

ABSTRACT
The ability of bacteria to metabolize glucosamine (GlcN) and N-acetyl-d-glucosamine (GlcNAc) is considered important for persistent colonization of the oral cavity. In the dental caries pathogen Streptococcus mutans, the NagR protein regulates the expression of glmS, which encodes a GlcN-6-P synthetase, and nagA (GlcNAc-6-P deacetylase) and nagB (GlcN-6-P deaminase), which are required for the catabolism of GlcNAc and GlcN. Two NagR-binding sites (dre) were identified in each of the promoter regions for nagB and glmS. Using promoter-reporter gene fusions, the role of each dre site was examined in the regulation of glmS and nagB promoter activities in cells grown with glucose, GlcNAc, or GlcN. A synergistic relationship between the two dre sites in the glmS promoter that required proper spacing was observed, but that was not the case for nagB. Binding of purified NagR to DNA fragments from both promoter regions, as well as to dre sites alone, was strongly influenced by particular sugar phosphates. Using a random mutagenesis approach that targeted the effector-binding domain of NagR, mutants that displayed aberrant regulation of both the glmS and nagAB genes were identified. Collectively, these findings provide evidence that NagR is essential for regulation of genes for both the synthesis and catabolism of GlcN and GlcNAc in S. mutans, and that NagR engages differently with the target promoter regions in response to specific metabolites interacting with the effector-binding domain of NagR.
IMPORTANCE Glucosamine and N-acetylglucosamine are among the most abundant naturally occurring sugars on the planet, and they are catabolized by many bacterial species as sources of carbon and nitrogen. Representing a group called lactic acid bacteria (LAB), the human dental caries pathogen Streptococcus mutans is shown to differ from known paradigm organisms in that it possesses a GntR/HutC-type regulator, NagR, that is required for the regulation of both catabolism of GlcN and biosynthesis. Results reported here reveal a simple and elegant mechanism whereby NagR differentially regulates two opposing biological processes by surveying metabolic intermediates. This study provides insights that may contribute to the development of novel therapeutic tools to combat dental caries and other infectious diseases.
INTRODUCTION
Amino sugars, including glucosamine (GlcN) and N-acetyl-d-glucosamine (GlcNAc or NAG), are some of the most abundant carbohydrates on earth. Since amino sugars can serve as sources of carbon and nitrogen, many bacteria have the capacity to utilize them for energy production and growth. We recently showed that the human dental pathogen Streptococcus mutans internalizes GlcN and GlcNAc via the phosphoenolpyruvate (PEP):sugar phosphotransferase system (PTS), which consists of a general enzyme I (EI) and a phospho-carrier protein (HPr) that transfer the phosphoryl group from PEP to a variety of substrate-specific EII enzymes responsible for the concurrent phosphorylation and internalization of their cognate sugars (1). S. mutans transports GlcN and GlcNAc primarily via the EIIMan permease, a fructose:mannose-type EII that is also the dominant transporter for glucose, mannose, and galactose (2–4). In fact, the PTS is the primary route for amino sugar uptake for most bacteria, providing cells with internalized GlcN-6-phosphate (GlcN-6-P) or GlcNAc-6-phosphate (GlcNAc-6-P) (5–7). To catabolize these phosphosugars, bacteria produce a GlcNAc-6-P deacetylase, encoded by nagA, that converts GlcNAc-6-P into GlcN-6-P. GlcN-6-P, generated either from GlcNAc by NagA or by PTS transport of GlcN, is deaminated by a GlcN-6-P deaminase (nagB) and concurrently isomerized to fructose-6-P (F-6-P), which can enter the Embden-Meyerhof-Parnas pathway (5). When these amino sugars are not available in the environment, most bacteria synthesize GlcN-6-P from F-6-P and glutamine to provide the building blocks for peptidoglycan and other cellular constituents. The gene encoding GlcN-6-P synthetase, glmS, and the catabolic genes, nagA and nagB, often are regulated oppositely in response to the availability of exogenous amino sugars (8), with the catabolic system being active when sufficient exogenous GlcNAc or GlcN is present. However, bacteria maintain a requisite balance between synthesis and catabolism of these amino sugars through a variety of mechanisms exerted at the transcriptional and posttranscriptional levels.
In Escherichia coli, the ROK (repressor, open reading frame, and kinase)-type transcription regulator NagC negatively regulates the transcription of the nagAB operon and a divergently transcribed PTS permease (nagE). The repressor activity of NagC depends on its binding to palindromic dre (for DasR responsive element) (9, 10) sites in the promoter regions of the target genes. When GlcNAc is present in the environment, intracellular GlcNAc-6-P accumulates and activates the expression of genes for GlcNAc and GlcN catabolism by disrupting the interaction between NagC and dre sites (5, 11–13). In the Gram-positive bacterium Bacillus subtilis, a GntR/HutC-type regulator, designated NagR (formerly YvoA), binds to nonhomologous operator sites, also called dre (14, 15), to control the transcription of nagA, nagB, and nagP, which codes for a GlcNAc-specific PTS permease. Unlike in E. coli, though, the allosteric effector(s) that alleviates repression of these target genes by NagR remains undetermined (14, 16, 17). Recently, an additional system encoded by ybgBA-gamAP and the GntR-type regulator GamR were discovered in B. subtilis and shown to encode products that regulate and effect the catabolism of GlcN (7, 17).
For the biosynthesis of GlcN-6-P, much of the regulation occurs at the posttranscriptional level. In E. coli and related species, the translational efficiency of the glmS mRNA is controlled by two small regulatory RNAs, GlmZ and GlmY, in response to the intracellular levels of GlcN-6-P (11, 18). In B. subtilis, however, the stability of the glmS transcript is controlled by a ribozyme structure formed in the 5′ untranslated region of the glmS mRNA, and the degradative activity of the ribozyme is activated by GlcN-6-P.
Despite advances in understanding the regulation of GlcN/GlcNAc metabolism in these model organisms, significant gaps remain in the understanding of how these pathways are governed in those lactic acid bacteria (LAB) that play significant roles in dental caries and other common infectious diseases (19). Recently, it was shown that the PTS was responsible for GlcN and GlcNAc internalization by the dental caries pathogen S. mutans, and that the loss of NagB or GlmS significantly affected the expression of virulence attributes in this organism (3, 8). Importantly, catabolism of GlcNAc or GlcN also yields ammonia, which can increase ΔpH (i.e., the difference in pH inside and outside the cell) and be utilized for the production of amino acids and other essential compounds (3). Some preliminary insights into molecular mechanisms by which S. mutans regulates GlcNAc/GlcN metabolism via NagA, NagB, and GlmS indicate that there are substantial differences from model organisms. In particular, we recently reported that the loss of an apparent homolog of NagR in S. mutans led to the derepression not only of nagA and nagB but also of glmS (3). Moreover, (i) the glmS gene of S. mutans lacks the ribozyme sequences conserved in many Gram-positive bacteria, (ii) dre sites could be identified upstream of the glmS coding sequence, and (iii) purified NagR of S. mutans could bind to the glmS promoter region in vitro (3). Here, we report a more detailed characterization of NagR with respect to its interaction with dre sites and the mechanisms by which it exerts control over the genes needed for the biosynthesis and catabolism of GlcN and GlcNAc.
MATERIALS AND METHODS
Strains and growth conditions.
S. mutans strains were routinely maintained on brain heart infusion (BHI; Difco Laboratories, Detroit, MI) agar plates. Antibiotics (Sigma, St. Louis, MO) were added to the agar plates as required at the following concentrations: kanamycin, 1 mg/ml; erythromycin, 10 μg/ml; and spectinomycin, 1 mg/ml. For enzymatic assays, S. mutans strains first were cultivated overnight in TV base medium (20) supplemented with specified carbohydrates (Thermo Fisher Scientific, Waltham, MA, and Sigma) and then diluted at a 1:50 ratio into the same, fresh TV medium and cultured until the optical density at 600 nm (OD600) reached around 0.5 (exponential phase). For growth monitoring using a Bioscreen C lab system (Helsinki, Finland), cells were cultured similarly overnight and subcultured until exponential phase in BHI or TV medium before being inoculated at a 1:150 dilution into fresh modified FMC medium containing only 20 mM GlcNAc (3).
DNA manipulation.
Standard molecular cloning protocols (21) were followed in the manipulation of various forms of DNA products, and transformation assays were performed primarily according to an established procedure that utilizes the natural competence phenotype displayed by S. mutans (22), with some modifications (23). All oligonucleotides used in this study were synthesized by Integrated DNA Technologies (Coralville, IA), and their sequences are listed in Table S1 in the supplemental material. To construct the nagR-ER mutant (which contains two amino acid replacements, i.e., E30A and R31A), a 1.5-kbp DNA fragment was generated using a recombinant PCR (or overlap extension PCR) to introduce into NagR amino acid replacements of E30A and R31A, which were engineered into the primer sequences. Specifically, a pair of primers, nagR-1 and nagRE30AR31A-3′, was used to generate the first half of the intended product, and another pair (nagRE30AR31A-5′ and busR-AS) for the second half was used, with a 41-bp sequence in overlap. These DNA fragments then served as templates in a recombinant PCR that used flanking primers nagR-1 and busR-AS for the amplification of the final product. Subsequently, this 1.5-kbp mutator DNA was used together with a helper plasmid, pLacG-em, in a cotransformation assay to introduce into UA159 background the nagR-ER mutation, a reversible lacG null mutation, and resistance to erythromycin (23). After screening by allele-specific PCR (with detection primer nagRE30AR31A-3′MAMA) (24) and confirmation of the nagR-ER mutation by sequencing, the pLacG-em plasmid was cured by passaging on lactose-based agar plates. Similarly, to introduce random nagR mutations into the background of UA159, a 500-bp DNA product containing only the putative effector-binding domain (from amino acids [aa] 88 to 226) of nagR was created using error-prone PCRs (25) and used along with a small amount of PglmS::lacZ fusion integration plasmid (about 1,000-fold less than the PCR product) in the transformation of strain UA159. Kanamycin-resistant transformants then were screened for the altered expression of the PglmS::lacZ fusion using X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; Sigma)-containing plates and β-galactosidase assays compared to that of the parental strain carrying the same reporter fusion.
Mutations of the NagR-binding sites (dre) upstream of the coding sequence of glmS, including Mdre1, Mdre2, and Mdre1/2, first were established on the chromosome of the wild-type strain UA159 by following the protocol used above. For example, a two-step recombinant PCR was carried out with primers (PglmSMdre1-5′ and PglmSMdre1-3′) containing an Mdre1 mutation that replaced the highly conserved site of dre (TATAC) with nucleotides GTGTA, as well as flanking primers (glmS-730up and glmS-700rev), resulting in a 2-kbp mutator DNA. This mutator DNA then was used in a cotransformation assay, along with the indicator plasmid pLacG-em, to mutagenize UA159. Positive clones were identified by screening with allele-specific PCR, using the primer PglmSMdre1-5′MAMA, and validated by sequencing. The other dre mutations of glmS were constructed similarly. In anticipation of altered GlcN-6-P synthetase activities in these mutants, 0 or 5 mM GlcNAc was included in the erythromycin-containing agar plates, despite the fact that none of these mutations led to a GlmS-negative phenotype. These mutants subsequently were used to provide template DNA in the construction of mutant versions of PglmS::cat fusions and the preparation of DNA probes for electrophoretic mobility shift assay (EMSA).
Promoter-reporter gene fusions were constructed by inserting a DNA fragment containing the promoter and the dre sequences, including the cognate ribosome-binding site, into an integration vector, pJL84 (26), in front of a promoterless cat gene or into a similar vector, pPMZ (27), that carries a promoterless lacZ gene originated from Streptococcus salivarius. Both vectors are suicide plasmids capable of delivering exogenous DNA into an unrelated phnA-mtlD locus of the chromosome. Notably, an alternative start site, GTG, for the nagA gene, 27 bp downstream from that of the current GenBank annotation, was used for creating the PnagA::cat fusion, as no activities were detected in the fusion constructed using the original start site.
Mutagenesis of the dre sites upstream of nagB was performed only on the plasmid vector that carries the PnagB::cat promoter fusion using the PCR-based procedure described above. DNA fragments subsequently were amplified from plasmid DNA carrying various derivatives of the nagB promoter and used as probes in EMSA.
Recombinant NagR preparation and protein-DNA interactions.
Recombinant NagR protein was overexpressed in E. coli and purified by following a procedure described elsewhere (3), with some modifications. Briefly, a maltose-binding protein (MBP) fusion of NagR was expressed from a vector plasmid, pMal-p2X (New England BioLabs, Ipswich, MA), carried by strain DH10B through induction with 0.05 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Purified MBP-NagR protein then was treated with factor Xa protease (NEB) in a buffer that contained 20 mM Tris-Cl, pH 8.0, 100 mM NaCl, and 2 mM CaCl2 at room temperature for at least 6 h, followed by incubation at 4°C overnight. As we have noted previously, the NagR protein, when overexpressed in vitro, tends to aggregate and precipitate out of solution. Taking advantage of this phenotype, aggregates of cleaved NagR protein were pelleted by brief centrifugation at 4°C, washed twice with the same factor Xa buffer, and then dissolved in 1× protein binding buffer (50 mM KCl, 1 mM EDTA, 5 mM dithiothreitol, 10 mM HEPES, pH 7.9, and 10% glycerol) supplemented with 300 mM NaCl.
Purified NagR protein was used in an EMSA by following a previously used protocol described elsewhere (3) and a fluorescence polarization (FP) assay (28). For EMSA, biotinylated probes were created through PCR amplification using primers that contain 5′-biotin modification and DNA templates carrying various alleles of the target promoters (described above). As an effort to reduce the aggregation of NagR, we have included 0.4 mg/ml bovine serum albumin (BSA) in all EMSAs in this report. For FP with NagR, a 45-nucleotide (nt), 5′-6-carboxyfluorescein (6FAM)-labeled oligonucleotide (6FAM-dre) was synthesized to form a stem-loop structure that contains the consensus binding site (dre) for NagR. Reactions were carried out in a 0.2-ml volume at room temperature in 1× protein binding buffer (described above) that also contained 10 ng/μl of poly(dI-dC), 5 mg/ml of MgCl2, 1 nM 6FAM-dre probe, and NagR protein at concentrations ranging from 5 nM to 1,000 nM. When specified, 1 to 5 mM NaCl and various sugar phosphates, including G-6-P, F-6-P, GlcNAc-6-P, and GlcN-6-P, also were added to the reaction mixtures. Measurements of the FP of each reaction was taken using a Synergy 2 (Biotek, Winooski, VT) plate reader, twice at room temperature, and a nonlinear fit binding curve was generated using GraphPad Prism version 5.00 for Macintosh (GraphPad Software, San Diego, CA) to determine the equilibrium dissociation constant (Kd). A single-site, saturation binding equation, Y = (Bmax · X)/(Kd + X) + NS · X, where Bmax represents the projected FP values under maximum specific binding and NS stands for the rate of nonspecific binding, was used to fit the curves. The goodness of fit was ≥0.95.
Enzymatic assays and quantitative RT-PCR.
Chloramphenicol acetyltransferase (CAT) (29) and β-galactosidase (30) assays were performed according to protocols routinely used in our laboratory, as described elsewhere (26, 27). For quantitative RT-PCR, total RNA was extracted from exponentially growing cells using an RNeasy minikit (Qiagen) (31) and then subjected to reverse transcription reactions using SuperScript III reverse transcriptase and random hexamers (Thermo Fisher). Subsequently, gene-specific primers were used to quantify each mRNA using the CFX96 Touch real-time PCR detection system (Bio-Rad, Hercules, CA) by following instructions from the supplier. A housekeeping gene (gyrA) served as an internal control for calculating fold change in transcript levels according to the ΔΔCq method, where Cq is quantification cycle (32).
RESULTS
Differential regulation of glmS and nagB by NagR requires dre.
A strain of S. mutans lacking the GntR/HutC-type regulator designated NagR showed aberrant expression of nagA, nagB, and glmS genes in response to exogenous GlcN and GlcNAc, and a purified recombinant NagR protein could bind to DNA fragments containing the promoter regions of all three genes (3). Since nagA and nagB are regulated oppositely to glmS in response to amino sugar availability, a mechanism must exist for NagR to interact differently with its target genes depending on the amount of GlcNAc and/or GlcN or their phosphorylated derivatives. We first evaluated the requirement for NagR and the conserved cis elements (dre) in the regulation of nagAB and glmS genes. As shown in Fig. 1A, two regions with sequence similarities to known dre sites can be identified near the promoters of glmS and nagB. Promoter-cat fusions were constructed using DNA fragments containing these promoter regions and introduced into the wild-type strain UA159, into an otherwise isogenic nagR null mutant, and into a strain carrying a mutated nagR, designated nagR-ER, which contains two amino acid replacements (E30A and R31A) (Fig. 1B). Glu30 and Arg31 are conserved residues that were shown to be required for DNA-binding activities of a homologous, GntR-type regulator, YvoA/NagR of Bacillus subtilis (15). CAT assays were carried out using these strains that had been cultivated in TV medium supplemented with various carbohydrates.
FIG 1.

Schematic of the upstream regions of the glmS and nagB genes (A) and alignment of sequences of NagR from S. mutans and YvoA/NagR from B. subtilis (B). (A) The locations of the predicted promoter elements are underlined and labeled as −35, −10, and +1 (transcription initiation site), and the start codon of each gene is represented by an arrow. The putative binding sites (dre1 and dre2) (see Table S2 in the supplemental material for the consensus sequence) for NagR are shown in boxes and labeled, with engineered modifications described below in italics. Also delineated are deletions and insertions introduced to alter the phasing of the dre sites. (B) Underlined are sites of mutations engineered in the DNA-binding domain (aa 2 to 69) near the N terminus of NagR and random mutations isolated in the putative effector-binding domain (aa 88 to 226) near the C terminus.
In wild-type S. mutans (Fig. 2), the expression of nagA and especially nagB was higher in cells growing on GlcN than on glucose, whereas glmS expression was much higher in glucose-grown cells than in GlcN-grown cells, confirming previous results (3, 8). However, in the nagR deletion mutant, glmS expression was greatly elevated regardless of the carbohydrate source, nagB expression was actually higher in glucose-grown cells than in GlcN-grown cells of the mutant, and nagA expression was elevated in cells lacking NagR compared with the wild-type strain grown on glucose. When the same experiments were performed with the nagR-ER strain, in which two amino acid substitutions would be predicted to render NagR incapable of binding to target sites, the pattern and magnitude of changes in gene expression was very similar to that seen with the nagR deletion mutant (Fig. 2).
FIG 2.

CAT assays measuring the promoter activities of glmS, nagB, and nagA, with each fused to a promoterless cat gene, in the backgrounds of wild-type UA159, a nagR null mutant, and the nagR-ER (E30A, R31A) mutant. Bacterial strains were cultivated in TV medium supplemented with glucose (A) or GlcN (B) as sole carbohydrates and harvested and processed for CAT assays as detailed in Materials and Methods. sp., specific. The bars represent the average activities (expressed in nanomoles per milligram of protein per minute) of three independent cultures, and the error bars denote standard deviations.
To further establish that NagR exerts its regulatory functions through interactions with conserved dre sites, we constructed mutant versions of the PnagB::cat and PglmS::cat promoter fusions by replacing the conserved 5-nucleotide dre sequence (TAT/GAC) of the various predicted dre sites with the sequence GTGTA (designated Mdre) (Fig. 1A). Since there are two putative dre sites in each of the nagB and glmS promoter regions (3), Mdre replacements were created individually and in combinations, resulting in various derivatives of the reporter fusions (Fig. 1A, 3, and 4). CAT assays were performed using promoter fusion strains created in the UA159 (wild-type) genetic background and the nagR deletion mutant that were grown with glucose or GlcN as the primary carbohydrate source. When the expression of an Mdre1 mutant of the PglmS::cat fusion (PglmSMdre1::cat) was measured, there was nearly complete derepression of the modified glmS promoter in both the wild-type and nagR mutant genetic backgrounds, with CAT activities of TV-glucose cultures being comparable to that of TV-GlcN cultures and expression levels similar to that in the nagR deletion mutant carrying a wild-type PglmS::cat fusion (Fig. 3). Second, when dre2 was mutated in the glmS promoter fusion strain (PglmSMdre2::cat), results nearly identical to those of PglmSMdre1::cat were obtained. Intriguingly, when both dre sites were mutated (PglmSMdre1/2::cat) in the wild-type genetic background, 80 to 100% higher CAT activity was expressed in cells cultured in glucose and GlcN, respectively, than what was measured in the single dre mutants grown under identical conditions. Interestingly, this increase in promoter activity in the double dre mutant required an intact nagR gene, as expression levels of the strain carrying mutations in both dre sites was comparable to that of the dre single mutants in the nagR deletion strain (Fig. 3).
FIG 3.

CAT specific activities (expressed in nanomoles of chloramphenicol acetylated per milligram of protein per minute) of fusions constructed using wild-type (W.T.) and various mutant versions of the glmS promoter in the backgrounds of UA159 and a nagR null mutant. Mutations in Mdre1, Mdre2, or Mdre1/2 are replacements of a conserved TATAC sequence in corresponding dre sites with GTGTA; Δ5nt and Δ10nt are truncations of a 5- or 10-nt sequence located between two dre sites without compromising the −10 promoter structure; 5ntRDM is a result of sequence randomization of the same 5 nucleotides in Δ5nt. Bacteria were cultured in TV base medium supplemented with 0.5% glucose (Glc) or GlcN and then harvested and processed for CAT assays as detailed in Materials and Methods. Results are derived from triplicate assays from three independent cultures and are presented as averages ± standard deviations.
FIG 4.

CAT specific activities (expressed in nanomoles per milligram of protein per minute) of fusions constructed with wild-type (W.T.) and mutant versions of the nagB promoter region in the background of UA159 and its otherwise-isogenic nagR null mutant. Mutation in Mdre1 is a replacement of a conserved TATAC sequence in dre1 with GTGTA, while Mdre2 is a replacement of similarly conserved TAGACC in dre2 with GTGTAA. +5nt and +10nt are insertions of a 5- and 10-nt random sequence, respectively, between two dre sites. Bacteria were cultured in TV base medium supplemented with 0.5% glucose (Glc) or GlcN and then harvested for CAT assays as detailed in Materials and Methods. Results are derived from three independent cultures assayed in triplicate and are presented as averages ± standard deviations.
Since dre1 in the nagB promoter overlaps the presumptive −10 element and dre2 overlaps the transcription initiation site (Fig. 1A), Mdre1 and Mdre2 mutations likely had adverse effects on the overall function of the promoter. This was reflected in the fact that all dre mutants of the PnagB::cat fusion expressed lower levels of CAT than did the wild-type promoter fusion in the nagR deletion mutant (Fig. 4). Still, the patterns of expression were clearly different from those observed for dre mutations in the glmS promoter fusion strain. First, unlike the case for PglmS::cat, the introduction of the Mdre1 mutation into the PnagB::cat fusion resulted in only partial derepression of the nagB promoter in glucose-grown cells, and the promoter remained responsive to induction by GlcN. Also of note, the loss of NagR resulted in further derepression of the Mdre1 mutant in the nagB promoter fusion strain, but there was no further induction by GlcN. Also, when the second dre was mutated (PnagBMdre2::cat), only partial derepression in glucose-grown cells again was observed, although the magnitude of derepression was lower than that for the Mdre1 mutation. Still, the behavior of the strains carrying individual Mdre1 and Mdre2 mutations in the nagR deletion mutant were very similar. Finally, when both dre sites were mutated, the fusion, PnagBMdre1/2::cat, expressed lower overall activities than either single mutant, with higher expression in glucose-grown cells than those grown in GlcN, regardless of whether an intact copy of nagR was present.
Therefore, a fundamental difference between nagB and glmS regulation is that individual dre sites play a much greater role in the regulation of glmS, and the double deletion of dre in the glmS promoter yielded much higher activity than the single mutants in the wild-type genetic background able to produce a functional NagR protein. In contrast, the loss of individual dre sites in the nagB promoter caused only partial derepression, and the loss of Mdre2 had less impact than the loss of Mdre1 in cells growing on glucose. Further, for nagB, the loss of both dre sites did not lead to further alleviation of repression and instead yielded much lower expression levels than the wild-type fusion found in cells lacking NagR, likely due to the overlap of the dre site with RNAP binding sites. It is also notable that the two dre sites in the nagB region are positioned with no space between them (20 bp in center-to-center distance), whereas those in the glmS promoter region are 57 bp apart (77 bp center to center).
The interaction of NagR with two dre sites differs for glmS and nagB.
When cooperative binding (or synergism) by a transcriptional regulator occurs, the spacing between the binding sites often is critical for proper control of gene expression. This is often a result of constraints imposed by the helical periodicity of DNA. To investigate this possibility in the functioning of NagR, additional mutations were introduced into the promoter fusions described above (Fig. 1A). Specifically, a 5-nucleotide deletion (Δ5nt) was engineered in the region between the −10 and dre2 of the glmS promoter to create a loss of one-half rotation of the DNA helix, while a 10-nucleotide deletion (Δ10nt) was introduced in the same region to maintain helical periodicity, resulting in strains PglmSΔ5nt::cat and PglmSΔ10nt::cat, respectively (Fig. 3). The expression of cat in these mutant derivatives was measured in the wild-type strain and nagR deletion mutant grown in TV medium supplemented with glucose or GlcN. As shown in Fig. 3, the PglmSΔ5nt::cat fusion in the wild-type genetic background expressed CAT at levels that were substantially higher than those of the single dre mutants but not quite as high as those of the double dre mutants, whether cells were grown on glucose or GlcN. In the nagR deletion strain growing on glucose, the PglmSΔ5nt::cat fusion expressed CAT at levels 50% higher than those when the dre sites were intact; CAT levels were lower in the Δ5nt mutant in cells growing on GlcN than on glucose but were the same between the Δ5nt mutant and the intact dre construct in the nagR mutant. As an additional control, another mutant of the PglmS::cat fusion was engineered by replacing the same 5-nt sequence deleted above with a random 5-nt sequence (Fig. 1A). The resultant fusion, PglmS5ntRDM::cat, produced CAT activities nearly identical to those expressed from the wild-type fusion under all conditions and genetic backgrounds tested (Fig. 3). Thus, it appears that binding by NagR to the neighboring dre sites plays critical roles in the regulation of glmS in response to carbohydrate source. In all cases, the Δ5nt deletion caused derepression and a loss of sensitivity to the presence of GlcN. Conversely, the PglmSΔ10nt::cat fusion in the wild-type background behaved more like the intact dre promoter fusion, with higher expression in glucose and repression by GlcN, apparently mediated by NagR, as evidenced by the constitutively high expression from this promoter fusion in the nagR mutant background. The lower levels of CAT expressed from the PglmSΔ10nt::cat fusion in the wild-type background growing on glucose likely was associated with the mutation impacting the −10 element of the promoter.
To determine if the interaction of NagR with the nagB promoter was similarly dependent on the phasing of dre, 5-nt and 10-nt insertions (+5nt and +10nt, respectively) (Fig. 1A and 4) were introduced between the closely situated dre sites within the nagB promoter. Unfortunately, these insertions could not be made in a way that did not perturb the promoter, so promoter activities of the modified PnagB::cat fusions were reduced dramatically relative to that of the wild-type fusion. Notwithstanding this finding, and similar to the wild-type strain, both the +5nt and +10nt insertion mutants, when present in the wild-type genetic background, produced higher activities in cells growing on GlcN than cells growing on glucose. Importantly, this response to GlcN disappeared when nagR was deleted. Collectively, the results add further support that fundamental differences exist in the ways in which NagR binds to the dre sites in the glmS and nagB promoters, with evidence supporting that cooperative binding is part of glmS, but not of nagB, regulation.
Effects of sugar metabolites on NagR binding activity.
We previously showed the ability of a recombinant NagR protein to bind to the promoter sequences of nagA, nagB, and glmS in vitro. In light of new genetic evidence suggesting the synergistic effects of neighboring dre sites in glmS, we performed a variety of NagR-DNA interaction assays to help correlate the phenotypes of certain genetically modified strains with NagR-DNA binding behaviors. To confirm that the dre sites functioned as true binding sites for NagR, the same Mdre mutations (Mdre1, Mdre2, and Mdre1/2) that replaced TAT/GAC with GTGTA in various CAT fusions (Fig. 3 and 4) were included in DNA probes of nagB and glmS promoters and used in electrophoretic mobility shift assays (EMSA) with purified NagR protein. The percentage of the probe shifted in each reaction then was calculated after measuring the intensity of the band that represented unbound DNA. The data presented here are based on the results from two repeats for each assay (Fig. 5). Compared to the wild-type probe for glmS and NagR, which form a band at the top of the polyacrylamide gel, the use of the probe with the Mdre1 mutation under the same condition caused a slight but consistent reduction in the amount of probe that was shifted (76% for the wild type, 70% for the Mdre1 mutant) (Fig. 5A). A more significant reduction in shift (down to 54%) was noted when only the second dre was mutated (Fig. 5A). Subsequently, when the probe glmSMdre1/2, which contained both Mdre1 and Mdre2 mutations, was used in binding with NagR, only 28% of the probe was shifted. Together, these results suggest that both dre sites found upstream of the glmS coding sequence are binding sites for NagR.
FIG 5.

Electrophoretic mobility shift assay (EMSA). One femtomole of biotinylated DNA probe containing regions upstream of the glmS (A) or nagB (B) coding sequences, including the respective dre mutants, was used in each reaction with 0, 1.3 μM (A), or 0.3 μM (B) NagR protein. When indicated, 20 mM glucose-6-P (G6P), GlcN-6-P (GlcN6P), or GlcNAc-6-P (NAG6P) also was added to the reaction mixture. Indicated below each lane are the average percentages and standard deviations (S.D.) of probe being shifted relative to the control lane of the same gel, calculated using the results from two independent repeats.
Similarly, the wild-type and three Mdre mutant versions of nagB probes (Mdre1, Mdre2, and Mdre1/2) were used in the binding assays with NagR protein (Fig. 5B). While mutation of dre1 abolished most of the shift of the probe by NagR, reducing the shift rate from 98% for the wild type to 56% for Mdre1, mutation of dre2 had only a minor impact (94% shifted) on the migration of the nagB probe. The Mdre1/2 probe that had both dre sites of the nagB promoter mutated showed the least amount (45%) of shift in signal. Therefore, while both NagR binding sites in the nagB promoter contribute to regulation by NagR, dre1 appears to play a dominant role, consistent with the results of the promoter mutagenesis analysis (Fig. 4).
In order for NagR to achieve differential regulation over the nagB and glmS genes, information on the availability of GlcN or GlcNAc needs to be transmitted, directly or indirectly, to NagR. To test whether NagR had the capacity to have its binding behaviors affected by amino sugars, we conducted EMSAs in the presence of glucose-6-P (G-6-P), GlcN-6-P, and GlcNAc-6-P, which are the products of the bacterial PTS transport of glucose, GlcN, and GlcNAc, respectively. As we reported previously (3), the inclusion of GlcN-6-P resulted in a clear reduction in the ability of NagR protein to bind to the glmS, and especially the nagB, probes (Fig. 5B). We also noted clear patterns of mobility shifting for both probes, indicating that GlcN-6-P reduces the intermolecular interactions that are characteristic of NagR proteins (17) and that lead to aggregation of the DNA-protein complexes at the origin of the gel. Such dissociation would allow for the interactions between NagR and two dre sites to manifest as two distinct bands, with the upper band resulting from both dre sites being occupied (Fig. 5). Furthermore, when G-6-P was included in the binding assays, a different impact was noted on the affinity of NagR for the glmS and nagB probes. Specifically, a significantly greater percentage of the glmS probe (up from 76% to 96%) showed a shift in the presence of G-6-P, whereas under the same conditions a consistently smaller proportion of the nagB probe was shifted, as indicated by the free probe now visible on the gel (Fig. 5B). Thus, the NagR protein possesses the ability to interact with these promoter regions differently in response to the presence of metabolic intermediates that appear to regulate the expression of these genes in vivo.
To confirm the EMSA results and provide more quantitative information about the effects of certain metabolites on the affinity of NagR for its targets, a fluorescence polarization (FP) assay was utilized. Fluorescence polarization, a measurement of the free rotation of the DNA probe in solution, increases when the DNA is bound by protein. A single 5′-6FAM-labeled, 45-nt DNA oligonucleotide (named 6FAM-dre) that contained the inverted repeats of the consensus dre sequence in S. mutans (AAA TTG GTC TAT ACC AAT TA) (3), separated by 5 cytosines, was synthesized to form a double-stranded target (see Fig. S1A in the supplemental material) for binding by NagR. When used alone, NagR protein bound to 6FAM-dre with high affinity, yielding a calculated dissociation constant (Kd) of 22.4 ± 2.1 nM (Fig. 6A). Consistent with the results of EMSA, the addition of 5 mM GlcN-6-P to the same assay significantly decreased the affinity of NagR for 6FAM-dre (Kd = 76.3 ± 12.7 nM) (Fig. 6B). Surprisingly, the addition of 1 mM GlcN-6-P slightly increased the affinity of NagR for 6FAM-dre to a Kd of 19.3 ± 1.5 nM (Fig. 6B), while the addition of 2 mM and 3 mM GlcN-6-P resulted in intermediate Kd values. Further, when the concentration of NagR was added in the 60 to 80 nM range, there was a consistent small and abrupt drop in FP values, and the remainder of the curve extended along a lower trajectory at higher NagR concentrations (Fig. 6A; also see Fig. S1B). Interestingly, increasing the concentration of GlcN-6-P appeared to mitigate this effect, resulting in better-fit curves and increased Bmax values (representing the FP values at maximum binding). This was most clear when the curves resulting from 4 different concentrations of GlcN-6-P were graphed together (see Fig. S1C). When G-6-P was added to the FP assays, however, NagR showed a significantly decreased affinity for 6FAM-dre at both 1 mM and 5 mM concentrations, yielding Kd values of 55.9 ± 7.7 nM and 90.9 ± 13.4 nM, respectively (Fig. 6C). The addition of F-6-P or GlcNAc-6-P at similar levels had no impact on the binding affinity of NagR to dre (data not shown). Finally, we used an unrelated probe carrying the consensus sequence of the catabolite response element (cre) recognized by the CcpA protein (28) as a control, and no specific binding by NagR was detected (Fig. 6D). These results provide direct evidence for the ability of G-6-P and GlcN-6-P to impact the binding affinity of NagR to a model dre.
FIG 6.

NagR binds specifically to a DNA probe containing a consensus dre sequence. Fluorescence polarization (FP) assays were performed using a fluorescein-labeled dre probe and purified NagR protein in the presence of additional NaCl (A), GlcN-6-P (B), or G-6-P (C). (D) As a negative control, a similarly labeled DNA oligonucleotide containing a known catabolite response element (cre) also was used in binding with NagR.
Point mutations in the putative effector-binding domain of NagR cause aberrant expression of glmS and nagAB.
According to computer predictions, NagR possesses an N-terminal, winged helix-turn-helix domain capable of DNA binding and a chorismate lyase-like domain near its C terminus, which has been suggested to be involved in both effector binding and dimerization in NagR of B. subtilis (15) (Fig. 1B). We hypothesized that NagR could recognize intracellular signals of phosphosugar metabolites via its effector-binding domain, which in turn would affect its DNA-binding characteristics. To test this hypothesis, we employed a random mutagenesis approach (23) that involved the transformation of the bacterium with a mutator DNA fragment along with an integration plasmid that delivers a PglmS::lacZ promoter fusion and kanamycin resistance determinant. The PCR product contained sequences from the effector-binding portion of NagR (aa 88 to 226) (Fig. 1B) with random mutations generated through error-prone PCRs. After obtaining kanamycin-resistant transformants, individual colonies were patched onto agar plates that contained 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) for chromogenic reactions mediated by the LacZ enzyme, with or without the addition of 5 mM GlcNAc. Colonies that produced altered intensities of blue color on these plates compared to the wild-type strain containing the same PglmS::lacZ fusion were analyzed by sequencing of the nagR alleles, and β-galactosidase activities in these isolates were measured after growth in various sugars of interest.
After several rounds of transformation assays and screening of more than 300 colonies, a number of interesting mutants were identified, each with altered expression of nagA, nagB, and glmS. As presented in Fig. 1B, most of the isolates had replacement mutations in a few isolated regions close to the C terminus of NagR, but three mutants had truncated NagR proteins (see Fig. S2 in the supplemental material). β-Galactosidase assays indicated that in all isolates listed, there was a significant increase in glmS promoter activity in cells growing on glucose (Fig. 7). Most of these isolates also showed high β-galactosidase expression when grown on GlcN. Interestingly, one isolate, EP4-71, contained four replacement mutations (K161R/H166Q/K172S/L201M) and produced increased β-galactosidase activities on glucose but nearly wild-type levels of activities on GlcN. We also measured transcripts levels of nagA, nagB, and glmS genes in selected isolates (EP4-45, EP4-50, and EP4-71) (see Table S3) growing on glucose. There was a general derepression of all three genes in isolates EP4-45 and EP4-50. However, EP4-71, when growing on glucose, showed slightly lower transcript levels of nagA (0.75- ± 0.05-fold) and nagB (0.63- ± 0.13-fold) than the wild-type strain but a significant increase in glmS mRNA (2.73- ± 0.41-fold). Consistent with the qRT-PCR data, most of these mutants showed an increased ability to grow on GlcNAc, whereas EP4-71 grew at a much lower rate than the parental strain (see Fig. S3). Another mutant, EP4-52, which bears A182V/E214G mutations, also presented significant growth deficiency on GlcNAc. Thus, mutant EP4-71 appears to be producing a derivative of NagR that is hypersuppressive for nagAB expression yet is less efficient at repressing the expression of glmS. While further characterization of these EP4 mutants is still under way, these data add support to the notion that the putative effector-binding domain of NagR is critical to differential regulation of nagA, nagB, and glmS in response to carbohydrate signals.
FIG 7.
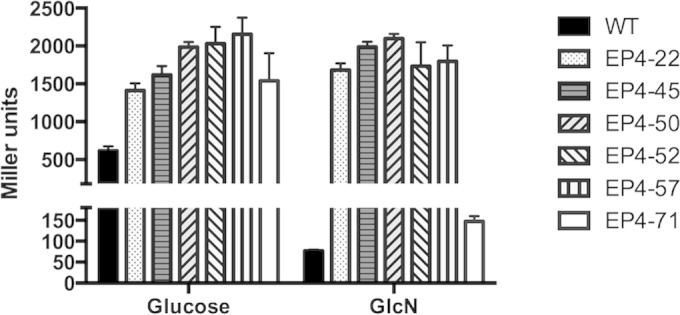
β-Galactosidase activities of the wild type (WT) and various nagR point mutants (EP4-22 to EP4-71) carrying the PglmS::lacZ fusion. Strains were grown to the exponential phase (OD600 of ≈0.5) in TV-glucose or TV-GlcN medium before being harvested, permeabilized by treatment with toluene-acetone (1:9, vol/vol), and subjected to Miller assays. Results here each represent the averages and standard deviations (error bars) from three biological repeats.
Sugar-specific effects on regulation of glmS and nagAB.
In the oral environment, bacteria must respond to frequent changes in carbohydrate sources and availability by adjusting gene regulation. Here, we monitored the transcription of the glmS and nagAB genes by cultivating UA159 carrying respective promoter-cat fusions in TV medium containing various hexoses and amino sugars, alone or in combinations. As shown in Table 1, when tested using TV with glucose, galactose, or fructose alone, no apparent differences were detected in nagA promoter activities, whereas growth on GlcN or GlcNAc alone resulted in high levels of CAT activities. When this analysis was repeated using a UA159 derivative carrying the PnagB::cat fusion, the difference among these three hexoses remained negligible, in contrast to the substantially enhanced nagB promoter activities in cells growing on amino sugars. Also, the expression of the PglmS::cat fusion was significantly lower when cells were growing on amino sugars, as opposed to cells growing on glucose, fructose, and especially galactose (Table 1).
TABLE 1.
Effects of hexoses (glucose, fructose, and galactose) and amino sugars on the expression of glmS, nagA, and nagB promoter fusionsa
| Sugar(s) | PnagA::cat |
PnagB::cat |
PglmS::cat |
|||
|---|---|---|---|---|---|---|
| CAT sp act | FOR | CAT sp act | FOR | CAT sp act | FOR | |
| GlcN | 79.8 ± 7.3 | 813.6 ± 71 | 24.7 ± 1.0 | |||
| GlcNAc | 57.7 ± 6.7 | 464.0 ± 27 | 11.5 ± 1.6 | |||
| Glucose | 1.0 ± 0.2 | 3.0 ± 0.9 | 269.6 ± 5.4 | |||
| Glucose and GlcN | 2.3 ± 0.6 | 34.2 | 1.4 ± 0.8 | 590.3 | 10.01 ± 4.2 | 26.9 |
| Glucose and GlcNAc | 16.3 ± 0.7 | 3.5 | 330.1 ± 33 | 1.4 | 31.2 ± 1.4 | 8.6 |
| Fructose | 0.5 ± 0.3 | 9.0 ± 0.7 | 362.6 ± 6.1 | |||
| Fructose and GlcN | 37.0 ± 1.8 | 2.2 | 224.3 ± 37 | 3.6 | 17.8 ± 1.5 | 20.4 |
| Fructose and GlcNAc | 15.1 ± 0.5 | 3.8 | 90.5 ± 14 | 5.1 | 10.5 ± 5.0 | 34.5 |
| Galactose | 1.1 ± 0.2 | 6.8 ± 0.2 | 483.0 ± 40 | |||
| Galactose and GlcN | 58.6 ± 6.0 | 1.4 | 751.4 ± 15 | 1.1 | 23.03 ± 2.6 | 21.0 |
| Galactose and GlcNAc | 56.9 ± 1.2 | 1.0 | 492.6 ± 53 | 0.9 | 24.0 ± 1.6 | 20.1 |
After measuring the CAT activities (expressed as the average number of nanomoles per milligram of protein per minute ± standard deviation) in the background of UA159, the fold of repression (FOR) by a specific hexose (for nagA and nagB) was calculated as the ratio between activities from cells growing on one amino sugar alone and those on two sugars, and the fold of repression by an amino sugar (for glmS) is the ratio between activities from cells growing on one hexose and those on two sugars. Each carbohydrate was present at a concentration of 0.5%.
The same strains then were cultivated in TV containing a mixture of equal concentrations of one amino sugar and one hexose. The resultant CAT activities derived from cells cultured on a single carbohydrate, amino sugars for nagAB and hexoses for glmS, and that of two mixed carbohydrates then were used to calculate the fold of repression by hexoses (for nagA and nagB) or by amino sugars (for glmS). As shown in Table 1, glucose strongly suppressed the expression of both nagA (34-fold) and nagB (590-fold) in the presence of GlcN, but much less repression was evident (3.5-fold and 1.4-fold for nagA and nagB, respectively) in cultures growing with glucose and GlcNAc. In contrast, fructose generally showed a lower capacity to repress nag gene expression when GlcN or GlcNAc was present, although the repression was more evident for GlcNAc than for GlcN. Finally, galactose showed little or no effect on the ability of GlcN or GlcNAc to induce nag gene expression. For glmS, the ability of amino sugars to repress its expression under different hexoses also varied. Specifically, glmS expression was more sensitive to repression by GlcN than GlcNAc in the presence of glucose, with CAT activities being repressed by 26.9- and 8.6-fold, respectively. However, in the presence of fructose, glmS appeared slightly more prone to repression by GlcNAc than GlcN, as evidenced by the fold of repression being 34.5 for GlcNAc and 20.4 for GlcN. When galactose was tested in combination with either amino sugar, glmS showed sensitivities comparable to that with the other two hexoses, and there was no difference whether GlcN or GlcNAc was present. Collectively, these results suggest that S. mutans differentially regulates amino sugar metabolism by directly sensing the presence of various species of carbohydrates.
DISCUSSION
The goal of this study was to begin to explore the molecular mechanisms that are responsible for the differential regulation by S. mutans of the genes for two enzymes that synthesize or degrade GlcN-6-P, GlmS (GlcN-6-P synthetase) and NagB (GlcN-6-P deaminase), so that cells can balance the requirement for this compound in biosynthetic processes with the opportunity to utilize GlcN and GlcNAc for energy production and pH homeostasis. Our previous work showed that the loss of NagR leads to the elevated expression of glmS, nagA, and nagB (3). Results from this study provide strong evidence for the hypothesis that NagR differentially regulates the transcription of all three genes in response to the availability of particular carbohydrates.
Repression of nagB and activation by exogenous amino sugars.
A combination of in vivo and in vitro experiments described here demonstrate that dre sites predicted by computer algorithms can function as sites for NagR binding (Fig. 3 to 6). The promoters of glmS and nagB each contain two dre sites, and there are significant differences in the locations of these dre sites with respect to promoter elements and in the spacing of the dre sites within these promoter regions (Fig. 1A). The close proximity of the dre sites and their overlap of the promoter elements for nagB should result in the inhibition of transcription initiation when NagR is active. Results from our EMSA have indicated that NagR has significantly greater affinity for the nagB probe than for the glmS or nagA promoter region (3); for the nagB promoter specifically, dre overlapping the −10 element (dre1) appears to play a dominant role in NagR binding (Fig. 5B). In the absence of any influence from allosteric factors, it is likely that NagR preferentially binds to the nagB promoter. Conversely, results of our fluorescence polarization (FP) assays have consistently shown that GlcN-6-P at concentrations of <2 or 3 mM appear to stimulate the binding of dre by NagR, whereas this effect is reversed at higher concentrations. On the basis of these results, it is likely that nagB promoter activity is repressed by NagR in the absence of exogenous GlcNAc or GlcN. When exogenous amino sugar is present, with intracellular GlcN-6-P reaching a certain threshold, conformational change in NagR and/or its dissociation from the nagB promoter would result in the derepression of the gene.
Regulation of glmS requires cooperative binding of NagR and interaction with metabolic effectors.
The organization of dre in the glmS promoter and the way in which NagR appears to interact with this promoter region differ substantially from that observed for NagR-nagB promoter interactions. The two dre sites near the glmS promoter flank the promoter elements and are separated by 57 bp, and NagR showed substantially lower affinity for these dre sites. Importantly, our results show strong evidence for cooperativity (or synergism) in the interaction of NagR with the two dre sites flanking the glmS promoter, requiring both cis elements in proper phasing for regulation of glmS (Fig. 3). In contrast, there is no indication of a similar mechanism influencing the expression of the nagB gene (Fig. 4). Taking into consideration the position of the two dre sites with respect to the glmS promoter, we hypothesize that intermolecular interactions of the NagR protein, when bound to both dre sites, result in looping in the DNA structure that prevents the loading of RNA polymerase complex and initiation of glmS transcription, a scenario similar in part to the model proposed for AraC of E. coli (33). Since NagR appears to preferentially bind to the nagB promoter, when cells are growing on glucose (or fructose and galactose) the dre sites near the glmS promoter may be unoccupied (or underoccupied, as described below), allowing the sufficient expression of GlmS and synthesis of GlcN-6-P for bacterial growth. However, in cells growing on amino sugars, the high affinity of NagR for the nagB promoter should be abolished due to increases in intercellular GlcN-6-P levels. If so, a temporary increase in free NagR protein could allow for cooperative binding to dre by NagR in the glmS promoter region and occupancy of both dre sites, a scenario that has been suggested in similar biological systems (33, 34). In support of this model, we have identified a nagR point mutant (EP4-71) that showed reduced expression of nagAB but at the same time had increased expression of glmS, indicative of opposing modes of regulation by the same regulator.
Also different from nagB, which is readily induced by amino sugars to levels comparable to that of a nagR null mutant (Fig. 2 and 4), the expression levels of the wild-type glmS promoter invariably showed further enhancement as a result of the deletion of nagR, even for cells growing on glucose or galactose (Fig. 3 and data not shown). These observations lead us to propose that NagR does not completely dissociate from the glmS promoter in vivo; rather, its cooperative binding to this promoter is subject to modulation by allosteric effectors. Precedence can be found in the activities of homologous regulators that suggest similar modes of interaction. In the nag system from B. subtilis, it was reported that NagR/YvoA normally binds to dre as a dimer in vitro but switches to a tetrameric state when present in excess (15). Also reported in the same study was the formation of a linear polymer consisting of alternating double-stranded DNA molecules and NagR/YvoA dimers that were locked in the induced conformation, a scenario potentially favored when NagR/YvoA is binding to two dre sites separated by a significant distance. Another recent study employing crystallography has suggested that conformational fine-tuning through interaction with putative metabolic intermediates (GlcNAc-6-P and GlcN-6-P), rather than complete removal from its cognate sites, is critical to the function of NagR/YvoA as a repressor (16). Results from our NagR-DNA interaction study instead have indicated that GlcN-6-P reduces protein-protein interactions in EMSAs (Fig. 5) as well as in FP assays. Specifically for FP, NagR protein consistently showed a slight drop in its binding capacity to dre at higher concentrations of NagR (60 to 80 nM). This result could be interpreted as NagR beginning to form tetramers at higher concentrations that in turn manifest as a reduced effective concentration and lower binding affinity. Interestingly, when GlcN-6-P was added to the reactions, this effect was alleviated. Therefore, it is conceivable that high levels of GlcN-6-P present in cells growing on amino sugars not only release NagR from the nagB promoter but also modulate the degree of oligomerization and conformation of NagR so that favorable binding sites are dre sites that are farther apart, such as those upstream of glmS.
G-6-P as an effector of NagR and more.
Besides GlcN-6-P, we have provided clear evidence supporting the potential of G-6-P to influence the activity of NagR by reducing the affinity of NagR to the consensus dre (Fig. 6) and differentially impacting the binding of NagR to glmS and nagB promoters (Fig. 5). These findings strongly suggest that G-6-P is another allosteric effector sensed by NagR, a feature potentially unique to S. mutans. However, as the addition of G-6-P led to both enhanced formation of NagR-DNA complex containing the glmS promoter (EMSA) and reduced affinity of NagR for consensus dre (FP), the exact role of G-6-P in the function of NagR remains to be elucidated. It also remains to be explored if additional cellular metabolites exist to modulate the function of NagR in concert with compounds tested in this study. Other possible mechanisms contributing to the regulation of the nagAB and glmS genes include the existence of additional regulators (protein or RNA) and/or binding sites for NagR (outside the core dre nucleotides) within the same promoter. Also, in the absence of GlcN-6-P, NagR that is bound to the dre1 site upstream of the glmS promoter could serve as an activator by recruiting RNA polymerase. Evidence in support of these notions includes the significant loss of CAT activity associated with deletion of nagR in the PglmS::cat fusion containing mutations in both dre sites (Fig. 3) coupled with the fact that the nagR mutant harboring a PnagB::cat fusion, or its +5nt and +10nt derivatives, consistently showed substantially higher CAT activities in glucose than GlcN-based medium (Fig. 4).
It is well established that bacteria prioritize carbohydrate metabolism through carbon catabolite repression (CCR), a phenomenon that is frequently affected by the global transcriptional regulator CcpA in low-G+C Gram-positive bacteria (35). While previous work has indicated the lack of a direct role by CcpA in regulating nagB and glmS (8), we have demonstrated here that common hexoses do show various degrees of the capacity to repress the expression of the nagA and nagB genes and also, interestingly, different sensitivities to suppression by amino sugars in their ability to enhance glmS expression (Table 1). Notably, the fact that galactose lacks the ability to repress the induction of nagAB genes by either amino sugar implies that certain metabolic intermediates, which are at lower levels in or absent from cells growing on galactose, as opposed to glucose or fructose, are critical to such inhibition, or that regulation is mediated by sugar-specific PTS complexes. Since the deletion of NagR invariably leads to the largely derepressed expression of these promoters under all mixed-sugar conditions (data not shown), these results lend further support to the central hypothesis that sugar-specific regulation of the nag pathway depends on NagR and its ability to respond to various metabolic signals.
According to a bioinformatic analysis using RegPrecise (see Table S2 in the supplemental material for details) (36), putative dre sites have been predicted near homologous genes, including glmS, required for amino sugar metabolism in related lactic acid bacteria (LAB). For 12 streptococci and one Lactococcus lactis strain, each bacterium contains at least two apparent dre sites upstream of a glmS gene, with significant spacing between the dre sites, similar to the case for S. mutans. In many members of the family Lactobacillaceae, there also are multiple dre sites upstream of the glmS and nagB homologues. Many of these LAB are important human pathogens that occupy similar niches of the host and are metabolically similar to S. mutans and one another, and these similarities may account for the apparent conservation in the regulatory pathways for amino sugar metabolism. Knowledge gained by studying NagR in the model bacterium S. mutans could be more broadly relevant to other important microbial pathogens, beneficial commensals, and industrially important bacteria.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants DE024782 (to L.Z.) and DE12236 (to R.A.B.) from the National Institute of Dental and Craniofacial Research.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00606-15.
REFERENCES
- 1.Postma PW, Lengeler JW, Jacobson GR. 1993. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev 57:543–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abranches J, Chen YY, Burne RA. 2003. Characterization of Streptococcus mutans strains deficient in EIIABMan of the sugar phosphotransferase system. Appl Environ Microbiol 69:4760–4769. doi: 10.1128/AEM.69.8.4760-4769.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moye ZD, Burne RA, Zeng L. 2014. Uptake and metabolism of N-acetylglucosamine and glucosamine by Streptococcus mutans. Appl Environ Microbiol 80:5053–5067. doi: 10.1128/AEM.00820-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeng L, Das S, Burne RA. 2010. Utilization of lactose and galactose by Streptococcus mutans: transport, toxicity, and carbon catabolite repression. J Bacteriol 192:2434–2444. doi: 10.1128/JB.01624-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plumbridge JA. 1991. Repression and induction of the nag regulon of Escherichia coli K-12: the roles of nagC and nagA in maintenance of the uninduced state. Mol Microbiol 5:2053–2062. doi: 10.1111/j.1365-2958.1991.tb00828.x. [DOI] [PubMed] [Google Scholar]
- 6.Marion C, Stewart JM, Tazi MF, Burnaugh AM, Linke CM, Woodiga SA, King SJ. 2012. Streptococcus pneumoniae can utilize multiple sources of hyaluronic acid for growth. Infect Immun 80:1390–1398. doi: 10.1128/IAI.05756-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaugue I, Oberto J, Putzer H, Plumbridge J. 2013. The use of amino sugars by Bacillus subtilis: presence of a unique operon for the catabolism of glucosamine. PLoS One 8:e63025. doi: 10.1371/journal.pone.0063025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawada-Matsuo M, Mazda Y, Oogai Y, Kajiya M, Kawai T, Yamada S, Miyawaki S, Oho T, Komatsuzawa H. 2012. GlmS and NagB regulate amino sugar metabolism in opposing directions and affect Streptococcus mutans virulence. PLoS One 7:e33382. doi: 10.1371/journal.pone.0033382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rigali S, Nothaft H, Noens EE, Schlicht M, Colson S, Muller M, Joris B, Koerten HK, Hopwood DA, Titgemeyer F, van Wezel GP. 2006. The sugar phosphotransferase system of Streptomyces coelicolor is regulated by the GntR-family regulator DasR and links N-acetylglucosamine metabolism to the control of development. Mol Microbiol 61:1237–1251. doi: 10.1111/j.1365-2958.2006.05319.x. [DOI] [PubMed] [Google Scholar]
- 10.Plumbridge J, Kolb A. 1995. Nag repressor-operator interactions: protein-DNA contacts cover more than two turns of the DNA helix. J Mol Biol 249:890–902. doi: 10.1006/jmbi.1995.0346. [DOI] [PubMed] [Google Scholar]
- 11.Plumbridge J. 1995. Co-ordinated regulation of amino sugar biosynthesis and degradation: the NagC repressor acts as both an activator and a repressor for the transcription of the glmUS operon and requires two separated NagC binding sites. EMBO J 14:3958–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alvarez-Anorve LI, Calcagno ML, Plumbridge J. 2005. Why does Escherichia coli grow more slowly on glucosamine than on N-acetylglucosamine? Effects of enzyme levels and allosteric activation of GlcN6P deaminase (NagB) on growth rates. J Bacteriol 187:2974–2982. doi: 10.1128/JB.187.9.2974-2982.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarez-Anorve LI, Bustos-Jaimes I, Calcagno ML, Plumbridge J. 2009. Allosteric regulation of glucosamine-6-phosphate deaminase (NagB) and growth of Escherichia coli on glucosamine. J Bacteriol 191:6401–6407. doi: 10.1128/JB.00633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bertram R, Rigali SB, Wood N, Lulko AT, Kuipers OP, Titgemeyer F. 2011. Regulon of the N-acetylglucosamine utilization regulator NagR in Bacillus subtilis. J Bacteriol 193:3525–3536. doi: 10.1128/JB.00264-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Resch M, Schiltz E, Titgemeyer F, Muller YA. 2010. Insight into the induction mechanism of the GntR/HutC bacterial transcription regulator YvoA. Nucleic Acids Res 38:2485–2497. doi: 10.1093/nar/gkp1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fillenberg SB, Grau FC, Seidel G, Muller YA. 2015. Structural insight into operator dre-sites recognition and effector binding in the GntR/HutC transcription regulator NagR. Nucleic Acids Res 43:1283–1296. doi: 10.1093/nar/gku1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaugué I, Oberto J, Plumbridge J. 2014. Regulation of amino sugar utilization in Bacillus subtilis by the GntR family regulators, NagR and GamR. Mol Microbiol 92:100–115. doi: 10.1111/mmi.12544. [DOI] [PubMed] [Google Scholar]
- 18.Gopel Y, Luttmann D, Heroven AK, Reichenbach B, Dersch P, Gorke B. 2011. Common and divergent features in transcriptional control of the homologous small RNAs GlmY and GlmZ in Enterobacteriaceae. Nucleic Acids Res 39:1294–1309. doi: 10.1093/nar/gkq986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amon J, Titgemeyer F, Burkovski A. 2010. Common patterns–unique features: nitrogen metabolism and regulation in Gram-positive bacteria. FEMS Microbiol Rev 34:588–605. doi: 10.1111/j.1574-6976.2010.00216.x. [DOI] [PubMed] [Google Scholar]
- 20.Burne RA, Wen ZT, Chen YY, Penders JE. 1999. Regulation of expression of the fructan hydrolase gene of Streptococcus mutans GS-5 by induction and carbon catabolite repression. J Bacteriol 181:2863–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 22.Lau PC, Sung CK, Lee JH, Morrison DA, Cvitkovitch DG. 2002. PCR ligation mutagenesis in transformable streptococci: application and efficiency. J Microbiol Methods 49:193–205. doi: 10.1016/S0167-7012(01)00369-4. [DOI] [PubMed] [Google Scholar]
- 23.Zeng L, Das S, Burne RA. 2011. Genetic analysis of the functions and interactions of components of the LevQRST signal transduction complex of Streptococcus mutans. PLoS One 6:e17335. doi: 10.1371/journal.pone.0017335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cha RS, Zarbl H, Keohavong P, Thilly WG. 1992. Mismatch amplification mutation assay (MAMA): application to the c-H-ras gene. PCR Methods Appl 2:14–20. doi: 10.1101/gr.2.1.14. [DOI] [PubMed] [Google Scholar]
- 25.Cadwell RC, Joyce GF. 1992. Randomization of genes by PCR mutagenesis. PCR Methods Appl 2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 26.Zeng L, Wen ZT, Burne RA. 2006. A novel signal transduction system and feedback loop regulate fructan hydrolase gene expression in Streptococcus mutans. Mol Microbiol 62:187–200. doi: 10.1111/j.1365-2958.2006.05359.x. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Zeng L, Burne RA. 2009. AguR is required for induction of the Streptococcus mutans agmatine deiminase system by low pH and agmatine. Appl Environ Microbiol 75:2629–2637. doi: 10.1128/AEM.02145-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shelburne SA, Olsen RJ, Suber B, Sahasrabhojane P, Sumby P, Brennan RG, Musser JM. 2010. A combination of independent transcriptional regulators shapes bacterial virulence gene expression during infection. PLoS Pathog 6:e1000817. doi: 10.1371/journal.ppat.1000817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw WV. 1975. Chloramphenicol acetyltransferase from chloramphenicol-resistant bacteria. Methods Enzymol 43:737–755. doi: 10.1016/0076-6879(75)43141-X. [DOI] [PubMed] [Google Scholar]
- 30.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 31.Ahn SJ, Lemos JA, Burne RA. 2005. Role of HtrA in growth and competence of Streptococcus mutans UA159. J Bacteriol 187:3028–3038. doi: 10.1128/JB.187.9.3028-3038.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 33.Schleif R. 2010. AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol Rev 34:779–796. doi: 10.1111/j.1574-6976.2010.00226.x. [DOI] [PubMed] [Google Scholar]
- 34.Cournac A, Plumbridge J. 2013. DNA looping in prokaryotes: experimental and theoretical approaches. J Bacteriol 195:1109–1119. doi: 10.1128/JB.02038-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deutscher J. 2008. The mechanisms of carbon catabolite repression in bacteria. Curr Opin Microbiol 11:87–93. doi: 10.1016/j.mib.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Novichkov PS, Kazakov AE, Ravcheev DA, Leyn SA, Kovaleva GY, Sutormin RA, Kazanov MD, Riehl W, Arkin AP, Dubchak I, Rodionov DA. 2013. RegPrecise 3.0–a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics 14:745. doi: 10.1186/1471-2164-14-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.