

Significance
Chaperones in the endoplasmic reticulum (ER) are essential for protein folding and for the maintenance of an efficient secretory pathway. These chaperones can also accompany their substrates during transit from the ER to the Golgi. The prototypical mammalian KDEL receptor (KDELR1) functions by returning chaperones and other proteins to the ER. We show that a recessive missense mutation of Kdelr1 in mice is associated with low numbers of lymphocytes in the blood (lymphopenia), reduced expression of the T-cell receptor, and compromised antiviral immunity.
Keywords: T-cell development, T-cell survival, positive selection, N-ethyl-N-nitrosourea, lymphocytes
Abstract
Endoplasmic reticulum (ER)-resident proteins are continually retrieved from the Golgi and returned to the ER by Lys-Asp-Glu-Leu (KDEL) receptors, which bind to an eponymous tetrapeptide motif at their substrate's C terminus. Mice and humans possess three paralogous KDEL receptors, but little is known about their functional redundancy, or if their mutation can be physiologically tolerated. Here, we present a recessive mouse missense allele of the prototypical mammalian KDEL receptor, KDEL ER protein retention receptor 1 (KDELR1). Kdelr1 homozygous mutants were mildly lymphopenic, as were mice with a CRISPR/Cas9-engineered frameshift allele. Lymphopenia was cell intrinsic and, in the case of T cells, was associated with reduced expression of the T-cell receptor (TCR) and increased expression of CD44, and could be partially corrected by an MHC class I-restricted TCR transgene. Antiviral immunity was also compromised, with Kdelr1 mutant mice unable to clear an otherwise self-limiting viral infection. These data reveal a nonredundant cellular function for KDELR1, upon which lymphocytes distinctly depend.
A substantial portion of newly synthesized proteins are retained in the endoplasmic reticulum (ER). A prototypical example is the chaperone BiP (encoded by HSPA5), which binds to newly synthesized Ig heavy chain (1). Following assembly of Ig heavy and light chains, the Ig complex is secreted from the cell, whereas BiP remains in the ER (2).
The retention of BiP and other ER-resident proteins is controlled by a C-terminal tetrapeptide motif: typically KDEL in mammalian cells (3) and HDEL in yeast (4). In yeast, this motif is recognized by the ERD2 protein, and ERD2 mutant strains secrete HDEL-bearing proteins that would otherwise be retained in the ER (1, 3, 5). The mammalian counterpart of ERD2, KDEL ER protein retention receptor 1 (KDELR1) (6), is normally resident in the cis-Golgi and is redistributed to the ER in the presence of large quantities of ligand (7). Once bound to ligand, KDELR1 is sorted into COPI vesicles (8) and transported back to the ER where a pH imbalance is predicted to cause ligand release (9).
Although KDELR1 has been well studied in vitro, almost nothing is known about its physiological role. BiP and other KDELR1 ligands are expressed in many cells, yet the proteins to which they bind differ in each cell type and may be differentially sensitive to KDELR1 activity. One such protein is T-cell receptor α (TCRα), which has been shown to interact with BiP when expressed in fibroblasts (10). TCRα is one of seven subunits of the TCR complex, along with TCRβ, CD3ε, γ, δ, and ζ (2), which collectively deliver vital signals for the development, survival and function of T cells. Before expression on the T-cell surface, the TCR complex must be assembled in the ER (11). Transfection of cell lines with only a single TCR subunit, such as TCRα, is insufficient for cell surface expression, and the isolated chain is rapidly degraded through an ER-associated degradation pathway (4, 12). Before this, a portion of the unassembled TCRα chains pass into the Golgi, from which they are retrieved by a KDEL-dependent pathway (10). Saturation of KDEL receptors with a cotransfected ligand allows TCRα chains to escape ER retention and reach the cell surface, thus avoiding ER-associated degradation (10).
Whether this or other KDELR1-dependent pathways are of any direct physiological relevance has not been examined. Here, we identify a chemically induced mouse germline Kdelr1 mutation and investigate its physiological consequences.
Results
Identification of a Kdelr1 Mutation.
During a large-scale N-ethyl-N-nitrosourea mouse mutagenesis operation (13), we observed several third-generation progeny with increased frequencies of CD8+CD44hi and CD4+CD44hi T cells (Fig. 1A). The frequency of CD44hi T cells in these mice increased with age and was most pronounced in the CD8+ compartment (Fig. 1B). This phenotype, named daniel gray, was also accompanied by hypopigmentation, although subsequent experiments suggested that the immune and pigmentation phenotypes had distinct genetic origins; the pigmentation phenotype will therefore not be addressed here.
Fig. 1.

An inherited phenotype associated with expansion of CD44hi T cells. (A) Flow cytometry plots of CD44 expression on CD4+ and CD8+ blood lymphocytes (Left) and frequencies of CD44hi cells in either population (Right). P values were determined by unpaired t test. (B) Frequencies of CD44hi cells among CD4+ and CD8+ populations as a function of age. Each symbol represents an individual mouse. Data are representative of 1 (B) or more than 10 (A) experiments (error bars represent SEM).
To isolate the genetic cause of the daniel gray T-cell phenotype, we performed genome-wide linkage analysis. A single linkage peak was observed on chromosome 7 [logarithm of odds score (LOD) = 5.42] between D7Mit76 and D7Mit317 (Fig. 2A), isolating the mutation to a ∼54-Mb critical region containing 791 protein-coding genes. To reduce this interval, F2 mice with a recombination event between D7Mit76 and D7Mit317 were genotyped at additional markers, reducing the mutation-containing interval to between D7Mit80 and D7Mit82 (containing 231 protein-coding genes and 13 miRNA genes) (Fig. 2B).
Fig. 2.
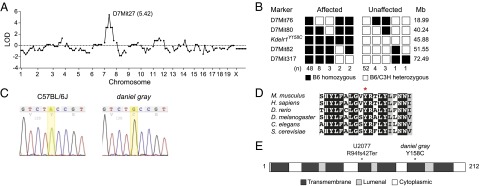
Identification of a missense mutation in Kdelr1. Chromosomal mapping (A) and fine mapping (B) of the daniel gray phenotype to mouse chromosome 7. Marker coordinates are based on the GRCm38 assembly. LOD, logarithm of odds score. (C) DNA sequence chromatogram of a mutation in Kdelr1. (D) Partial Clustal Omega alignment of mouse KDELR1 and its orthologs in human and model organisms. Y158 is indicated with an asterisk. Dark shading indicates full conservation, gray indicates partial structural conservation, and unshaded residues are not conserved. (E) Schematic of the 212-aa KDELR1 protein based on Uniprot annotations. Locations of the daniel gray Y158C variant and U2077 frameshift variant are indicated.
To identify the causative mutation within the fine-mapped interval, DNA from a single daniel gray male was subjected to exome sequencing. Two homozygous mutations were identified within the critical region, both of which were predicted to alter protein-coding sense. The first was in Kdelr1, whereas the second appeared to be related to the pigmentation phenotype. The Kdelr1 mutation (Kdelr1dgy) was characterized by an A-to-G transition at chr7:45881621 (GRCm38.p4), and was predicted to create a tyrosine-to-cysteine missense substitution at codon 158 (Y158C) (Fig. 2C). Of the three Kdelr1 coding splice variants annotated in GRCm38.p4, Y158 is present in all but one noncanonical variant (ENSMUST00000107719). Y158 is highly conserved throughout evolution (Fig. 2D), and the Y158C mutation was predicted to be “probably damaging” by the PolyPhen-2 HVAR algorithm (score of 0.996). KDELR1 is an integral membrane protein with seven transmembrane domains (Fig. 2E), and mutagenesis experiments have established that the sixth transmembrane domain of KDELR1, in which Y158 lies, is critical for KDEL recognition and ligand-induced redistribution to the ER (14). Expression of wild-type and Y158C mutant KDELR1 proteins was equivalent in transfected cell lines (Fig. 3 A and B), suggesting that the Y158C mutation alters the function of KDELR1, rather than its stability. Kdelr1 and its two paralogs (Kdelr2 and Kdelr3) were expressed in wide range of tissues, with no indication that Kdelr1 was preferentially expressed in lymphocytes (Fig. 3C).
Fig. 3.

Protein expression level and tissue expression pattern. (A) Intact expression of a KDELR1Y158C construct. Kdelr1WT or Kdelr1Y158C plasmids were cotransfected with a GFP-expressing control plasmid into mouse Neuro2A cells, and 24 h posttransfection lysates subjected to Western blotting with anti-FLAG, anti-GFP, and anti-GAPDH antibodies. Predicted molecular weight of full-length KDELR1 is 24.56 kDa (plus FLAG tag). The lower molecular-weight band (asterisk, ∼20 kDa) is suspected to represent KDELR1 following removal of an N-terminal signal peptide [28 aa, as predicted by SignalP 4.1 (B)]. Data are from a single experiment. (C) Relative expression of Kdelr1 and its paralogs, Kdelr2 and Kdelr3, in selected tissues in the mouse. Transcript data were obtained from ref. 35, with probe sets listed at the top of each column.
To confirm that the Y158C Kdelr1 variant was responsible for T lymphopenia, we used CRISPR/Cas9 mutagenesis to create a null allele of Kdelr1. We recovered a single founder (U2077) homozygous for a 4-bp deletion predicted to create a frameshift mutation (R94fs42Ter) (Fig. 4A). This mouse showed a similar degree of T lymphopenia and elevated CD44 expression as Kdelr1dgy/dgy homozygotes (Fig. 4B), suggesting that Y158C was also a null allele. Wild-type Kdelr1 was also introduced into the daniel gray germline via transgenesis. Compared with nontransgenic littermates, daniel gray mice carrying the wild-type Kdelr1 transgene had higher frequencies of CD4+ lymphocytes, although they did not show correction of the CD44hi phenotype (Fig. 4C), possibly due to partial silencing or suboptimal expression from a nonnative promoter. This combination of partial phenotypic correction with a wild-type transgene and a complete phenocopy with CRISPR/Cas9 mutagenesis supports Kdelr1dgy as the variant responsible for T lymphopenia in daniel gray mice.
Fig. 4.

Kdelr1 mutation is causative for daniel gray phenotype. (A) Schematic of the Cas9/sgRNA targeting site at the Kdelr1 locus. Boxes indicate exons, with dark shading indicating protein-coding sequence. The sgRNA targeting sequence is underlined, and the protospacer-adjacent motif (PAM) is labeled in red. (B) Frequencies and CD44 mean fluorescence intensity (MFI) of CD4+ and CD8+ blood lymphocytes, including those from a CRISPR/Cas9 engineered mutant of Kdelr1 (U2077). CRISPR WT mice represent nonmutant littermates of U2077. (C) Frequencies and CD44 MFI of CD4+ and CD8+ blood lymphocytes, including those from daniel gray mice with a wild-type Kdelr1 transgene [Tg(Kdelr1)]. P values were calculated by unpaired t test. Each symbol in B and C represents an individual mouse (error bars represent SEM).
Impaired T-Cell Development and Survival in Kdelr1 Mutants.
Thymic T-cell development was moderately impaired in Kdelr1dgy/dgy mutants from the CD4+CD8+ stage onward, culminating in a 75–85% reduction of single-positive CD8 and CD4 cells (Fig. 5A). CD4 and CD8 numbers were similarly low in the spleen, with a 75% reduction compared with heterozygous littermates (Fig. 5B). Splenic B-cell numbers were also reduced by ∼50% in both transitional (T1, T2) and follicular compartments, with marginal zone B-cell numbers mildly increased (Fig. 5C). Natural killer (NK) T cells were almost absent from the spleens of Kdelr1 homozygous mutants, whereas NK cell numbers were equivalent to those in heterozygotes (Fig. 5D).
Fig. 5.

Compromised lymphocyte development. Frequencies and absolute numbers of T, B, and NK subset were measured in thymus (A) and spleen (B–D) by flow cytometry. Thymic subsets were gated as follows: DN (CD4−CD8α−), DP (CD4+CD8α+), CD4SP (CD4+CD8α−), and CD8SP (CD4−CD8α+). Splenic subsets were as follows: T1 (B220+CD93+CD23−), T2 (B220+CD93+CD23+), Fo (follicular; B220+CD23+CD21/35int), MZ (marginal zone; B220+CD23−CD21/35hi), NK (NK1.1+CD3ε−), and NKT (NK1.1+CD3ε+). Asterisks represent P values less than 0.05, as determined by unpaired t test. Data are representative of two experiments, with three mice per genotype (error bars represent SEM).
To determine the developmental origin of the lymphocyte deficiency in Kdelr1 mutant mice, allelically marked mixtures of wild-type (CD45.1+) and heterozygous or homozygous mutant (CD45.2+) bone marrow were injected into irradiated Rag1 mutant recipients. Mutant precursor cells were outcompeted by wild type in all major T-cell populations, including αβ, γδ, and NKT cell subsets (Fig. 6A). Mutant B-cell precursors were at less of a disadvantage, apparent only in splenic and mature recirculating bone marrow subsets (Fig. 6A). CD44 expression was elevated on Kdelr1 mutant CD8+ and CD4+ T cells, indicating that this phenotype was also cell intrinsic (Fig. 6B). Similarly, surface TCR expression (as measured by TCRβ and CD3ε expression) was lower on Kdelr1 mutant cells. However, surface expression of IgM was not reduced on mutant CD19+ B cells (Fig. 6B). These findings collectively indicated that antigen receptor-bearing Kdelr1 mutant lymphocytes had an intrinsic developmental defect, associated with increased expression of CD44 on T cells. They also excluded the possibility that increased expression of CD44 and reduced surface TCR was a consequence of lymphopenia, or was due to a dysregulation of the ER chaperone-intensive assembly of MHC molecules.
Fig. 6.

Cell-intrinsic defects in lymphocyte development, surface marker expression, survival, and proliferation. (A) Lethally irradiated Rag1 mutant recipients (CD45.2+) were transplanted with an equal mixture of wild-type (CD45.1+) and heterozygous or homozygous Kdelr1 mutant (CD45.2+) bone marrow. Wild-type donor chimerism (CD45.1+) was measured 8 wk later in the spleen, bone marrow, and thymus. Bone marrow subsets were gated as follows: preproB (B220+IgM−), immature (B220intIgM+), mature (B220hiIgM+). Thymic subsets: ETP (Lin−CD44+CD25−CD117+), DN2 (Lin−CD44+CD25+), DN3 (Lin−CD44−CD25+), DN4 (Lin−CD44−CD25−), DP (CD4+CD8α+), CD4SP (CD4+CD8α−), and CD8SP (CD4−CD8α+). Lineage markers were CD11b, CD3ε, B220, Ter119, Ly6G, NK1.1, and CD8α. Spleen subsets: T1, T2, Fo, and MZ B cells were gated as in Fig. 5. γδT (CD3ε+γδTCR+), NKT (NK1.1+CD3ε+), NK (NK1.1+CD3ε−), B (CD19+), eosinophils (Lin−CD11b+F4/80+SSChi), macrophages (Lin−CD11b+F4/80+SSClo), and neutrophils (Lin−CD11b+F4/80−Ly6Ghi). Lineage markers included CD19, TCRβ, NK1.1, and the viability dye 7-AAD. (B) Geometric mean surface expression of CD44, CD3ε, and TCRβ on CD4+ and CD8+ cells in mixed bone marrow chimeras. Surface IgM was measured on CD19+ cells. Histogram overlays show relative expression of CD44 on CD4+ and CD8+ cells. (C) Thymectomized mice were bled at the indicated weeks after surgery, and frequencies of CD4+, CD8+, and CD19+ cells were normalized to the average value in nonthymectomized littermates (set as 1). (D) Mice were injected with the thymidine analog EdU 4 h before organ harvest, and the percentages of EdU+ cells within various T-cell subsets was quantified. Asterisks represent P values less than 0.05 as determined by unpaired t tests (only performed at week 8 time point in C). Each symbol in D represents an individual mouse. Data are representative of one (C and D) or two (A and B) experiments, with at least four mice per genotype (error bars represent SEM).
Peripheral T-cell lymphopenia in Kdelr1 mutant mice may have been the consequence of a partial block in T-cell differentiation, reduced T-cell survival, or a combination of both. To study T-cell numbers in the absence of new thymic emigrants, homozygous and heterozygous mutant mice were thymectomized (Fig. 6C). Frequencies of CD4+ cells in thymectomized Kdelr1dgy/dgy animals were reduced to a greater extent than they were in thymectomized Kdelr1+/dgy littermates, and remained that way until 8 wk postthymectomy. Despite the severe reduction in CD8+ numbers in Kdelr1dgy/dgy mutants, CD8+ frequencies were depleted to the same extent as thymectomized heterozygotes (Fig. 6C), which may reflect the increased homeostatic proliferation potential of CD8+ cells relative to CD4+ cells (15). Indeed, treatment of mice with the thymidine analog 5-ethynyl-2′-deoxyuridine (EdU) revealed that proliferation of Kdelr1 mutant T cells was increased in the thymus from the DP stage onward (Fig. 6D), and in the periphery. In summary, these data showed that both thymic and peripheral T-cell survival were impaired in Kdelr1dgy/dgy mutants.
To dissect the nature of the T-cell survival defect, the Kdelr1dgy mutation was studied in the context of T-cell–specific overexpression of BCL2 (EμBCL2-25), deficiency of the BH3-only protein Bim (Bcl2l11−/−), or mutation of the death receptor Fas (Faslpr/lpr). BCL2 overexpression and Bim deficiency showed almost identical effects, leading to a relative increase in T-cell numbers, yet not correcting the relative increase of CD44 surface expression or decrease of CD3ε expression (Table 1). Mutation of Fas, but not targeted mutation of Bcl2l11 or overexpression of BCL2, restored T-cell numbers in Kdelr1 homozygous mutants to that of their heterozygous littermates (Table 1). These data suggested that T-cell death in Kdelr1dgy/dgy mutants was likely to be mediated by Fas rather than Bim and reiterated that dysregulation of CD44 and CD3ε expression was not a consequence of lymphopenia.
Table 1.
Effects of BCL2 overexpression, or Bcl2l11 or Fas mutation on T-cell development
| T-cell subset | Kdelr1 | — | EμBCL2-25+ | Bcl2l11−/− | Faslpr/lpr |
| CD3ε+ (×10e6) | +/dgy | 9.1 ± 0.91 | 15.5 ± 1.6 | 17.5 ± 1.7 | 12.2 ± 3.1 |
| dgy/dgy | 3.0 ± 0.27 | 6.8 ± 0.76 | 11.1 ± 1.7 | 10.0 ± 1.3 | |
| P | 0.005 | 0.006 | 0.04 | 0.54 | |
| CD4+ (×10e6) | +/dgy | 4.5 ± 1.3 | 7.6 ± 0.9 | 8.0 ± 1.3 | 5.9 ± 1.3 |
| dgy/dgy | 1.3 ± 0.33 | 2.7 ± 0.35 | 4.8 ± 0.88 | 6.7 ± 1.1 | |
| P | 0.08 | 0.007 | 0.11 | 0.63 | |
| CD8+ (×10e6) | +/dgy | 4.3 ± 0.87 | 6.8 ± 0.59 | 6.0 ± 0.21 | 1.4 ± 0.47 |
| dgy/dgy | 1.5 ± 0.41 | 2.5 ± 0.19 | 2.9 ± 0.35 | 1.9 ± 0.19 | |
| P | 0.04 | 0.003 | 0.0007 | 0.35 | |
| CD4+CD44MFI | +/dgy | 214 ± 54 | 156 ± 5 | 121 ± 14 | 327 ± 33 |
| dgy/dgy | 264 ± 25 | 237 ± 6 | 199 ± 23 | 388 ± 15 | |
| P | 0.44 | 0.00008 | 0.04 | 0.17 | |
| CD4+CD3εMFI | +/dgy | 2,773 ± 250 | 3,189 ± 15 | 2,916 ± 55 | 1,760 ± 73 |
| dgy/dgy | 2,219 ± 85 | 2,181 ± 63 | 2,250 ± 177 | 2,025 ± 50 | |
| P | 0.11 | 0.0003 | 0.03 | 0.03 | |
| CD8+CD44MFI | +/dgy | 91 ± 6 | 98 ± 3 | 92 ± 8 | 247 ± 13 |
| dgy/dgy | 327 ± 73 | 391 ± 39 | 299 ± 40 | 395 ± 36 | |
| P | 0.048 | 0.005 | 0.01 | 0.02 | |
| CD8+CD3εMFI | +/dgy | 1,414 ± 141 | 1,712 ± 25 | 1,614 ± 9 | 910 ± 21 |
| dgy/dgy | 999 ± 39 | 977 ± 31 | 1,052 ± 93 | 873 ± 32 | |
| P | 0.06 | 0.000003 | 0.009 | 0.38 |
Splenic T cells from mice with a T-cell–expressed EμBCL2-25 transgene, Bcl2l11 deletion, or Faslpr/lpr mutation were analyzed by flow cytometry. MFI, median fluorescence intensity. Data are from one experiment, with four mice per genotype (SEM). P values below the Bonferroni correction threshold (0.0018) are in bold.
As a final measure of T-cell function, Kdelr1dgy/dgy mutant mice and littermate controls were infected with the Armstrong strain of lymphochoriomeningitis virus (LCMV). CD8+ T-cell priming was unchanged in infected Kdelr1 mutant mice, and antigen-specific CD8+ T cells expressed intracellular cytokines in response to ex vivo LCMV peptide stimulation (Fig. 7 A and B). However, virus-specific CD8+ T cells expressed less inflammatory cytokines than their wild-type counterparts (Fig. 7C). This functional deficiency was most apparent after infection with LCMV clone 13: a variant of LCMV Armstrong capable of establishing a persistent infection, which is common in the context of reduced T-cell activity (16). At 53 d after infection, all but one Kdelr1dgy/dgy mutant (10/11) was persistently infected with LCMV, as opposed to 4 of 13 heterozygous littermates (Fig. 7D). This sensitivity to chronic infection suggested that Kdelr1 mutant T cells were prone to immunological “exhaustion” (17).
Fig. 7.
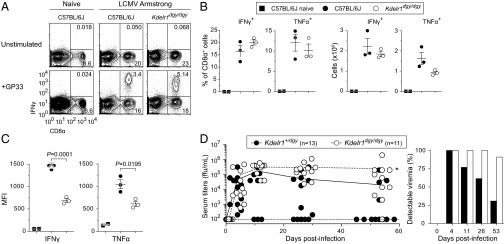
Kdelr1 is required to control chronic viral infection. Mice infected i.v. with 2 × 105 plaque-forming units (PFU) of LCMV Armstrong were killed 12 d after infection, and splenocytes were stimulated with the GP33-41 (KAVYNFATM) LCMV peptide. Frequencies (A and B), absolute numbers (B), and fluorescence intensities (C) of cells expressing intracellular cytokines were then measured by flow cytometry. MFI, median fluorescence intensity. (D) Mice were infected i.v. with 1 × 106 PFU of LCMV clone 13, and viral titers in the blood were measured at 4, 11, 26, and 53 d after infection. ffu, focus-forming units; *P = 0.005 (Mann–Whitney test at day 53). Dashed line at Y = 102 indicates the assay detection limit of 100 ffu/mL, which was used to calculate the proportion of mice with detectable viremia. Data are representative of one (A–C) or two (D) experiments, with 2–13 mice per group (error bars represent SEM).
Improvement of Positive Selection in the Presence of a Class I-Restricted TCR Transgene.
Given the importance of the TCR in T-cell homeostasis and function, we investigated the reduction of surface TCR expression on Kdelr1 mutant T cells by using a pair of TCR transgenes. These transgenes represented both low (HY) and high (OT-I) affinity MHC class I-restricted TCRs. The HY TCR, which can be detected by the T3.70 clonotypic antibody, is specific for a male-restricted minor histocompatibility antigen (18). In female mice, HY TCR-bearing thymocytes are positively selected by the MHC class I molecule H-2Db, whereas the same cells are deleted in males. The ovalbumin peptide-specific OT-I TCR, on the other hand, is positively selected in both males and females (19).
Unlike nontransgenic homozygous Kdelr1 mutants, HY and OT-I transgenic mutants expressed CD3ε on the surface of CD8SP thymocytes at levels equivalent to their heterozygous counterparts (Fig. 8A). Positive selection was also impaired in homozygous mutant HY transgenic females, with clonotype-positive CD8+ numbers reduced to an equivalent extent as in a polyclonal repertoire (Fig. 8 B and C, 8 × 106 vs. 2 × 106, compared with Fig. 5A, 1.5 × 106 vs. 0.4 × 106). Kdelr1 mutant T3.70+CD8+ cells were also fully deleted in male HY transgenics, indicating that negative selection was not compromised by the Kdelr1 mutation (Fig. 8 B and C). Importantly, the number of Vα2+ cells selected in the presence of the OT-I transgene was not significantly reduced, unlike the >90% reduction seen in the absence of the transgene (Fig. 8 B and C).
Fig. 8.

Selective correction of T lymphopenia by a high-affinity class I-restricted TCR transgene. (A) Median fluorescence intensity of CD3ε on the surface of polyclonal (−) or clonotype-positive (OT-I Vα2+ or HY T3.70+) CD8SP thymocytes. Frequencies (B) and absolute numbers (C) of thymic subsets in male and female mice with or without the HY or OT-I TCR transgenes. Only those cells that express the clonotypic TCR (T3.70+ or Vα2+) are displayed for the HY and OT-I transgenic animals. P values were determined by unpaired t test. Each symbol in A and C represents an individual mouse. Data are from one experiment, with two to six mice per group (error bars represent SEM).
Discussion
Our data reveal a nonredundant function for mammalian KDELR1 associated with B- and T-cell lymphopenia. With respect to the T-cell lymphopenia, multiple aspects of the phenotype were consistent with a defect in TCR activity. Positive selection of T cells, for example, is critically dependent upon the TCR, as is T-cell survival in the periphery (15). Similarly, control of LCMV clone 13 infection is especially sensitive to impaired T-cell function (17).
Expression of the effector/memory T-cell marker CD44, which is increased under lymphopenic conditions (20), was also increased on Kdelr1 mutant T cells. However, lymphopenia did not appear to be the cause, implying that cell-intrinsic pathways were responsible. High CD44 expression could also be functionally relevant in the context of viral infection, given the propensity for hyperactivated T cells to experience immune exhaustion (21).
Although TCR expression was lower in Kdelr1 mutant cells compared with controls, this may be explained by the imbalance between CD44hi and CD44lo populations. CD44 expression is inversely correlated with that of the TCR, such that an increase in the proportion of CD44hi cells (as seen in daniel gray) could explain a reduction in mean TCR expression. What is not clear is whether one precedes the other: i.e., does KDELR1 directly regulate expression of CD44 and/or the TCR? Or does it act elsewhere?
In one hypothetical scenario, mutation of Kdelr1 would disrupt retention of chaperones in the ER and compromise assembly of the TCR. The observation that the OT-I transgene expression could restore surface expression of CD3ε and correct positive selection would be consistent with this, given that TCR assembly may be less demanding on chaperone trafficking in the absence of error-prone recombination. However, the HY TCR transgene could also restore CD3ε expression but did not completely rescue positive selection. One possible explanation for this transgene-specific correction of positive selection is the relative affinity of the two receptors: the OT-I TCR is known to be at the higher end of the polyclonal affinity spectrum, whereas HY TCR is at the lower end (22). With the number of TCR complexes held equal, this would permit a limited amount of surface TCR to achieve a stronger positive selection signal in the presence of OT-I.
Another untested prediction of the TCR assembly hypothesis is that KDEL-bearing proteins destined for ER retention should be secreted when KDELR1 function is impaired. Even in the absence of a Kdelr1 mutation, ER chaperones can be detected on the surface of immature thymocytes (23), presumably due to saturation of the ER retention pathway. This indicates that the retention pathway is highly burdened during thymocyte development, and it is therefore not surprising that these cells are selectively sensitive to Kdelr1 mutation. Although polyclonal TCR assembly may be one pathway affected by this mutation, it is clearly not the only one, and expression of a high-affinity TCR transgene may serve to shorten or even bypass these sensitive developmental phases.
Early B-cell development may place similar demands on the ER chaperone network. BiP itself was discovered as a heavy chain binding protein (1) and associates with the CH1 domain until replaced by correctly folded light chains (24). The fact that IgM expression is not impaired suggests that KDELR1 has different effects upon TCR versus BCR surface expression and that other KDELR1-dependent pathways may be important for promoting B-cell development.
During the preparation of this manuscript, Kamimura et al. (25) reported an independent mouse mutant of Kdelr1 (S123P) with a strikingly similar phenotype to the one presented here. Kamimura et al. attribute the T-cell phenotype to an increased integrated stress response due to reduced activation of protein phosphatase 1 (PP1), which was associated with reduced dephosphorylation of eIF2a, and a subsequent increase of stress responsive gene expression including the proapoptotic Bim (Bcl2l11) (25). It is clear from our Kdelr1dgy/dgy;Bcl2l11−/− double mutants that increased Bim was not sufficient to explain T lymphopenia, although it may still be necessary. Although impaired assembly of the TCR may be one explanation, the precise mechanism of T lymphopenia in Kdelr1 mutants remains to be determined, as do the redundant and nonredundant functions of Kdelr2 and Kdelr3.
Materials and Methods
Mice.
Animals were housed at The Scripps Research Institute or University of Texas (UT) Southwestern Medical Center, and all procedures were in accordance with guidelines of the institutional animal care and use committees. The Kdelr1daniel gray allele (C57BL/6-Kdelr1m1Btlr; Mouse Genome Informatics ID 4843278) was generated on a pure C57BL/6J background by N-ethyl-N-nitrosourea mutagenesis, as previously described (26), and maintained through heterozygote by homozygote matings. The daniel gray strain is available from the Mutant Mouse Regional Resource Center (034372-JAX). The Rag1maladaptive strain (Rag1m1Btlr; Mouse Genome Informatics ID 3851764) was developed in-house (27), whereas Tg(BCL2)25Wehi (28), Bcl2l11tm1.1Ast (29), C57BL/6.MRL-Faslpr/J (30), and C57BL/6.SJL- PtprcaPepcb/BoyJ (CD45.1) strains were obtained from The Jackson Laboratory. OT-I TCR transgenic mice were provided by C. Surh (The Scripps Research Institute). C3H/HeN females were obtained from Taconic, C57BL/6J males used for mutagenesis were obtained from The Jackson Laboratory, and all other C57BL/6J mice were obtained from The Scripps Research Institute or UT Southwestern Medical Center breeding colonies.
Linkage Analysis.
The index daniel gray mutant (C57BL/6J) was outcrossed to C3H/HeN females, and F1 daughters were backcrossed to their father. F2 mice were grouped into mutant and wild-type cohorts based upon their T-cell phenotype. Individual mice were typed at 128 microsatellite markers spaced across the genome (31), with expected and observed genotype frequencies used to calculate LODs at each position. Having established a single critical interval, F2 mice were genotyped at additional markers within the interval.
Sequencing.
Genomic DNA from a single male daniel gray homozygote was used to create a fragment library for whole-exome sequencing using oligonucleotide probes from Life Technologies’ TargetSeq Custom Enrichment Kit and modified to run on the HiSeq 2500 platform (Illumina). Paired-end 2 × 100-bp sequencing was performed using an Illumina HiSeq 2500 instrument to detect heterozygous autosomal and hemizygous X-linked mutations. Reads were demultiplexed using CASAVA according to their index sequence and lane numbers. Reads were mapped to the GRCm38 mouse reference genome sequence (C57BL/6J) using Burrows-Wheeler Aligner (BWA), version 0.6.2. Duplicate reads were removed by SAMtools and indel regions left aligned by Genome Analysis Toolkit (GATK). Coverage was calculated over targeted regions using BEDTools. Variants were called and annotated by a combination of SAMtools, SnpEff, SnpSift, and ANNOVAR. Variants present in dbSNP (build 137) were removed, as were variants observed in a total of 40 previously sequenced mice with unshared G0 sires. Synonymous mutations, and mutations not predicted to affect splicing or coding sense, were also eliminated. Coding region coverage in the daniel gray homozygote sample was calculated at 95.5% for 10× coverage and 90.3% for 20× coverage. Kdelr1 PCR amplicons from wild-type and daniel gray genomic DNA were sequenced with an ABI 3730xl capillary sequencer.
Western Blotting.
Mouse Kdelr1 was cloned into the pCMV14/3×FLAG vector to add a C-terminal 3×FLAG tag for detection by Western blotting. The Y158C daniel gray mutation was incorporated into the expression construct by site-directed mutagenesis, and all plasmids were verified by capillary sequencing. Kdelr1WT or Kdelr1Y158C were cotransfected with a GFP-expressing plasmid into mouse Neuro2A cells. Twenty-four hours posttransfection, cells were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer, and lysates were subjected to gel electrophoresis and Western blotting. Anti-FLAG M2 antibody (Sigma; F3165) and anti-GFP JL8 antibody (Clontech; 632380) were used at a dilution of 1:2,000.
CRISPR/Cas9 Mutagenesis.
DNA oligo pairs corresponding to the Kdelr1 CRISPR target (5′-GAACGACGAGGAACTCCACC-3′, selected using CRISPR Design [crispr.mit.edu]) were synthesized, annealed, and cloned into plasmid pX330 (Addgene) (32). Neuro-2A cells transfected with CRISPR target plasmids were subjected to the surveyor assay to validate CRISPR activity (33). For in vitro transcription of CRISPR small-guide RNA (sgRNA) and Cas9 mRNA, the T7 promoter was first added to template by PCR. PCR products were purified using QiaQuick Purification Kit (Qiagen) and served as the templates for in vitro transcription using T7 Quick High Yield RNA Synthesis Kit (New England Biolabs) for sgRNA, or using mMESSAGE mMACHINE T7 ULTRA kit (Life Technologies) for Cas9 mRNA. Both Cas9 mRNA and sgRNA were purified using MEGAclear kit (Life Technologies) and eluted in RNase-free water for zygote microinjection. Mice were genotyped using PCR primers (Kdelr1-F: 5′-CCTACCTGTGAGCTGAGATGCC-3′; Kdelr1-R: 5′-TCCTGTCAGATACTCTGCCTCTACC-3′) and a sequencing primer (Kdelr1-SEQ: 5′-CTGTTACCCTCTTATCAGGTGG-3′) flanking the CRISPR target site.
Transgenesis.
The mammalian expression plasmid pSI (Promega) was modified to replace the SV40 enhancer/early promoter with the promoter of the mouse Rosa26 locus (from pROSA26-promoter; Addgene) to generate the vector pRosa26-TG. The coding sequence of Kdelr1 was then cloned into pRosa26-TG to create pRosa26-Kdelr1. Purified plasmid DNA was microinjected into the male pronucleus of fertilized Kdelr1dgy/dgy oocytes. A total of 294 embryos was transferred into 19 pseudopregnant CD-1 females, which gave birth to 41 pups. Five transgenic founders were identified among weanlings by sequencing genomic DNA amplified across the daniel gray mutation site, four of which were bred to daniel gray homozygotes for complementation experiments.
Zygote Microinjection.
Female C57BL/6J or Kdelr1dgy/dgy mice were superovulated by injecting 6.5 U of pregnant mare’s serum gonadotropin (Millipore; 367222) and 6.5 U of human chorionic gonadotropin (Sigma-Aldrich; C1063) 48 h later. Superovulated females were subsequently paired overnight with C57BL/6JJcl or Kdelr1dgy/dgy males (The Jackson Laboratory). The following day, fertilized eggs were collected from female oviducts, and in vitro transcribed Cas9 mRNA (20 ng/μL) and sgRNA (10 ng/μL), or pRosa26-Kdelr1 plasmid DNA, were injected into the male pronucleus or cytoplasm of the fertilized eggs. The injected embryos were cultured in M16 medium (Sigma-Aldrich; M7292) at 37 °C and 95% air/5% CO2. For the production of mutant mice, two-cell stage embryos were transferred into the ampulla of the oviduct (10–20 embryos per oviduct) of pseudopregnant Hsd:ICR (CD-1) females (Harlan Laboratories).
Flow Cytometry.
Blood from the retroorbital plexus of isoflurane-anesthetized mice was collected in cluster tubes (Costar) containing 20 μL of 6% (wt/vol) EDTA in water. Fifty microliters of blood was subjected to two rounds of red blood cell lysis with ammonium chloride before staining. Lymphocyte suspensions from blood, bone marrow (femurs and tibias from one hind leg), spleen, and thymus were counted (Z2 Coulter Counter; Beckman Coulter) and stained with a combination of the following mouse-reactive antibodies: FITC-conjugated IgM (goat polyclonal; 1020-02; Southern Biotech); FITC-conjugated F4/80 (BM8); PE-conjugated IgD (11-26); PerCP-Cy5.5–conjugated TCRβ (H57-597); APC-conjugated CD8α (53-6.7), CD11b (M1/70), CD93 (AA4.1), IgM (II/41), Ly6G (RB6-8C5), NK1.1 (PK136), TER119 (TER-119; eBioscience); FITC-conjugated CD4 (GK1.5), CD23 (B3B4), HY-TCR (T3.70), PE-conjugated CD11b (M1/70), CD19 (1D3), CD21/35 (7G6), CD44 (IM7), γδ TCR (GL3), Vα2 TCR (B20.1), PerCP-Cy5.5–conjugated B220 (RA3-6B2), CD8α (53-6.7), CD19 (1D3), NK1.1 (PK136); CD44 (IM7; Horizon V500; BD); APC-conjugated CD3ε (145-2C11); APC-Cy7–conjugated CD45.2 (104), PE-Cy7–conjugated CD19 (6D5), CD45.1 (A20; Pacific blue), and CD16/32 (93; purified; BioLegend). 7-Aminoactinomycin D (7-AAD) was purchased from eBioscience, and EdU labeling and staining were performed according to the manufacturer’s instructions (Invitrogen) 4 h after a single i.p. injection of 100 mg/kg EdU. Samples were acquired on a FACSCalibur or LSRFortessa (BD), and data were analyzed with FlowJo software (Tree Star).
Hematopoietic Chimeras.
Rag1 mutant recipient mice were gamma-irradiated with a split dose of 11 Gy (2 × 5.5 Gy, 137Cs source). The next day, mice were injected with 2 × 106 bone marrow cells via the retroorbital plexus and were maintained on trimethoprim/sulfamethoxazole antibiotic water until killed for analysis 8 wk later.
Thymectomy.
Thymi were removed by suction from 4- to 6-wk-old mice anesthetized with ketamine (100 mg per kg body weight) and xylazine (10 mg/kg body weight), and were bled at 2, 4, and 8 wk after surgery.
Viral Infection.
Mice were infected i.v. with 1 × 106 plaque-forming units (PFU) of clone 13 LCMV. Viral stocks were prepared and viral titers determined from serial dilutions of serum by a focus-forming assay on VeroE6 cells, as previously described (16). LCMV Armstrong was administered i.v. at 2 × 105 PFU per mouse. Mice were killed 12 d later, and splenocytes stimulated for 5 h with 10−7 M LCMV GP33 (KAVYNFATM) peptide as described previously (34). Stimulated cells were then stained for CD3ε and CD8α, fixed, permeabilized, and stained with antibodies against IFN-γ (XMG1.2) or TNF-α (MP6-XT22). GP33-induced cytokine production was calculated by subtracting the frequency of cytokine-positive cells in unstimulated samples.
Acknowledgments
We thank Mercedes Gutierrez for animal care, and Eva Moresco, Peter Jurek, and Anne Murray for assistance with manuscript preparation. This work was supported by the Bill and Melinda Gates Foundation, NIH/National Institute of Allergy and Infectious Diseases Grant U19AI100627 (to B.B.), and Wellcome Trust Grant 100083/Z/12/Z (to O.M.S.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The data reported in this paper have been deposited in the Mouse Genome Informatics database (Kdelr1daniel gray [C57BL/6-Kdelr1m1Btlr]; Mouse Genome Informatics ID 4843278).
References
- 1.Haas IG, Wabl M. Immunoglobulin heavy chain binding protein. Nature. 1983;306(5941):387–389. doi: 10.1038/306387a0. [DOI] [PubMed] [Google Scholar]
- 2.Bole DG, Hendershot LM, Kearney JF. Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J Cell Biol. 1986;102(5):1558–1566. doi: 10.1083/jcb.102.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48(5):899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- 4.Pelham HR. Evidence that luminal ER proteins are sorted from secreted proteins in a post-ER compartment. EMBO J. 1988;7(4):913–918. doi: 10.1002/j.1460-2075.1988.tb02896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Semenza JC, Hardwick KG, Dean N, Pelham HR. ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell. 1990;61(7):1349–1357. doi: 10.1016/0092-8674(90)90698-e. [DOI] [PubMed] [Google Scholar]
- 6.Lewis MJ, Pelham HR. A human homologue of the yeast HDEL receptor. Nature. 1990;348(6297):162–163. doi: 10.1038/348162a0. [DOI] [PubMed] [Google Scholar]
- 7.Lewis MJ, Pelham HR. Ligand-induced redistribution of a human KDEL receptor from the Golgi complex to the endoplasmic reticulum. Cell. 1992;68(2):353–364. doi: 10.1016/0092-8674(92)90476-s. [DOI] [PubMed] [Google Scholar]
- 8.Orci L, et al. Bidirectional transport by distinct populations of COPI-coated vesicles. Cell. 1997;90(2):335–349. doi: 10.1016/s0092-8674(00)80341-4. [DOI] [PubMed] [Google Scholar]
- 9.Wilson DW, Lewis MJ, Pelham HR. pH-dependent binding of KDEL to its receptor in vitro. J Biol Chem. 1993;268(10):7465–7468. [PubMed] [Google Scholar]
- 10.Yamamoto K, et al. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 2001;20(12):3082–3091. doi: 10.1093/emboj/20.12.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klausner RD, Lippincott-Schwartz J, Bonifacino JS. The T cell antigen receptor: Insights into organelle biology. Annu Rev Cell Biol. 1990;6:403–431. doi: 10.1146/annurev.cb.06.110190.002155. [DOI] [PubMed] [Google Scholar]
- 12.Lippincott-Schwartz J, Bonifacino JS, Yuan LC, Klausner RD. Degradation from the endoplasmic reticulum: Disposing of newly synthesized proteins. Cell. 1988;54(2):209–220. doi: 10.1016/0092-8674(88)90553-3. [DOI] [PubMed] [Google Scholar]
- 13.Arnold CN, et al. ENU-induced phenovariance in mice: Inferences from 587 mutations. BMC Res Notes. 2012;5:577. doi: 10.1186/1756-0500-5-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Townsley FM, Wilson DW, Pelham HR. Mutational analysis of the human KDEL receptor: Distinct structural requirements for Golgi retention, ligand binding and retrograde transport. EMBO J. 1993;12(7):2821–2829. doi: 10.1002/j.1460-2075.1993.tb05943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29(6):848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Evans CF, Borrow P, de la Torre JC, Oldstone MB. Virus-induced immunosuppression: Kinetic analysis of the selection of a mutation associated with viral persistence. J Virol. 1994;68(11):7367–7373. doi: 10.1128/jvi.68.11.7367-7373.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 18.Kisielow P, Blüthmann H, Staerz UD, Steinmetz M, von Boehmer H. Tolerance in T-cell-receptor transgenic mice involves deletion of nonmature CD4+8+ thymocytes. Nature. 1988;333(6175):742–746. doi: 10.1038/333742a0. [DOI] [PubMed] [Google Scholar]
- 19.Hogquist KA, et al. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76(1):17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 20.Sprent J, Surh CD. Normal T cell homeostasis: The conversion of naive cells into memory-phenotype cells. Nat Immunol. 2011;12(6):478–484. doi: 10.1038/ni.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zajac AJ, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188(12):2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kieper WC, Burghardt JT, Surh CD. A role for TCR affinity in regulating naive T cell homeostasis. J Immunol. 2004;172(1):40–44. doi: 10.4049/jimmunol.172.1.40. [DOI] [PubMed] [Google Scholar]
- 23.Wiest DL, et al. Incomplete endoplasmic reticulum (ER) retention in immature thymocytes as revealed by surface expression of “ER-resident” molecular chaperones. Proc Natl Acad Sci USA. 1997;94(5):1884–1889. doi: 10.1073/pnas.94.5.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hendershot L, Bole D, Köhler G, Kearney JF. Assembly and secretion of heavy chains that do not associate posttranslationally with immunoglobulin heavy chain-binding protein. J Cell Biol. 1987;104(3):761–767. doi: 10.1083/jcb.104.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kamimura D, et al. KDEL receptor 1 regulates T-cell homeostasis via PP1 that is a key phosphatase for ISR. Nat Commun. 2015;6:7474. doi: 10.1038/ncomms8474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Georgel P, Du X, Hoebe K, Beutler B. ENU mutagenesis in mice. Methods Mol Biol. 2008;415:1–16. doi: 10.1007/978-1-59745-570-1_1. [DOI] [PubMed] [Google Scholar]
- 27.Siggs OM, et al. The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat Immunol. 2011;12(5):434–440. doi: 10.1038/ni.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strasser A, Harris AW, Cory S. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell. 1991;67(5):889–899. doi: 10.1016/0092-8674(91)90362-3. [DOI] [PubMed] [Google Scholar]
- 29.Bouillet P, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286(5445):1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356(6367):314–317. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 31.Siggs OM, Li X, Xia Y, Beutler B. ZBTB1 is a determinant of lymphoid development. J Exp Med. 2012;209(1):19–27. doi: 10.1084/jem.20112084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guschin DY, et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- 34.Krebs P, Crozat K, Popkin D, Oldstone MBA, Beutler B. Disruption of MyD88 signaling suppresses hemophagocytic lymphohistiocytosis in mice. Blood. 2011;117(24):6582–6588. doi: 10.1182/blood-2011-01-329607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu C, Macleod I, Su AI. BioGPS and MyGene.info: Organizing online, gene-centric information. Nucleic Acids Res. 2013;41(Database issue):D561–D565. doi: 10.1093/nar/gks1114. [DOI] [PMC free article] [PubMed] [Google Scholar]