

Abstract
Types 1 and 2 diabetes are on the rise worldwide. Although the treatment of hyperglycemia has benefited from recent advances, aggressive efforts to maintain euglycemia may be fraught with risk, especially in older subjects or in subjects vulnerable to hypoglycemic unawareness. Hence, strategies to prevent and treat the complications of hyperglycemia are essential. In this review we summarize recent updates on the biology of the receptor for advanced glycation endproducts (RAGE) in the pathogenesis of both micro- and macrovascular complications of diabetes, insights from the study of mouse models of obesity and diabetic complications, and from associative studies in human subjects. The study of the mechanisms and consequences of the interaction of the RAGE cytoplasmic domain with the formin, mDia1, in RAGE signal transduction, will be discussed. Lastly, we review the “state-of-the-art” on RAGE-directed therapeutics. Tackling RAGE/mDia1 may identify a novel class of therapeutics preventing diabetes and its complications.
Obesity, insulin resistance, hyperglycemia, diabetes, and diabetic complications represent a growing health epidemic in the United States and the world at large.1,2 Attempts to keep glycemic levels in tight control can lead to hypoglycemia and lethality, especially in elderly patients. New strategies are needed to treat and prevent the progression of diabetic complications of hyperglycemic conditions and diabetes.
Diabetes encompasses two major forms, including types 1 and 2 diabetes. Other less common forms of diabetes include gestational diabetes and Maturity Onset Diabetes of the Young (MODY). Type 1 diabetes (T1D) is an autoimmune disease where the body destroys its pancreatic β-cells, and therefore has no or reduced capacity to produce insulin. Type 2 diabetes (T2D) is characterized by a state of insulin resistance, which reflects the inability of cells to properly respond to insulin. The clinical diagnosis of diabetes includes determination of the levels of hemoglobin A1c (HgbA1c), which gives an overview of blood glucose levels over a ~3 month period. Generally, nondiabetic subjects have HgbA1c under 5.7, prediabetic subjects have HgbA1c between 5.7 and 6.4, and diabetic subjects have HgbA1c over 6.5. Increasingly, diabetes is diagnosed in young people under the age of 20 years. The SEARCH for Diabetes in Youth multicenter study of the Centers for Disease Control (CDC) and National Institutes of Health (NIH) reported that in the years 2008–2009 ~18,436 people under 20 years of age were diagnosed annually with T1D and 5,089 people under age 20 years were diagnosed with T2D.3
Despite the different etiologies of T1D and T2D, the defining feature of both forms of diabetes is the ensuing hyperglycemia, or pathologically induced high blood glucose levels.4 Physiologically and pathologically increased blood glucose levels create an environment conducive to increased production of advanced glycation endproducts (AGEs), which are the irreversible products of nonenzymatic reactions between amine groups of proteins and carbonyl groups of sugars. Reversible intermediates of these reactions include Schiff bases and Amadori products. HbgA1c is an example of an Amadori product.5–7 In a prediabetic and diabetic environment in humans and animals, AGEs can crosslink with both intracellular and extracellular proteins as well as bind to their central cell surface receptor RAGE (receptor for advanced glycation endproducts) in circulation and in a variety of tissues.8 AGE-RAGE interactions activate gene expression of adhesion molecules and proinflammatory cytokines, which upregulates local inflammation and oxidative stress.9 Expression of RAGE increases in the presence of increased AGEs, such as in hyperglycemic and diabetic conditions in both clinical and nonclinical models.8–10 Deletion of Ager (Advanced glycation endproduct receptor, gene encoding RAGE) in mouse models prevents the development of obesity and insulin resistance.11 Targeting RAGE and its stimulated signaling pathways could lead to the development of novel drug targets to treat diabetes and its complications. In this review we detail the evidence highlighting RAGE as an attractive target for antidiabetes pathogenesis and antidiabetes complications drug development.
BIOLOGY AND STRUCTURE OF RAGE
RAGE is a 35 kDa transmembrane protein that is remarkably conserved among humans, swine, rats, and mice, as examples. Its N-terminal extracellular portion has one V-type immunoglobulin domain important for ligand binding and two C-type immunoglobulin domains. Of note, the N-terminus V-C1 is suggested to be the key motif required for RAGE ligand binding. RAGE’s carboxy-terminal cytoplasmic domain is short but imperative for signaling.12 RAGE is normally expressed at low levels in a variety of cell types including monocytes/macrophages, lymphocytes, endothelial cells, smooth muscle cells, neurons, hepatocytes, cardiomyocytes, and podocytes.13 However, in the healthy lung RAGE is expressed at higher levels.14 AGEs are not the exclusive ligands of RAGE. Other ligands include HMGB1,15 S100/calgranulins,16 amyloid-β peptide (Aβ) and other forms of amyloid,17 lysophosphatidic acid (LPA),18 and macrophage adhesion ligand-1 (MAC-1).19 As AGEs are among the most commonly studied of the RAGE ligands, it is currently under investigation as to how each of these non-AGE RAGE ligands changes over the course of diabetes and diabetic complications. RAGE ligands are multivalent and therefore many RAGE receptors may cluster together to bind one ligand, which induces downstream signaling. Different downstream signaling pathways may be induced by RAGE ligands via RAGE, depending on the specific cell type and the duration of stimulation (that is, acute vs. chronic stresses). These signaling pathways include JAK/STAT, PKC/PI3K/Akt, MAPK/Erk, and Src/RhoA/Cdc42, which activate various transcription factors including NFκB and Egr1. NFκB leads to the transcription of genes involved in inflammation and Egr1 leads to the transcription of genes involved in cell motility, adhesion, and structure (Figure 1). Currently, the field is investigating which signaling pathway each specific RAGE ligand activates and what are the downstream physiological effects of these ligands.
Figure 1.

Overview of RAGE signaling pathways. RAGE ligands induce a variety of signaling cascades, which lead to upregulation of inflammation and cellular mechanics. The PI3K/Akt and the Src/RhoA pathways may signal through mDia1 to increase NFκB and Egr-1 induced transcription. Downstream signaling of RAGE also includes the JAK/STAT and MAPK/Erk pathways, which also lead to upregulation of NFκB and Egr-1 and other transcriptional mechanisms. NFκB transcribed genes upregulate inflammation, and Egr-1 transcribed genes are involved in cell structure, motility, and adhesion and in tissue injury.
One critical signaling effector molecule for RAGE is diaphanous1 (mDia1).20 mDia1 is a formin family adaptor protein that is a downstream effector of Rho GTPases. mDia1 nucleates actin polymerization at the barbed end of growing actin filaments through interactions with profilin-bound actin monomers. mDia1 is involved in cell migration and motility, axonal outgrowth, focal adhesion, and cell polarization.21 mDia1 is the mouse ortholog to Drosophila Diaphanous and human Dia1, Drf1, or DIAPH1.22,23
RAGE has various splice variants and isoforms,24 including sRAGE, which is not membrane bound. There are two forms of sRAGE: cell surface cleaved RAGE (also known as soluble RAGE and denoted as sRAGE) and endogenous secretory RAGE (esRAGE), also known as RAGEv1. sRAGE is formed by cleavage from full-length RAGE by matrix metalloproteinases (MMPs) and ADAM10. Therefore, sRAGE lacks both the transmembrane domain and cytoplasmic tail. esRAGE (RAGEv1) is an alternatively spliced form and is secreted by the cell.25 Both forms of sRAGEs circulate in human plasma along with other splice variants of RAGE.26,27
Genome-wide association studies (GWAS) identified a variety of single nucleotide polymorphisms (SNPs) in human RAGE. Two SNPs, rs2071288 and rs2070600, are associated with lower amounts of both sRAGE and esRAGE in patients with emphysema and chronic obstructive pulmonary disease (COPD).28 The rs2070600 SNP is also associated with circulating sRAGE levels,29 increased risk of developing diabetic nephropathy in T1D patients,30 and with retinopathy.31,32 The rs2070600 SNP is a −374 thiamine to adenine change, which causes the glycine at position 82 to change to a serine in the ligand binding V-domain of RAGE. Therefore, rs2070600 is also referred to as G82S. G82S increases proinflammatory signaling pathways leading to increased TNF-α secretion as studied in mononuclear phagocytes from human subjects.16 Physiologically, this suggests that G82S may enhance ligand binding to RAGE and thereby upregulate RAGE signaling.
RAGE AND DIABETIC COMPLICATIONS
Hyperglycemic conditions cause diabetic complications that affect the entire body. These diabetic complications are not limited to but include neuropathies, retinopathies, nephropathies, cardiomyopathies, atherosclerosis, and delayed wound healing.
Diabetic neuropathies are peripheral nervous system diseases characterized by loss of myelinated fibers and atrophy of axons followed by an inflammatory response. Clinical symptoms include pain and loss of perception, which can lead to amputations. RAGE is increased in skin biopsies of patients with diabetic neuropathy, as characterized by lower intraepidermal nerve-fiber density, and dyslipidemia.33 RAGE ligands are also increased in the peripheral nerves of diabetic mice as compared to Ager null mice. Ager null mice are partially protected from the loss of pain perception.34 Ager null diabetic mice are also protected from suppressed axonal regeneration. After peripheral nerve injury, Ager null diabetic mice display higher myelinated nerve densities and conduction velocities as compared to wildtype diabetic mice. Lethal irradiation of wildtype mice followed by bone marrow transplantation of Ager null bone marrow into wildtype mice improved axonal regeneration, as well as increased content of antiinflammatory, M2 macrophages into the injured area.35
Diabetic retinopathy is a disease of the capillaries and neural retina in the eye. Hyperglycemic conditions lead to increased vascular permeability, bleeding, secretion of lipids from blocked vessels, and blood vessel formation on the retina. The clinical endstage of diabetic retinopathy is blindness. Diabetic Ager null mice have decreased retinal capillary degradation and decreased adherent leukocytes in the retinas as compared to wildtype diabetic mice. These diabetic Ager null mice are also protected from an increase in F4/80+ microglia in the retina, as seen in the wildtype diabetic mice. Furthermore, diabetic Ager null mice have increased glyoxalase-1 (GLO-1) activity. GLO-1 detoxifies methylglyoxal (MG), which is a precursor of the RAGE ligand, AGE.36
Diabetic nephropathy is characterized by a thickened glomerular basement membrane, podocyte loss, mesangial proliferation in the glomerulus, fibrosis, protein-rich urine due to increased permeability, and a suppressed glomerular filtration rate. These pathologies are a result of the diabetic hyperglycemic environment, and eventually lead to kidney failure in human and animal models. When Ager is deleted in OVE26 mice, a T1D mouse model of severe diabetic nephropathy, the mice are protected from tubular atrophy, interstitial fibrosis, glomerulosclerosis, and mesangial matrix expansion (Figure 2). Further, electron microscopy studies revealed that podocyte loss and glomerular basement membrane thickening were significantly attenuated in OVE26 mice devoid of Ager. At the functional level, mice devoid of Ager displayed significant protection against albuminuria and from reduction in glomerular filtration rate. Therefore, diabetic Ager null mice display significant protection against reduced renal function in diabetes as compared to diabetic controls.37
Figure 2.

Deletion of Ager in OVE26 mice imparts partial protection from the structural abnormalities of diabetic nephropathy (DN) at age 7 months. (a) No histologic abnormalities were detected in FVB RKO mice (not shown). By contrast, OVE26 mice display well developed features of diabetic nephropathy including diffuse and global mesangial sclerosis and focal hyaline casts (b). OVE26 RKO mice are markedly protected from the development of mesangial sclerosis and tubular cast formation (c). There were significant differences between OVE26 mice and OVE26 RKO mice with respect to percent cortical area occupied by casts (d) and the severity of mesangial sclerosis (e), where 0 =no mesangial sclerosis; 1 =mild; 2 =moderate; 3 =severe (*P <0.05). Original magnifications are marked above each image. Semiquantitative scoring (d,e) was performed on n =7 OVE26 RKO and n =13 OVE26 mice. (Note: RKO =mice devoid of Ager). Republished from Reiniger, N., et al. Diabetes 59, 2043–2054 (2010).
Cardiomyopathies from diabetic conditions are defined as changes in the structure and function of the myocardium. These structural changes include increased left ventricular mass, lipo-toxicity, oxidative stress, necrosis, and interstitial fibrosis. The functional changes include diastolic, systolic, contractile, and mitochondrial dysfunction and exist in both clinical and non-clinical models.38 RAGE expression is increased in the diabetic hearts of db/db mice. This increase in RAGE results in myocardial fibrosis and an altered extracellular matrix structure, which leads to diastolic dysfunction.39 AGE/RAGE signaling also leads to systolic dysfunction through its effects on the ryanodine receptor (RyR). AGE/RAGE signaling causes an increase in oxidative stress in the RyR, which leads to a decrease in sarcoplasmic reticulum calcium content. This decreased SR calcium reserve results in decreased systolic function.40 In ischemia/reperfusion (I/R) injury in diabetes, deletion of Ager is highly protective against loss of cardiac functional myocardium, at least in part due to I/R-mediated generation of RAGE ligand, AGEs.
Atherosclerosis is a macrovascular complication of diabetes and a leading cause of morbidity and mortality in humans and animals with diabetes. It results in the narrowing of arterial walls through the formation of atherosclerotic lesions. Hyperglycemic conditions induce chronic inflammation and injury to the arterial wall, which then leads to oxidized low-density lipoprotein (oxLDL) accumulation in the endothelium. Monocytes infiltrate the area, differentiate into macrophages, and absorb the oxLDL. These processes lead to the formation of foam cells. This cascade of events induces smooth muscle cell proliferation and collagen accumulation, which results in the formation of a lipid-filled lesion with a fibrous cap. These lesions accumulate and can rupture, resulting in a blockade of the blood vessel. RAGE ligands are increased in the diabetic atherosclerotic Apoe null mouse model, as compared to nondiabetic Apoe null controls. Diabetic Ager null/Apoe null mice display decreased leukocyte recruitment, decreased proinflammatory markers, and decreased markers of oxidative stress. Furthermore, diabetic Ager null/Apoe null mice demonstrate reduced atherosclerotic plaque lesion size as compared to diabetic Apoe null controls.41 Further, in distinct studies, deletion of Ager in Apoe null mice rendered diabetic with streptozotocin resulted in reduced lesional macrophage, T cell, and smooth muscle cell content, in parallel with increased lesional collagen (Figure 3).42 Putative roles for the ROCK1 branch of the transforming growth factor-beta (TGF-β) signaling pathway were illustrated by unbiased transcriptomic profiling of aortic tissue from the mice under study. In vitro, RAGE ligand stimulated migration and proliferation of vascular smooth muscle cells isolated from mouse aorta was dependent on ROCK, as illustrated by cellular treatment with inhibitors of this pathway.
Figure 3.
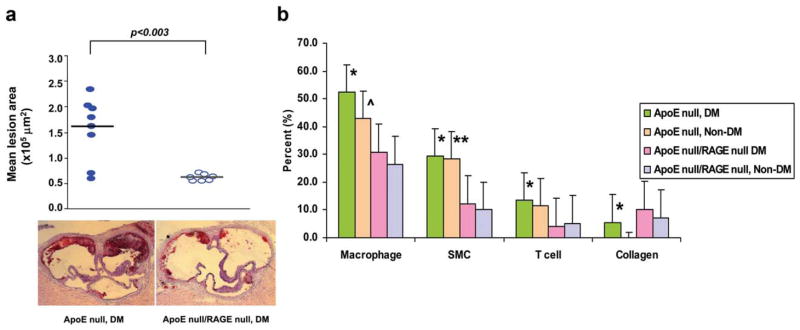
Deletion of Ager suppresses diabetes-accelerated atherosclerosis and effect of diabetes and Ager deficiency on atherosclerotic lesion content. (a) Male Apoe null (n =8) and Apoe null/Ager null mice (n =7) were rendered diabetic with streptozotocin at age 6 weeks. Mice were sacrificed at age 14 weeks and aortas were retrieved. Mean atherosclerotic lesion area at the aortic sinus is reported; statistical considerations are indicated in the text. (b) Immunostaining and picrosirius red staining of atherosclerotic lesions from the indicated Apoe null mice was performed for detection of macrophages, smooth muscle cells, T cells, and collagen (the latter using picrosirius red and polarizing microscopy) at age 24 weeks. Note that the lesions in the non-diabetic or any Ager-deficient mice were quite small at age 14 weeks; hence, detailed lesion characterization was performed at age 24 weeks (after 18 weeks of established hyperglycemia). Lesion areas at the aortic sinus were 6.8 × 105 vs. 0.9 × 105 μm2 in Apoe null diabetic vs. nondiabetic Apoe null mice, respectively; P <0.02. Lesions areas in nondiabetic Apoe null/Ager null mice were 0.5 × 105 μm2; P <0.05 vs. nondiabetic Apoe null mice. Lesion areas in diabetic Apoe null/Ager null mice were 3.8 × 105 μm2; P <0.05 vs. diabetic Apoe null mice. Once we established these relationships at 24 weeks of age, we then proceeded to analyze lesion composition. In all cases, the percent of cell type/lesion area, or %collagen/lesion area, was then calculated using the Zeiss imaging analysis program. n =5 mice/group. Error bars are standard deviations. Statistical considerations are indicated in the figure. In b, each distinct set of bars represents the cell type or collagen content. The four bars in each set represent the genotype and the diabetic state (*P <0.03 vs. Apoe null/Ager null diabetic. **P <0.01 vs. Apoe null/Ager null nondiabetic. ^P <0.02 vs. Apoe null Ager null nondiabetic). Republished from Bu, D.X. et al. Circ. Res. 106, 1040–1051 (2010).
Diabetic patients display markers of delayed wound healing upon injury. After injury, a blood clot is formed, followed by infiltration of inflammatory cells into the wound area to fight infection and digest necrotic tissue. Then new granulation tissue is produced to repair the wound, and is filled with new blood vessels to provide enough nutrients for the new tissue to grow and remain healthy. Epithelial cells subsequently form a cap over the new tissue until the wound matures. Keratinocytes, fibroblasts, macrophages, neutrophils, endothelial cells, and rearrangement of the extracellular matrix (ECM) all play a role in this process. Diabetic hyperglycemic conditions cause abnormalities in these cell types and cell processes including decreased migration, proliferation, and general dysfunction. In a model of diabetic rat skin injury, streptozotocin-induced diabetic rats have increased amounts of AGEs in their skin as compared to nondiabetic rats before injury. After a burn injury, diabetic rats have increased RAGE at the wound site as compared to their nondiabetic control, in which RAGE is barely detectable.43 Nadatani et al. showed that RAGE mRNA expression increases after gastric ulcer healing, and Ager null mice or sRAGE-treated mice display enhanced healing as compared to wildtype (WT) mice.44
SOLUBLE RAGE, DIABETES, AND DIABETIC COMPLICATIONS
Soluble RAGE is thought to act as a decoy to the RAGE receptor in that it binds to and sequesters RAGE ligands, thereby competing with the ability of RAGE ligands to bind to cell surface RAGE and initiate signal transduction. Levels of sRAGEs have been measured in human subjects with obesity and diabetes. Although reports are somewhat conflicting on the directionality of the sRAGE patterns with respect to state or extent of disease, it has been shown, nevertheless, that relationships exist between levels of sRAGEs and disease. Many articles have been published in this area, examples of which are reviewed below.
Clinical studies
In humans, circulating sRAGE increases after bariatric surgery, which was postulated to contribute to lower insulin resistance and weight loss.45 Furthermore, sRAGE levels were shown to inversely correlate with body mass index (BMI),46 and treatment with the widely used class of antidiabetic drugs, thiazolidine-diones, increases sRAGE and esRAGE levels.47 All these studies suggest that increased sRAGE is associated with improved insulin sensitivity.
Circulating sRAGE decreases in patients with atherosclerosis,48 coronary artery disease,49 hypercholesterolemia,50 hypertension,49 vascular dementia, or Alzheimer’s.51 Circulating esRAGE levels decrease in heart failure patients.52 On the contrary, it was reported that in the serum of both T1D and T2D patients, sRAGE and esRAGE levels are increased as compared to nondiabetic controls.53 Similarly, reports from the Collaborative Atorvastatin Diabetes Study (CARDS) reported that levels of both sRAGE and esRAGE were higher in T2D patients with coronary heart disease but not stroke.54 It has been reported that sRAGE levels are also increased in renal dysfunction.55 It is unclear if and to what degree some of the discrepant findings on the directionality of sRAGE levels and disease status may reflect the state of renal function. In this context, Piarulli et al. proposed a model in which levels of sRAGE might be phasic. They suggested that in progression of atherosclerosis, levels of sRAGE gradually decline in parallel with increased levels of AGEs (and other inflammatory ligands). However, upon an acute superimposed event, levels of sRAGE rise.56 Taken together, it is unknown whether the absolute levels of sRAGE/esRAGE are a pathological consequence of the disease and/or if they reflect the body’s attempt to protect itself from the negative effects of RAGE ligand-induced signaling. Finally, in most studies reporting measurements of sRAGEs, the investigators did not report the prevalence of AGER SNPs known to affect levels of the sRAGEs. Hence, much work remains to be done to establish the timing and directionality of sRAGE values in human subjects, both in health and disease.
Many clinical studies measure both sRAGE and the widely used diabetes biomarker, glycosylated hemoglobin 1c (HbA1c), but most of them do not correlate the two. Some studies find sRAGE and HbA1c levels move in the same direction,57 some studies find changes in sRAGE but not in HbA1c,58 and some studies find sRAGE and HbA1c levels move in opposite directions.59 Therefore, if sRAGE correlates with HbA1c, the directionality of that relationship is unclear. These studies cover a wide range of diabetic conditions, complications, and treatments, so the relationship between sRAGE and Hba1c may change based on the specific patient’s condition.
Animal studies
Various animal studies provide evidence for pharmacological levels of sRAGE as a protective molecule from a variety of diabetic complications.
Atherosclerosis
Administration of sRAGE is shown to be protective in atherosclerosis. sRAGE treatment prevents the development of atherosclerosis60 and angiotensin II-mediated atherosclerosis in Apoe null mice.61 sRAGE can reverse atherosclerotic lesion size in diabetic Apoe null mice.62 Mechanistically, sRAGE may bind to hypochlorus acid-modified LDL (HOCL-LDL) in atherosclerotic plaques and prevent HOCL-LDL uptake by the CD36 receptor. HOCL-LDL uptake causes lipid accumulation in macrophages. Therefore, sRAGE may prevent formation of foam cells in atherosclerotic plaques.63
Wound healing
Topical administration of sRAGE to endothelial lesions in C57BL/6 db/db diabetic mice improves wound healing by increasing neovascularization and formation of granulation tissue, and by decreasing the size of the endothelial cap.64 Intraperitoneal injection of sRAGE also improved wound closure in db/db mice in a time-dependent manner. In that setting, sRAGE causes the mice to exhibit an earlier influx of inflammatory cells and cytokines, followed by a later period of healing and repair. In contrast, diabetic mice that did not receive sRAGE had a sustained presence of inflammatory cells and cytokines at the same time the sRAGE-treated mice showed healing and resolution.65
Nephropathy
Wendt et al.66 showed that intraperitoneal injections of sRAGE into diabetic db/db mice is protective in diabetic nephropathy. sRAGE treatment decreases expression of RAGE and the RAGE ligands, S100/calgranulins, in the renal cortex of db/db mice. sRAGE treatment also preserves renal function as sRAGE-treated mice displayed less leakage of albumin in their urine, less expansion of the mesangium, and less thickening of the glomerular basement membrane. This decrease in mesangial area and glomerulosclerosis is mediated, at least in part, by a decrease in TGF-β expression. sRAGE treatment in these mice prevents increases in expression of VEGF and VCAM-1 in renal cortex tissue. As VEGF and VCAM-1 are important mediators of inflammatory cell adhesion and migration, sRAGE treatment prevents the influx of inflammatory cells such as mononuclear phagocytes into glomeruli. sRAGE also inhibits VEGF expression in S100B-treated podocytes in culture.
Retinopathy
A critical factor in the development of diabetic retinopathy is the adhesion of leukocytes to vessels in the retina. This attachment contributes to the breakdown of the retina-blood vessel barrier and prevents blood in the small retinal arteries from reaching the retinal capillaries. The presence of AGEs increases adhesion of leukocytes to endothelial cells in culture. Coincubation of sRAGE and AGEs diminishes the AGE-induced adhesion of leukocytes and endothelial cells as compared to cells coincubated with nonimmunized mouse IgG (a generic protein) and AGE.67
Neuropathy
Increased NFκB signaling is associated with diabetic neuronal dysfunction, and sRAGE treatment of WT diabetic mice prevents the upregulation of NFκB. sRAGE treatment also prevents the increase of interleukin (IL)–6, a proinflammatory cytokine that is regulated by NFκB. AGEs can induce NFκB expression in euglycemic mice, while cotreatment of sRAGE and AGE prevents this upregulation. Functionally, sRAGE restores pain perception in diabetic mice as determined by a hot plate test.34
COMING FULL CIRCLE: RAGE AND THE PATHOGENESIS OF OBESITY
It is now well recognized that AGEs are not simply products of high glucose environments. Hyperlipidemia and increased oxidative stress may also result in the formation of AGEs.5 It was recently shown that the RAGE ligand carboxy methyl lysine (CML)-AGE is highly significantly expressed in human obese vs. lean adipose tissue, in parallel with increased expression of RAGE.68
In animal models, the effect of high-fat diet-induced obesity was tested. Similar to results in human subjects, RAGE ligand content in the adipose tissue increased shortly after hyperglycemia. To test the role of RAGE, mice devoid of Ager and their littermate controls were fed a high-fat diet or low-fat diet. Genetic deletion of Ager results in highly significant protection against high-fat diet-induced obesity and metabolic dysfunction (Figure 4). Energy expenditure was higher in mice devoid of Ager fed a high-fat diet, with no differences noted in food consumption.11 Glucose and insulin tolerance tests showed significantly higher sensitivity to both factors in Ager null mice vs. WT controls fed a high-fat diet. These effects were mediated in part by myeloid RAGE, as lethal irradiation of WT mice followed by reconstitution with Ager null vs. WT bone marrow resulted in partial protection against high-fat diet-induced obesity and metabolic dysfunction. To avoid the potential impact of Ager deletion throughout development and early life, WT mice were treated with sRAGE, either at the time of high-fat diet feeding or 3 weeks after initiation of a high-fat diet. In both cases, sRAGE treatment significantly slowed the increase in body mass observed in the vehicle-treated animals. These studies, for the first time, implicated ligand-RAGE interaction in the pathogenesis of obesity and suggested that the RAGE axis contributed both to the causes of T2D as well as its complications.
Figure 4.
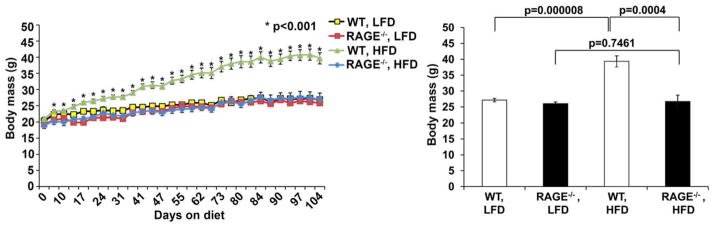
Mice devoid of Ager are resistant to high-fat diet-induced obesity. WT and Ager null male mice were fed low-fat diet or high-fat diet and body mass measured. Time course studies were performed (left panel) and in the right panel the mean body mass at the end of the study is shown. n =7–8 mice per group. *P <0.001 comparing WT vs. Ager null mice fed high-fat diet. Republished from Song, F. et al. Diabetes 63, 1948–1965 (2014).
RAGE SIGNALING AND THE FORMIN mDia1: DECIPHERING THE MECHANISMS
A critical challenge in the biology of RAGE was to decipher the molecular mechanisms by which RAGE signaling is stimulated upon ligand engagement. Our studies using the RAGE cytoplasmic tail (ctRAGE) as “bait” in a yeast-two-hybrid assay identified mDia1 as a putative binding partner.20 mDia1 is a formin molecule adaptor protein that regulates the actin cytoskeleton and is required for RAGE signaling. mDia1 nucleates actin polymerization at the barbed end of the forming actin filament. This nucleation is mediated by profilin bound actin monomers, which prevents spontaneous nucleation and localizes the nucleation to the barbed end.
mDia1 interacts with the cytoplasmic tail domain of RAGE through its FH1 domain to induce cell motility in C6 rat glioma cells. The induced cell motility by RAGE-mDia1 interaction is dependent on the activation of the Rho GTPases Rac1 and Cdc42. It is important to note that this RAGE-mDia1 signaling mechanism is independent of RhoA, a known activator of mDia1.20
Several amino acids in mDia1, such as Arg-5 and Gln-6, are critical for binding to ctRAGE. When vascular smooth muscle cells were transfected with a plasmid that contains mutations to alanine at these two amino acid residues and then stimulated by RAGE ligand S100B, vascular smooth muscle cells lose the ability to phosphorylate AKT, to migrate, and to proliferate. Therefore, RAGE-mDia1 signaling is involved in smooth muscle cell migration and proliferation.69 In guide-wire injury to the femoral artery in mice, deletion of Drf1 (diaphanous related formin, gene encoding mDia1) prevents vascular remodeling.70
Inflammation is a hallmark in the development of insulin resistance and T2D, processes that involve immune cell migration and cell adhesion. mDia1 is required for cell migration in rat C6 glioma cells,20 the macrophage and microglia inflammatory response in human and mouse cell lines,71,72 and cell–cell adhesion, as Dia1 stabilizes adherens junctions in colon carcinoma cells.73
Although more work remains to be done to identify the extent to which RAGE signaling is dependent on mDia1, the studies to date suggest that in a range of cell types and stress settings, RAGE/mDia1 cooperation is important to transduce the downstream effects of RAGE ligands.
TARGETING RAGE AND THERAPEUTIC APPROACHES
Based on the compelling links of RAGE to diabetes pathogenesis and its complications, efforts to target the receptor therapeutically have been launched.
The first agent tested in animal models was sRAGE, the ligand decoy. Experiments in animal models suggested the benefits of therapeutic administration of the agent to animals with diabetic complications and in high-fat diet-fed mice vulnerable to obesity. Studies in human subjects, monitoring the levels of sRAGE, suggested links between the levels and disease activity. However, as noted above, the directionality of the findings was not always consistent. Further, it is necessary to note that as the ligands of RAGE might interact with multiple receptors, sRAGE, although beneficial in animals, might block access to other cellular pathways. Besides sRAGE, other forms of therapies to specifically target RAGE have been developed.
A review of other forms of RAGE therapies reveals a diverse array of approaches (summarized in Figure 5). For example, it has been reported that anti-RAGE antibodies display activity in diabetic nephropathy and other complications.74 RAGE peptide aptamers have been suggested as a means to block RAGE signaling.75 Small-molecule antagonists of RAGE were shown to block immune activation in islet allografts76 in mice and Alzheimer’s disease pathology in mouse models.77 Nanocarriers bearing RAGE siRNAs were tested in animal models of myocardial infarction.78 Finally, it is plausible that blockade of the interaction of ctRAGE with the FH1 domain of mDia1 may represent a logical and targeted means to block RAGE signaling. Given evidence for mDia1 roles in the pathobiologies of RAGE reported to date, it is quite conceivable that such an approach might be beneficial.
Figure 5.

Strategies for targeting RAGE. Experimental evidence from animal models as well as associative evidence in human studies provides strong support for the role of this pathway in the pathogenesis of diabetes and its complications. Indeed, a number of therapeutic strategies targeting RAGE and its signaling pathways are in active therapeutic development. Examples of such strategies are shown in this figure, including sRAGE, the extracellular ligand binding domain of RAGE that sequesters RAGE ligands and blocks their activation of cell surface receptors (I); anti-RAGE antibodies (II); small molecules that block the binding of RAGE ligands to the receptor (III); RAGE peptide aptamers that block ligand binding to the various domains of RAGE (IV); nanocarriers delivering siRNAs to knockdown RAGE expression (V); and small molecules (or other forms of agents) that block the binding of the RAGE tail to the FH1 domain of mDia1 (VI).
SUMMARY AND FUTURE DIRECTIONS
In summary, the evolving biology of RAGE places the receptor and its ligand families in the midst of mechanisms that contribute to the pathogenesis of diabetes and to its complications. Given these considerations, it is reasonable to deduce that fundamental inflammatory signaling cascades go awry when the ligands of RAGE accumulate to pathological levels in obesity and high-fat feeding, autoimmunity, and oxidative stress, even well before the onset of hyperglycemia. Indeed, once hyperglycemia ensues, end-organ damage appears to accelerate. Evidence to date, both in diabetic micro- and macrovascular complications, implicates contributions of inflammatory mechanisms. Stopping the cycle of RAGE ligand, RAGE-mediated cellular stress may be essential in preventing the chronic consequences of hyperglycemia and end-organ failure.
What are the prospects for long-term antagonism of the RAGE axis? Evidence indicates that the prospects may be good. Studies in mice devoid of Ager suggested no apparent abnormalities in homeostasis. However, in stress settings the mice appear to exhibit protection against end-organ injuries. Indeed, the subtle but significant benefits of improved insulin sensitivity in mice devoid of Ager and fed a low-fat diet11 suggest that blocking RAGE may optimize metabolic function by affording protection against cues in the environment that trigger microinflammation and energy conservation.
Acknowledgments
The authors gratefully acknowledge funding for this work from the US Public Health Service, the JDRF, and the ADA. The authors acknowledge the expert assistance of Ms Latoya Woods in the preparation of this article.
Footnotes
CONFLICT OF INTEREST
The authors report no conflicts of interest.
References
- 1.WHO. [Accessed 9 February 2015];Obesity and Overweight. 2015 < http://www.who.int/mediacentre/factsheets/fs311/en/>.
- 2.CDC. [Accessed 9 February 2015];Adult Obesity Facts. 2014 < http://www.cdc.gov/obesity/data/adult.html>.
- 3.CDC. National Diabetes Statistics Report. 2014 < http://www.cdc.gov/diabetes/pubs/statsreport14/national-diabetes-report-web.pdf>.
- 4.Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabetic Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 5.Gaens KH, Stehouwer CD, Schalkwijk CG. Advanced glycation endproducts and its receptor for advanced glycation endproducts in obesity. Curr Opin Lipidol. 2013;24:4–11. doi: 10.1097/MOL.0b013e32835aea13. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt AM, Hori O, Brett J, Yan SD, Wautier JL, Stern D. Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler Thromb. 1994;14:1521–1528. doi: 10.1161/01.atv.14.10.1521. [DOI] [PubMed] [Google Scholar]
- 7.Wu CH, Huang SM, Lin JA, Yen GC. Inhibition of advanced glycation endproduct formation by foodstuffs. Food Funct. 2011;2:224–234. doi: 10.1039/c1fo10026b. [DOI] [PubMed] [Google Scholar]
- 8.Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. 2006;114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rogge MM. The role of impaired mitochondrial lipid oxidation in obesity. Biol Res Nurs. 2009;10:356–373. doi: 10.1177/1099800408329408. [DOI] [PubMed] [Google Scholar]
- 11.Song F, et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes. 2014;63:1948–1965. doi: 10.2337/db13-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmidt AM, et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. 1992;267:14987–14997. [PubMed] [Google Scholar]
- 13.Brett J, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 14.Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006;323:475–488. doi: 10.1007/s00441-005-0069-0. [DOI] [PubMed] [Google Scholar]
- 15.Taguchi A, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 16.Hofmann MA, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 17.Yan SD, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 18.Rai V, et al. Lysophosphatidic acid targets vascular and oncogenic pathways via RAGE signaling. J Exp Med. 2012;209:2339–2350. doi: 10.1084/jem.20120873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chavakis T, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med. 2003;198:1507–1515. doi: 10.1084/jem.20030800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hudson BI, et al. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem. 2008;283:34457–34468. doi: 10.1074/jbc.M801465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallar BJ, Alberts AS. The formins: active scaffolds that remodel the cytoskeleton. Trends Cell Biol. 2003;13:435–46. doi: 10.1016/s0962-8924(03)00153-3. [DOI] [PubMed] [Google Scholar]
- 22.Kuhn S, Geyer M. Formins as effector proteins of Rho GTPases. Small GTPases. 2014;5:e29513. doi: 10.4161/sgtp.29513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thumkeo D, Watanabe S, Narumiya S. Physiological roles of Rho and Rho effectors in mammals. Eur J Cell Biol. 2013;92:303–315. doi: 10.1016/j.ejcb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Lopez-Diez R, Rastrojo A, Villate O, Aguado B. Complex tissue-specific patterns and distribution of multiple RAGE splice variants in different mammals. Genome Biol Evol. 2013;5:2420–2435. doi: 10.1093/gbe/evt188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tam XH, Shiu SW, Leng L, Bucala R, Betteridge DJ, Tan KC. Enhanced expression of receptor for advanced glycation end-products is associated with low circulating soluble isoforms of the receptor in Type 2 diabetes. Clin Sci (Lond) 2011;120:81–89. doi: 10.1042/CS20100256. [DOI] [PubMed] [Google Scholar]
- 26.Koyama H, et al. Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:2587–2593. doi: 10.1161/01.ATV.0000190660.32863.cd. [DOI] [PubMed] [Google Scholar]
- 27.Geroldi D, Falcone C, Emanuele E. Soluble receptor for advanced glycation end products: from disease marker to potential therapeutic target. Curr Med Chem. 2006;13:1971–1978. doi: 10.2174/092986706777585013. [DOI] [PubMed] [Google Scholar]
- 28.Cheng DT, et al. Systemic soluble receptor for advanced glycation endproducts is a biomarker of emphysema and associated with AGER genetic variants in patients with chronic obstructive pulmonary disease. Am J Resp Crit Care Med. 2013;188:948–957. doi: 10.1164/rccm.201302-0247OC. [DOI] [PubMed] [Google Scholar]
- 29.Gaens KH, et al. Association of polymorphism in the receptor for advanced glycation end products (RAGE) gene with circulating RAGE levels. J Clin Endocrinol Metab. 2009;94:5174–5180. doi: 10.1210/jc.2009-1067. [DOI] [PubMed] [Google Scholar]
- 30.Prevost G, et al. Polymorphisms of the receptor of advanced glycation endproducts (RAGE) and the development of nephropathy in type 1 diabetic patients. Diabetes Metab. 2005;31:35–39. doi: 10.1016/s1262-3636(07)70164-7. [DOI] [PubMed] [Google Scholar]
- 31.Ramprasad S, Radha V, Mathias RA, Majumder PP, Rao MR, Rema M. Rage gene promoter polymorphisms and diabetic retinopathy in a clinic-based population from South India. Eye (Lond) 2007;21:395–401. doi: 10.1038/sj.eye.6702239. [DOI] [PubMed] [Google Scholar]
- 32.Balasubbu S, et al. Association analysis of nine candidate gene polymorphisms in Indian patients with type 2 diabetic retinopathy. BMC Med Genet. 2010;11:158. doi: 10.1186/1471-2350-11-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park SY, Kim YA, Hong YH, Moon MK, Koo BK, Kim TW. Up-regulation of the receptor for advanced glycation end products in the skin biopsy specimens of patients with severe diabetic neuropathy. J Clin Neurol. 2014;10:334–341. doi: 10.3988/jcn.2014.10.4.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bierhaus A, et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J Clin Invest. 2004;114:1741–1751. doi: 10.1172/JCI18058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juranek JK, et al. RAGE deficiency improves postinjury sciatic nerve regeneration in type 1 diabetic mice. Diabetes. 2013;62:931–943. doi: 10.2337/db12-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McVicar CM, et al. Role of the receptor for advanced glycation endproducts (RAGE) in retinal vasodegenerative pathology during diabetes in mice. Diabetologia. 2015;58:1129–1137. doi: 10.1007/s00125-015-3523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reiniger N, et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes. 2010;59:2043–2054. doi: 10.2337/db09-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–39. doi: 10.1007/s11154-010-9131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hutchinson KR, Lord CK, West TA, Stewart JA., Jr Cardiac fibroblast-dependent extracellular matrix accumulation is associated with diastolic stiffness in type 2 diabetes. PloS One. 2013;8:e72080. doi: 10.1371/journal.pone.0072080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan D, et al. Effects of advanced glycation end products on calcium handling in cardiomyocytes. Cardiology. 2014;129:75–83. doi: 10.1159/000364779. [DOI] [PubMed] [Google Scholar]
- 41.Soro-Paavonen A, et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bu DX, et al. Activation of the ROCK1 branch of the transforming growth factor-beta pathway contributes to RAGE-dependent acceleration of atherosclerosis in diabetic ApoE-null mice. Circ Res. 2010;106:1040–1051. doi: 10.1161/CIRCRESAHA.109.201103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tian M, et al. The relationship between inflammation and impaired wound healing in a diabetic rat burn model. J Burn Care Res. 2014 doi: 10.1097/BCR.0000000000000171. e-pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 44.Nadatani Y, et al. High-mobility group box 1 inhibits gastric ulcer healing through Toll-like receptor 4 and receptor for advanced glycation end products. PloS One. 2013;8:e80130. doi: 10.1371/journal.pone.0080130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brix JM, Hollerl F, Kopp HP, Schernthaner GH, Schernthaner G. The soluble form of the receptor of advanced glycation endproducts increases after bariatric surgery in morbid obesity. Int J Obes (Lond) 2012;36:1412–1417. doi: 10.1038/ijo.2012.107. [DOI] [PubMed] [Google Scholar]
- 46.Norata GD, et al. Circulating soluble receptor for advanced glycation end products is inversely associated with body mass index and waist/hip ratio in the general population. Nutr Metab Cardiovasc Dis NMCD. 2009;19:129–134. doi: 10.1016/j.numecd.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 47.Tan KC, et al. Thiazolidinedione increases serum soluble receptor for advanced glycation end-products in type 2 diabetes. Diabetologia. 2007;50:1819–1825. doi: 10.1007/s00125-007-0759-0. [DOI] [PubMed] [Google Scholar]
- 48.Hudson BI, et al. Association of serum soluble receptor for advanced glycation end-products with subclinical cerebrovascular disease: the Northern Manhattan Study (NOMAS) Atherosclerosis. 2011;216:192–198. doi: 10.1016/j.atherosclerosis.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Falcone C, et al. Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arterioscler Thromb Vasc Biol. 2005;25:1032–1037. doi: 10.1161/01.ATV.0000160342.20342.00. [DOI] [PubMed] [Google Scholar]
- 50.Santilli F, et al. Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radic Biol Med. 2007;43:1255–1262. doi: 10.1016/j.freeradbiomed.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 51.Emanuele E, et al. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol. 2005;62:1734–1736. doi: 10.1001/archneur.62.11.1734. [DOI] [PubMed] [Google Scholar]
- 52.Wang LJ, Lu L, Zhang FR, Chen QJ, De Caterina R, Shen WF. Increased serum high-mobility group box-1 and cleaved receptor for advanced glycation endproducts levels and decreased endogenous secretory receptor for advanced glycation endproducts levels in diabetic and non-diabetic patients with heart failure. Eur J Heart Fail. 2011;13:440–449. doi: 10.1093/eurjhf/hfq231. [DOI] [PubMed] [Google Scholar]
- 53.Prasad K. Low levels of serum soluble receptors for advanced glycation end products, biomarkers for disease state: myth or reality. Int J Angiol. 2014;23:11–16. doi: 10.1055/s-0033-1363423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Colhoun HM, et al. Total soluble and endogenous secretory receptor for advanced glycation end products as predictive biomarkers of coronary heart disease risk in patients with type 2 diabetes: an analysis from the CARDS trial. Diabetes. 2011;60:2379–2385. doi: 10.2337/db11-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vazzana N, Santilli F, Cuccurullo C, Davi G. Soluble forms of RAGE in internal medicine. Intern Emerg Med. 2009;4:389–401. doi: 10.1007/s11739-009-0300-1. [DOI] [PubMed] [Google Scholar]
- 56.Piarulli F, Sartore G, Lapolla A. Glyco-oxidation and cardiovascular complications in type 2 diabetes: a clinical update. Acta Diabetol. 2013;50:101–110. doi: 10.1007/s00592-012-0412-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yanagisawa K, et al. Switching to multiple daily injection therapy with glulisine improves glycaemic control, vascular damage and treatment satisfaction in basal insulin glargine-injected diabetic patients. Diabetes Metab Res Rev. 2014;30:693–700. doi: 10.1002/dmrr.2537. [DOI] [PubMed] [Google Scholar]
- 58.Koyama H, et al. Comparison of effects of pioglitazone and glimepiride on plasma soluble RAGE and RAGE expression in peripheral mononuclear cells in type 2 diabetes: randomized controlled trial (PioRAGE) Atherosclerosis. 2014;234:329–334. doi: 10.1016/j.atherosclerosis.2014.03.025. [DOI] [PubMed] [Google Scholar]
- 59.Amin MN, Mosa AA, El-Shishtawy MM. Clinical study of advanced glycation end products in ogyptian diabetic obese and non-obese patients. Int J Biomed Sci IJBS. 2011;7:191–200. [PMC free article] [PubMed] [Google Scholar]
- 60.Park L, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 61.Lee D, et al. The effect of soluble RAGE on inhibition of angiotensin II-mediated atherosclerosis in apolipoprotein E deficient mice. PloS One. 2013;8:e69669. doi: 10.1371/journal.pone.0069669. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Bucciarelli LG, et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106:2827–2835. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- 63.Marsche G, Weigle B, Sattler W, Malle E. Soluble RAGE blocks scavenger receptor CD36-mediated uptake of hypochlorite-modified low-density lipoprotein. FASEB J. 2007;21:3075–3082. doi: 10.1096/fj.07-8316com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wear-Maggitti K, Lee J, Conejero A, Schmidt AM, Grant R, Breitbart A. Use of topical sRAGE in diabetic wounds increases neovascularization and granulation tissue formation. Ann Plast Surg. 2004;52:519–521. doi: 10.1097/01.sap.0000122857.49274.8c. discussion 22. [DOI] [PubMed] [Google Scholar]
- 65.Goova MT, et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159:513–525. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wendt TM, et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol. 2003;162:1123–1137. doi: 10.1016/S0002-9440(10)63909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moore TC, et al. The role of advanced glycation end products in retinal microvascular leukostasis. Invest Ophthalmol Visual Sci. 2003;44:4457–4464. doi: 10.1167/iovs.02-1063. [DOI] [PubMed] [Google Scholar]
- 68.Gaens KH, et al. Nepsilon-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler Thromb Vasc Biol. 2014;34:1199–1208. doi: 10.1161/ATVBAHA.113.302281. [DOI] [PubMed] [Google Scholar]
- 69.Rai V, et al. Signal transduction in receptor for advanced glycation end products (RAGE): solution structure of C-terminal rage (ctRAGE) and its binding to mDia1. J Biol Chem. 2012;287:5133–5144. doi: 10.1074/jbc.M111.277731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toure F, et al. Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ Res. 2012;110:1279–1293. doi: 10.1161/CIRCRESAHA.111.262519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu Y, et al. Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J Biol Chem. 2010;285:23233–23240. doi: 10.1074/jbc.M110.117457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bianchi R, Kastrisianaki E, Giambanco I, Donato R. S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J Biol Chem. 2011;286:7214–7226. doi: 10.1074/jbc.M110.169342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol. 2002;4:408–415. doi: 10.1038/ncb796. [DOI] [PubMed] [Google Scholar]
- 74.Jensen LJ, Denner L, Schrijvers BF, Tilton RG, Rasch R, Flyvbjerg A. Renal effects of a neutralising RAGE-antibody in long-term streptozotocin-diabetic mice. J Endocrinol. 2006;188:493–501. doi: 10.1677/joe.1.06524. [DOI] [PubMed] [Google Scholar]
- 75.Reverdatto S, Rai V, Xue J, Burz DS, Schmidt AM, Shekhtman A. Combinatorial library of improved peptide aptamers, CLIPs to inhibit RAGE signal transduction in mammalian cells. PloS One. 2013;8:e65180. doi: 10.1371/journal.pone.0065180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen Y, et al. RAGE ligation affects T cell activation and controls T cell differentiation. J Immunol. 2008;181:4272–4278. doi: 10.4049/jimmunol.181.6.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deane R, et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. 2012;122:1377–1392. doi: 10.1172/JCI58642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ku SH, et al. Deoxycholic acid-modified polyethylenimine based nanocarriers for RAGE siRNA therapy in acute myocardial infarction. Arch Pharm Res. 2015 doi: 10.1007/s12272-014-0527-x. e-pub ahead of print. [DOI] [PubMed] [Google Scholar]