

Significance
Chronic hepatitis B virus (HBV) infection is maintained by the persistence of episomal HBV closed circular DNA (cccDNA) in infected hepatocytes. Current therapeutic regimes have no or limited impact on cccDNA, and the development of cccDNA-targeted therapies is complicated by our limited understanding of cccDNA regulation. We present a novel approach and first detailed analysis to our knowledge of cccDNA chromatin from de novo infected cells and infected liver tissue and reveal general features of cccDNA chromatin organization, and features that are unique to each source of cccDNA. We show that cccDNA chromatin is modulated by innate immunity and manipulated with an epigenetic agent, thereby establishing the importance of chromatin for cccDNA regulation and as a potential target for therapy of chronic HBV infection.
Keywords: hepatitis B virus, HBV, cccDNA, chromatin, epigenetics
Abstract
Chronic hepatitis B virus (HBV) infection affects 240 million people worldwide and is a major risk factor for liver failure and hepatocellular carcinoma. Current antiviral therapy inhibits cytoplasmic HBV genomic replication, but is not curative because it does not directly affect nuclear HBV closed circular DNA (cccDNA), the genomic form that templates viral transcription and sustains viral persistence. Novel approaches that directly target cccDNA regulation would therefore be highly desirable. cccDNA is assembled with cellular histone proteins into chromatin, but little is known about the regulation of HBV chromatin by histone posttranslational modifications (PTMs). Here, using a new cccDNA ChIP-Seq approach, we report, to our knowledge, the first genome-wide maps of PTMs in cccDNA-containing chromatin from de novo infected HepG2 cells, primary human hepatocytes, and from HBV-infected liver tissue. We find high levels of PTMs associated with active transcription enriched at specific sites within the HBV genome and, surprisingly, very low levels of PTMs linked to transcriptional repression even at silent HBV promoters. We show that transcription and active PTMs in HBV chromatin are reduced by the activation of an innate immunity pathway, and that this effect can be recapitulated with a small molecule epigenetic modifying agent, opening the possibility that chromatin-based regulation of cccDNA transcription could be a new therapeutic approach to chronic HBV infection.
Hepatitis B virus (HBV) infection is widespread in humans and is a major public health concern. Primary infection outside the newborn period is usually self-limited, but a subset of infected individuals does not eliminate the virus and goes on to a lifelong persistent infection. Worldwide, at least 240 million people are persistently infected, many of whom develop chronic liver injury (chronic hepatitis B or CHB) (1). CHB often progresses to cirrhosis and liver failure, and is also strongly linked to the development of hepatocellular carcinoma (HCC). It is estimated that CHB accounts for more than 80% of HCC cases in areas of high HBV incidence (2).
HBV belongs to the family of Hepadnaviridae, a group of small DNA viruses that infect hepatocytes and replicate through the reverse transcription of an RNA intermediate (3). The 3.2-kb HBV genome in viral particles is in a circular, partially double-stranded DNA conformation (relaxed circular DNA or rcDNA), a result of the unusual replication mechanism of HBV. rcDNA is transcriptionally inert and must be converted into covalently closed circular DNA (cccDNA) in the nucleus of infected cells before viral RNAs can be transcribed. cccDNA is the only template for HBV transcription and, because HBV RNA templates genomic reverse transcription, its persistence is required for persistent infection. HBV replication itself is noncytolytic, but it induces an immune response that in the case of CHB leads to persistent liver inflammation. Suppression of HBV reverse transcription with nucleos(t)ide analogs (NA) reduces viral load and liver damage, but has little effect on the nuclear cccDNA pool. As a result, it is not curative and must be taken continuously; withdrawal of therapy leads to prompt relapse of HBV replication (4, 5). The only other therapy for CHB that is clinically approved is treatment with IFN-α. In addition to its important immunomodulatory effects, IFN-α treatment has antiviral effects that have been attributed to transcriptional down-regulation of cccDNA (6, 7), and potentially also destabilization of cccDNA (8); however, this finding remains to be confirmed. Unfortunately, only a small subset of patients responds to IFN-α treatment, which is moreover generally associated with poor tolerability (9). Given the limitations of NA and IFN-α treatment, novel approaches to therapy of CHB are needed. Among the attractive potential approaches would be ones that either deplete or transcriptionally silence cccDNA. Unfortunately, developing these new approaches has been hampered by our limited understanding of the molecular processes involved in cccDNA formation, maintenance, and transcriptional regulation.
Previous studies show that HBV cccDNA is assembled together with cellular histone proteins into episomal chromatin (10–12) and that transcription of cccDNA depends on the cellular transcriptional machinery (13). It has been well established for cellular DNA that its assembly and compaction into chromatin controls the accessibility of DNA to the transcriptional machinery (14), and that this is dynamically regulated by posttranslational modifications (PTMs) of histone proteins within chromatin (15, 16). Although it is reasonable to assume that cccDNA chromatin and transcription could be regulated by PTMs in a similar manner, it has not been investigated in detail yet which PTMs are present in cccDNA chromatin and how they are distributed across the HBV genome. Hence, it is unclear to what extent cccDNA chromatin might follow the “rules” that have been established for the regulation of cellular chromatin and whether inducing transcriptional silencing of cccDNA by chromatin-mediated mechanisms might be a feasible approach to treatment of chronic HBV infection. Here, we present, to our knowledge, the first detailed analysis of cccDNA chromatin using a novel HBV cccDNA chromatin immunoprecipitation followed by massive parallel sequencing approach (cccDNA ChIP-Seq). We find that despite the overlapping transcription units and high density of regulatory elements within the small HBV genome, cccDNA chromatin is highly organized and modified with PTMs at specific regions. By comparing the chromatin landscape of cccDNA between model systems of de novo HBV infection and liver tissue from chronically infected patients, we demonstrate that PTMs in cccDNA chromatin accurately predict the transcriptional state of the viral promoters within these different contexts, strongly suggesting that chromatin contributes to the transcriptional regulation of cccDNA. Lastly, we demonstrate that IFN-α treatment down-regulates HBV transcription by reducing active PTMs in cccDNA chromatin, and that this effect can be recapitulated with a small molecule epigenetic modifying agent, suggesting that silencing cccDNA epigenetically might be a viable therapeutic approach.
Results
Experimental System: De Novo Infection of HepG2 Cells with HBV and cccDNA-Containing Chromatin Enrichment.
HepG2 is a hepatoblastoma-derived cell line that supports HBV replication (17) following transfection with HBV DNA, but lacks the entry pathway for HBV and, thus, cannot support virion infection. Available stably transfected HepG2 cells lines can build a pool of cccDNA by reverse transcription of pregenomic RNA originally emanating from an integrated copy of the transfected HBV genome (18, 19). However, because chromatin derived from the episomal cccDNA cannot be distinguished from chromatin derived from the integrated HBV genome, these cells cannot be used to unambiguously assign PTMs to episomal viral genomes. Accordingly, we chose to establish cccDNA more naturally by infecting cells de novo with HBV virions. To this end, we established a HepG2 cell line susceptible to de novo HBV infection by stably overexpressing the recently identified HBV receptor NTCP1 (sodium taurocholate cotransporting peptide 1) (20). HBV transcription in de novo infected HepG2-NTCP1 cells was detectable from 3 dpi (days after infection) onwards and reached steady-state levels at approximately 7–9 dpi, suggesting that infection was fully established at this point (Fig. 1A). Using viral particles concentrated from cell culture supernatant of a stable HBV-producing cell line, more than 50% of the cells of the HepG2-NTCP1 monolayer could be infected, as detected by HBV core protein immunostaining (Fig. 1B), and both HBV eAg and sAg secretion could be detected at 7 dpi under these conditions (Fig. 1C). The presence of cccDNA at 7 dpi was confirmed by Southern blot analysis (Fig. 1D and Fig. S1A) and quantified by cccDNA-specific quantitative PCR (qPCR) assay, and we estimate that ∼2.4 copies of cccDNA were formed per cell (Fig. 1E). Whereas cccDNA can be selectively detected by PCR with specific primers to a certain degree of confidence (Fig. S1B), unbiased DNA detection methods such as massive parallel sequencing cannot discriminate between cccDNA and other HBV DNA species such as rcDNA. We therefore developed a protocol to prepare chromatin that contains only HBV DNA from cccDNA for our cccDNA ChIP-Seq assay (summarized in Fig. 1F). We cross-linked infected cells with formaldehyde and pelleted nuclei to enrich for chromatin and remove the bulk of encapsidated cytosolic replicative HBV DNA intermediates. The nuclei were then digested with micrococcal nuclease (Mnase) to obtain mononucleosomes (Fig. S1C). These mononucleosomes were further purified by sucrose density gradient centrifugation, thereby removing most remaining nonnucleosomal DNA fragments from our preparation (Fig. S1D). Deep sequencing of a purified mononucleosome fraction (Mono-Seq) identified both human and HBV DNA fragments of ∼160 bp (Fig. 1G), a size typical for mononucleosomes from formaldehyde–cross-linked chromatin (21), confirming that our preparation indeed contained HBV DNA originating from nucleosomal cccDNA (Fig. S1 E and F). Furthermore, Mono-Seq detected a fraction of HBV-specific reads equating to ∼2.2 copies of cccDNA per cell in the mononucleosome fraction, comparable to the number detected by cccDNA-specific qPCR (Fig. 1E). Having established that the purified mononucleosomes contained HBV DNA from cccDNA, we next performed ChIP-Seq with antibodies specific for different PTMs. Although we were able to detect HBV-specific reads in ChIP-Seq pilot experiments, a significant number of total sequencing reads would be required to obtain acceptable sequencing coverage of the HBV genome (Fig. S2A). To increase the sequencing coverage for HBV, we introduced a target enrichment step specific for HBV DNA before sequencing using overlapping biotinylated HBV-specific oligonucleotides that cover the whole HBV genome multiple times. Importantly, target enrichment did not significantly alter the ratio of HBV-specific reads between the different sequencing libraries, or the distribution of those reads along the HBV genome (Fig. S2 B and C).
Fig. 1.
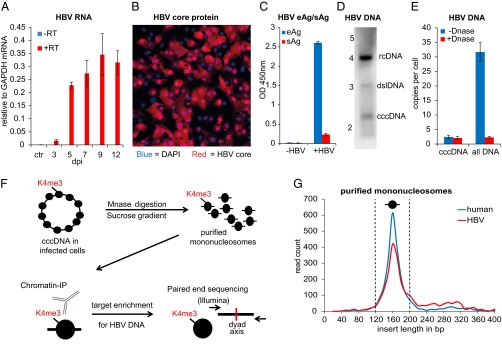
De novo infection of HepG2-NTCP1 cells and preparation of cccDNA chromatin. (A) Quantitative RT-PCR (qRT-PCR) of HBV RNA collected 3 dpi to 12 dpi (with or without reverse transcription in blue and red, respectively) normalized to GAPDH mRNA (n = 2, ±SD). Uninfected HepG2-NTCP1 cells served as the control (ctr). (B) HBV core protein immunostaining of HepG2-NTCP1 cells at 7 dpi showing an infection rate of ∼73%. HBV core (HBc) protein is shown in red, cell nuclei stained with DAPI in blue. (C) eAg and sAg levels in supernatant of cells infected with or without HBV at 7 dpi measured by ELISA. (D) Southern blot analysis of HBV DNA in HIRT extract at 7 dpi. Migration of a DNA standard is indicated in kilobases on the left; position of relaxed circular (rcDNA), double-stranded linear (dslDNA) and cccDNA is indicated on the right. (E) qPCR of HBV DNA at 7 dpi with primers specific for cccDNA or total HBV DNA. Shown is the average copy number of HBV molecules per cell. Samples were treated with or without Plasmid-Safe DNase as indicated (n = 2 ±SD). (F) Schematic of cccDNA ChIP-Seq assay. Nucleosomes in cccDNA (and cellular chromatin) are marked with PTMs, e.g., H3K4me3, at specific positions. HBV-infected cells were digested with micrococcal nuclease and resulting mononucleosomes purified by sucrose gradient centrifugation. Nucleosomes with PTM were enriched with specific antibodies by ChIP and the associated DNA enriched for HBV-specific sequences. Paired-end sequencing was used to determine the insert size and middle point (nucleosome dyad axis) of each DNA fragment. (G) Length distribution of cellular and HBV DNA from isolated mononucleosomes. The insert size distribution of 2,400 paired-end reads aligning to either human (blue) or HBV (red) genome is shown. The vertical dashed lines indicate the insert size window that was used for computational analysis.
Fig. S1.

Control experiments for Southern blot, cccDNA-specific qPCR, and mononucleosome preparation from HBV-infected HepG2-NTCP1 cells. (A) Southern blot of HIRT extracts from cells that were mock infected (-HBV) or infected and cultured in the absence or presence of the HBV replication inhibitor entecavir (+HBV and +HBV +ETV, respectively). The position of relaxed circular (rcDNA), double-stranded linear (dslDNA), single-stranded DNA (ssDNA), and cccDNA is indicated on the right. The presence of cccDNA was verified by heating the sample to 85 °C (+HBV +85 °C), which denatures rcDNA and dslDNA but not cccDNA into ssDNA. Digestion of this sample with EcoRI (+HBV +85 °C/EcoRI) converts cccDNA into dslDNA. (B) qPCR analysis of HBV DNA at 7 dpi. DNA was extracted from whole cells and nuclear and cytoplasmic fractions and treated with Plasmid-Safe DNase (+PsD), or extracted from cells infected and cultured in the presence of ETV (+ETV). Equal amounts of each cellular fraction were analyzed with primers specific for cccDNA or total HBV DNA, and normalized to a HBV plasmid standard. (C) Nuclei isolated from infected cells were incubated with increasing amounts of MNase. Samples were reverse cross-linked, separated by agarose gel electrophoresis. and visualized by ethidium bromide staining. The position of mononucleosomal DNA is indicated. (D) Sucrose gradient centrifugation of MNase-digested chromatin. An aliquot of each fraction (bottom) was reverse cross-linked, separated by agarose gel electrophoresis, and visualized by ethidium bromide staining. The peak fractions were pooled and used for cccDNA ChIP-Seq assay. (E) HBV eAg was quantified by ELISA at 7 dpi from supernatant of cells that were mock infected (-HBV) or infected and cultured in the absence or presence of ETV (+HBV and +HBV +ETV, respectively). Comparable levels of eAg suggest that cccDNA levels established upon de novo infection are not affected by ETV treatment (see also Fig. S1 A and B). (F) Mononucleosomes were isolated from cells infected and cultured in the presence of ETV. Equal amounts of mononucleosomal DNA were analyzed by qPCR with primer pair “HBV 3” and HBV DNA quantified relative to a plasmid standard. ETV treatment has no impact on the amount of nucleosome-associated HBV DNA.
Fig. S2.
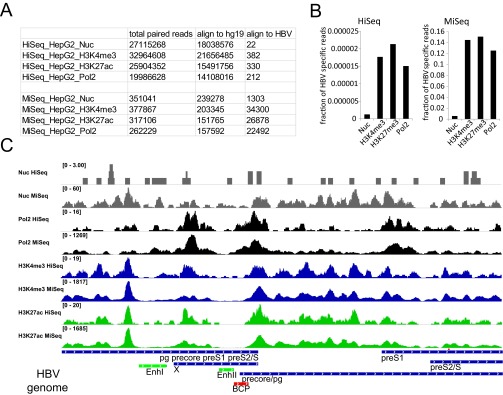
Controls for HBV target enrichment steps in HepG2-NTCP1 cccDNA ChIP-Seq assay. (A) Total number of paired reads from mononucleosomes (Nuc), H3K4me3, H3K27ac, and Polymerase 2 (Pol2) ChIP that align to human (hg19) and HBV genomes. Samples that were sequenced on a HiSeq2500 (HiSeq_X) instrument were not enriched for HBV DNA, whereas samples sequenced on a MiSeq instrument (MiSeq_X) were enriched for HBV DNA by target enrichment before sequencing. The same data are presented in B for HiSeq (Left) and MiSeq (Right) as the fraction of all mapped paired reads that align to the HBV genome. For sufficient sequencing coverage 1,455 paired mapped reads would be required (SI Materials and Methods). (C) Distribution of nucleosome, RNA Polymerase 2, H3K4me3, and H3K27ac read density along the HBV genome. The difference in the total number of reads is indicated in the upper left corner of each sequencing track.
cccDNA Chromatin in HepG2-NTCP1 Cells Has High Levels of Active PTMs Enriched at Specific Regions of the HBV Genome.
PTMs associated with both active and repressive transcriptional states in cellular chromatin were chosen for cccDNA evaluation (Fig. 2A): We selected four PTMs associated with active transcription, H3K4me3, H3K27ac, H3K122ac, and H3K36me3. H3K4me3 is found at the transcription start site (TSS) of actively transcribed genes (22, 23), whereas histone acetylation marks H3K27ac and H3K122ac indicate active TSS and also active gene enhancers (24–26). H3K36me3 is associated with active transcription as well, but is enriched in the gene body toward the 3′ end of transcribed genes in the host genome (27, 28). We also probed for the presence of two PTMs associated with transcriptional silencing, H3K9me3 and H3K27me3. H3K9me3 can be found predominantly in transcriptionally silent heterochromatin and H3K27me3 at transcriptionally repressed genes (29). To determine the relative abundance of these marks in cccDNA, we compared the fraction of HBV-specific sequencing reads in the Mono-Seq library (serving as input) to the fraction of reads mapping to HBV in the different ChIP-Seq libraries (Fig. 2B). The fraction of reads mapping to HBV for active marks H3K4me3, H3K27ac, and H3K122ac was significantly higher than in the Mono-Seq input sample (26.6×, 27.7×, and 13.5×, respectively), indicating that these marks are highly enriched in cccDNA chromatin. In contrast to the other active PTMs, the fraction of HBV-specific reads for H3K36me3 was lower than for mononucleosomes (0.16×), indicating that H3K36me3 was not enriched in cccDNA. Both PTMs associated with transcriptional silencing, H3K9me3 and H3K27me3, were underrepresented in cccDNA (0.07× and 0.02×).
Fig. 2.
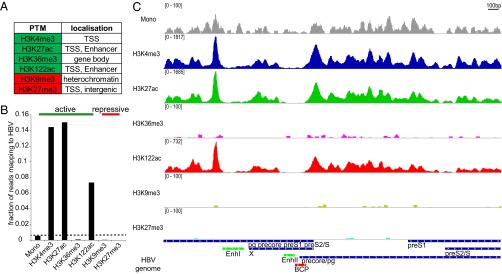
ChIP-Seq analysis of HBV-infected HepG2-NTCP1 cccDNA. (A) Histone posttranslational modifications (PTMs) used in this study and their primary location in cellular chromatin. PTM associated with active and repressed transcription are highlighted in green and red, respectively. (B) The fraction of all mapped paired reads in each sequencing library that align to HBV. “Mono” refers to the Mono-Seq library and indicates what fraction of reads could be expected to map to HBV by chance in the other ChIP-Seq libraries (dashed line). PTMs associated with active and repressed transcription are indicated by the green and red bar, respectively. (C) Distribution of nucleosomes and PTMs along the HBV genome. Read density for each track is represented by height on the y axis scaled to a minimum of 100 reads. HBV transcripts are shown in blue, Enhancer elements in green and the basal core promoter (BCP) in red and represented on the x axis. Transcription occurs from left to right. Note that the 3′ region of pg, precore, preS1, and preS2 mRNA are represented in one transcript only (“pg precore preS1 preS2/S”).
Because PTMs appear in specific patterns, e.g., at promoters and enhancers in human chromatin (30), we next investigated the distribution of these PTMs relative to the regulatory regions of the HBV genome (Fig. 2C). The HBV genome has four major transcription units encoding for precore/pg, preS1, preS2, and X mRNA, as well as two enhancer regions, one of which (Enhancer II) overlaps with the basal core promoter (BCP) (Fig. 2C, bottom row). Our results show that the EnhancerII/BCP region was largely devoid of nucleosomes and PTMs followed by a peak of H3K4me3, H3K27ac, and H3K122ac downstream at the TSS of precore/pg. A nucleosome-depleted region followed by a strong signal of active PTMs resembles the pattern that can be observed at the TSS of many transcribed genes in human chromatin (25, 31). We did not detect additional peaks of comparable strength at the TSS of preS1, preS2, or X, which possibly indicates that the precore/pg HBV promoter is the strongest in de novo infected HepG2-NTCP1 cells. Another region with high levels of H3K4me3, H3K27ac, and H3K122ac was located just upstream of Enhancer I. There is no TSS in immediate vicinity, suggesting that this peak of active PTM might be linked to the enhancer function of Enhancer I. Overall, our ChIP-Seq data indicates that cccDNA chromatin in HepG2-NTCP1 cells is heavily enriched in active PTMs H3K4me3, H3K27ac, and H3K122ac but not H3K36me3, at specific regions in the HBV genome, and is low or depleted in repressive PTMs H3K27me3 and H3K9me3.
Shared and Unique Features of cccDNA Chromatin-Infected HepG2-NTCP1 Cells, Primary Hepatocytes, and Liver Tissue.
Although HepG2 cells are a valuable in vitro model system for HBV infection, they differ in their transcriptional profile (32) and other aspects such as innate immune signaling (33, 34) from primary liver cells. It is therefore important to understand to what extent cccDNA chromatin in HepG2 cells is representative of cccDNA in vivo. To approach this question, we infected primary human hepatocytes (PHH) with HBV and obtained HBV positive (HBV+) liver tissue to analyze cccDNA chromatin as described for HepG2-NTCP1 cells (Fig. 1F). Globally, cccDNA chromatin in both PHH and HBV+ liver tissue displayed relatively high levels of the active marks H3K4me3, H3K27ac, and H3K122ac and low levels of the repressive mark H3K27me3 (Fig. 3A and Fig. S3A), similar to what we observed for HepG2-NTCP1 cccDNA (Fig. 2B). We next compared the genomic distribution of the highly enriched PTMs between all three samples (Fig. 3B). The distribution of H3K4me3, H3K27ac, and H3K122ac revealed common features of cccDNA chromatin and features unique to each sample. A feature that is shared between all three sources of cccDNA is a distinct peak of active PTMs upstream of Enhancer I, followed by a nucleosome-depleted region. Another region of low occupancy was at the Enhancer II/BCP region upstream of the precore/pg TSS. Among the three samples, HepG2-NTCP1 cccDNA had the strongest peak for the active PTMs H3K4me3, H3K27ac, and H3K122ac at the precore/pg TSS, whereas PHH cccDNA showed high levels of active PTMs throughout most of HBV genome with a more pronounced enrichment of active PTMs within X and close to the preS1 TSS. In the HBV+ liver sample, active PTMs were relatively low in the region surrounding the precore/pg TSS, but displayed high levels of enrichment at the TSS of preS2 and within the X gene. Overall, the profiles of H3K4me3, H3K27ac, and H3K122ac within each sample were quite similar, with the exception of HBV+ liver cccDNA with a unique peak of H3K122ac at preS1 TSS. This comparative analysis of PTMs suggests that the distribution of PTMs in cccDNA chromatin can vary substantially between cell culture models and infected tissue, and that cccDNA chromatin is therefore not exclusively determined by viral components, but can adapt to different cellular environments.
Fig. 3.
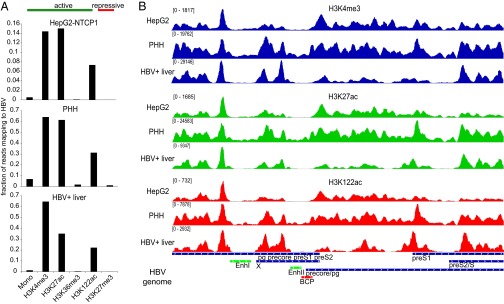
Shared and unique features of cccDNA chromatin in HepG2-NTCP1, PHH, and HBV+ liver tissue. (A) The fraction of all mapped paired reads in each sequencing library that align to HBV for HepG2-NTCP1 (Top), PHH (Middle), and HBV+ liver (Bottom). (B) Distribution of H3K4me3 (Top, in blue), H3K27ac (Middle, in green) and H3K122ac (Bottom, in red) read density along the HBV genome (Very Bottom) in HepG2-NTCP1, PHH, and HBV+ liver chromatin.
Fig. S3.
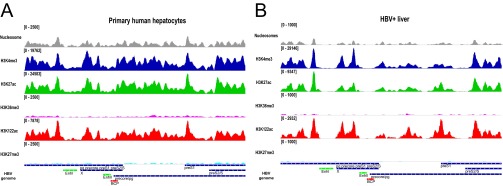
Distribution of nucleosome and PTM density along the HBV genome in PHH (A) and HBV+ liver cccDNA (B) as shown for HepG2-NTCP1 in Fig. 2C.
PTM Distribution at HBV Promoters Is Comparable to Human Chromatin and Predicts Transcriptional Activity.
In cellular chromatin, PTMs such as H3K4me3, H3K27ac, and H3K122ac indicate transcriptionally active promoters. To determine whether in cccDNA chromatin enrichment of active PTMs at HBV promoters is predictive for transcription as well, we looked at their overlap with RNA Polymerase II (Pol2) occupancy and the relative levels of HBV RNA species in the two samples with the most different PTM profiles, HepG2-NTCP1 cells and HBV+ liver tissue. In HepG2-NTCP1, cccDNA Pol2 enrichment was found at three different locations: in a region between EnhancerI and EnhancerII, at the TSS of precore/pg, and close to the preS1 TSS (Fig. 4A). Interestingly, only at the TSS of precore/pg Pol2 aligned to the +1 nucleosome (marked by high levels of H3K4me3) in a manner that is characteristic of actively transcribed human promoters (30) (Fig. S4A). Consistent with this observation, Northern blot analysis (Fig. 4B) confirmed that precore/pgRNA were the most prominent HBV RNA species in de novo infected HepG2-NTCP1 cells. In HBV+ liver cccDNA, however, Pol2 occupancy was low close to the precore/pg TSS and enriched close to the TSS of preS2 (Fig. 4A). The peak of Pol2 enrichment aligned only at the preS2/S TSS with the +1 nucleosome as observed in actively transcribed human genes, and Northern blot analysis showed that preS2/S RNA was by far the most abundant HBV transcript in the HBV+ liver sample (Fig. 4B). Analysis of eAg and sAg secretion, encoded by precore RNA and preS2/S RNA, respectively, confirmed our Northern blot analysis (Fig. S4B). Together this data suggests that the distribution of active PTMs and Pol2 in cccDNA predicts HBV promoter use.
Fig. 4.
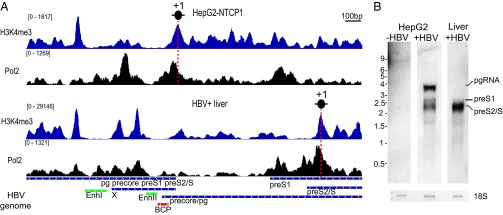
Differences in Pol2 binding and transcriptional profile between infected HepG2-NTCP1 cells and HBV+ liver. (A) H3K4me3 and Pol2 read density along the HBV genome in HepG2-NTCP1 and HBV+ liver. The vertical red dashed line indicates the position of the +1 nucleosome. (B) Northern blot analysis of 1 μg of RNA extracted at 7 dpi from uninfected HepG2-NTCP1 cells, HBV-infected HepG2-NTCP1 cells, and HBV+ liver sample. The bottom row shows 18S rRNA loading control visualized by methylene blue staining. Migration of the RNA ladder in kilobases is indicated on the left; the migration of the different HBV RNA species is indicated on the right.
Fig. S4.
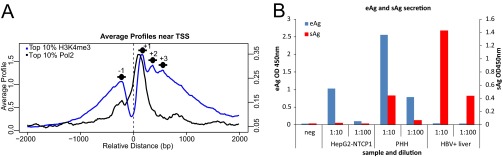
H3K4me3 and Pol2 distribution at human TSS and eAg/sAg secretion patterns in HepG2-NTCP1, PHH, and HBV+ liver. (A) H3K4me3 and Pol2 read density in HBV+ liver sample was plotted in a window of ±2 kb around the TSS of RefSeq genes (dotted vertical line). The signal from the 10% highest transcribed genes is shown (63). (B) Detection of HBV eAg and sAg in the supernatant of HBV-infected HepG2-NTCP1 or PHH at 7 dpi, or from serum matching the HBV+ liver sample shown in A by ELISA.
The Levels of Active PTMs in cccDNA Are Comparable to Transcriptionally Active Human Chromatin.
Our data shows that active PTMs in cccDNA are distributed relative to HBV promoters in a manner that is comparable to human chromatin. We next assessed the level of PTMs enrichment (or lack thereof) in cccDNA in context to the levels of PTMs that can be observed at transcriptionally active and silent loci in human chromatin directly by ChIP-qPCR. For human chromatin, the promoters of β-actin (ACTB) and Nanog (a stem cell-specific gene) were used as references for actively transcribed and transcriptionally repressed genes, respectively. Specific H3K36me3 enrichment at 3′ end of genes was tested at the GAPDH locus. As expected, levels of H3K4me3, H3K27ac, and H3K122ac were high at the ACTB promoter and low at the Nanog promoter, whereas H3K27me3 was enriched at the Nanog promoter and H3K36me3 was enriched at the 3′ end of the GAPDH locus (Fig. S5A). HBV cccDNA was analyzed at four different loci (position 1–4) for PTM levels (Fig. 5A). Interestingly, we found variations in the absolute levels of the H3K4me3, H3K27ac, and H3K122ac between the three sources of cccDNA analyzed (Fig. 5B): In HepG2-NTCP1 cccDNA, the enrichment of H3K4me3, H3K27ac, and H3K122ac was overall comparable to the levels observed at the active ACTB promoter. In PHH cccDNA, however, H3K4me3, and especially H3K27ac, levels were significantly higher than in HepG2-NTCP1 cccDNA (and the ACTB promoter), whereas H3K122ac levels remained comparable to HepG2-NTCP1 cccDNA. In HBV+ liver cccDNA, H3K4me3 levels were as high as in PHH cccDNA, but H3K27ac levels were not elevated relative to HepG2-NTCP1 cccDNA. H3K122ac levels in HBV+ liver cccDNA were slightly lower than those observed in HepG2-NTCP1 cccDNA, PHH cccDNA, and at the ACTB promoter. As indicated by the ChIP-Seq data, H3K27me3 levels at the four different HBV loci were, if detectable by ChIP-qPCR, in all three samples significantly lower than at the Nanog promoter (Fig. 5B). H3K36me3 levels in HepG2-NTCP1 and HBV+ liver cccDNA might increase toward the 3′ end of HBV transcripts (position 2), but are overall significantly lower than observed at the 3′ end of GAPDH. These observations are summarized in Fig. 5C and show that the level of active, promoter (and enhancer) specific PTMs H3K4me3, H3K27ac, and H3K122ac in cccDNA chromatin reaches or exceeds the levels observed at a highly transcribed human promoter, and that the repressive PTM H3K27me3 is present only at low levels in cccDNA.
Fig. S5.
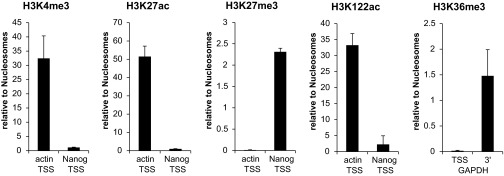
Validation of specific ChIP enrichment for the different PTMs analyzed. Enrichment for different histone modifications at different human loci in HepG2-NTCP1 cells is shown relative to mononucleosome input material.
Fig. 5.

Quantification of PTM levels in cccDNA chromatin relative to human chromatin. (A) Localization of amplicons 1–4 across the HBV genome. H3K4me3 enrichment from HepG2-NTCP1 cells (Fig. 2C) is shown as reference. (B) PTM enrichment in cccDNA and human chromatin from HepG2-NTCP1, PHH, and HBV+ liver analyzed by ChIP-qPCR. ChIP signal for each amplicon was normalized to the mononucleosome input material. Enrichment of PTM in cccDNA was compared with positive control loci in human chromatin: ACTB TSS for H3K4me3, H3K27ac, and H3K122ac; Nanog TSS for H3K27me3; and GAPDH 3′region for H3K36me3 (n = 2, ±SD). (C) Summary of the relative levels of PTM in cccDNA chromatin from HepG2-NTCP1, PHH, and HBV+ liver. -, very low/barely detectable; (+), low; +, comparable. +++, >2x higher relative to cellular control loci.
The Suppressive Effect of IFN-α on Active PTMs and Transcription of cccDNA Can Be Recapitulated with a Small Molecule Epigenetic Modifying Agent.
Treatment with the antiviral cytokine IFN-α reduces HBV RNA accumulation (6, 35, 36), and limited literature suggests that this reduction in transcription might involve changes of PTMs on cccDNA (6). However, this hypothesis has not been tested for specific PTMs in the context of de novo infected cells. To test whether transcriptional down-regulation of cccDNA by IFN-α is linked to changes in cccDNA chromatin, we used de novo infected PHH because HepG2 cells are partially defective in their innate immune response (33, 34). As expected, HBV RNA levels were reduced upon IFN-α treatment of infected PHH (Fig. 6A), whereas transcription of the IFN-stimulated gene IFIT1 was strongly induced (Fig. S6A). ChIP-qPCR analysis of HBV-infected PHH revealed that IFN-α treatment reduced the levels of the active PTMs H3K4me3, H3K27ac, and particularly H3K122ac in cccDNA (Fig. 6B and Fig. S6B). Interestingly, we did not detect an increase in the level of the repressive PTM H3K27me3, which is hence not likely to contribute to the transcriptional down-regulation of cccDNA upon IFN-α treatment that we detect. To confirm that lower levels of active PTMs on cccDNA and reduced HBV transcription are functionally linked, we next used a small molecule epigenetic agent to manipulate the active PTMs in cccDNA chromatin more specifically. Toward this end, we used the small molecule C646 that specifically inhibits p300/CBP (37), the histone acetyltransferases for H3K27ac and H3K122ac (26, 38). C646 treatment reduced HBV transcription in a dose-dependent manner (Fig. 6C), and in the absence of measurable toxicity under the assay conditions (Fig. 6D), supporting that HBV transcription and active PTM levels in cccDNA (Fig. S6D) are functionally linked. The lack of IFIT1 induction shows that the transcriptional down-regulation of cccDNA was independent of the IFN-α pathway (Fig. 6C). IFN-α treatment has been suggested to be linked to a reduction of cccDNA (8); however, cccDNA levels were not affected by our IFN-α treatment (Fig. S6C). Likewise, cccDNA levels were not affected by C646 treatment (Fig. 6E). This data shows that the targeted manipulation of PTM levels can recapitulate the effect of IFN-α on cccDNA transcription, supporting the importance of chromatin for the regulation of cccDNA transcription.
Fig. 6.
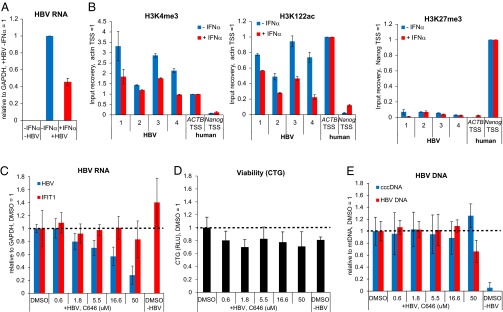
IFN-α treatment reduces HBV transcription and active PTMs on cccDNA and can be recapitulated with p300/CBP inhibitor C646. (A) PHH were infected with HBV and treated with or without IFN-α for 72 h from 4 dpi to 7 dpi. HBV mRNA levels were measured by qRT-PCR, normalized to GAPDH mRNA and plotted relative to the untreated sample (-IFNα) (n = 2, ±SD) (B) Changes in PTM levels in PHH ± IFN-α measured by ChIP-qPCR. Level of enrichment relative to nucleosomes was normalized to a cellular control locus as described for Fig. 5B. (C) p300 inhibition by C646 reduces HBV transcription. PHH were infected with HBV and treated with various concentrations of C646 for 72 h from 4 dpi to 7 dpi. Total HBV RNA and IFIT1 mRNA was measured by qRT-PCR and normalized to GAPDH mRNA. The values for both transcripts are plotted relative to the DMSO control sample (n = 3, ±SD). (D) Cell viability was measured by CellTiter-Glo assay (CTG); the relative luminescence units (RLU) are shown relative to the DMSO control (n = 3, ±SD). (E) Total HBV DNA and cccDNA levels were analyzed by qRT-PCR, normalized to mitochondrial DNA and plotted relative to DMSO control sample (n = 3, ±SD).
Fig. S6.
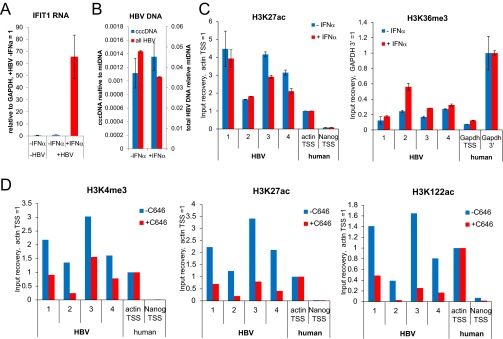
Global effect of C646 treatment on PTM levels. (A) PHH were infected with HBV and treated with or without IFN-α for 72 h from 4 dpi to 7 dpi. IFIT1 mRNA levels were measured by qRT-PCR, normalized to GAPDH mRNA, and plotted relative to the infected, untreated sample (-IFNα) (n = 2, ±SD). (B) PHH were infected with HBV and treated with or without IFN-α for 72 h from 4 dpi to 7 dpi. Total HBV DNA and cccDNA levels were analyzed by qRT-PCR and normalized to mitochondrial DNA (n = 2, ±SD). (C) PHH were infected with HBV and treated with or without IFN-α for 72 h from 4 dpi to 7 dpi. Changes in H3K27ac and H3K36me3 levels with or without IFN-α were measured by ChIP-qPCR. The level of enrichment relative to nucleosomes was normalized to a cellular control locus as described for Fig. 5B (n = 2, ±SD). (D) PHH were infected with HBV and treated ±25 μM C646 for 72 h from 4 dpi to 7 dpi. Enrichment of H3K4me3, H3K27ac, and H3K122ac relative to nucleosomes was measured by ChIP-qPCR and normalized for ±25 μM C646 treatment to β-actin transcription start site as an cellular control locus as described for Fig. 5B (n = 1).
Discussion
A tremendous amount of research in the past years has been devoted to the genomewide mapping of PTMs in cellular chromatin of numerous cells types and tissues. From this body of work, we have learned that PTMs are distributed in specific patterns, e.g., relative to gene promoters or enhancers (30), where PTMs can regulate transcription and other processes either by recruiting PTM-specific binding proteins (16), or by directly altering the physical property of individual nucleosomes (39) and the chromatin fiber (40). Although HBV cccDNA is assembled into chromatin as well, its circular conformation, small genome size, and compact coding and transcript organization are remarkably different from the cellular genome. It is therefore open to question whether within this context the typical PTM patterns and regulatory mechanisms that apply to cellular chromatin are maintained. Previously, cccDNA chromatin was analyzed by ChIP of complete cccDNA molecules followed by qPCR with cccDNA-specific primers (12). Although this approach has proven useful to probe for the general association of proteins and PTMs with cccDNA, the distribution of PTMs and other factors along the HBV genome has remained elusive. Understanding how PTMs are organized relative to genetic elements within HBV genome is crucial to understanding the chromatin-based regulation of cccDNA.
In this study, we overcame previous technical limitations and present, to our knowledge, the first genome-wide maps of PTMs (and Pol2) in HBV cccDNA chromatin at high resolution. Our HBV cccDNA ChIP-Seq assay reveals that PTMs are distributed nonrandomly across the HBV genome, strongly suggesting that PTMs in chromatinized cccDNA were specifically introduced following histone assembly on the viral genome. Our analysis reveals several key features common to all of the infected cells that we examined. In all three infected contexts, we detected high levels of H3K4me3, H3K27ac, and H3K122ac. In cellular chromatin, H3K4me3 and H3K27ac enrichment at promoters is known to stimulate transcription by recruiting components of the preinitiation complex and other transcriptional activators (41–43). Because H3K4me3 (and H3K27ac) is enriched at HBV promoters as well, and because H3K4me3 enrichment at the +1 nucleosome of transcribed HBV promoters aligns with Pol2 occupancy in a manner that is reminiscent of active promoters in human chromatin, we conclude that active transcription in cccDNA is regulated by similar chromatin-based mechanisms as human chromatin. The observation that H3K4me3 and H3K27ac levels can be significantly higher in cccDNA compared with, e.g., the ACTB promoter (especially in PHH and HBV+ liver) suggests that they might even be of particular importance for HBV transcription.
Separate from the mechanisms of transcriptional activation achieved by H3K4me3 and H3K27ac, H3K122ac can activate transcription by directly increasing nucleosome mobility (26). Although H3K122ac is enriched in cccDNA chromatin as well, the enrichment is lower than for H3K4me3 and H3K27ac. This data could indicate that the small circular cccDNA tolerates only lower levels of H3K122ac, maybe because the unmodified state of H3K122 is important for transcriptional regulation in the context of overlapping transcription units (44) that are present in the HBV genome. Surprisingly, we detected relatively low levels of H3K36me3, a PTM that is usually enriched in the gene body of transcribed genes to suppress aberrant transcription initiation in the wake of Pol2 transcription (45). The low levels of H3K36me3 are possibly due to the negative regulation by the highly enriched mark H3K4me3 (46) present throughout cccDNA chromatin. It remains to be determined whether higher levels of H3K36me3 would interfere with the regulation of cccDNA transcription. In sharp contrast to most activating marks, the repressive mark H3K27me3 is strikingly underrepresented in HBV chromatin, and this feature too was preserved in all three contexts of HBV infection. Because the levels of H3K27me3 in cccDNA are dramatically lower than at the H3K27me3-regulated Nanog promoter, we suggest that there is limited H3K27me3-mediated repression in cccDNA. Interestingly, Riviere et al. recently reported comparably minute levels of H3K27me3 in cccDNA established in the hepatoma cell line HepaRG (47), supporting our finding. Although cccDNA chromatin appears to be generally in an open, transcriptionally permissive conformation throughout the HBV genome, the relative transcription of the four viral promoters differed substantially between the different samples analyzed. HBV transcription can be modulated by different cellular (transcription) factors (48), and different expression levels especially of liver-specific transcription factors among HepG2, PHH, and liver tissue (49) might likely contribute to the selective promoter activation or silencing. However, because we do not detect significant levels of repressive H3K27me3 even at silent HBV promoters (e.g., precore/pg in HBV+ liver), selective silencing of HBV promoters seems to be independent of canonical, H3K27me3-mediated gene repression. Differential DNA methylation could contribute to promoter-specific silencing as well; however, even artificially induced, complete methylation of CpG islands in HBV, cccDNA reduces precore/pg RNA only by ∼50% (50). Therefore, it remains to be determined exactly how selected HBV promoters are silenced. Overall, our analysis of HBV-infected cells and HBV+ liver tissue shows that HBV chromatin shares some basic features with cellular chromatin, especially those linked to active transcription, but lacks significant levels of PTMs associated with transcriptional repression, which, in cellular chromatin, are indispensable for transcriptional control. We would like to point out, however, that the HBV+ liver sample that we analyzed is not representative for the full complexity of chronic HBV infection, because both global and gene-specific HBV RNA levels can vary substantially between different patients and the different phases of chronic HBV infection (51–53). Hence, an extension of our study to additional individuals and different phases of the chronic HBV infection will be required to get a more complete picture of cccDNA chromatin regulation in vivo.
Interestingly, even near-complete transcriptional silencing of cccDNA seems to occur in vivo, because it has been observed that patients who overcame HBV infection can still carry intact, but apparently transcriptionally largely silent, cccDNA molecules in their liver (54). One intracellular pathway that has been shown to control HBV transcription and replication is the type I IFN response pathway (7, 55, 56). Inhibition of this pathway by a Jak1 inhibitor has been connected to reactivation of HBV in previously “cured” patients (57), in whom gene expression had been dramatically attenuated to below detectable levels. This finding supports that the IFN response pathway likely contributes to transcriptional control of cccDNA. Our experimental data confirms that stimulation of the IFN response with IFN-α not only reduces HBV RNA levels, but also modulates cccDNA chromatin. Importantly, we did not detect an increase of H3K27me3 upon IFN-α treatment, despite the report of increased binding of PRC2 (the enzyme complex responsible for H3K27 trimethylation), to cccDNA upon IFN-α treatment (6). Our data indicate that the initial effect of IFN-α signaling on cccDNA seems to be the reduction of active PTMs rather than the deposition of repressive PTMs. High levels of H3K27ac, possibly in concert with other active marks, have been shown to antagonize H3K27me3 deposition in cellular chromatin (58); therefore, removal of active PTMs seems a logic first step toward the transcriptional silencing of cccDNA. Indeed IFN-α–dependent recruitment of histone deacetylase 1 (HDAC1) to cccDNA has been reported (6) and might contribute to the reduction of H3K27ac and H3K122ac that we detect, and it is likely that this and additional PTM modifying enzymes lead to a transcriptional less permissive cccDNA chromatin state in the presence of IFN-α.
IFN-α treatment is a clinically approved therapy for chronic HBV infection, but it benefits only a subset of patients and is generally poorly tolerated (9). It would therefore be highly desirable to be able to recapitulate the changes in cccDNA chromatin induced by IFN-α more efficiently and specifically. Our finding that the epigenetic inhibitor C646 inhibits HBV transcription in a dose-dependent manner in the absence of measurable toxicity provides a proof of concept that epigenetic manipulation of cccDNA can lead to substantial reduction of HBV transcription and, hence, could present a novel approach to the therapy of chronic HBV infection. Realizing such an approach is likely to require considerable additional effort, including the analysis of additional PTMs, additional viral strains, clinical samples, and signaling pathways to further elucidate the role of modulation of HBV chromatin structure in the regulation of viral infection. Our data and cccDNA ChIP-Seq assay described here should facilitate additional investigations aimed at exploring the therapeutic potential of this approach. Such an approach will further reduce viral particle and antigen production and could facilitate (alone or in combination with other manipulations) the ability of the immune system to reestablish control over or even clear chronic HBV infection (59).
Materials and Methods
For the cccDNA ChIP-Seq assay HBV-infected cells were harvested with Accutase (Millipore) at 7 dpi and cross-linked in 1× PBS with 1% freshly cracked formaldehyde (Pierce) for 5 min. Fixation was quenched by adding 125 mM glycine final concentration for 2 min. Next, cells were washed with ice cold nuclei isolation buffer [1× PBS with 0.1% Triton X-100, 0.1% Nonidet P-40, 1 mM DTT, 10 mM sodium butyrate, 1× protease inhibitor (Roche)] once and resuspended in the same buffer for 10-min incubation on ice with occasional mixing. Nuclei were pelleted and resuspended in Mnase digestion buffer [50 mM Tris pH 7.5, 4 mM MgCl2, 1 mM CaCl2, 10% (vol/vol) glycerol, 10 mM sodium butyrate, 1× protease inhibitor] with 500 U/mL Mnase (Fermentas). HBV+ liver tissue was minced in small pieces and fixed as described as above. Cross-linked liver tissue was washed in ice cold nuclei isolation buffer once, resuspended in the same buffer, and homogenized with 10 strokes of a dounce homogenizer. After 10 min on ice, the homogenized sample was passed through a cell strainer to remove debris, the released nuclei pelleted and resuspended in Mnase digestion buffer. Nuclei in Mnase digestion buffer were digested for 10 min at 37 °C to mainly mononucleosomes before digestion was quenched with 10 mM EGTA final concentration on ice. The digested nuclei were spun at 6,500 × g for 5 min and the supernatant (S1) saved into a fresh tube. The pellet was resuspended in Mnase buffer with 10 mM EGTA and 300 mM NaCl and mildly sonicated on ice four times for 10 pulses in Heat Systems W-375 sonicator at 50% duty cycle and power setting 3. The sample was spun at 6,500 × g for 5 min, and the supernatant (S2) combined with the S1 fraction. S1 and S2 fraction were mixed with an equal volume of 0% sucrose buffer (50 mM Tris pH 7.5, 50 mM NaCl, 5 mM EDTA pH 8.0, 0.01% Nonidet P-40, 10 mM sodium butyrate, 1× protease inhibitor) and concentrated in a Vivaspin 4 spin column (Sartorius) with 100K cutoff membrane to ∼250 μL. The concentrated sample was applied to a 5–30% (wt/vol) sucrose gradient and centrifuged in a SW41Ti (Beckman Coulter Inc.) rotor with 40,000 rpm at 4 °C for 4 h. Mononucleosome containing fractions were pooled, concentrated to ∼500 μL and 100 ng/μL BSA added. For ChIP, antibodies were prebound in ChIP dilution buffer (20 mM Tris pH 8.0, 150 mM NaCl, 2 mM EDTA pH 8.0, 1% Triton X-100, 10 mM sodium butyrate, 1× protease inhibitor) to 20- to 30-μL Protein G Dynabeads (Life Technologies) on a rotator at 4 °C for 2 h. The antibodies used were as follows: 0.5 μg of H3K4me3 (Abcam; ab8580), 2 μg of H3K9me3 (Abcam; ab8898), 1 μg of H3K27me3 (Millipore; 07–449), 2 μg of H3K36me3 (Millipore; ABE435), 0.6 μg of H3K27ac (Abcam; ab4729), 1.2 μg of H3K122ac (Abcam; ab33309), 2 μg of RNA Polymerase 2 (Abcam; ab817). One microgram of purified mononucleosomes (for histone modifications) or 20 μg of Mnase-digested chromatin (for Polymerase 2 ChIP) were diluted in 400 μL of ChIP dilution buffer, added to the bead–antibody complex and incubated on a rotator at 4 °C overnight. Samples were washed in LiCl was buffer (500 mM LiCl, 50 mM Tris pH 8.0, 1 mM EDTA pH 8.0, 1% Nonidet P-40, 0.7% sodium deoxycholate) five times and eluted in elution buffer (1% SDS, 100 mM NaHCO3). The samples were decross-linked at 65 °C for 5–6 h in the presence of Proteinase K (Ambion) and purified with Qiagen PCR purification columns. ChIP DNA yield was quantified by using QuBit Fluorometric Quantitation (Life Technologies). One to two nanograms of DNA was used to prepare sequencing libraries with the KAPA Hyper Prep Kit (KAPA Biosystems) according to the manufacturer's instructions. For barcoding, 2 μL of adaptors from a TruSeq ChIP Sample Preparation Kit (Illumina) were used. Libraries were amplified with 13 PCR cycles and sequenced on a HiSeq2500 instrument (Illumina) in rapid run mode with 2 × 33 bp paired-end reads. For target enrichment of HBV DNA, custom-designed xGen Lockdown probes (Integrated DNA Technology) of 60 bp each tiling the entire HBV genome with twofold coverage were used. For the target enrichment reaction, equal amounts of barcoded sequencing libraries were pooled (500–1,000 ng of DNA total amount) and enriched for HBV DNA according to the manufacturer's xGen rapid capture protocol v2 with slight modifications: Hybridization was performed at 50 °C, and 50 μL of Dynabeads MyOne Streptavidin C1 (Life Technologies) were used per capture reaction. HBV DNA-enriched libraries were subsequently amplified with eight PCR cycles (KAPA HIFI HotStart ReadyMix), sequenced on a MiSeq instrument (Illumina) with 2 × 33 bp paired-end reads and analyzed as described in SI Materials and Methods. Raw and processed sequencing data were deposited in Gene Expression Omnibus under accession no. GSE68402. Additional details of cell culture, HBV infection, and additional techniques that were used to detect HBV DNA, RNA and proteins by qPCR, immunofluorescence, and ELISA are described in SI Materials and Methods (also see Tables S1 and S2 and Dataset S1).
Table S1.
Primers used throughout this study
| Application and amplicon | Sequence (5′→3′) |
| RNA (cDNA) | |
| GAPDH_RT_For | CTGCACCACCAACTGCTTAG |
| GAPDH_RT_Rev | ACAGTCTTCTGGGTGGCAGT |
| HBV_RT_For | GTGCACTTCGCTTCACCTCT |
| HBV_RT_Rev | TTGACATTGCTGAGAGTCCAA |
| IFIT1_RT_For | ATCTCAGAGGAGCCTGGCTAA |
| IFIT1_RT_Rev | GCTCCAGACTATCCTTGACCTG |
| DNA | |
| cccDNA_For | CGTCTGTGCCTTCTCATCTGC |
| cccDNA_Rev | GCACAGCTTGGAGGCTTGAA |
| totalHBVDNA_For | ACTCACCAACCTCCTGTCCT |
| totalHBVDNA_Rev | GACAAACGGGCAACATACCT |
| mitochondrial_For | CCGTCTGAACTATCCTGCCC |
| mitochondrial_Rev | GCCGTAGTCGGTGTACTCGT |
| ChIP, human | |
| actin_TSS_For | TAGAAGTCGCAGGACCACACT |
| actin_TSS_Rev | TGGGTAGGTTTGTAGCCTTCAT |
| Nanog_TSS_For | TGGGTTTGTCTTCAGGTTCTG |
| Nanog_TSS_Rev | CTACTGACCCACCCTTGTGAA |
| GAPDH_TSS_For | CACTAGGCGCTCACTGTTCTC |
| GAPDH_TSS_Rev | GTTGACTCCGACCTTCACCTT |
| GAPDH_3′_For | GACCTGCCGTCTAGAAAAACC |
| GAPDH_3′_Rev | ACCACCTGGTGCTCAGTGTAG |
| ChIP, HepG2 and PHH | |
| HBV_1_For | TTAACAGGCCTATTGATTGGAAA |
| HBV_1_Rev | TCAACGCAGGATAACCACATT |
| HBV_2_For | AGCAGGCTTTCACTTTCTCG |
| HBV_2_Rev | AACGGGGTAAAGGTTCAGGTA |
| HBV_3_For | GCATGGACATCGACCCTTA |
| HBV_3_Rev | GGAAAGAAGTCAGAAGGCAAAA |
| HBV_4_For | AACACATAGCGCCTCATTTTG |
| HBV_4_Rev | AGGATTGCTGGTGGAAAGATT |
| ChIP, HBV+ liver | |
| HBV_1_liver_For | GGAAAGTATGTCAAAGGATTGTGG |
| HBV_1_liver_Rev | GCAGGATAGCCACACTGTGTAA |
| HBV_2_liver_For | TCACTTTCTCGCCAACTTACAA |
| HBV_2_liver_Rev | CAACGGGGTAAAGGTTCAGAT |
| HBV_3_liver_For | CATGGACATTGACCCGTATAAA |
| HBV_3_liver_Rev | GGAAAGAAGTCAGAAGGCAAAA |
| HBV_4_liver_For | TGGGAACAAGAGCTACAGCAT |
| HBV_4_liver_Rev | TGGGAACAGAAAGATTTGTCC |
Table S2.
Sequencing statistics
| Platform_source_sample | Total paired reads | Align to hg19 | Align to HBV |
| HiSeq_HepG2_Nuc | 27,115,268 | 18,038,576 | 22 |
| HiSeq_HepG2_H3K4me3 | 32,964,608 | 21,656,485 | 382 |
| HiSeq_HepG2_Pol2 | 19,986,628 | 14,108,016 | 212 |
| HiSeq_HepG2_H3K27ac | 25,904,352 | 15,491,756 | 330 |
| MiSeq_HepG2_Nuc | 351,041 | 239,278 | 1,303 |
| MiSeq_HepG2_H3K4me3 | 377,867 | 203,345 | 34,300 |
| MiSeq_HepG2_H3K27ac | 317,106 | 151,765 | 26,878 |
| MiSeq_HepG2_H3K36me3 | 319,947 | 187,676 | 159 |
| MiSeq_HepG2_H3K122ac | 288,700 | 123,689 | 9,818 |
| MiSeq_HepG2_H3K9me3 | 304,878 | 173,216 | 73 |
| MiSeq_HepG2_H3K27me3 | 332,437 | 222,860 | 24 |
| MiSeq_HepG2_Pol2 | 262,229 | 157,592 | 22,492 |
| MiSeq_PHH_Nuc | 766,274 | 433,188 | 32,293 |
| MiSeq_PHH_H3K4me3 | 1,756,738 | 384,035 | 686,929 |
| MiSeq_PHH_H3K27ac | 1,963,980 | 468,012 | 745,339 |
| MiSeq_PHH_H3K36me3 | 907,288 | 458,702 | 8,801 |
| MiSeq_PHH_H3K122ac | 1,193,158 | 444,859 | 202,305 |
| MiSeq_PHH_H3K27me3 | 957,448 | 528,078 | 6,471 |
| MiSeq_PHH_Pol2 | 656,525 | 398,908 | 57,379 |
| MiSeq_Liver_Nuc | 567,496 | 336,414 | 4,629 |
| MiSeq_Liver_H3K4me3 | 1,217,139 | 240,467 | 440,262 |
| MiSeq_Liver_H3K27ac | 607,543 | 224,061 | 121,036 |
| MiSeq_Liver_H3K36me3 | 580,042 | 293,675 | 1,083 |
| MiSeq_Liver_H3K122ac | 474,828 | 184,868 | 52,746 |
| MiSeq_Liver_H3K27me3 | 380,035 | 224,116 | 274 |
| MiSeq_Liver_Pol2 | 486,928 | 259,406 | 23,175 |
DNA libraries were sequenced on a HiSeq2500 instrument (HiSeq_X_X) without or MiSeq instrument (MiSeq_X_X) with prior enrichment of HBV DNA by target enrichment. Shown is the total number of paired sequencing reads and the number of paired reads aligning concordantly to the human genome (hg19) or hepatitis B virus genome (HBV).
SI Materials and Methods
HepG2 Cell Culture and Infection.
HepG2 cells (obtained from ATCC) were cultured in DMEM/F12, GlutaMAX (Life Technologies), 10% (vol/vol) FBS (Gibco), and 1× Penicillin/Streptomycin (Gibco) at 37 °C and 5% CO2. For generating the stable HepG2-NTCP1 cell line, human SLC10A (NTCP1) cDNA (Origene) was cloned into pLenti6/V5-D-TOPO (Invitrogen), and lentiviral particles were produced with the virapower lentiviral expression system (Invitrogen) by following manufacturer’s instructions. HepG2 cells were transduced and selected in the presence of 20 μg/mL Blasticidin (Gibco). Several clones were evaluated in HBV infection assays, and the clone with the highest infection rate (based on eAg secretion at 7 dpi) was used throughout this study. For HBV infection, HepG2-NTCP1 cells were seeded at 90% confluence on collagen I-coated 96-well plates (Life Technologies), six-well plates (Life Technologies) or T-75 flasks (BD Biosciences) in hepatocyte maintenance media (HMM) comprising of Williams’ E media (Gibco) with Primary Hepatocyte Maintenance Supplements (Life Technologies), 18 μg/mL Hydrocortisone (Sigma), and 2% (vol/vol) DMSO (Sigma). Cells were infected with 1,000 genome equivalents per cells (GE) HBV in HMM containing 4% (wt/vol) PEG 8000 (Sigma) 48 h after seeding. After 16 h, virus containing media was removed, the cells washed four times, and further incubated in HMM. Media was changed every other day. In some experiments, cells were infected and cultured in the presence of 100 nM Entecavir.
Primary Human Hepatocyte Culture and Infection.
Primary human hepatocytes (PHH) were obtained from Gibco. PHH were thawed and plated on collagen I-coated 96-well plates (Life Technologies) or T-75 flasks (BD Biosciences) as recommended by Life Technologies. Briefly, cells were thawed in a water bath at 37 °C and spun at 100 × g for 10 min in Cryopreserved Hepatocytes Recovery Medium (AP Sciences). Cells were resuspended and plated in Williams’ Medium E media supplemented with Plating Supplement Pack (Life Technologies) at 8 × 104 or 1.5 × 107 cells in 96-well format or T-75 flask, respectively. PHH were infected with 1000GE HBV 5 h after seeding in HMM in the presence of 4% (wt/vol) PEG 8000 (Sigma). After 16 h, virus containing media was removed, the cells washed four times, and further incubated in HMM. Media was changed every other day. PHH were treated with 1000 IU/mL human recombinant IFN-α (Gibco), C646 (Sigma), or vehicle (DMSO, Sigma) as indicated.
Liver Tissue and Serum.
Nonmalignant, HBV-positive frozen liver tissue (pathology status = normal) and matching serum was obtained from Analytical Biological Services (case no. 7213) and originated from a 53-y-old female patient.
HBV Viral Particle Preparation.
HBV viral particles were concentrated from cell culture supernatant of a HepG2 cell line with a stably integrated 1.3mer copy of the HBV ayw strain (GenBank accession no. V01460). HBV viral particles were precipitated from cell culture supernatant with PEG-it (System Biosciences) following manufacturer’s instructions. Precipitated viral particles were resuspended in PBS with 20% (vol/vol) glycerol and stored at −80 °C, or further purified by ultracentrifugation in a sucrose step gradient with 2 mL of 70% (wt/vol) sucrose in PBS bottom fraction overlaid with 3 mL of 20% (wt/vol) sucrose in PBS and 8 mL of viral particle solution. Samples were centrifuged at 4 °C and 38.000 rpm for 3 h in SW41Ti rotor (Beckman Coulter Inc.). Viral particles were recovered from the interface between 20% and 70% sucrose layers, aliquoted, and stored at −80 °C. Viral titers were determined by qPCR relative to a HBV DNA plasmid standard.
ELISA.
HBV eAg ELISA was performed with HBe ELISA kit (International Immuno Diagnostics) and sAg ELISA kit (Alpha Diagnostic International) according to manufacturer’s instructions. Absorbance was measured at 450 nm with a Pherastar FS microplate reader (BMG Labtech).
Immunofluorescence.
For HBV core protein staining, cells were fixed with 4% (wt/vol) paraformaldehyde in PBS at room temperature (RT) for 15 min. After washing with PBS three times, cells were incubated with blocking buffer [3% (wt/vol) BSA, 0.5% Triton X-100, 10% (vol/vol) FBS in 1× PBS] for 30 min. The HBV core protein antibody (DAKO B0586) was diluted in incubation buffer [3% (wt/vol) BSA, 0.5% Triton X-100 in 1× PBS) 1:500 and added to cells at 4 °C overnight. On the next day, the cells were washed four times with incubation buffer and stained with DAPI (Calbiochem) and secondary antibody goat anti-rabbit IgG conjugated with Alexa Fluor 594 (Life Technologies) 1:2,000 in incubation buffer at RT in the dark for 45 min. After washing three times in PBS with 0.1% Tween-20, images were acquired with Floid Imaging Station (Life Technologies).
RNA Extraction and cDNA Synthesis.
RNA was extracted with RNAeasy Mini Kit (Qiagen) and DNase digested according to manufacturer’s instructions. For cDNA synthesis, 250–500 ng of total RNA was reverse transcribed with SuperScript VILO MasterMix (Life Technologies) at 42 °C for 1 h and the reverse transcriptase heat inactivated at 85 °C for 5 min. cDNA was diluted 1:10 in 10 mM Tris pH 8, 2 μL per reaction were used for qPCR analysis.
Northern Blot.
One microgram of RNA samples and 4 μL of Millenium RNA ladder (Ambion) was separated by denaturizing 1.2% (wt/vol) agarose gel electrophoresis with reagents from the Northern Max Kit (Ambion) and transferred to HyBond XL nylon membranes (Amersham) by using the TurboBlotter system (GE Healthcare). A DIG-labeled (-) strand HBV RNA probe was transcribed from ScaI-linearized pGEM3Z-HBV plasmid with DIG Northern Starter Kit (Roche) according to manufacturer’s instructions. Hybridization at 68 °C, washes, and detection with CDP-star (Roche) was carried out according to manufacturer’s instructions. Images were acquired with the Molecular Imager Gel Doc XR+ System (Bio-Rad).
HIRT Extraction and Southern Blot.
Protein-free HBV DNA from at least 2 × 106 HepG2-NTCP1 cells (two wells of a six-well plate) was prepared by HIRT extraction at 7 dpi exactly as described in detail by Cai et al (60). For Southern blot analysis, 0.5 μL of Quick-Load 1-kb DNA ladder (New England Biolabs) and 15 μg (Fig. S1A) or 25 μg (Fig. 1D) of HIRT-extracted DNA were loaded per lane and separated by electrophoresis in a 1.2% (wt/vol) agarose gel in 1× Tris-Acetate-EDTA buffer. After electrophoresis the DNA was depurinated, denatured, and neutralized exactly as described by Cai et al. (60) and transferred onto Hybond XL membrane (Amersham) by using the TurboBlotter system (GE Healthcare). (-) strand HBV DNA was detected with a DIG-labeled (+) strand HBV RNA probe transcribed from a ScaI-linearized pGEM3Z-HBV plasmid with DIG Northern Starter Kit (Roche) according to manufacturer’s instructions. Hybridization at 50 °C, washes, and detection with CDP-star (Roche) were carried out according manufacturer’s instructions. Images were acquired with the Molecular Imager Gel Doc XR+ System (Bio-Rad).
DNA Extraction and Plasmid-Safe DNase Digestion.
DNA was extracted with DNeasy Blood and Tissue Kit according to manufacturer’s instructions, except for Fig. S1B where whole-cell extracts, nuclear fraction, and cytoplasmic fraction were adjusted to 1% SDS, reverse cross-linked at 65 °C for 5 h and, subsequently, digested with 1 μL of RNase Mixture (Ambion) at 37 °C for 1 h and 40 μg of Proteinase K (Ambion) at 56 °C for 2 h. DNA was purified by Phenol/Chloroform extraction and EtOH precipitation. For cccDNA analysis, DNA was treated for 3 h at 37 °C with ±10 U plasmid-safe DNase I (Epicentre). The DNase was inactivated at 70 °C for 30 min and 2 μL (40 ng of input DNA) used for qPCR analysis.
qPCR Analysis.
For qPCR, Fast SYBR Green Master Mix (Applied Biosciences) was used on a 7900HT Fast Real-Time PCR machine (Applied Biosciences). PCR conditions were 20-s initial denaturation at 95 °C followed by 40 cycles of 95 °C denaturation for 1 s and annealing/elongation at 60 °C for 20 s. For cccDNA detection, the annealing/elongation steps was extended to 35 s. Primers used throughout this study are listed in Table S1. cccDNA copy number was quantified relative to a HBV plasmid DNA standard. The cccDNA copies per cell in Fig. 1E were estimated based on the total amount of DNA used for the each PCR (40 ng) under the assumption that one cell contains ∼7 pg of DNA (40 ng of DNA = 5,714 cells).
Bioinformatic Analysis.
Raw sequencing files were demultiplexed and converted to Fastq format by using CASAVA, version 1.8.2 (Illumina). Paired-end sequencing reads in fastq format were mapped to the HBV or human genome with Bowtie2 (61) in no-discordant and no-mixed mode. For histone modifications and Polymerase 2, the insert sizes were specified to be between 120 and 200 bp and 30 and 300 bp, respectively. Sequencing reads from HepG2-NTCP1 cells and PHH were aligned to the HBV strain ayw (GenBank V01460.1). For the HBV+ liver sample, the HBV genome reference sequence was assembled de novo from the sequencing data by using Samtools mpileup (62). Both HBV DNA sequences were shifted by −500 nt relative to the HBV sequence published in GenBank V01460.1 and are provided below. We defined 30× coverage of the HBV genome as a minimum baseline for each sample that we would like to achieve (63). Such coverage required 1,455 paired reads (∼3,200 bp HBV genome × 30 fold coverage/2 × 33 bp for each mapped pair). A custom python script was used to determine the middle point (dyad axis of nucleosome) of each mapped pair of reads by using the alignment output files (SAM format). ChIP-seq density profiles (BedGraph format) were generated by using the IGV genome browser (64, 65). The average profiles of H3K4me3 and Polymerase 2 between ±2 kb of TSS of human RefSeq genes were produced with CEAS 1.0.2 (66), a list of the 10% most expressed genes in liver tissue was obtained from ref. 32. Sequencing and alignment statistics are listed in Table S2.
Supplementary Material
Acknowledgments
We thank Amy Kistler for technical support with Illumina sequencing.
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE68402).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1518090112/-/DCSupplemental.
References
- 1.Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30(12):2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 2.Di Bisceglie AM. Hepatitis B and hepatocellular carcinoma. Hepatology. 2009;49(5, Suppl):S56–S60. doi: 10.1002/hep.22962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganem D, Varmus HE. The molecular biology of the hepatitis B viruses. Annu Rev Biochem. 1987;56:651–693. doi: 10.1146/annurev.bi.56.070187.003251. [DOI] [PubMed] [Google Scholar]
- 4.Jones SA, Hu J. Hepatitis B virus reverse transcriptase: Diverse functions as classical and emerging targets for antiviral intervention. Emerg Microbes Infect. 2013;2(9):e56. doi: 10.1038/emi.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chang J, Guo F, Zhao X, Guo J-T. Therapeutic strategies for a functional cure of chronic hepatitis B virus infection. Acta Pharm Sin B. 2014;4(4):248–257. doi: 10.1016/j.apsb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belloni L, et al. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest. 2012;122(2):529–537. doi: 10.1172/JCI58847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allweiss L, et al. Immune cell responses are not required to induce substantial hepatitis B virus antigen decline during pegylated interferon-alpha administration. J Hepatol. 2014;60(3):500–507. doi: 10.1016/j.jhep.2013.10.021. [DOI] [PubMed] [Google Scholar]
- 8.Lucifora J, et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343(6176):1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perrillo R. Benefits and risks of interferon therapy for hepatitis B. Hepatology. 2009;49(5, Suppl):S103–S111. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- 10.Bock C-T, Schranz P, Schröder CH, Zentgraf H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes. 1994;8(3):215–229. doi: 10.1007/BF01703079. [DOI] [PubMed] [Google Scholar]
- 11.Bock CT, et al. Structural organization of the hepatitis B virus minichromosome. J Mol Biol. 2001;307(1):183–196. doi: 10.1006/jmbi.2000.4481. [DOI] [PubMed] [Google Scholar]
- 12.Pollicino T, et al. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology. 2006;130(3):823–837. doi: 10.1053/j.gastro.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Rall LB, Standring DN, Laub O, Rutter WJ. Transcription of hepatitis B virus by RNA polymerase II. Mol Cell Biol. 1983;3(10):1766–1773. doi: 10.1128/mcb.3.10.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128(4):707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 15.Voss TC, Hager GL. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet. 2014;15(2):69–81. doi: 10.1038/nrg3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang CM, et al. Production of hepatitis B virus in vitro by transient expression of cloned HBV DNA in a hepatoma cell line. EMBO J. 1987;6(3):675–680. doi: 10.1002/j.1460-2075.1987.tb04807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ladner SK, et al. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother. 1997;41(8):1715–1720. doi: 10.1128/aac.41.8.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci USA. 1987;84(4):1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan H, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnakumar R, Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell. 2010;39(5):736–749. doi: 10.1016/j.molcel.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Santos-Rosa H, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419(6905):407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 23.Schneider R, et al. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6(1):73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- 24.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107(50):21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40(7):897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tropberger P, et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell. 2013;152(4):859–872. doi: 10.1016/j.cell.2013.01.032. [DOI] [PubMed] [Google Scholar]
- 27.Bannister AJ, et al. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J Biol Chem. 2005;280(18):17732–17736. doi: 10.1074/jbc.M500796200. [DOI] [PubMed] [Google Scholar]
- 28.Barski A, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129(4):823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Beisel C, Paro R. Silencing chromatin: Comparing modes and mechanisms. Nat Rev Genet. 2011;12(2):123–135. doi: 10.1038/nrg2932. [DOI] [PubMed] [Google Scholar]
- 30.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 31.Cairns BR. The logic of chromatin architecture and remodelling at promoters. Nature. 2009;461(7261):193–198. doi: 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- 32.Hart SN, et al. A comparison of whole genome gene expression profiles of HepaRG cells and HepG2 cells to primary human hepatocytes and human liver tissues. Drug Metab Dispos. 2010;38(6):988–994. doi: 10.1124/dmd.109.031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tnani M, Bayard BA. Evidence for IRF-1-dependent gene expression deficiency in interferon unresponsive HepG2 cells. Biochim Biophys Acta. 1999;1451(1):59–72. doi: 10.1016/s0167-4889(99)00089-0. [DOI] [PubMed] [Google Scholar]
- 34.Li K, Chen Z, Kato N, Gale M, Jr, Lemon SM. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-β production in hepatocytes. J Biol Chem. 2005;280(17):16739–16747. doi: 10.1074/jbc.M414139200. [DOI] [PubMed] [Google Scholar]
- 35.Uprichard SL, Wieland SF, Althage A, Chisari FV. Transcriptional and posttranscriptional control of hepatitis B virus gene expression. Proc Natl Acad Sci USA. 2003;100(3):1310–1315. doi: 10.1073/pnas.252773599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rang A, Günther S, Will H. Effect of interferon alpha on hepatitis B virus replication and gene expression in transiently transfected human hepatoma cells. J Hepatol. 1999;31(5):791–799. doi: 10.1016/s0168-8278(99)80279-7. [DOI] [PubMed] [Google Scholar]
- 37.Bowers EM, et al. Virtual ligand screening of the p300/CBP histone acetyltransferase: Identification of a selective small molecule inhibitor. Chem Biol. 2010;17(5):471–482. doi: 10.1016/j.chembiol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin Q, et al. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011;30(2):249–262. doi: 10.1038/emboj.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tropberger P, Schneider R. Scratching the (lateral) surface of chromatin regulation by histone modifications. Nat Struct Mol Biol. 2013;20(6):657–661. doi: 10.1038/nsmb.2581. [DOI] [PubMed] [Google Scholar]
- 40.Shogren-Knaak M, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–847. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 41.Vermeulen M, et al. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131(1):58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 42.Lauberth SM, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152(5):1021–1036. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stasevich TJ, et al. Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature. 2014;516(7530):272–275. doi: 10.1038/nature13714. [DOI] [PubMed] [Google Scholar]
- 44.Hainer SJ, Martens JA. Identification of histone mutants that are defective for transcription-coupled nucleosome occupancy. Mol Cell Biol. 2011;31(17):3557–3568. doi: 10.1128/MCB.05195-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smolle M, Workman JL. Transcription-associated histone modifications and cryptic transcription. Biochim Biophys Acta. 2013;1829(1):84–97. doi: 10.1016/j.bbagrm.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pedersen MT, et al. The demethylase JMJD2C localizes to H3K4me3-positive transcription start sites and is dispensable for embryonic development. Mol Cell Biol. 2014;34(6):1031–1045. doi: 10.1128/MCB.00864-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rivière L, et al. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J Hepatol. 2015:S0168-8278(15)00450-X. doi: 10.1016/j.jhep.2015.06.023. [DOI] [PubMed] [Google Scholar]
- 48.Quasdorff M, Protzer U. Control of hepatitis B virus at the level of transcription. J Viral Hepat. 2010;17(8):527–536. doi: 10.1111/j.1365-2893.2010.01315.x. [DOI] [PubMed] [Google Scholar]
- 49.Quasdorff M, et al. A concerted action of HNF4alpha and HNF1alpha links hepatitis B virus replication to hepatocyte differentiation. Cell Microbiol. 2008;10(7):1478–1490. doi: 10.1111/j.1462-5822.2008.01141.x. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Y, et al. Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PLoS One. 2014;9(10):e110442. doi: 10.1371/journal.pone.0110442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ganem D. Persistent infection of humans with hepatitis B virus: Mechanisms and consequences. Rev Infect Dis. 1982;4(5):1026–1047. doi: 10.1093/clinids/4.5.1026. [DOI] [PubMed] [Google Scholar]
- 52.McMahon BJ. The natural history of chronic hepatitis B virus infection. Hepatology. 2009;49(5, Suppl):S45–S55. doi: 10.1002/hep.22898. [DOI] [PubMed] [Google Scholar]
- 53.Su Q, et al. Circulating hepatitis B virus nucleic acids in chronic infection: Representation of differently polyadenylated viral transcripts during progression to nonreplicative stages. Clin Cancer Res. 2001;7(7):2005–2015. [PubMed] [Google Scholar]
- 54.Yang H-C, Kao J-H. Persistence of hepatitis B virus covalently closed circular DNA in hepatocytes: Molecular mechanisms and clinical significance. Emerg Microbes Infect. 2014;3(9):e64. doi: 10.1038/emi.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tur-Kaspa R, et al. Alpha interferon suppresses hepatitis B virus enhancer activity and reduces viral gene transcription. J Virol. 1990;64(4):1821–1824. doi: 10.1128/jvi.64.4.1821-1824.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shlomai A, et al. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc Natl Acad Sci USA. 2014;111(33):12193–12198. doi: 10.1073/pnas.1412631111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Caocci G, et al. Reactivation of hepatitis B virus infection following ruxolitinib treatment in a patient with myelofibrosis. Leukemia. 2014;28(1):225–227. doi: 10.1038/leu.2013.235. [DOI] [PubMed] [Google Scholar]
- 58.Tie F, et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136(18):3131–3141. doi: 10.1242/dev.037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guidotti LG, Isogawa M, Chisari FV. Host-virus interactions in hepatitis B virus infection. Curr Opin Immunol. 2015;36:61–66. doi: 10.1016/j.coi.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cai D, et al. A southern blot assay for detection of hepatitis B virus covalently closed circular DNA from cell cultures. Methods Mol Biol. 2013;1030:151–161. doi: 10.1007/978-1-62703-484-5_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, et al. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: Key considerations in genomic analyses. Nat Rev Genet. 2014;15(2):121–132. doi: 10.1038/nrg3642. [DOI] [PubMed] [Google Scholar]
- 64.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shin H, Liu T, Manrai AK, Liu XS. CEAS: Cis-regulatory element annotation system. Bioinformatics. 2009;25(19):2605–2606. doi: 10.1093/bioinformatics/btp479. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.