metabolic similarities to uninfected mice. DON appears to reverse brain metabolism associated with disease or to prevent the metabolic perturbation, presumably acting through its immunomodulatory effects or acting directly on brain metabolism. Although these data do not identify the specific metabolites promoting disease, they do identify a set of metabolites which are specifically associated with ECM and are selectively blocked by treatment with DON.
Fig. S5.

The effect of PbA infection and DON treatment during PbA infection on the glutaminolysis pathway in the brain, liver, and serum. (A) DON blocks the first step of glutaminolysis by inhibiting glutaminase. (B) PbA infection and DON treatment of PbA-infected mice affect the glutaminolysis pathway differently in the brain than in the liver and serum. Shown are absolute fold change and FDRs by Welch’s t test for the indicated two-way comparisons. Positive fold changes (red shading) represent significantly increased (FDR <0.05) metabolite abundance in the first group compared with the second group. Negative fold changes (blue shading) represent significantly decreased (FDR <0.05) metabolite abundance in the first group compared with the second group.
Fig. S6.

DON significantly alters citrulline, urea, and nitric oxide metabolism in the brain during ECM. (A and B) Pathways analysis of differentially abundant metabolites for each two-way comparison (shown in Fig. 4D) demonstrated several significantly overrepresented pathways that are shared between the two comparisons. Shown are the ratio of overlap of metabolites in the dataset with metabolites in the pathway and the log(P value) of the overlap by Fisher’s exact test. Metabolites discussed in the text are shown in bold.
Table S2.
The 81 metabolites reversed by DON treatment during PbA infection
| Metabolite | Main pathway | Subpathway | Mass | Pubchem ID | HMDB ID | Comparison groups (Welch's t tests) | |||
| PbA-infected, untreated vs. uninfected, untreated | PbA-infected, DON-treated, low clinical score (<6) vs. PbA-infected, untreated | ||||||||
| Fold change | FDR | Fold change | FDR | ||||||
| 1-arachidonoylglycerophosphoethanolamine | Lipid | Lysolipid | 500.27826 | 42,607,465 | HMDB11517 | 1.56 | 0.0176 | −2.04 | 0.0036 |
| 1-linoleoylglycerophosphoethanolamine | Lipid | Lysolipid | 476.27826 | 52,925,130 | HMDB11507 | 2.87 | 0.000066561 | −2.63 | 0.0006 |
| 1-oleoylglycerophosphoethanolamine | Lipid | Lysolipid | 478.29391 | 9,547,071 | HMDB11506 | 2.35 | 0.0007 | −2.27 | 0.0031 |
| 1-oleoylglycerophosphoinositol | Lipid | Lysolipid | 597.30453 | 1.57 | 0.0421 | −3.03 | 0.0005 | ||
| 1-oleoylglycerophosphoserine | Lipid | Lysolipid | 522.28374 | 9,547,099 | 2.58 | 0.0002 | −2.94 | 0.0005 | |
| 1-oleoylplasmenylethanolamine | Lipid | Lysolipid | 462.29899 | 2.58 | 0.0002 | −2.04 | 0.0032 | ||
| 1-palmitoylglycerophosphoethanolamine | Lipid | Lysolipid | 452.27826 | 9,547,069 | HMDB11503 | 1.90 | 0.0004 | −2.13 | 0.0006 |
| 1-palmitoylglycerophosphoinositol | Lipid | Lysolipid | 571.28888 | HMDB61695 | 1.51 | 0.0317 | −2.38 | 0.0006 | |
| 1-palmitoylglycerophosphoserine | Lipid | Lysolipid | 496.26809 | 9,547,100 | 2.99 | 0.0188 | −3.23 | 0.0127 | |
| 1-palmitoylplasmenylethanolamine | Lipid | Lysolipid | 436.28334 | 2.30 | 0.0003 | −2.27 | 0.0021 | ||
| 1-stearoylglycerophosphoethanolamine | Lipid | Lysolipid | 480.30956 | 9,547,068 | HMDB11130 | 2.48 | 0.0005 | −2.63 | 0.0027 |
| 1-stearoylglycerophosphoglycerol | Lipid | Lysolipid | 511.30414 | 2.94 | 0.0012 | −3.23 | 0.0022 | ||
| 1-stearoylglycerophosphoinositol | Lipid | Lysolipid | 599.32018 | HMDB61696 | 1.95 | 0.0013 | −2.50 | 0.0005 | |
| 1-stearoylglycerophosphoserine | Lipid | Lysolipid | 524.29939 | 9,547,101 | 2.28 | 0.0004 | −2.94 | 0.0005 | |
| 1-stearoylplasmenylethanolamine | Lipid | Lysolipid | 464.31464 | 2.36 | 0.0024 | −3.33 | 0.0009 | ||
| 2-aminobutyrate | Amino Acid | Methionine, cysteine, SAM, and taurine metabolism | 130 | 439,691 | HMDB00650 | 18.07 | 3.4872E-06 | −2.38 | 0.0129 |
| 2-hydroxy-3-methylvalerate | Amino acid | Leucine, isoleucine, and valine metabolism | 131.07136 | 164,623 | HMDB00317 | 7.66 | 0.0000878 | −3.45 | 0.0031 |
| 2-hydroxybutyrate (AHB) | Amino acid | Methionine, cysteine, SAM, and taurine metabolism | 103.04006 | 440,864 | HMDB00008 | 12.68 | 0.000008827 | −2.38 | 0.0168 |
| 2-methylbutyrylcarnitine (C5) | Amino acid | Leucine, isoleucine, and valine metabolism | 246.16999 | 6,426,901 | HMDB00378 | 5.17 | 0.0001 | −2.00 | 0.048 |
| 2-stearoylglycerophosphoethanolamine | Lipid | Lysolipid | 480.30956 | 2.42 | 0.0047 | −2.86 | 0.001 | ||
| 2'-deoxycytidine | Nucleotide | Pyrimidine metabolism, cytidine containing | 226.08333 | 13,711 | HMDB00014 | 2.01 | 0.001 | −1.75 | 0.0113 |
| 2'-deoxyuridine | Nucleotide | Pyrimidine metabolism, uracil containing | 111.01997 | 13,712 | HMDB00012 | 1.68 | 0.0006 | −1.49 | 0.0109 |
| 3-hydroxybutyrate (BHBA) | Lipid | Ketone bodies | 103.04007 | 441 | HMDB00357 | 3.76 | 0.000012987 | −3.45 | 0.0002 |
| 3-hydroxydecanoate | Lipid | Fatty acid, monohydroxy | 187.13396 | 26,612 | HMDB02203 | 2.78 | 0.0005 | −2.33 | 0.0049 |
| 3-hydroxyisobutyrate | Amino acid | Leucine, isoleucine, and valine metabolism | 103.04007 | 87 | HMDB00336 | 3.47 | 0.00007885 | −2.56 | 0.0021 |
| 3-hydroxylaurate | Lipid | Fatty acid, monohydroxy | 215.16526 | 94,216 | HMDB00387 | 3.32 | 0.0002 | −2.00 | 0.0146 |
| 3-Hydroxyoctanoate | Lipid | Fatty acid, monohydroxy | 159.10266 | 26,613 | HMDB01954 | 2.68 | 0.0001 | −2.38 | 0.0014 |
| 3-indoxyl sulfate | Amino acid | Tryptophan metabolism | 212.0023 | 10,258 | HMDB00682 | 3.53 | 0.0042 | −3.03 | 0.0224 |
| 3-methylglutarylcarnitine | Amino acid | Lysine metabolism | 290.15982 | 128,145 | HMDB00552 | 20.85 | 0.000003694 | −1.52 | 0.0295 |
| 4-hydroxyphenylpyruvate | Amino acid | Phenylalanine and tyrosine metabolism | 179.03498 | 979 | HMDB00707 | −3.13 | 0.000027222 | 1.96 | 0.0047 |
| 5-dodecenoate (12:1n7) | Lipid | Medium-chain fatty acid | 197.1547 | 5,312,378 | HMDB00529 | 2.14 | 0.0023 | −1.89 | 0.014 |
| 5,6-dihydrouracil | Nucleotide | Pyrimidine metabolism, uracil containing | 115.05021 | 649 | HMDB00076 | −1.33 | 0.0322 | 1.93 | 0.0031 |
| cAMP | Nucleotide | Purine metabolism, adenine containing | 328.04524 | 6,076 | HMDB00058 | −2.94 | 3.4872E-06 | 1.76 | 0.0428 |
| Allantoin | Nucleotide | Purine metabolism, (hypo)xanthine/Inosine containing | 157.03671 | 204 | HMDB00462 | 8.34 | 9.2603E-06 | −2.04 | 0.0209 |
| Ascorbate (vitamin C) | Cofactors and vitamins | Ascorbate and aldarate metabolism | 332.1 | HMDB00044 | −2.44 | 0.0017 | 2.02 | 0.0302 | |
| Aspartate | Amino acid | Alanine and aspartate metabolism | 134.04479 | 5,960 | HMDB00191 | −1.43 | 0.0095 | 1.44 | 0.0064 |
| β-Alanine | Nucleotide | Pyrimidine metabolism, uracil containing | 174 | 239 | HMDB00056 | −1.45 | 0.0086 | 1.33 | 0.0473 |
| β-Hydroxyisovalerate | Amino acid | Leucine, isoleucine, and valine metabolism | 117.05572 | 69,362 | HMDB00754 | 3.27 | 0.0001 | −1.96 | 0.0047 |
| Betaine | Amino acid | Glycine, serine, and threonine metabolism | 118.08626 | 247 | HMDB00043 | 5.38 | 6.4137E-06 | −2.00 | 0.0031 |
| Butyrylcarnitine | Lipid | Fatty acid metabolism (also BCAA metabolism) | 232.15434 | 439,829 | HMDB02013 | 5.08 | 0.000055261 | −1.85 | 0.0473 |
| Choline | Lipid | Phospholipid metabolism | 104.10699 | 305 | HMDB00097 | −1.49 | 0.0011 | 1.28 | 0.0048 |
| Citrulline | Amino acid | Urea cycle; arginine and proline metabolism | 176.10297 | 9,750 | HMDB00904 | 2.11 | 0.0004 | −1.96 | 0.0031 |
| Corticosterone | Lipid | Steroid | 347.22169 | 5,753 | HMDB01547 | 4.52 | 0.0017 | −2.00 | 0.0031 |
| Cytidine 5′-diphosphocholine | Lipid | Phospholipid metabolism | 533.10553 | 13,804 | HMDB01413 | −1.75 | 0.0045 | 1.71 | 0.0224 |
| Cytidine-5′-diphosphoethanolamine | Lipid | Phospholipid metabolism | 445.0531 | 123,727 | HMDB01564 | −1.27 | 0.025 | 1.33 | 0.0334 |
| Ethanolamine | Lipid | Phospholipid metabolism | 174.1 | 700 | HMDB00149 | −1.59 | 0.0032 | 1.57 | 0.0141 |
| Ethylmalonate | Amino acid | Leucine, isoleucine, and valine metabolism | 131.03498 | 11,756 | HMDB00622 | 2.44 | 0.0001 | −1.67 | 0.0109 |
| γ-Glutamylglutamate | Peptide | γ-glutamyl amino acid | 277.10303 | 92,865 | HMDB11737 | −1.61 | 0.0009 | 1.50 | 0.0098 |
| Glutamate | Amino acid | Glutamate metabolism | 148.06044 | 611 | HMDB00148 | −1.49 | 0.0004 | 1.29 | 0.0205 |
| Glutamine | Amino acid | Glutamate metabolism | 147.07642 | 5,961 | HMDB00641 | 1.42 | 0.0000878 | −1.25 | 0.0051 |
| Glycerophosphorylcholine (GPC) | Lipid | Phospholipid metabolism | 258.1101 | 71,920 | HMDB00086 | −5.88 | 9.4428E-06 | 2.10 | 0.0109 |
| Heptanoate (7:0) | Lipid | Medium-chain fatty acid | 129.0921 | 8,094 | HMDB00666 | −3.70 | 7.8856E-07 | 1.96 | 0.013 |
| Hexanoylcarnitine | Lipid | Fatty acid metabolism (acyl carnitine) | 260.18564 | 6,426,853 | HMDB00705 | 12.66 | 0.0007 | −3.85 | 0.02 |
| Histidine | Amino acid | Histidine metabolism | 154.0622 | 6,274 | HMDB00177 | 2.14 | 0.000071544 | −1.39 | 0.0295 |
| Hydroxybutyrylcarnitine | Lipid | Fatty acid metabolism (acyl carnitine) | 248.14925 | 53,481,617 | HMDB13127 | 10.86 | 0.000003694 | −3.57 | 0.0008 |
| Imidazole propionate | Amino acid | Histidine metabolism | 141.06586 | 70,630 | HMDB02271 | 3.13 | 0.0361 | −4.76 | 0.0189 |
| Isobutyrylcarnitine | Amino acid | Leucine, isoleucine, and valine metabolism | 232.15434 | 168,379 | HMDB00736 | 7.99 | 0.000042116 | −2.33 | 0.0288 |
| Isoleucine | Amino acid | Leucine, isoleucine, and valine metabolism | 132.10191 | 6,306 | HMDB00172 | 1.59 | 0.0015 | −1.35 | 0.0428 |
| Isovalerylcarnitine | Amino acid | Leucine, isoleucine, and valine metabolism | 246.16999 | 6,426,851 | HMDB00688 | 11.45 | 0.000017712 | −2.56 | 0.0109 |
| Kynurenine | Amino acid | Tryptophan metabolism | 209.09207 | 161,166 | HMDB00684 | 32.79 | 2.3284E-07 | −2.04 | 0.0097 |
| Linoleate (18:2n6) | Lipid | Polyunsaturated fatty acid (n3 and n6) | 279.23295 | 5,280,450 | HMDB00673 | 1.95 | 0.0005 | −1.45 | 0.0064 |
| Linoleoylcarnitine | Lipid | Fatty acid metabolism (acyl carnitine) | 424.34214 | 6,450,015 | HMDB06469 | 140.92 | 0.000054612 | −6.67 | 0.0408 |
| Lysine | Amino acid | Lysine metabolism | 147.11281 | 5,962 | HMDB00182 | 1.84 | 0.0002 | −1.37 | 0.0223 |
| Myristoleate (14:1n5) | Lipid | Long-chain fatty acid | 225.186 | 5,281,119 | HMDB02000 | 1.87 | 0.001 | −1.54 | 0.0249 |
| N-acetylleucine | Amino acid | Leucine, isoleucine, and valine metabolism | 172.09791 | 70,912 | HMDB11756 | 6.25 | 0.0002 | −3.45 | 0.0034 |
| N-acetylphenylalanine | Amino acid | Phenylalanine and tyrosine metabolism | 208.09682 | 74,839 | HMDB00512 | 20.58 | 0.000003694 | −3.70 | 0.0022 |
| N-acetyltyrosine | Amino acid | Phenylalanine and tyrosine metabolism | 222.07718 | 68,310 | HMDB00866 | 1.71 | 0.0203 | −2.13 | 0.016 |
| N-Δ-acetylornithine | Amino acid | Urea cycle; arginine and proline metabolism | 175.10772 | 9,920,500 | 2.87 | 0.0023 | −2.04 | 0.0473 | |
| N1-methyl-2-pyridone-5-carboxamide | Cofactors and vitamins | Nicotinate and nicotinamide metabolism | 153.06586 | 69,698 | HMDB04193 | 3.56 | 0.0025 | −25.00 | 7.7062E-06 |
| Orotidine | Nucleotide | Pyrimidine metabolism, orotate containing | 287.05209 | 92,751 | HMDB00788 | −1.52 | 0.0086 | 1.52 | 0.036 |
| Palmitoyl-linoleoyl-glycerophosphocholine | Lipid | Lysolipid | 802.56049 | 2.18 | 5.1316E-06 | −1.33 | 0.0098 | ||
| Pipecolate | Amino acid | Lysine metabolism | 130.08626 | 849 | HMDB00070 | 32.00 | 0.000003694 | −2.33 | 0.014 |
| Ribonate | Carbohydrate | Pentose metabolism | 165.04046 | 5,460,677 | HMDB00867 | −4.35 | 0.0002 | 1.85 | 0.0097 |
| S-methylcysteine | Amino acid | Methionine, cysteine, SAM, and taurine metabolism | 119.01613 | 24,417 | HMDB02108 | 4.34 | 0.0071 | −2.70 | 0.0473 |
| Tartronate (hydroxymalonate) | Xenobiotics | Bacterial/fungal | 118.99859 | 45 | HMDB35227 | −2.50 | 9.4428E-06 | 1.68 | 0.0034 |
| Tauro-β-muricholate | Lipid | Primary bile acid metabolism | 514.28439 | 168,408 | HMDB00932 | 25.51 | 0.0009 | −11.11 | 0.0077 |
| Taurocholate | Lipid | Primary bile acid metabolism | 514.28439 | 6,675 | HMDB00036 | 9.56 | 0.0004 | −5.88 | 0.0031 |
| Tiglyl carnitine | Amino acid | Leucine, isoleucine, and valine metabolism | 244.15434 | 22,833,596 | HMDB02366 | 88.06 | 2.3284E-07 | −2.78 | 0.0349 |
| Urea | Amino acid | Urea cycle; arginine and proline metabolism | 121.07201 | 1,176 | HMDB00294 | 1.89 | 0.0067 | −1.89 | 0.0212 |
| Valerate | Lipid | Short-chain fatty acid | 101.0608 | 7,991 | HMDB00892 | −3.85 | 7.8856E-07 | 2.05 | 0.0109 |
| Valine | Amino acid | Leucine, isoleucine, and valine metabolism | 118.08626 | 6,287 | HMDB00883 | 2.42 | 0.0003 | −1.72 | 0.0171 |
Shown are 81 metabolites that were differentially abundant in both the PbA-infected, untreated vs. uninfected, untreated comparison and the PbA-infected, DON-treated, low clinical score vs. PbA-infected, untreated comparison. Only metabolites meeting differential abundance thresholds of an absolute fold change ≥1.2 and an FDR <5% in both comparisons are shown. BCAA, branch-chain amino acids; HBMD, Human Metabolome Database; SAM, S-adenosyl methionine.
Fig. 5.

Recovery from ECM with DON treatment is associated with normal brain metabolites. Clustering heat map showing the normalized abundance of the 81 metabolites that were shared between the two main comparisons in Fig. 4D. Each row is a metabolite, and each column represents brain tissue from a single mouse. Unsupervised hierarchical clustering of samples was performed using Ward’s method for linkage analysis and Pearson’s dissimilarity as distance measure. Metabolites were grouped according to pathway annotations (labels at right).
Discussion
HCM is a deadly complication of P. falciparum malaria despite treatment with effective antimalarial drugs. That is, even in when the replication of P. falciparum is effectively suppressed, there is no effective adjunctive treatment for HCM once signs of neurologic disease have commenced, and overall mortality remains high. Therefore a treatment that can arrest disease and promote healing in the late stages is urgently needed. Here we report that the Gln analog DON is an effective therapy for ECM even when treatment is first initiated after infected animals show neurological signs of disease. This clinical response was accompanied by the ability of DON to inhibit pathology as measured by decreases in BBB dysfunction, brain swelling, and degranulation of parasite-specific CD8+ T cells that accumulated in the brain. Furthermore, DON is able to reverse or prevent metabolic changes associated with the disease state.
Previously, DON was shown to have antiparasitic activity, albeit weak, both in vitro and in vivo (29, 30). Although the antiparasitic activity of DON may contribute to its ability to arrest ECM and promote healing, we do not believe plays a critical role. Indeed, DON had little effect on the parasite load in the brains of PbA-infected mice during the critical period when the BBB was restored and brain swelling decreased in DON-treated mice.
On the other hand, CD8+ effector T cells that release perforin have been shown to play a critical role in promoting the pathogenesis that constitutes ECM (16). CD8+ T-cell activation results in a dramatic shift from oxidative metabolism to aerobic glycolysis and glutaminolysis necessary for T-cell expansion and effector function (18–20). Both T-cell cytokine production and proliferation are blocked by restricting the availability of extracellular Gln (21) or by treating T cells with DON (22). DON treatment of PbA-infected mice did not prevent or decrease the accumulation of CD8+ T cells in the brain. Rather, the number of CD8+ T cells that degranulated was decreased following DON treatment. These observations suggest that DON’s protective effect is mediated not by preventing the proliferation and generation of CD8+ effector T cells but rather by blocking CD8+ T-cell effector function.The ability of DON to block CD8+ T-cell function may account, in part, for the remarkably fast kinetics by which DON mediates its protective effect. The ability of DON to promote survival at such a late stage of the disease when animals are suffering from BBB dysfunction and brain swelling distinguishes our findings from all other attempts to treat and reverse ECM.
Although our studies were motivated by the ability of DON to inhibit immune function, we also investigated the effect of DON treatment on infected brain metabolism by interrogating the brain for known metabolites. To this end, we observed more than 81 metabolic changes in the brains of infected mice compared with uninfected mice that were either reversed or blocked by DON. Unequivocally, such findings do not reveal the mechanism by which DON mediates its effects. The complexity of the fluctuations in Gln and Glu levels in the brain during infection and upon treatment most likely reflects DON’s inhibition not only of glutaminase activity but also of Gln transport and other Gln-using enzymes (24). Rather, the changes we identified provide a metabolic profile, a biomarker for diseased brains in ECM. Notably, although these 81 metabolites correlated with DON treatment and disease state in the brain, such correlations were not observed in the serum or the livers of the DON-treated mice.
In summary, we demonstrate, for the first time to our knowledge, the ability of a pharmacologic intervention to arrest disease and promote survival in the late stages of ECM. Because of the high mortality in HCM once neurologic symptoms are manifested, our findings have relevant and immediate clinical implications. Furthermore, our studies reveal a potentially selective metabolic signature for ECM and the subsequent reversal of diseases. It will be of interest to determine if this signature is applicable to other neurologic disorders and whether DON might be capable of arresting and/or reversing a broad spectrum of neuroinflammatory diseases.
Materials and Methods
All experiments were approved by the National Institute of Allergy and Infectious Diseases Animal Care and Use Committee (NIAID ACUC). The NIAID ACUC approved the Animal Study Proposal identification number LIG-1E, which adheres to the regulations of the Animal Welfare Regulations and Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Detailed materials and methods can be found in SI Materials and Methods. C57BL/6 mice were infected with PbA and monitored for peripheral blood parasitemia (31) and ECM clinical scores (25). DON (1.3 mg/kg) was administered i.p. Parasite loads were quantified by quantitative PCR in brain tissue taken from mice that were anesthetized and transcardially perfused (32). Terminal assays also were carried out to quantify brain pathology. BBB integrity was assessed by quantifying EB in brains of mice that were injected with EB intraorbitally 3 h before the mice were anesthetized and transcardially perfused and the brains were removed (33). Brain swelling was measured by weighing brains before and after desiccation. Brain hemorrhages were quantified by histological examination of H&E-stained brain sections. To characterize leukocytes that accumulated in brains, single-cell suspensions were prepared of brains from anesthetized and transcardially perfused mice. Cells were analyzed by flow cytometry using appropriately labeled antibodies specific for cell-surface markers and a CD8+ T-cell–specific GAP50-tetramer (28). Single-cell suspensions of spleens were analyzed similarly. To quantify degranulation of T cells in vivo, mice were given phycoerythrin (PE)-conjugated antibodies specific for CD107a and CD107b i.v. 1 h before mice were killed and brains and spleens were removed for analysis by flow cytometry (34). Metabolic profiling of brain, liver, and serum was performed at Metabolon (35).
SI Materials and Methods
Animal and Malaria Infections.
C57BL/6 female mice (7–10 wk old) were obtained from The Jackson Laboratories. Mice were infected with PbA by injecting i.p. 1 × 106 PbA-infected RBC obtained from infected C57BL/6 mice. Peripheral blood parasitemia was determined by flow cytometry as described below. Infected mice were monitored for the progression of ECM using a 10-point clinical scoring system that rates mice from a score of 0 (no signs) to 10 (moribund) as previously described (25). Briefly, animals were scored by testing five categories: interactions/reflex, cage grasp, visual placing, gait/posture/appearance, and capacity to hold their body weight on a baton. Each category was scored from 0 to 2, where 0 represented normal individuals, 1 intermediate, and 2 the worst case for that particular parameter. Clinical scores have not been correlated directly with brain pathology.
DON Treatment.
For each DON treatment, mice weighing ∼25 g were injected i.p. with 1.3 mg/kg DON (Sigma; catalog no. D2141) in 200 μL PBS.
Quantification of Peripheral Blood Parasitemia by Flow Cytometry.
Parasitemia was determined by flow cytometry using a modification of a previously described method (31). Briefly, blood was obtained from mouse tail veins, fixed with 0.025% aqueous glutaradehyde solution, washed with 2 mL PBS, resuspended, and stained with the following: the DNA dye Hoechst 33342 (Sigma) (8 μM), the DNA and RNA dye dihydroethidium (diHEt) (10 μg/mL), the pan C57BL/6 lymphocyte marker allophycocyanin (APC)-conjugated Ab specific for CD45.2 (BioLegend), and the RBC marker APC-Cy7–conjugated Ab specific for Ter119 (BD Pharmingen). Cells were analyzed on a BD LSRII flow cytometer equipped with UV (325 nm), violet (407 nm), blue (488 nm), and red (633 nm) lasers. Data were analyzed using FlowJo software (Tree Star Technologies). iRBCs were CD45.2−, Ter119+, Hoechst+ and diHEt+. Parasitemia was calculated as the number of iRBCs divided by the total number of RBCs.
Quantification of Parasite Loads in Brains.
Mice were anesthetized and transcardially perfused with cold PBS, and brains were removed and immediately frozen in liquid nitrogen. Brains were thawed, and 1 mL Qiagen RNeasy lysis buffer was added immediately. Brains were homogenized, and RNA was extracted from the homogenate using a Qiagen RNeasy Mini kit according to the manufacturer’s instructions. Genomic DNA was digested on a column using RNase free DNase set (Qiagen), and the elimination of genomic DNA was confirmed using no reverse transcriptase controls. cDNA was generated using the iScript cDNA synthesis kit (Bio-Rad Laboratories). SYBR Green PCR master mix (Bio-Rad) was used to determine the relative expression of parasite 18s ribosomal RNA and of three mouse housekeeping genes, hprt, gapdh, and actb. The primer sequences were Pb 18s 5′-AAGCATTAAATAAAGCGAATACATCCTTAC-3′ and 5′-GGAGATTGGTTTTGACGTTTATGTG-3′; mouse hprt 5-TGCTCGAGATGTGATGAAGG-3′ and 5′-TCCCCTGTTGACTGGTCATT-3′; mouse gaphd 5′-GTGGAGTCATACTGGAACATGTAG-3′ and 5′-AATGGTGAAGGTCGGTGTG-3′; and mouse actb 5′-TCC GGC ATG TGC AAA GC-3′ and 5′-TCC TTC TGA CCC ATT CCC A-3′.
The geometrical mean of the threshold cycle (Ct) values of mouse housekeeping genes was first determined to create a normalized base line for the brain to allow a comparison of the ΔCt values of 18s gene amplification. The fold changes in gene expression then were calculated by comparing the ΔCt values of PbA-infected mice on day 5 p.i. with the values on day 6 p.i. for mice treated with DON on day 5p p.i. or mice left untreated.
Assessment of BBB Integrity.
EB (20 mg/kg) was injected intraorbitally on the specified day; 3 h later the mice were anesthetized and perfused with saline, and their brains were removed and frozen at immediately −80 °C for later processing (33). EB was extracted using N, N-dimethylformamide and was quantified using a Varioskan Flash fluorometer (620 nm excitation; 695 nm emission).
Assessment of Brain Swelling.
Brains were removed from animals and weighed. Brains then were desiccated at 80 °C for 12 h and weighed again. Percent water content was calculated using this decrease in weight.
Quantification of Brain Hemorrhages.
Brain samples were fixed in 10% buffered formalin, embedded in paraffin, and sectioned. Sections were stained with H&E for ultrastructural examination and detection of hemorrhages. For quantification of hemorrhages, 10 microscopic 40×-power fields were examined, and hemorrhages were counted.
Flow Cytometry.
For quantification of brain-infiltrating leukocytes, mice were anesthetized with ketamine/xylazine at the specified times and were transcardially perfused with cold PBS; then the brains and spleens were removed. Brains were dissected, minced, and digested with 1 mg/mL collagenase for 30 min at 37 °C. After the tissue was passed through 70-µm nylon mesh, homogenates were placed on a 90–60–40% discontinuous Percoll gradient and centrifuged for 18 min at 1,000 × g; then the cells at the 40–60% interface containing leukocytes were collected for analysis. Spleens were mashed using a 70-µm nylon mesh, and RBCs were lysed. The cells were washed and resuspended in FACS buffer consisting of PBS and 1% FBS. The following fluorescent dye-conjugated antibodies specific for the following cell-surface markers were used for staining: Brilliant Violet (BV) 421–NK1.1 (BioLegend), BV605–CD4 (BioLegend), BV785–CD8 (BioLegend), PE–Ly6G (BD Pharmingen), PE-Cy7–CD3 (eBioscience), APC–Ly6C (BD Pharmingen), APC-Cy7–CD45.2 (BD Pharmingen), and LIVE/DEAD Fixable Aqua Dead Cell Stain (Life Technologies). Gating of subsets is depicted in Fig. S1. Cell acquisition data were obtained on a BD LSRII flow cytometer. Data were analyzed with FlowJo software (Tree Star Technologies).
To quantify GAP50-tetramer–binding CD8+ T cells (28) in tissues from uninfected mice, GAP50-tetramer–binding CD8+ T cells were enriched by magnetic bead separation as described (36). Briefly, a single-cell suspension was prepared from spleen and peripheral lymph nodes (axillary, brachial, inguinal, and cervical) and incubated with 0.5 mg GAP50-tetramer (obtained from the NIH Tetramer Core Facility, Emory University, Atlanta, GA) conjugated to APC for 30 min on ice. Cells were washed and incubated for 40 min at 4 °C with anti-APC beads (Miltenyi Biotec). The cells were washed and applied to magnetic columns (Miltenyi). Both the bound GAP50-tetramer–positive and unbound cells were incubated for 30 min at 4 °C with antibodies specific for CD8, CD3, CD44, MHC class II, and F4/80 obtained from BioLegend, eBioscience, and Tonbo. Cells were washed, analyzed on an LSRFortessa flow cytometer (BD Biosciences), and the acquired data were analyzed using FlowJo software. To quantify GAP50-tetramer–binding CD8+ T cells in the tissues of PbA-infected mice, brain and spleen were collected, and single-cell suspensions prepared as described above. Cells were stained for 30 min at 4 °C with the following fluorescent dye-conjugated antibodies specific for the following cell-surface markers: BV785-CD8 (BioLegend); BV421-CD44 (BioLegend); BV605-CD4 (BioLegend); BV711-CD11b (BioLegend); PE-CD107a (BioLegend); PE-CD107b (BioLegend); PerCP-Cy5.5-Thy1.2; Db:GAP50-Bio; APC-streptavidin (Life Technologies); Aqua Dead Cell Stain (Life Technologies); Alex Fluor 700-Ly6C (BioLegend); and APC-Cy7–CD45.2 (BD Pharmingen). Cell-acquisition data were obtained on a BD LSRII flow cytometer and were analyzed with FlowJo software (Tree Star Technologies).
For the detection of in vivo degranulation, mice were given PE-conjugated antibodies specific for CD107a (12.5 µg) and for CD107b (12.5 µg) (BioLegend) by i.v. injection 1 h before the mice were killed and their brains and spleens were removed, as previously described (34).
Metabolic/Metabolomic Profiling.
On day 6a p.i., mice from all experimental groups were killed to collect brain, liver, and serum. Tissue samples were snap frozen in liquid nitrogen and stored at −80 °C until sample preparation. Automated tissue processing and metabolomic profiling by ultra-HPLC-MS/MS and GC-MS were performed at Metabolon using multiple quality control standards as described (35). After extraction of raw data, peaks were identified by comparison with known purified standards and recurrent unknown entities present in Metabolon’s reference library. Peak quantification was performed using area-under-the-curve with data normalization to correct for day-to-day variation in studies spanning multiple days. Statistical analyses of metabolomics data were performed in ArrayStudio 5.0 and R 3.1.1 (www.R-project.org/). Two-way comparisons between experimental groups were analyzed by Welch’s t test for each of the three tissue types using log-transformed data in R. To correct for multiple testing, false-discovery rates (FDR) were estimated using the q-value method (37), and only metabolites with an FDR <5% were considered significant. Canonical pathways analysis was applied to metabolites meeting a fold-change threshold of > |1.2| and an FDR <5% for each two-way comparison and for each tissue using Ingenuity Pathway Analysis (Qiagen).
Acknowledgments
This study was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases and by NIH Grant R01AI077610 (to J.D.P.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1516544112/-/DCSupplemental.
References
- 1.World Health Organization . World Malaria Report 2014. WHO; Geneva: 2014. [Google Scholar]
- 2.Molyneux ME, Taylor TE, Wirima JJ, Borgstein A. Clinical features and prognostic indicators in paediatric cerebral malaria: A study of 131 comatose Malawian children. Q J Med. 1989;71(265):441–459. [PubMed] [Google Scholar]
- 3.Taylor TE, et al. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat Med. 2004;10(2):143–145. doi: 10.1038/nm986. [DOI] [PubMed] [Google Scholar]
- 4.Potchen MJ, et al. Acute brain MRI findings in 120 Malawian children with cerebral malaria: New insights into an ancient disease. AJNR Am J Neuroradiol. 2012;33(9):1740–1746. doi: 10.3174/ajnr.A3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seydel KB, et al. Brain swelling and death in children with cerebral malaria. N Engl J Med. 2015;372(12):1126–1137. doi: 10.1056/NEJMoa1400116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shikani HJ, et al. Cerebral malaria: We have come a long way. Am J Pathol. 2012;181(5):1484–1492. doi: 10.1016/j.ajpath.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grau GE, Craig AG. Cerebral malaria pathogenesis: Revisiting parasite and host contributions. Future Microbiol. 2012;7(2):291–302. doi: 10.2217/fmb.11.155. [DOI] [PubMed] [Google Scholar]
- 8.Engwerda C, Belnoue E, Grüner AC, Rénia L. Experimental models of cerebral malaria. Curr Top Microbiol Immunol. 2005;297:103–143. [PubMed] [Google Scholar]
- 9.Nacer A, et al. Neuroimmunological blood brain barrier opening in experimental cerebral malaria. PLoS Pathog. 2012;8(10):e1002982. doi: 10.1371/journal.ppat.1002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Promeneur D, Lunde LK, Amiry-Moghaddam M, Agre P. Protective role of brain water channel AQP4 in murine cerebral malaria. Proc Natl Acad Sci USA. 2013;110(3):1035–1040. doi: 10.1073/pnas.1220566110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penet MF, et al. Imaging experimental cerebral malaria in vivo: Significant role of ischemic brain edema. J Neurosci. 2005;25(32):7352–7358. doi: 10.1523/JNEUROSCI.1002-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baptista FG, et al. Accumulation of Plasmodium berghei-infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect Immun. 2010;78(9):4033–4039. doi: 10.1128/IAI.00079-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grau GE, et al. Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J Infect Dis. 2003;187(3):461–466. doi: 10.1086/367960. [DOI] [PubMed] [Google Scholar]
- 14.Dorovini-Zis K, et al. The neuropathology of fatal cerebral malaria in malawian children. Am J Pathol. 2011;178(5):2146–2158. doi: 10.1016/j.ajpath.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pai S, et al. Real-time imaging reveals the dynamics of leukocyte behaviour during experimental cerebral malaria pathogenesis. PLoS Pathog. 2014;10(7):e1004236. doi: 10.1371/journal.ppat.1004236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howland SW, Claser C, Poh CM, Gun SY, Renia L. Pathogenic CD8+ T cells in experimental cerebral malaria. Semin Immunopathol. 2015;37(3):221–223. doi: 10.1007/s00281-015-0476-6. [DOI] [PubMed] [Google Scholar]
- 17.Nitcheu J, et al. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J Immunol. 2003;170(4):2221–2228. doi: 10.4049/jimmunol.170.4.2221. [DOI] [PubMed] [Google Scholar]
- 18.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259–283. doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. 2015;36(2):71–80. doi: 10.1016/j.it.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol. 2014;14(7):435–446. doi: 10.1038/nri3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carr EL, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. 2010;185(2):1037–1044. doi: 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang R, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakaya M, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40(5):692–705. doi: 10.1016/j.immuni.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thangavelu K, Chong QY, Low BC, Sivaraman J. Structural basis for the active site inhibition mechanism of human kidney-type glutaminase (KGA) Sci Rep. 2014;4:3827. doi: 10.1038/srep03827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waisberg M, Vickers BK, Yager SB, Lin CK, Pierce SK. Testing in mice the hypothesis that melanin is protective in malaria infections. PLoS One. 2012;7(1):e29493. doi: 10.1371/journal.pone.0029493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yen LF, Wei VC, Kuo EY, Lai TW. Distinct patterns of cerebral extravasation by Evans blue and sodium fluorescein in rats. PLoS One. 2013;8(7):e68595. doi: 10.1371/journal.pone.0068595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nag S. Blood-brain barrier permeability using tracers and immunohistochemistry. Methods Mol Med. 2003;89:133–144. doi: 10.1385/1-59259-419-0:133. [DOI] [PubMed] [Google Scholar]
- 28.Howland SW, et al. Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol Med. 2013;5(7):916–931. doi: 10.1002/emmm.201202273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plaimas K, et al. Computational and experimental analysis identified 6-diazo-5-oxonorleucine as a potential agent for treating infection by Plasmodium falciparum. Infect Genet Evol. 2013;20:389–395. doi: 10.1016/j.meegid.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Queen SA, Jagt DL, Reyes P. In vitro susceptibilities of Plasmodium falciparum to compounds which inhibit nucleotide metabolism. Antimicrob Agents Chemother. 1990;34(7):1393–1398. doi: 10.1128/aac.34.7.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malleret B, et al. A rapid and robust tri-color flow cytometry assay for monitoring malaria parasite development. Sci Rep. 2011;1(118):14. doi: 10.1038/srep00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon EB, et al. Inhibiting the Mammalian target of rapamycin blocks the development of experimental cerebral malaria. MBio. 2015;6(3):00725–00715. doi: 10.1128/mBio.00725-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim JV, Kang SS, Dustin ML, McGavern DB. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature. 2009;457(7226):191–195. doi: 10.1038/nature07591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuzefpolskiy Y, Baumann FM, Kalia V, Sarkar S. Early CD8 T-cell memory precursors and terminal effectors exhibit equipotent in vivo degranulation. Cell Mol Immunol. 2015;12(4):400–408. doi: 10.1038/cmi.2014.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin SY, et al. Multiple Tissue Human Expression Resource (MuTHER) Consortium An atlas of genetic influences on human blood metabolites. Nat Genet. 2014;46(6):543–550. doi: 10.1038/ng.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haluszczak C, et al. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J Exp Med. 2009;206(2):435–448. doi: 10.1084/jem.20081829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]