

Abstract
Toxin–antitoxin (TA) systems are implicated in the downregulation of bacterial cell growth associated with stress survival and latent tuberculosis infection, yet the activities and intracellular targets of these TA toxins are largely uncharacterized. Here, we use a specialized RNA-seq approach to identify targets of a Mycobacterium tuberculosis VapC TA toxin, VapC-mt4 (also known as VapC4), which have eluded detection using conventional approaches. Distinct from the one other characterized VapC toxin in M. tuberculosis that cuts 23S rRNA at the sarcin–ricin loop, VapC-mt4 selectively targets three of the 45 M. tuberculosis tRNAs (tRNAAla2, tRNASer26 and tRNASer24) for cleavage at, or adjacent to, their anticodons, resulting in the generation of tRNA halves. While tRNA cleavage is sometimes enlisted as a bacterial host defense mechanism, VapC-mt4 instead alters specific tRNAs to inhibit translation and modulate growth. This stress-linked activity of VapC-mt4 mirrors basic features of eukaryotic tRNases that also generate tRNA halves and inhibit translation in response to stress.
Mycobacterium tuberculosis has adapted to survive a wide range of assaults—from our immune response to antimicrobial therapeutics—intended to eradicate the organism. However, we lack a full understanding of the molecular switches that enable M. tuberculosis to endure these stresses, to slow replication or to become dormant as a latent tuberculosis infection. Emerging studies on the molecular underpinnings of stress survival in Escherichia coli generally point to a major role for chromosomal toxin–antitoxin (TA) systems, which are operons comprising adjacent genes encoding two small (~10 kDa) proteins—a toxin and its cognate antitoxin that inhibits toxin activity in the TA protein–protein complex. The first described chromosomal TA system was E. coli mazEF, which was found to be induced by stress1,2. E. coli mazEF causes growth arrest and eventually cell death1,2. These studies and those on other TA systems in E. coli are consistent with a general role for TA systems in cell survival during periods of stress3. Observations in M. tuberculosis suggest that TA systems are also important for stress survival in this organism. Several TA loci in M. tuberculosis are induced during heat shock4, hypoxia5,6, DNA damage7, nutrient starvation8, macrophage infection5,9,10 and antibiotic treatment11,12. Most recently, RNA-seq analysis of M. tuberculosis cells subjected to starvation revealed that the majority of TA systems, 75%, were upregulated to some degree with 25% upregulated twofold or higher13.
The M. tuberculosis genome contains an estimated 48 members5,14 of the VapBC (virulence-associated protein) family, the highest number of VapBC TA systems among free-living bacteria. VapC toxins are characterized by the presence of a PIN (PilT amino-terminal) domain. The 48 M. tuberculosis VapC toxins all share protein sequence similarity and possess a PIN domain containing a conserved quartet of acidic residues and a fifth invariant serine or threonine residue, which are responsible for coordinating divalent cation(s) in the catalytic centre15. The presence of a PIN domain suggests a role for the VapC toxins as ribonucleases, yet the body of literature on the enzymatic activity of VapC toxins has been inconsistent and contradictory. In our opinion, the most consistent data are derived from five recent reports. First, the solitary VapC toxin in Shigella or Salmonella cleaves tRNAfMet at a single, identical site in the anticodon stem loop (ASL)16. The solitary VapC in the spirochaete Leptospira interrogans also cleaves tRNAfMet (ref. 17). Second, the VapC from M. smegmatis cleaves synthetic ssRNAs at a short consensus sequence with some dependence on secondary structure18. In vivo, this VapC cleaves mRNA transcripts to downregulate M. smegmatis glycerol uptake and metabolism18. Third, we determined that a representative M. tuberculosis VapC toxin, VapC-mt4 (Rv0595c, also known as VapC4), recognizes ACGC or AC(A/U)GC in RNA19. Fourth, another M. tuberculosis VapC toxin, VapC20 (VapC-mt20 using our convention), cleaves at the highly conserved sarcin–ricin loop of 23S rRNA in intact ribosomes only, with some sequence specificity20.
A detailed understanding of the properties of the 48 VapC paralogs is essential for the interpretation of their physiological role in M. tuberculosis and other pathogens. In this study, we identify the primary target of VapC-mt4 using a specialized RNA-seq approach. Distinct from all other VapC toxins, and TA toxins in general, VapC-mt4 arrests growth by translation inhibition resulting from selectively targeting three of the 45 tRNAs present in M. tuberculosis for cleavage at a single site in their anticodon loop. This highly selective tRNA substrate discrimination is contingent on recognition of the consensus sequence in an appropriate structural context. In agreement, VapC-mt4-tRNA-simulated docking experiments place the toxin active site in proximity to the cleavage site in the tRNA anticodon loop. Overall, these studies bring to light a common theme between the role of VapC-mt4 in this pathogen and stress responses in eukaryotic cells that also engage cleaved tRNAs in unconventional roles.
Results
RNA-seq reveals VapC-mt4 cleaves specific tRNA isoacceptors
Our earlier study of VapC-mt4 revealed that this toxin inhibits translation and cleaves RNA at a consensus sequence of ACGC or AC(U/A)GC and that the GC sequence within this motif is essential for cleavage19. However, we were unable to clearly identify a primary target for this toxin. Although our data suggested that tRNA(s) may be the primary targets for M. tuberculosis VapC-mt4, we were unable to establish this connection using a battery of conventional biochemical, genetic and molecular biological approaches19. To overcome this roadblock, we used a specialized RNA-seq method we recently developed to identify RNA targets of endoribonucleolytic toxins21. This RNA-seq method was designed to differentially detect RNA cleavage products that carry a 5′-hydroxyl (OH) or a 5′-monophosphate (P). Bacterial transcripts possess a 5′-triphosphate (mRNAs), 5′-monophosphate (rRNAs and tRNAs) or 5′-hydroxyl (noncoding RNA intermediates and products cleaved by certain endoribonucleolytic toxins such as MazF or RNases such as RNase A and T1 (ref. 22)). Another important feature of this RNA-seq method is its use of E. coli as a surrogate host, which unlike M. tuberculosis, does not contain a 5′-to-3′ exoribonuclease. Thus, the 5′ ends generated by VapC-mt4 cleavage can be readily detected because they will be stable.
Using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, McKenzie et al.23 reported that VapC toxins generate a 5′-P cleavage product. However, Winther and Gerdes suggested that the ends were instead marked with a 5′-OH because the 5′ terminus of the 3′ VapC RNA product could be phosphorylated without a prior dephosphorylation step16. Therefore, we constructed both 5′-P and 5′-OH cDNA libraries from RNA harvested from exponentially growing E. coli cells expressing VapC-mt4 from an arabinose-inducible vector. We did not identify any RNAs enriched by 10-fold or more in the 5′-P library relative to the uninduced control. In contrast, in the 5′-OH library, a cleavage product derived from three identical tRNAs—tRNAAla2, tRNAAla41 and tRNAAla58—exhibited a 171-fold increase in transcript abundance, relative to the uninduced control (Fig. 1a). For simplicity, we will refer to these three identical tRNAs as tRNAAla2 from this point forward (numbering based on the GtRNAdb Genomic tRNA Database24). The sequence immediately upstream and downstream of the 5′ end of the cleavage site, A37C38–G39C40 (where–indicates the position of cleavage) was a 100% match to the recognition sequence deduced from our published in vivo and in vitro primer extension experiments19. Generation of the putative tRNAAla2 cleavage product was specific to VapC-mt4 induction; we did not detect any other tRNA fragment that displayed more than a 10-fold increase in abundance in the comparison of 5′-OH libraries derived from VapC-mt4-induced cells relative to uninduced cells (Supplementary Fig. 1).
Figure 1. VapC-mt4 Cleaves tRNAAla2 in E. coli.

(a) Histogram representing the fold change in 5′-OH ends observed along the length of the tRNAAla2 upon comparison of cells with VapC-mt4 to those without VapC-mt4. The sequence surrounding the site of cleavage in tRNAAla2 is shown on the right; red arrow denotes position of cleavage. (b) Primer extension analysis with total E. coli RNA following the induction of VapC-mt4 for the times indicated. The RNA sequence (ACGC consensus in red) and positions of cleavage (yellow arrow) shown below the gel image. G, A, T and C lanes correspond to DNA-sequencing ladders using the same primer and a tRNAAla2 DNA template. The major primer extension band migrates between the G and Tresidues in the sequencing ladder instead of aligning exactly to the G residue. We attribute this to the repeatable aberrant migration of the sequencing ladder below this tRNA sequence. (c) Illustration of the RNA-seq cleavage site in the tRNAAla2 ASL, yellow arrow; ACGC consensus sequence in red, anticodon shaded in grey. (d,e) Cleavage assay with in vivo purified tRNAAla2 (d) or in vitro-synthesized tRNAAla2 (e) and increasing amounts of VapC-mt4 (ratios of toxin to RNA were 0:1, 1.25:1, 2.5:1 and 5:1). Control reactions on the right contained the highest concentration of VapC-mt4 preincubated with VapB antitoxin or EDTA before addition of the respective tRNAs. Reactions were incubated at 37 °C for 3 h. Sizes of full-length and cleaved tRNA products on the left. Complete gel images for (b,d,e) are shown in Supplementary Fig. 2.
Validation of VapC-mt4 specificity for E. coli tRNAAla2
We proceeded to confirm our RNA-seq result (plotted in Fig. 1a) suggesting that tRNAAla2 is a major target for cleavage by VapC-mt4 using two approaches. In the first approach, we performed primer extension on the RNA samples from E. coli cells 0, 20, 40, 60, 80 and 100 min after induction. A strong cleavage product accumulated with time at the same site in tRNAAla2 identified by RNA-seq (Fig. 1b and Supplementary Fig. 2). The validated RNA-seq cleavage site mapped to the junction of the stem and loop in the ASL (Fig. 1c).
For our second approach, we purified tRNAAla2 from total E. coli RNA using a biotinylated oligonucleotide complementary to a sequence in the ASL unique to this isoacceptor. Incubation of pure VapC-mt4 with this tRNAAla2 led to complete cleavage with increasing toxin concentration, generating products consistent with cleavage at the A37C38–G39C40 site in the ASL identified by RNA-seq and primer extension (Fig. 1d). Cleavage was abolished when VapC-mt4 was preincubated with its cognate antitoxin VapB-mt4 and when EDTA was added to the reaction to chelate Mg2 + (Fig. 1d).
Although none of the bases comprising the ACGC sequence in E. coli tRNAAla2 cleaved by VapC-mt4 are modified, modifica-tions in proximal bases may enhance cleavage (for example, there is a 7-methylguanosine at position 46 of tRNAAla2). To address this, we compared the efficiency of cleavage of tRNAAla2 produced in vivo containing modified bases (Fig. 1d) with that synthesized in vitro lacking modified bases (Fig. 1e). There was no difference in the efficiency of cleavage between the two versions of tRNAAla2, indicating that base modifications in this tRNA are not required for VapC-mt4 cleavage. Cleavage by VapC-mt4 was also specific for tRNAAla2 since non-ACGC-containing tRNAs (tRNAfMet (ref. 19)) or other ACGC-containing tRNAs (for example, tRNAAsp3) subjected to the same reaction as Fig. 1e were not cleaved. Therefore, our RNA-seq approach was a valid predictor of the VapC-mt4 RNA target and site of cleavage in E. coli cells because tRNAAla2, but no other tRNA (Supplementary Fig. 1), was cleaved at the same site both in vivo and in vitro.
VapC-mt4 cleaves three M. tuberculosis tRNAs within the ASL
Our RNA-seq analysis suggested that VapC-mt4 generally targets tRNA and not rRNA as recently reported for VapC-mt20 (ref. 20). Since VapC-mt4 cleavage of tRNAAla2 did not require base modifications (Fig. 1d,e), we used synthetic tRNAs to test toxin specificity for all M. tuberculosis tRNAs containing a consensus sequences. ACGC was the predominant in vivo cleavage site identified by our earlier in vivo primer extension experiments19 and was also represented in the highest ranking RNA-seq target, tRNAAla2. We surveyed all 45 tRNAs in M. tuberculosis (all but three—two tRNAMet and one tRNAfMet—have unique anticodons) and identified 13 distinct tRNAs with ACGC or the related ACAGC or ACUGC cleavage consensus sequences19. We synthesized each of these 13 M. tuberculosis tRNAs and subjected them to the same VapC-mt4 in vitro cleavage assay used in Fig. 1d,e. VapC-mt4 exhibited a strong preference for three of the 13 tRNAs (that is, complete cleavage of full-length tRNA), tRNAAla2 (Fig. 2a), tRNASer26 (Fig. 2b) and tRNASer24 (Fig. 2c). Preincubation of VapC-mt4 with either its cognate antitoxin VapB-mt4 or EDTA prevented tRNA cleavage. tRNAGlu33, representative of the 10 tRNAs that were either weakly cut or not cut at all, is shown in Fig. 2d. The entire set of 10 consensus site-containing tRNAs that were tested for cleavage and not classified as a VapC-mt4 target is shown in Supplementary Fig. 3). Therefore, VapC-mt4 efficiently cleaved only 3 of 13 M. tuberculosis tRNAs harbouring the cleavage consensus sequence. Although VapC-mt4 cleaved tRNAs with the same name (tRNAAla2 from E. coli and M. tuberculosis), these tRNAs are not identical (71% identical overall among their 76 nts). However, there is high identity in the most relevant portion of these two tRNAs: the anticodon loop. The first 7 nts (which includes the anticodon) of the 9 nt loop are identical.
Figure 2. VapC-mt4 targets a specific subset of tRNAs in M. tuberculosis.
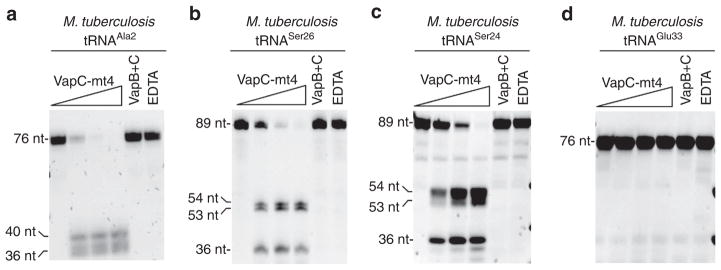
(a–c) In vitro VapC-mt4 cleavage assays showing the three tRNAs from among the 13 consensus-containing M. tuberculosis tRNAs tested that are cleaved to completion: tRNAAla2 (a), tRNASer26 (b) and tRNASer24 (c). (d) tRNAGlu33 is shown as a representative example of a tRNA not cut by VapC-mt4 even though it contains the cleavage consensus sequence. In vitro-synthesized tRNAs were incubated with increasing amounts of VapC-mt4 (ratios of toxin to RNA were 0:1, 1.25:1, 2.5:1, and 5:1). Control reactions on the right contained the highest concentration of VapC-mt4 preincubated with VapB antitoxin or EDTA before addition of the respective tRNAs. Reactions were incubated at 37 °C for 3 h. Sizes of full-length and cleaved tRNA products are indicated on the left. Note that in some cases extra bands are visible for the cleavage products because the T7 RNA polymerase used to synthesize the tRNAs frequently leads to 3′ end heterogeneity (usually ±1 nt)61,62. Complete gel images for a–c are shown in Supplementary Fig. 3. Complete gel images for d along with full gels images of all 10 tRNAs that were weakly cut or not cut at all are shown in Supplementary Fig. 4.
We then used primer extension to identify the position of VapC-mt4 cleavage in the synthetic tRNAs shown in Fig. 2. Using this approach, all three tRNA cleavage sites mapped within the anticodon loop (Fig. 3a,c,e and Supplementary Fig. 4). Finally, we confirmed that VapC-mt4 efficiently cleaves tRNAs from M. tuberculosis cells. We isolated total RNA from M. tuberculosis and incubated it with VapC-mt4 alone or VapC-mt4 pre-incubated with its cognate antitoxin VapB-mt4 as a control. We then performed northern analysis with an isoacceptor-specific oligonucleotide complementary to the ASL for tRNASer26 or tRNASer24 (Fig. 4a,b); we also performed a similar experiment for E. coli tRNAAla2 as a control (Fig. 4c). Addition of VapC-mt4 resulted in the loss of hybridization, indicating that the respective tRNAs were cleaved (Fig. 4a–c). In the case of M. tuberculosis tRNAAla2, sequence similarity among alanine isoacceptor tRNAs was too high to enable selective hybridization for only tRNAAla2. In summary, VapC-mt4 cleaves three M. tuberculosis tRNAs within their anticodons (tRNASer24 and tRNASer26; when referring to both of these tRNAs they will subsequently be called tRNASer24/6) or immediately 3′ of the anticodon (tRNAAla2). As with E. coli tRNAAla2, this cleavage appears to be efficient in the presence or absence of tRNA modifications.
Figure 3. VapC-mt4 cleaves three M. tuberculosis tRNAs near consensus sequences.

(a–f) Primer extension analysis with in vitro-synthesized M. tuberculosis tRNAAla2 (a,b), tRNASer26 (c,d) and tRNASer24 (e,f). The RNA sequence (ACGC or ACUGC consensus in red) and positions of cleavage (yellow arrow) shown below gel images in left panels. G, A, T and C lanes correspond to DNA-sequencing ladders using the same primer and matched tRNA DNA template as the corresponding primer extension. To the right of each primer extension are illustrations highlighting the cleavage site (yellow arrow) relative to the consensus sequence (red) and anticodon (grey shaded). tRNAs were incubated with increasing amounts of VapC-mt4 (ratios of toxin to RNA were 0:1, 2.5:1 and 5:1) for 3 h at 37 °C. Convention when numbering tRNA bases dictates that the anticodon is designated as numbered positions 34–36. This convention is followed in b,d,f. Note, however, that for tRNASer26 (d) and tRNASer24 (f) the actual base numbers for the anticodon are 35–37. Complete gel images are shown in Supplementary Fig. 5.
Figure 4. VapC-mt4 also cleaves modified M. tuberculosis tRNAs.
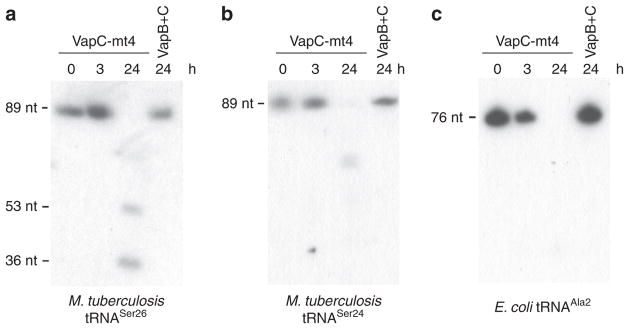
(a,b) Total RNA (2 μg) was incubated with VapC-mt4 (20 pmol) for the times indicated, and northern analysis was performed using isoacceptor-specific oligonucleotides complementary to each ASL. VapB + C samples denote preincubation of the VapC-mt4 toxin (20 pmol) with VapB-mt4 antitoxin (120 pmol) before addition of total RNA. (c) Analogous experiment for E. coli tRNAAla2 using E. coli total RNA and an isoacceptor-specific oligonucleotide complementary to the ASL. Complete gel images are shown in Supplementary Fig. 6.
VapC-mt4 cleavage requires an ACGC in the proper context
VapC-mt4 appears to require more than the consensus sequence alone since only tRNAAla2 was cleaved even though there are 12 E. coli tRNAs containing a VapC-mt4 cleavage consensus sequence. Therefore, to better characterize the properties of RNA recognition and cleavage by this toxin, we first tested if VapC-mt4 cleavage at these consensus RNA sequences required single-stranded (ss) or double-stranded (ds) RNA. As with the MazF toxin25, VapC-mt4 also requires that the cleavage consensus sequence resides within ssRNA, since an RNA fragment containing the ACGC consensus sequence could only be cleaved as a ssRNA (Fig. 5, lanes 2,4). Increasing concentrations of a complementary RNA fragment (leading to the formation of dsRNA) resulted in a gradual decrease in cleavage (Fig. 5, lanes 4–9).
Figure 5. VapC-mt4 cleavage requires a single-stranded RNA template.

A 20-nt 5′ end-labelled RNA containing an ACGC consensus sequence alone (lane 1), after incubation with VapC-mt4 (lane 2), or after incubation; with VapB-mt4 and VapC-mt4 (lane 3). This 20-nt RNA was also preincubated with increasing amounts of an RNA complement lacking an ACGC consensus (lanes 4–10) followed by incubation with VapC-mt4. The positions of the dsRNA, ssRNA and cleaved ssRNA are shown on the left. The ratio of toxin to RNA was 64.2:1 and the assay was incubated at 37 °C for 3 h. A complete gel image is shown in Supplementary Fig. 7.
We next enlisted E. coli tRNAAsp3 as a tool to study the importance of context. tRNAAsp3 has the ACGC sequence located in the identical position as in E. coli tRNAAla2, yet VapC-mt4 only cleaved tRNAAla2 (Fig. 6 compare ‘wild-type’ tRNA red sequences). Interestingly, although these two tRNAs are 66% identical overall they differ at only three positions in their entire anticodon loop, two bases in the anticodon and one base just 5′ of the anticodon (Fig. 6 compare sequences in ‘wild-type’ illustrations). To establish if the sequence of the anticodon influenced cleavage, we mutated the tRNAAla2 anticodon to a GUC (the anticodon for tRNAAsp3) and the tRNAAsp3 anticodon to a UGC (the anticodon for tRNAAla2; Fig. 6 ‘mutant’ panels). Wild-type tRNAAla2 was efficiently cleaved, but the anticodon mutant version was no longer cleaved (Fig. 6a ‘mutant’ panel). Conversely, tRNAAsp3 with a wild-type anticodon was not cleaved, while the mutated version was efficiently cleaved (Fig. 6b ‘mutant’ panel). Therefore, the ACGC sequence is not the only determinant for RNA cleavage by VapC-mt4; the consensus sequence must be single-stranded and positioned in the tRNA such that all required conditions for proper recognition and cleavage are met.
Figure 6. VapC-mt4 cleavage requires an ACGC in the proper context.

(a) Left panel, complete cleavage of wild-type tRNAAla2 by VapC-mt4; right panel, no cleavage after mutation of two bases (blue) in the tRNAAla2 anticodon to match those in tRNAAsp3. (b) Left panel, no cleavage of wild-type tRNAAsp3 by VapC-mt4; right panel, complete cleavage after mutation of two bases (blue) in the tRNAAsp3 anticodon to match those in tRNAAla2. Cleavage site (yellow arrow), consensus sequence (red), mutated bases (blue), anticodon (grey shaded), base pairing represented by black dots (●). In vitro-synthesized tRNAs were incubated with increasing amounts of VapC-mt4 (ratios of toxin to RNA were 0:1, 1.25:1, 2.5:1 and 5:1). Control reactions on the right contained the highest concentration of VapC-mt4 preincubated with VapB antitoxin or EDTA before addition of the respective tRNAs. Reactions were incubated at 37 °C for 3 h. Sizes of full-length and cleaved tRNA products on the left. The sizes of the tRNAAsp3 mutant cleavage products are predicted based on the site of cleavage in tRNAAla2. Convention when numbering tRNA bases dictates that the anticodon bases are numbered 34–36. This convention is followed in a,b. Note, however, that in the case of tRNAAsp3 (b) the actual base numbers for the anticodon are 35–37. Note that in some cases extra bands are visible for the cleavage products because the T7 RNA polymerase used to synthesize the tRNAs frequently leads to 3′ end heterogeneity (usually ±1 nt)61,62. Complete gel images are shown in Supplementary Fig. 8.
VapC-mt4 recognizes sequence and structural determinants
Having established that sequences in the anticodon immediately adjacent to a consensus sequence influence E. coli tRNA cleavage by VapC-mt4, we performed more detailed studies to further interrogate the importance of sequence and secondary structure elements within a biologically relevant subset of VapC-mt4 targets. We chose M. tuberculosis tRNASer24 from the three tRNAs cleaved by VapC-mt4 because it also contains an ACGC consensus site that bridges the junction between the anticodon stem and the anticodon loop, analogous to E. coli tRNAAla2 (Fig. 7).
Figure 7. tRNA cleavage by VapC-mt4 requires both structure and sequence determinants.

(a–e) Secondary structure outline of M. tuberculosis tRNASer24 (top) and cleavage assays comparing wild-type VapC-mt4 cleavage (a) with mutants that alter the consensus sequence but not ASL base pairing (b,c), a mutant that alters the consensus and disrupts one base pair in the stem (d), a mutant that retains the consensus sequence but removes all base pairing in the stem (e). Mutated bases (blue), cleavage site (yellow arrow), consensus sequence (red), anticodon (grey shaded), base pairing represented as black dots (●). In vitro-synthesized tRNAs were incubated with increasing amounts of VapC-mt4 (ratios of toxin to RNA were 0:1, 1.25:1, 2.5:1 and 5:1). Control reactions on the right contained the highest concentration of VapC-mt4 preincubated with VapB antitoxin or EDTA before addition of the respective tRNAs. Reactions were incubated at 37 °C for 3 h. Sizes of full-length and cleaved tRNA products on the left. Convention when numbering tRNA bases dictates that the anticodon bases are numbered 34–36. This convention is followed in a–e. Note however that in the case of tRNASer24 (a–e) the actual base numbers for the anticodon are 35–37. Complete gel images are shown in Supplementary Fig. 9.
We had previously determined that the GC residues in the ACGC recognition sequence are essential for VapC-mt4 cleavage of a synthetic 20-nt RNA template19. To test the importance of the first two AC residues, we mutated each individually, incubated with VapC-mt4, and assessed the extent of cleavage (Fig. 7a–c). First, we synthesized tRNASer24 with the first A in the ACGC sequence mutated to a C. In comparison with the complete cleavage of wild-type tRNASer24 using the highest concentration (10 pmol) of VapC-mt4, an ACGC→CCGC mutant was cleaved less efficiently (Fig. 7a,b). Second, we synthesized an ACGC→AAGC mutant and found that it was cleaved as efficiently as wild type (Fig. 7c). Third, we altered the ACGC consensus sequence to create minor or major changes in secondary or tertiary structure of the ASL. An ACGC→ACAC mutation, now lacking the G–C base pair at the stem of the ASL as well as one of the two critical consensus residues, severely impaired cleavage (Fig. 7d). Therefore, three of the four bases in the VapC-mt4 ACGC cleavage consensus sequence are important for cleavage. Cleavage was less efficient when the first A of the consensus was mutated, and mutation of the G or terminal C (previously reported by Sharp et al.19) abolished cleavage. Finally, introducing mutations in the ASL that disrupt base pairing while keeping the ACGC consensus intact, yielded a marginal level of cleavage but at a different position (Fig. 7e). We conclude that recognition of specific tRNA targets by VapC-mt4 requires proper sequence and structural context.
Simulated docking of VapC-mt4 and tRNA
Interestingly, in contrast to E. coli tRNAAla2 (whose cut site is A37C38↓G39C40), the cleavage sites for each of the three M. tuberculosis tRNAs are in proximity to, but not within their consensus sequences. To model how VapC-mt4 may potentially interact in distinct ways with either tRNAAla2 or tRNASer24/6, we performed protein–RNA docking simulations using the programme 3dRPC26 (Fig. 8). Since a structure of VapC-mt4 has not been determined, we generated a homology model of VapC-mt4 using the HHpred homology model workflow and Modeller27,28 from three published structures: M. tuberculosis VapBC5 (PDB accession code 3DBO29), Rickettsia VapBC2 (PDB accession code 3ZVK30) and FitAB (PDB accession code 2H1C31). On the basis of the published stoichiometry of VapC homologues32,33, a VapC-mt4 dimer was generated through simulated docking using GRAMM-X27,34 and then the interactions between the complex were optimized with RosettaDock35. There are also no structures for any of the three VapC-mt4 targets. But tRNAPhe and tRNALeu (PDB accession codes 4TNA, chain A and 3UZ3, chain B36,37) are close homologues of tRNAAla2 and tRNASer24/6, respectively. We used tRNALeu as a representative for both serine isotypes recognized by VapC-mt4 as they all contain an expanded variable loop. The position of a divalent cation in our model was extrapolated by superpositioning the structure of Pyrobaculum aerophilum VapC with a coordinated divalent cation with our VapC-mt4 model generated by DaliLite (PDB accession code 2FE1 (ref. 32))38. On the basis of the alignment we predict that VapC-mt4 residues Asn98 and Asn116 likely coordinate a metal ion in its active site due to the proximity of the P. aerophilum VapC equivalent.
Figure 8. VapC-mt4-tRNA docking simulations.

(a) Predicted binding of a (VapC-mt4)2 (blue and green) to tRNAAla2 (Type I tRNA homologue, tRNAPhe model shown in orange; PDB accession code 4TNA) using a protein–RNA docking simulation programme 3dRPC. The consensus sequence 5′-ACUGC-3′ is highlighted in red and boxed. The upper right boxed magnification shows a 90°rotated view highlighting the proximity of the tRNA consensus sequence to the VapC monomer not thought to be involved in activity (green). The lower right boxed magnification is a detailed view of the cleavage site. The VapC-mt4 key residues, tRNA consensus sequence and the target nucleotides are shown as sticks, with a black triangle indicating the cleavage site. (b) Predicted binding of a (VapC-mt4)2 (blue and green) to tRNASer24/Ser26 (Type II tRNA homologue, tRNALeu model shown in orange; PDB accession code 3UZ3). The expanded variable loop of tRNASer26 is depicted in orange outline and the consensus sequence 5′-ACUGC-3′ is highlighted in red. The boxed magnification is a detailed view of the cleavage site showing the VapC-mt4 key residues, the tRNALeu consensus sequence and the target nucleotides are shown in sticks, with a black triangle indicating the cleavage site. The extrapolated position of a divalent cation is shown as a purple sphere in both a,b. (c) A comparison of (VapC-mt4)2 binding, highlighting the positional shift in tRNA binding depending on the target tRNA type (grey, tRNAAla2; red, tRNASer24/Ser26). The presence of expanded variable loop in tRNASer26 (shown in red outline) influences the binding of VapC-mt4 (positions lower in anticodon loop) possibly explaining the difference observed in target cleavage sites. Convention when numbering tRNA bases dictates that the anticodon bases are numbered 34–36. This convention is followed in a–c. Note however that in the case of tRNASer26/Ser24 (b,c) the actual base numbers for the anticodon are 35–37.
While both docked models indicate that one active site from each VapC-mt4 dimer is positioned adjacent to the experimentally determined tRNA cleavage site located in the anticodon loop, the overall (VapC-mt4)2 orientations differ depending on the tRNA it recognizes. In the VapC-mt4 dimer-tRNAPhe/Ala2 model, one monomer of VapC-mt4 is predicted to interact with the base paired anticodon stem of the tRNA with potential additional contacts with the D loop (Fig. 8a). The other monomer of VapC and its active site is modelled adjacent to the predicted cleavage site between residues 36 and 37 (Fig. 8a, right panel). However in the VapC-mt4 dimer-tRNALeu/Ser26 model, the large expanded variable loop likely causes the protein/toxin to recognize the anticodon stem closer toward the anticodon (Fig. 8b). Large expanded variable loops are present in both tRNALeu and tRNASer; therefore, we predict this influences the location of the cleavage site. Also in this case, the active site in one VapC monomer is positioned adjacent to the tRNA backbone near cleavage site residue 36 (Fig. 8b, right panel). Interestingly, the tRNAAla model shows significant interactions with the 5′-ACUGC-3′ consensus sequence in contrast to the tRNASer model, which shows minimal interactions (Fig. 8c).
Discussion
VapBC modules are among the most prevalent TA systems in the genomes of a variety of pathogens, comprising >40% of the ~700 TA modules identified in 126 complete genomes of free-living bacteria39. Therefore, when organisms carry multiple members of a TA system family, VapBC systems are inexplicably the most abundant. The multiple iterations of VapBC TA systems (though none are identical) are associated with increased virulence in the pathogens that contain them, accounting for the origin of the name Vap, for virulence-associated protein40. A detailed understanding of the properties of M. tuberculosis VapC paralogs is essential for interpretation of their physiological role in M. tuberculosis and other pathogens. We studied a representative family member, M. tuberculosis VapC-mt4 (aka VapC4), to obtain a better understanding of the RNA targets and molecular mechanism of VapC toxin recognition and cleavage. In contrast to VapC-mt20, which cleaves 23S rRNA at the sarcin–ricin loop20, VapC-mt4 specifically targeted just three of the 45 total M. tuberculosis tRNA isotypes—tRNAAla2, tRNASer26 and tRNASer24. All three of these tRNAs contain one of the cleavage consensus sequences for this toxin identified in our earlier report, namely ACGC or ACUGC. Two tRNAs contained the ACUGC recognition sequence, which was the strongest cleavage site among 12 sites in the ~3.5-kb bacteriophage MS2 RNA template that were cut by VapC-mt4 (ref. 19). Thus, cleavage of tRNA involves sequence recognition by VapC-mt4. This contrasts with 23S rRNA cleavage by VapC-mt20, which appears to be more dependent on recognition of the stem loop structure of the sarcin–ricin loop20. In fact, VapC-mt4 differs from all other TA system toxins, the majority of which (1) predominantly cleave mRNA independent of the ribosome and at all cleavage consensus sequences (MazF family) or (2) cleave mRNA in a ribosome-dependent manner with no clear sequence specificity (RelE family) or some degree of sequence specificity (HigB, YafQ toxins).
Tertiary fold or recognition of determinants in the sugar-phosphate backbone appears to also contribute to the selectivity of VapC-mt4, given that many other M. tuberculosis tRNAs also harbor the same recognition sequence yet only a few are efficiently cleaved. While tRNAs generally adopt L-shaped three-dimensional structures to enable entry into the ribosome, there may be some subtle variation in the overall structure that can be distinguished by VapC-mt4. In fact, while all tRNAs possess three stem-loops (D, anticodon, TΨC), the cloverleaf secondary structures of tRNA do vary slightly. Type II tRNAs (tRNASer26 and tRNASer24 in our case) also contain an extended variable arm between the anticodon and TΨC stem loop (Fig. 3). Also, although the D arm usually has a 4-bp stem, tRNASer26 and tRNASer24, have a 3-bp stem. In fact, our simulated docking experiments indicate that the variable loop influences the positioning of the VapC-mt4 dimer on the tRNA, which we propose alters the location of the cleavage site (Fig. 6). Independent of tertiary fold, the interaction of VapC-mt4 with the sugar-phosphate backbone may contribute to cleavage specificity. On the basis of studies of tRNA recognition for aminoacylation of tRNAGln by glutaminyl-tRNA synthetase, class II tRNA synthetase interaction with the sugar-phosphate backbone is an important recognition determinant in addition to base recognition at the anticodon loop, at the acceptor end, and with the interior base pair of the D stem41.
Strikingly, VapC-mt4 cuts all three M. tuberculosis tRNA targets precisely within the anticodon loop. In the case of tRNAAla2, VapC-mt4 cleaves at the position immediately 3′ adjacent to the anticodon (Fig. 3b). For tRNASer26/4, VapC-mt4 cleavage occurs within the anticodon itself between the second and the third nucleotides (Fig. 3d,f). However, there was no correlation between the tRNA isoacceptors targeted for cleavage and their corresponding codon usage preferences. tRNAAla2 decodes the lowest percentage of alanine codons (10%) in M. tuberculosis, while tRNASer26 (26%) and tRNASer24 (21%) decode intermediate percentages of serine codons (with tRNASer28 decoding the highest number of serine codons, 35%). Since the ASL of tRNA interacts with the decoding centre of the 30S subunit, cleavage at the anticodon sequence of the ASL by VapC-mt4 is consistent with the known inhibition of translation by VapC-mt4 on overexpression in E. coli using an arabinose-inducible plasmid (<10% of wild type19).
In some strains of E. coli, colicins are secreted into the environment to kill the neighbouring bacterial cells and reduce competition42. Colicins D and E5 enlist cleavage of tRNA at a single site within the ASL to inhibit translation and cause cell death. The lethality of colicin D is due to specific cleavage of all four tRNAArg isoacceptors between positions 38 and 39, corresponding to the junction of the loop and stem on the 3′ side of the anticodon43. Likewise, Salmonella/Shigella VapC cleaves tRNAfMet at the same position as colicin D16. As with VapC-mt4, colicin E5 cleaves within the anticodon loop, at positions 34 and 35 of the anticodon44. Also serving a defensive role, the E. coli endoribonuclease PrrC triggers cell suicide by cleaving tRNALys between anticodon positions 33 and 34 in response to bacteriophage T4 infection of E. coli cells45–47. Therefore, colicins D, E5 and PrrC all cause cell death. In the case of colicins D and E5, death is undoubtedly due to the absence of their cognate immunity proteins in neighbouring cells that take up the colicins alone42,43. For PrrC-mediated cell death, it is thought that after T4 phage infection, the Stp phage protein alters the association of the ‘antitoxin’ EcoprrI with PrrC, releasing free PrrC and leading to unchecked cleavage of lysine tRNA45–47.
In contrast, the general physiological role of most TA systems, including VapC-mt4, appears to involve stress survival and not cell death due to the presence of a cognate antitoxin that dynamically regulates toxin activity in response to stress3. In fact, as demonstrated for the MazF toxin, cell death can be precluded following toxin activation when the cognate antitoxin is also expressed2. Cell death only occurs after prolonged activation of the toxin, when the cell reaches ‘the point of no return’2. Our earlier study showed that VapC-mt4 overexpression results in translation inhibition with accumulation of polysomes, indicative of a block at the elongation step19. Possibly, ribosomal stalling may result from a non-productive interaction of the cleaved ASL with the A site of the ribosome. In addition, two studies have demonstrated that cleavage or alteration of the ASL leads to defects in aminoacylation. First, a break in the phosphodiester backbone of the ASL between position 34 and 35 of yeast tRNAMet inhibits aminoacylation48. Second, aminoacylation by E. coli glutaminyl-tRNA synthetase requires recognition of the sugar-phosphate backbone at positions 34–38 (ref. 41). Therefore, disruption of the backbone by cleavage between position 36 and 37 by VapC-mt4 would also likely disrupt aminoacylation by class II synthetases, potentiating its effect on translation and growth.
VapC-mt4 targets a small subset of M. tuberculosis tRNAs with exquisite specificity. It not only distinguishes between tRNA isotypes, it selectively cleaves some, but not all, isoacceptors. More specifically, VapC-mt4 cleaves one isoacceptor of tRNAAla (tRNAAla2) of the three represented in M. tuberculosis and two of the four isoacceptors of tRNASer (tRNASer26 and tRNASer24) are cleaved efficiently. Although colicins D and E5 exhibit specificity for tRNAs, their tRNA targets are either limited to all four tRNAArg isoacceptors (colicin D43) or extend to four isotypes (tRNAAsn, tRNAAsp, tRNATyr and tRNAHis for colicin E5 (ref. 44)). Since our data identifies the presence of the cleavage consensus sequence ACGC or ACUGC as one major determinant for tRNA cleavage, some of the selectively of VapC-mt4 is attributed to this requirement since not all isoacceptors of tRNASer harbour this essential sequence. In contrast, the sequence requirements for colicins appear to be less specific than for VapC-mt4—colicin E5 cleaves at (C/T)GU49, while the interrogation of sequence requirements for colicin D has not been reported43. No atomic level structures of colicins bound to tRNAs have been reported. Structural studies of VapC-mt4 bound to tRNA will be instrumental in pinpointing additional determinants (tertiary fold and/or sugar-phosphate backbone interactions) that guide toxin specificity for a handful of tRNA targets.
Although the precise physiological triggers for VapC-mt4 have not yet been identified, we know that the expression of this toxin leads to growth inhibition in M. tuberculosis14,19. In fact, with few exceptions, the unifying feature of TA systems is their role as stress responsive growth regulators. Consistent with this role, there is mounting evidence linking increased expression of TA systems with several stresses in M. tuberculosis, especially those encountered during latent tuberculosis4–13. As a complement to transcript-oriented studies, recent proteomic profiles generated from parallel analyses by mass spectrometry and two-dimensional differential gel electrophoresis reinforced the role of TA systems in stress50. Culture filtrates from M. tuberculosis cells after nutrient starvation contained elevated levels of 13 TA toxins, including a >10-fold increase in VapC-mt4 (ref. 50).
The need for the high number of VapC toxins in M. tuberculosis (48 predicted) is not yet clear. All VapC toxins are expected to function as nucleases due to the presence of a PIN domain. However, our findings reported here coupled with those of the only other well-characterized M. tuberculosis VapC toxin—VapC20, which targets the 23S rRNA sarcin–ricin loop20—suggest that the VapC family members will not be functionally redundant. Instead, each VapC may cleave unique substrates or sites within a given target, thus allowing a defined molecular response to one or more stresses. Given that M. tuberculosis cells can remain dormant yet viable for many years in latent tuberculosis in a granuloma, the resulting activity of each toxin may impart some degree of growth downregulation that is then lifted before cells reach the point of no return. Thus, through activation of a shifting array of VapC toxins in a calibrated, asynchronous manner, M. tuberculosis may be able to limit growth while never reaching the point of cell death.
Interestingly, the presence of cleaved tRNA halves and smaller fragments is also emerging as a hallmark of stress responses in many eukaryotes (reviewed in refs 51–53). In this rapidly evolving area, tRNA halves or other tRNA-derived fragments have been associated with a range of functions, including translational control, apoptosis and RNA interference regulation. Therefore, our results uncover a new parallel between the VapC-mt4 toxin-mediated stress response in M. tuberculosis and post-transcriptional tRNA cleavage at the ASL during stress in eukaryotes. Since the activities of only two VapC toxins have been characterized, it is highly likely that more VapC toxins will also target tRNAs. This would further expand the repertoire of tRNAs acting in a non-conventional role, shed light on the conundrum of why M. tuberculosis cells have so many VapBC TA systems and strengthen our understanding of how their orchestrated expression impacts stress survival, growth and virulence.
Methods
Strains and plasmids
The E. coli strain BL21(DE3) (F-ompT hsdSβ(rβ-mβ) dcm gal (DE3) tonA) (Novagen) was used for protein expression The E. coli strain BW25113Δ6 (F− lacIq rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 ΔmazEF ΔchpBIK ΔrelBE ΔyefM-yoeB ΔdinJ-yafQ)21 was used for the isolation of RNA. E. coli K-12 Mach1 T1 cells (ΔrecA1398 endA1 tonA Φ80ΔlacM15 ΔlacX74 hsdR(rk+ mk+); Invitrogen) were used for all cloning experiments. The pBAD33-VapC-mt4 and pET28a-His6-VapC-mt4 was described in our earlier work19. All E. coli liquid cultures were grown in M9 minimal media supplemented with 0.2% (w/v) casamino acids, 1 mM MgSO4, 0.001% (w/v) thiamine and either 0.2% glucose or 0.2% glycerol at 37 °C. 1 mM isopropyl 1-thio-β-D-galactopyranoside, 0.2% arabinose, 45 μg ml−1 kanamycin and 25 μg ml−1 chloramphenicol were added to culture medium as required.
Purification of recombinant VapC-mt4-His6
E. coli BL21(DE3)pLysE was transformed with the pET28a-His6-VapC-mt4 plasmid. Transformants were selected and grown in 1 l of M9 minimal media containing 0.2% glucose at 37°C to an OD600 of 0.6–0.8 and toxin expression induced with isopropyl 1-thio-β-D-galactopyranoside for 4 h. Recombinant VapC-mt4 was then purified as previously described19. In brief, the cells were lysed by sonication. Cell lysates were centrifuged at 32,000g for 10 min to remove cell debris. Protein was then purified from the cleared cell lysates by nickel–nitrilotriacetic acid affinity chromatography (Qiagen).
Double-stranded RNA cleavage
A 20-base ACGC-containing RNA oligo-nucleotide, NWO1430 (5′-AGGAAGAUACGCGAUAUGAA-3′), was 5′ end labelled with [γ-32P]-ATP using T4 polynucleotide kinase (NEB) following the manufacturer’s protocol. The labelled oligonucleotide was diluted with diethylpyrocarbonate (DEPC) -treated water to yield a final concentration of 0.2 pmol μl−1. The labelled RNA was then mixed with different amounts (at ratios of 0:1, 0.1:1, 0.2:1, 0.4:1, 0.8:1 and 1:1) of a complementary RNA oligo, NWO1541(5′-UUCAUAUCGCGUAUCUUCCU-3′). The two oligonucleotides were incubated at 70 °C for 3 min and then allowed to cool slowly to room temperature (~30 min). Samples were adjusted to 10 mM Tris pH 7.5, 150 mM NaCl, 10 mM MgCl2; 20 units of RNase inhibitor (NEB) was then added to each reaction followed by 17 pmol of VapC-mt4 (when indicated) in a final volume of 10 μl. The samples were incubated at 37 °C for 1 h and reactions stopped by adding 10 μl of sequence loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol FF). Samples were then heated to 95 °C for 5 min before electrophoresis on a 15% polyacrylamide, 7 M urea gel.
In vitro synthesis of tRNA
E. coli and M. tuberculosis tRNAs were synthesized in vitro following the method described by Sisido et al.54 with minor modifications. In brief, a synthetic DNA oligonucleotide containing the T7 RNA polymerase promoter and the 5′ end of the tRNA gene of interest was annealed to a second oligonucleotide corresponding to the 3′ end of the tRNA gene. The annealed oligonucleotides were then extended using Taq DNA polymerase to create dsDNA containing the entire tRNA gene preceded by the T7 promoter. The product was then resolved on a 2% agarose gel to confirm its size and purified using the QIAquick Gel Extraction Kit (Qiagen). The sequence of the product was verified by automated DNA sequence analysis. Two-hundred ng of the tRNA gene was then transcribed in vitro using the RiboMAX Large Scale RNA Production System (Promega) as recommended by the manufacturer. The transcription reaction was separated on a 9% polyacrylamide, 7 M urea gel and visualized by staining with EtBr to confirm the size and purity of the transcribed tRNA. The tRNA transcript was then excised from the gel and incubated for 18 h at 37 °C in elution buffer (1 mM EDTA, 0.5 M ammonium acetate, 10 mM magnesium acetate, 0.1% SDS). The eluate was then collected, and the gel pieces were washed in fresh elution buffer. The elution and wash were combined, the tRNA was ethanol precipitated and resuspended in nuclease-free water.
Purification of tRNA from E. coli cell extracts
Total E. coli RNA was isolated using the hot phenol method. A culture of E. coli BW25113Δ6 was grown in M9 medium until the OD600 reached a value of ~0.6. The cells were pelleted and resuspended in 500 μl Buffer A (0.5% SDS, 20 mM sodium acetate, pH 4.0; 10 mM EDTA) and 500 μl phenol, pH 4.0. After vortexing to completely resuspend the cell pellet, the samples were heated to 60 °C for 10 min and centrifuged to separate the phases. The RNA-containing aqueous phase was removed, ethanol precipitated and resuspended in nuclease-free water. Individual tRNAs were then isolated from the total RNA using a biotinylated oligonucleotide that specifically annealed to tRNAAla2 adapted from the procedure of Yokogawa et al.55. We first washed 450 μl of MagneSphere magnetic beads (Promega) three times (we used a magnetic stand to recover the beads after all wash and hybridization steps) with 600 μl of wash buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA and 1 M NaCl). We then added 600 pmol of the 5′ biotinylated oligonucleotide for tRNAAla2 isolation, NWO1974 (5′-Biotin-AGACCTCCTGCGTGCAAAGCAGGC-3′), adjusted the volume to 300 μl with 10 mM Tris-HCl pH 7.5 and incubated at room temperature for 15 min with gentle inversion every 3 min. After binding of the tRNAAla2-specific oligonucleotide, the beads were washed three times with wash buffer. After the final wash, the beads were resuspended in 50 μl hybridization buffer (20 mM Tris-HCl pH 7.6, 1.8 M tetramethylammonium chloride (TMA-Cl), 0.2 mM EDTA). One mg of total RNA in 50 μl nuclease-free water was then added to the beads. Samples were then gently inverted, heated to 65 °C for 3 min and placed at 60 °C for 20 min with inversion every 4 min. The supernate containing unbound RNA was separated from the beads and transferred to a fresh tube and saved for rebinding. The beads containing the bound tRNAs were washed six times with 800 μl Tris-HCl, pH 7.6. The target tRNA was then eluted from the beads with 150 μl of Tris-HCl, pH 7.6 heated to 75 °C for 5 min and transferred to another tube containing 2 μl of 1 M MgCl2 to assist in proper folding. To maximize recovery of tRNA, we repeated the last step with the saved aliquot and combined elutions.
In vitro tRNA cleavage
Cleavage of tRNAs produced via T7 transcription or purified from total RNA was performed in vitro following the assay described by Winther and Gerdes16 with slight modifications. In brief, 2 pmol of tRNA was incubated with 2.5, 5 or 10 pmol of VapC-mt4 at 37 °C for 3 h in 10 mM HEPES pH 7.5, 15 mM KCl, 3 mM MgCl2, 10% glycerol in a final volume of 10 μl. To check for cleavage inhibition by the cognate antitoxin, VapB-mt4 was preincubated with VapC-mt4 for 15 min at 37 °C before the RNA substrate was added. In samples containing EDTA, 1 μl of 125 mM EDTA was added to the reaction before the addition of the RNA substrate. The products of these reactions were separated on a 9% polyacrylamide, 7 M urea gel and visualized by SYBR Gold (Invitrogen) staining.
In vivo primer extension
E. coli BW25113Δ6 cells carrying pBAD33-VapC-mt4 were grown in M9 minimal media containing 0.2% glycerol at 37 °C to an OD600 of 0.3–0.4. Arabinose was then added to a final concentration of 0.2%. Aliquots were removed 0, 20, 40, 60, 80 and 100 min post induction and total RNA was extracted as described in the ‘Purification of tRNA from E. coli cell extracts’ section above. The primer NWO1976 (5′-GGGATCGAACCGCAGACC-3′) was 5′ end labelled with [γ-32P]ATP using T4 polynucleotide kinase (NEB). Five μg of total RNA was mixed with 40 pmol of the labelled oligonucleotide in a 6.5-μl reaction containing annealing buffer for the SuperScript III reverse transcriptase (Invitrogen). The samples were heated to 80 °C for 3 min and allowed to cool slowly to room temperature over 30 min. First Strand Reaction Mix (3.5 μl; Invitrogen) and 2 μl SuperScript III Enzyme Mix (Invitrogen) were then added. The reactions were incubated at 52 °C for 90 min, then heat inactivated at 70 °C for 10 min followed by addition of 12 μl of sequence loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue and 0.05% xylene cyanol). The DNA-sequencing ladders for primer extension reactions were prepared using the Sequenase Version 2.0 DNA-Sequencing Kit (Agilent) as recommended.
In vitro primer extension
T7-transcribed M. tuberculosis tRNAs were cleaved as described in the ‘In vitro tRNA cleavage’ section, purified by phenol:chloroform extraction and ethanol precipitated. Primer extension reactions were then performed as described in the previous section using 2 pmol of cleaved tRNA. Primers: tRNAAla2, NWO2164 (5′-GGAGCCTAGGGGACTCGAA-3′); tRNASer26, NWO2165 (5′-GAGGCGAGAGGATTTGAACCTCC-3′); and tRNASer24, NWO2166 (5′-GGAGGATGCGGGATTTGAACCC-3′).
RNA-seq of E. coli RNA
E. coli BW25113Δ6 cells carrying pBAD33-VapC-mt4 were grown in M9 minimal media containing 0.2% glycerol at 37 °C to OD600 0.3–0.4. The culture was split in half and arabinose was then added to a final concentration of 0.2% to one of the cultures. Aliquots from induced and uninduced cultures were removed 0, 20, 40, 60, 80 and 100 min post induction. Total RNA was extracted as described in the ‘Purification of tRNA from E. coli cell extracts’; 5′ RNA-seq was performed using total RNA from the 20-min induced and unin-duced cultures. The 20-min induction time was selected because it coincided with the initiation of toxicity, ensuring that the cleavage site being documented was the direct consequence of toxin induction and not due to secondary effects. Two pools of RNA, those possessing a 5′-P and those possessing a 5′-OH. After removal of 5′-P RNA from the 5′-OH pool, the resulting 5′-OH RNA was converted to 5′-P RNA via phosphorylation by 50 U OptiKinase. A 5′ SOLiD RNA adaptor was then ligated onto RNA from both pools. Reverse transcription and PCR was then performed based on the methods outlined in Schifano et al.21 and Vvedenskaya et al.56.
tRNA northern analysis
Total RNA was isolated from E. coli as described above. Total RNA from M. tuberculosis H37Rv was isolated as previously described57. In brief, M. tuberculosis cells were grown to exponential phase and then pelleted. Cell pellets were resuspended in TRIzol Reagent (Invitrogen). RNA was treated with TURBO DNase (Invitrogen). Total RNA (2 μg) from each organism was incubated with 20 pmol of VapC-mt4 in a 10-μl reaction containing 10 mM HEPES pH 7.5, 15 mM KCl, 3 mM MgCl2, 10% glycerol at 37 °C for 0, 3 or 24 h. To check for cleavage inhibition by the cognate antitoxin, 120 pmol VapB-mt4 was preincubated with VapC-mt4 for 15 min at 37 °C before the RNA substrate was added and incubated at 37 °C for 24 h. The products of these reactions were separated on a 9% polyacrylamide, 7 M urea gel and visualized by SYBR Gold (Invitrogen) staining. RNA was transferred to nitrocellulose and hybridized with oligonucleotides specific for a given tRNA species. The oligonucleotides used were: E. coli tRNAAla2, NWO2268, 5′-CGTGCAAAGCAGGCGCTC-3′; M. tuberculosis tRNASer26, NWO2277, 5′-GCAGTGAGCCCCATTCG-3′; and M. tuberculosis tRNASer24, NWO2270, 5′-CCCTTGAAGGGGGACAACTCATTA-3′.
VapC-mt4-tRNA molecular modelling
All protein–RNA docking simulations were performed with the programme 3dRPC26. The homology model of VapC-mt4 was generated by the HHpred homology model workflow and Modeller using the optimal multiple template function27,28. The structural templates used in Modeller to generate the VapC-mt4 homology model were Rickettsia felis VapC (from VapBC2 complex; PDB accession code 3ZVK, chain A30; 19% identity, 28.7% similarity to VapC-mt4), M. tuberculosis VapC (from VapBC5 complex; PDB accession code 3DBO, chain B29; 36% identity, 53.7% similarity) and Neisseria gonorrhoeae FitB (from FitAB complex; PDB accession code 2H1C, chain A31; 27% identity, 28.8% similarity). The VapC-mt4 homology dimer models were first generated by the global search programme GRAMM-X27,34, followed by further refinement using the programme RosettaDock35. The manganese atom was positioned in the VapC-mt4 homology model using an alignment of Pyrobaculum aerophilum VapC (PDB accession code 2FE1)32 generated by DaliLite38. The homologue of tRNAAla2 used in this study was Saccharomyces cerevisiae tRNAPhe (PDB accession code 4TNA, chain A)37 and the homologue for the tRNASer26 was Escherichia coli tRNALeu (PDB accession code 3UZ3, chain B)36. The expanded variable loop of M. tuberculosis tRNASer26 was generated by the programme 3dRNA using the secondary structure predicted by NAVIEW58,59 and extrapolated onto the tRNALeu variable loop by least-squares fitting using Coot60. With both tRNA models, the changes in the target anticodon loop residues corresponding to tRNAAla2 or tRNASer26 sequences were manually introduced using Coot. The proposed models were selected based on the proximity between the targeted region of the tRNA ASL and the proposed VapC catalytic residues.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health under award numbers R21 AI072399 (to N.A.W. and R.N.H.) and R01 GM095693 (to N.A.W.), R01 GM088343 (to B.E.N.), R21 AI097881 (to R.N.H.) and T32 AI007403 (to J.W.C.); a National Science Foundation CAREER award MCB 0953714 (to C.M.D.). C.M.D is a Pew Scholar in the Biomedical Sciences.
Footnotes
Author contributions
J.W.C., J.D.S., I.O.V., R.N.H., B.E.N., C.M.D. and N.A.W. designed the research. J.W.C., J.D.S., E.D.H., T.M., I.O.V. and A.K. performed the research. J.W.C., J.D.S., E.D.H., T.M., R.N.H., B.E.N., C.M.D. and N.A.W. analysed the data. J.W.C., E.D.H., T.M., B.E.N., C.M.D. and N.A.W. wrote the paper.
Accession codes: The RNA sequencing data have been deposited in the NCBI Sequence Read Archive under accession code PRJNA277981.
Competing financial interests: The authors declare no competing financial interests.
Supplementary Information accompanies this paper at http://www.nature.com/naturecommunications
References
- 1.Aizenman E, Engelberg-Kulka H, Glaser G. An Escherichia coli chromosomal ‘addiction module’ regulated by guanosine [corrected] 3′,5′-bispyrophosphate: a model for programmed bacterial cell death. Proc Natl Acad Sci USA. 1996;93:6059–6063. doi: 10.1073/pnas.93.12.6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hazan R, Sat B, Engelberg-Kulka H. Escherichia coli mazEF-mediated cell death is triggered by various stressful conditions. J Bacteriol. 2004;186:3663–3669. doi: 10.1128/JB.186.11.3663-3669.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamaguchi Y, Inouye M. Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat Rev Microbiol. 2011;9:779–790. doi: 10.1038/nrmicro2651. [DOI] [PubMed] [Google Scholar]
- 4.Stewart GR, et al. Dissection of the heat-shock response in Mycobacterium tuberculosis using mutants and microarrays. Microbiology. 2002;148:3129–3138. doi: 10.1099/00221287-148-10-3129. [DOI] [PubMed] [Google Scholar]
- 5.Ramage HR, Connolly LE, Cox JS. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009;5:e1000767. doi: 10.1371/journal.pgen.1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rustad TR, Harrell MI, Liao R, Sherman DR. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE. 2008;3:e1502. doi: 10.1371/journal.pone.0001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rand L, et al. The majority of inducible DNA repair genes in Mycobacterium tuberculosis are induced independently of RecA. Mol Microbiol. 2003;50:1031–1042. doi: 10.1046/j.1365-2958.2003.03765.x. [DOI] [PubMed] [Google Scholar]
- 8.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol. 2002;43:717–731. doi: 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 9.Cappelli G, et al. Profiling of Mycobacterium tuberculosis gene expression during human macrophage infection: upregulation of the alternative sigma factor G, a group of transcriptional regulators, and proteins with unknown function. Res Microbiol. 2006;157:445–455. doi: 10.1016/j.resmic.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Korch SB, Contreras H, Clark-Curtiss JE. Three Mycobacterium tuberculosis Rel toxin-antitoxin modules inhibit mycobacterial growth and are expressed in infected human macrophages. J Bacteriol. 2009;191:1618–1630. doi: 10.1128/JB.01318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Provvedi R, Boldrin F, Falciani F, Palu G, Manganelli R. Global transcriptional response to vancomycin in Mycobacterium tuberculosis. Microbiology. 2009;155:1093–1102. doi: 10.1099/mic.0.024802-0. [DOI] [PubMed] [Google Scholar]
- 12.Singh R, Barry CE, 3rd, Boshoff HI. The three RelE homologs of Mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. J Bacteriol. 2010;192:1279–1291. doi: 10.1128/JB.01285-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cortes T, et al. Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Rep. 2013;5:1121–1131. doi: 10.1016/j.celrep.2013.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahidjo BA, et al. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PLoS ONE. 2011;6:e21738. doi: 10.1371/journal.pone.0021738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arcus VL, Rainey PB, Turner SJ. The PIN-domain toxin-antitoxin array in mycobacteria. Trends Microbiol. 2005;13:360–365. doi: 10.1016/j.tim.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 16.Winther KS, Gerdes K. Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc Natl Acad Sci USA. 2011;108:7403–7407. doi: 10.1073/pnas.1019587108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopes AP, et al. VapC from the leptospiral VapBC toxin-antitoxin module displays ribonuclease activity on the initiator tRNA. PLoS ONE. 2014;9:e101678. doi: 10.1371/journal.pone.0101678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKenzie JL, et al. A VapBC toxin-antitoxin module is a posttranscriptional regulator of metabolic flux in mycobacteria. J Bacteriol. 2012;194:2189–2204. doi: 10.1128/JB.06790-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharp JD, et al. Growth and translation inhibition through sequence-specific RNA binding by Mycobacterium tuberculosis VapC toxin. J Biol Chem. 2012;287:12835–12847. doi: 10.1074/jbc.M112.340109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winther KS, Brodersen DE, Brown AK, Gerdes K. VapC20 of Mycobacterium tuberculosis cleaves the sarcin-ricin loop of 23S rRNA. Nat Commun. 2013;4:2796. doi: 10.1038/ncomms3796. [DOI] [PubMed] [Google Scholar]
- 21.Schifano JM, et al. An RNA-seq method for defining endoribonuclease cleavage specificity identifies dual rRNA substrates for toxin MazF-mt3. Nat Commun. 2014;5:3538. doi: 10.1038/ncomms4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Zhang J, Hara H, Kato I, Inouye M. Insights into the mRNA cleavage mechanism by MazF, an mRNA interferase. J Biol Chem. 2005;280:3143–3150. doi: 10.1074/jbc.M411811200. [DOI] [PubMed] [Google Scholar]
- 23.McKenzie JL, et al. Determination of ribonuclease sequence-specificity using pentaprobes and mass spectrometry. RNA. 2012;18:1267–1278. doi: 10.1261/rna.031229.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan PP, Lowe TM. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–D97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, et al. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol Cell. 2003;12:913–923. doi: 10.1016/s1097-2765(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Liu S, Guo D, Li L, Xiao Y. A novel protocol for three-dimensional structure prediction of RNA-protein complexes. Sci Rep. 2013;3:1887. doi: 10.1038/srep01887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- 28.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miallau L, et al. Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. J Biol Chem. 2009;284:276–283. doi: 10.1074/jbc.M805061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mate MJ, et al. Crystal structure of the DNA-bound VapBC2 antitoxin/toxin pair from Rickettsia felis. Nucleic Acids Res. 2012;40:3245–3258. doi: 10.1093/nar/gkr1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mattison K, Wilbur JS, So M, Brennan RG. Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J Biol Chem. 2006;281:37942–37951. doi: 10.1074/jbc.M605198200. [DOI] [PubMed] [Google Scholar]
- 32.Bunker RD, McKenzie JL, Baker EN, Arcus VL. Crystal structure of PAE0151 from Pyrobaculum aerophilum, a PIN-domain (VapC) protein from a toxin-antitoxin operon. Proteins. 2008;72:510–518. doi: 10.1002/prot.22048. [DOI] [PubMed] [Google Scholar]
- 33.Xu K, et al. Protein expression, crystallization and preliminary X-ray crystallographic analysis of the isolated Shigella flexneri VapC toxin. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013;69:762–765. doi: 10.1107/S1744309113014012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tovchigrechko A, Vakser IA. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006;34:W310–W314. doi: 10.1093/nar/gkl206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lyskov S, Grey JJ. The RosettaDock server for local protein-protein docking. Nucleic Acids Res. 2008;36:W233–W238. doi: 10.1093/nar/gkn216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demeshkina N, Jenner L, Westhof E, Yusupov M, Yusupova G. A new understanding of the decoding principle on the ribosome. Nature. 2012;484:256–259. doi: 10.1038/nature10913. [DOI] [PubMed] [Google Scholar]
- 37.Hingerty B, Brown RS, Jack A. Further refinement of the structure of yeast tRNAPhe. J Mol Biol. 1978;124:523–534. doi: 10.1016/0022-2836(78)90185-7. [DOI] [PubMed] [Google Scholar]
- 38.Hasegawa H, Holm L. Advances and pitfalls of protein structural alignment. Curr Opin Struct Biol. 2009;19:341–348. doi: 10.1016/j.sbi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 39.Pandey DP, Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–976. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katz ME, Strugnell RA, Rood JI. Molecular characterization of a genomic region associated with virulence in Dichelobacter nodosus. Infect Immun. 1992;60:4586–4592. doi: 10.1128/iai.60.11.4586-4592.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McClain WH, Schneider J, Bhattacharya S, Gabriel K. The importance of tRNA backbone-mediated interactions with synthetase for aminoacylation. Proc Natl Acad Sci USA. 1998;95:460–465. doi: 10.1073/pnas.95.2.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cascales E, et al. Colicin biology. Microbiol Mol Biol Rev. 2007;71:158–229. doi: 10.1128/MMBR.00036-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tomita K, Ogawa T, Uozumi T, Watanabe K, Masaki H. A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc Natl Acad Sci USA. 2000;97:8278–8283. doi: 10.1073/pnas.140213797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogawa T, et al. A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science. 1999;283:2097–2100. doi: 10.1126/science.283.5410.2097. [DOI] [PubMed] [Google Scholar]
- 45.Amitsur M, Levitz R, Kaufmann G. Bacteriophage T4 anticodon nuclease, polynucleotide kinase and RNA ligase reprocess the host lysine tRNA. EMBO J. 1987;6:2499–2503. doi: 10.1002/j.1460-2075.1987.tb02532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaufmann G. Anticodon nucleases. Trends Biochem Sci. 2000;25:70–74. doi: 10.1016/s0968-0004(99)01525-x. [DOI] [PubMed] [Google Scholar]
- 47.Meineke B, Shuman S. Structure-function relations in the NTPase domain of the antiviral tRNA ribotoxin Escherichia coli PrrC. Virology. 2012;427:144–150. doi: 10.1016/j.virol.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aphasizhev R, Senger B, Fasiolo F. Importance of structural features for tRNA(Met) identity. RNA. 1997;3:489–497. [PMC free article] [PubMed] [Google Scholar]
- 49.Ogawa T, Inoue S, Yajima S, Hidaka M, Masaki H. Sequence-specific recognition of colicin E5, a tRNA-targeting ribonuclease. Nucleic Acids Res. 2006;34:6065–6073. doi: 10.1093/nar/gkl629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Albrethsen J, et al. Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol Cell Proteomics. 2013;12:1180–1191. doi: 10.1074/mcp.M112.018846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gebetsberger J, Polacek N. Slicing tRNAs to boost functional ncRNA diversity. RNA Biol. 2013;10:1798–1806. doi: 10.4161/rna.27177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phizicky EM, Hopper AK. tRNA biology charges to the front. Genes Dev. 2010;24:1832–1860. doi: 10.1101/gad.1956510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thompson DM, Parker R. Stressing out over tRNA cleavage. Cell. 2009;138:215–219. doi: 10.1016/j.cell.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 54.Sisido M, Ninomiya K, Ohtsuki T, Hohsaka T. Four-base codon/anticodon strategy and non-enzymatic aminoacylation for protein engineering with non-natural amino acids. Methods. 2005;36:270–278. doi: 10.1016/j.ymeth.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 55.Yokogawa T, Kitamura Y, Nakamura D, Ohno S, Nishikawa K. Optimization of the hybridization-based method for purification of thermostable tRNAs in the presence of tetraalkylammonium salts. Nucleic Acids Res. 2010;38:e89. doi: 10.1093/nar/gkp1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vvedenskaya IO, Goldman SR, Nickels BE. Preparation of cDNA libraries for high-throughput RNA sequencing analysis of RNA 5′ ends. Methods Mol Biol. 2015;1276:211–228. doi: 10.1007/978-1-4939-2392-2_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schifano JM, et al. Mycobacterial toxin MazF-mt6 inhibits translation through cleavage of 23S rRNA at the ribosomal A site. Proc Natl Acad Sci USA. 2013;110:8501–8506. doi: 10.1073/pnas.1222031110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao Y, et al. Automated and fast building of three-dimensional RNA structures. Sci Rep. 2012;2:734. doi: 10.1038/srep00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruccoleri RE, Heinrich G. An improved algorithm for nucleic acid secondary structure display. Comput Appl Biosci. 1988;4:167–173. doi: 10.1093/bioinformatics/4.1.167. [DOI] [PubMed] [Google Scholar]
- 60.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pleiss JA, Derrick ML, Uhlenbeck OC. T7 RNA polymerase produces 5′ end heterogeneity during in vitro transcription from certain templates. RNA. 1998;4:1313–1317. doi: 10.1017/s135583829800106x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.