

ABSTRACT
Gammaherpesviruses (GHVs) carry homologs of cellular genes, including those encoding a viral cyclin that promotes reactivation from latent infection. The viral cyclin has reduced sensitivity to host cyclin-dependent kinase inhibitors in vitro; however, the in vivo significance of this is unclear. Here, we tested the genetic requirement for the viral cyclin in mice that lack the host inhibitors p27Kip1 and p18INK4c, two cyclin-dependent kinase inhibitors known to be important in regulating B cell proliferation and differentiation. While the viral cyclin was essential for reactivation in wild-type mice, strikingly, it was dispensable for reactivation in mice lacking p27Kip1 and p18INK4c. Further analysis revealed that genetic ablation of only p18INK4c alleviated the requirement for the viral cyclin for reactivation from latency. p18INK4c regulated reactivation in a dose-dependent manner so that the viral cyclin was dispensable in p18INK4c heterozygous mice. Finally, treatment of wild-type cells with the cytokine BAFF, a known attenuator of p18INK4c function in B lymphocytes, was also able to bypass the requirement for the viral cyclin in reactivation. These data show that the gammaherpesvirus viral cyclin functions specifically to bypass the cyclin-dependent kinase inhibitor p18INK4c, revealing an unanticipated specificity between a GHV cyclin and a single cyclin-dependent kinase inhibitor.
IMPORTANCE The gammaherpesviruses (GHVs) cause lifelong infection and can cause chronic inflammatory diseases and cancer, especially in immunosuppressed individuals. Many GHVs encode a conserved viral cyclin that is required for infection and disease. While a common property of the viral cyclins is that they resist inhibition by normal cellular mechanisms, it remains unclear how important it is that the GHVs resist this inhibition. We used a mouse GHV that either contained or lacked a viral cyclin to test whether the viral cyclin lost importance when these inhibitory pathways were removed. These studies revealed that the viral cyclin was required for optimal function in normal mice but that it was no longer required following removal or reduced function of a single cellular inhibitor. These data define a very specific role for the viral cyclin in bypassing one cellular inhibitor and point to new methods to intervene with viral cyclins.
INTRODUCTION
The gammaherpesviruses (GHVs) include the human pathogens Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), which are associated with multiple lymphoproliferative and inflammatory diseases (1). Gammaherpesvirus 68 (GHV68) (or murine herpesvirus type 4 [MuHV-4]) infects laboratory mice and provides a small-animal model for these infections and diseases, as it allows examination of all stages of infection, using both wild-type (WT) and mutant viruses and mice (2).
The GHVs have an intimate relationship with cells of the immune system. The GHVs establish lifelong infection in cells of the immune system, primarily B cells, and exploit the natural lifestyle of B cells for their maintenance and propagation (3). Primary GHV infection results in production of virus and lysis of many cells. Acute infection is controlled by the immune response in a healthy host, so that within 2 weeks postinfection (p.i.), only latent infection can be detected. Latent, or quiescent, infection is exquisitely attuned to the host cells and is typified by very low levels of viral gene expression in cells that harbor the viral genome. Latently infected cells retain the capacity to reactivate from latency, to express the full complement of viral genes, and to make new virus. Reactivation from latency is one mechanism contributing to virus transmission. The balance between latent infection and reactivation is constantly regulated by the immune response, with immune compromise and multiple cofactors influencing virus reactivation (2, 4).
Some of the GHVs, including KSHV, GHV68, and herpesvirus saimiri, encode a conserved viral cyclin (v-cyclin) that is homologous to host D-type cyclins (5–7). EBV does not encode a v-cyclin, but instead, at a similar position in the viral genome, carries genes that upregulate the expression of host D-type cyclins (8–10). Cyclins are the regulatory components of cyclin/cyclin-dependent kinase (CDK) complexes that directly promote cell cycle progression. Exogenous expression studies demonstrated that v-cyclins differ from host cyclins in that they are more promiscuous in their CDK partners and, when partnered with host CDKs, phosphorylate more potential substrates (11). V-cyclins differ from cellular cyclins in their relative insensitivity to host cyclin-dependent kinase inhibitors (CKIs) (12). A primary function of the CKIs, including both the INK4 and Cip/Kip proteins, is to limit cyclin/CDK activity and restrain cell cycle progression (13). The CKIs also regulate additional cellular processes, including development, senescence, differentiation, and restraint of malignant transformation (14). The specificity and redundancy of host CKIs remain ongoing areas of investigation, with cell-type- and context-specific roles identified for many CKIs.
V-cyclins have the capacity to interact with a wide range of proteins and can be oncogenic when constitutively expressed (15, 16). Despite this capacity of v-cyclins upon exogenous expression, in vivo analysis of v-cyclin-deficient viruses using the GHV68 system revealed that a primary function of the v-cyclin during GHV infection is to promote reactivation from latent infection (17, 18). Notably, the v-cyclin is required for reactivation from latency in multiple strains and genetic deficiencies. Furthermore, the v-cyclin is required for optimal chronic disease relative to WT GHV68, an observation that suggests that viral reactivation from latency is critical for promotion of chronic GHV disease (19–22). The conservation of the v-cyclin in many GHVs, combined with the strong association of the v-cyclin with GHV-associated disease, highlights the role of the v-cyclin in GHV pathogenesis. Despite these studies, the mechanisms of action of the v-cyclins in vivo remain poorly understood. Here, we made use of the GHV68 system to study the genetic requirement for the v-cyclin in reactivation from latency in the presence or absence of the host CKIs p27Kip1 and p18INK4c, two prominent CKIs in B cells. By using sensitive assays to detect latency and reactivation, these studies allowed us to study the genetic interplay between v-cyclin-dependent reactivation and host CKIs in the context of rare, latently infected primary cells. Based on these studies, we demonstrate that the v-cyclin, with all of its potential binding partners and activities, is required primarily to bypass a single host protein, p18INK4c. These studies define an unanticipated specificity of the v-cyclin and a single host CKI, identifying a new axis for modulation of chronic GHV pathogenesis.
MATERIALS AND METHODS
Viruses and tissue culture.
Viruses were passaged and grown and their titers were determined using two GHV68 viruses, (i) parental WT GHV68 (WUMS clone; ATCC VR1465) and (ii) a v-cyclin.Stop virus (vCycKO) containing a translation stop cassette, as described previously (17). 293T cells, NIH 3T12 cells, and murine embryonic fibroblasts (MEFs) were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with fetal bovine serum (FBS), 100 U penicillin/ml, 100 mg streptomycin/ml, and 2 mM l-glutamine. MEFs were generated from C57BL/6 embryos as described previously (23). 293T cells were CaPO4 transfected with pcDNA-p18INK4c (24) for whole-cell lysate preparation at 48 h.
Mice, infections, and organ harvest.
p18INK4c−/− (gene name, CDKN2C) and p27Kip1−/− (gene name, CDKN1B) mice on a C57BL/6 background (David Franklin, Purdue University) were previously described (25). All p18 and p27 knockout (p18KO and p27KO) and heterozygous mice were bred and maintained at the University of Colorado in accordance with all federal and university policies. C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME). The mice were age and gender matched for infection between 8 and 12 weeks, with isoflurane anesthetization prior to intraperitoneal (i.p.) injection with 106 PFU of virus in 0.5 ml of complete DMEM. Spleens and peritoneal cells (PerCs) were harvested as described previously (17). All the infected mice were housed in an animal biosafety level 2 facility in accordance with all university regulations.
Genotyping of p18 and p27 mice.
Genotypes were determined by tail DNA PCR. The primers used for PCR analysis were as follows: p18WT-F, 5′ AGCCATCAAATTTATTCATGTTGCAGG; p18MG-47, 5′ CCATTTGCCTCCATCAGGCTAATGACC; pGKNeo-R, 5′ CCAGCCTCTGAGCCCAGAAAGCGAAGG; p27KO-1, 5′ AGATGTCAAACGTGAGAGTGTCTAACG; p27KO-2, 5′ TCCAGGTTATGAGAGTGTCTAACG; p27Neo-F, 5′ CTTGGGTGGAGAGGCTATTC; and p27Neo-R, 5′ AGGTGAGATGACAGGAGATC. Each set of primers (both forward and reverse) was used to genotype both strains of mice, and each mouse genotype was determined in duplicate reactions. PCRs consisted of denaturation at 95°C for 5 min, followed by 40 cycles of denaturation (95°C; 45 s), annealing (57°C; 45 s), and elongation (72°C; 45 s). Samples were visualized on a 2% agarose gel using ethidium bromide to identify p18 WT (600 bp), p18 deficient (400 bp), and p18 heterozygous (both bands), and p27 WT (200 bp), p27 deficient (300 bp), and p27 heterozygous (both bands). Female p27−/− homozygous mice are infertile (26), and therefore, all p27-deficient mice were bred from heterozygotes.
Frequency of latently infected cells.
The frequency of cells harboring the GHV68 genome was determined by limiting-dilution nested PCR of the GHV68 gene 50 sequence, as described previously (19, 23). Twelve PCRs were analyzed for each cell dilution, with six dilutions per sample per experiment. Controls included uninfected cells alone or with the addition of 10 copies or 1 copy of gene 50 plasmid DNA.
Frequency of cells reactivating from latency.
The frequency of cells capable of reactivating was determined using a limiting-dilution reactivation assay, as described previously (19, 23). Briefly, at 42 days p.i., PerCs were harvested and plated on MEF monolayers, starting at 4 × 104 PerCs/well. Preformed virus was detected by parallel plating of mechanically disrupted cells (mechanical disruption with glass beads in a Fastprep Bio101 homogenizer [ThermoSavant] kills >99% of cells with no detectable inactivation of virus). Twelve dilutions were plated per sample, and 24 wells were plated per dilution. All wells were scored for viral cytopathic effect (CPE).
The limiting-dilution assay for ex vivo reactivation quantifies the frequency of virus-infected cells capable of reactivating from latency, as measured by virus reactivation and amplification on mouse embryonic fibroblast monolayers. To discriminate between virus produced by reactivation, a process that requires live cells, and virus resulting from preformed virus, which does not require intact cells, all reactivation assays included a parallel dilution series of infected cells that had been mechanically disrupted. These mechanically disrupted samples measured the amount of preformed virus. In all of the experiments described here, plating of mechanically disrupted cells resulted in no cytopathic effect. While we included mechanically disrupted samples in Fig. 1, we did not depict these uniformly negative values in all subsequent figures.
FIG 1.

The genetic requirement for the GHV68 v-cyclin in reactivation is relieved in p18INK4c-deficient mice. Shown are the frequencies of reactivation (A, C, E, G, and I) and viral-genome-positive cells (B, D, F, H, and J) in infected PerCs from mice infected with WT GHV68 or vCycKO. (A and B) Frequency of reactivation from latency (A) or viral-genome-positive cells (B), comparing WT GHV68 and vCycKO in C57BL/6 mice. (C and D) Frequency of reactivation (C) or viral-genome-positive cells (D), comparing WT GHV68 and vCycKO in p27Kip1+/− p18INK4c−/− mice. (E and F) Frequency of reactivation (E) or viral-genome-positive cells (F), comparing WT GHV68 and vCycKO in BKip1+/− mice. (G and H) Frequency of reactivation (G) or viral-genome-positive cells (H), comparing WT GHV68 and vCycKO in p27Kip1−/− mice. (I and J) Frequency of reactivation (I) or viral-genome-positive cells (J), comparing WT GHV68 and vCycKO in p18INK4c−/− mice. PerCs were collected 6 weeks p.i. and were limiting-dilution plated either onto indicator MEFs to measure the frequency of reactivating cells by CPE or for nested-PCR detection of the viral genome to measure latently infected cells. The data represent averages ± standard errors of the mean (SEM) of the results of 2 to 4 independent experiments, each with 4 or 5 pooled mice per sample. The frequency of reactivating or genome-positive cells was calculated by Poisson distribution, indicated by the dotted lines at 63%. Within each genotype, there was a statistically significant difference in reactivation between WT virus and vCycKO, as calculated by paired t test (P < 0.05). Using the same criteria, there was no statistically significant difference in the frequencies of genome-positive cells between WT virus and vCycKO. Mechanically disrupted cells were plated in parallel to measure the presence of preformed infectious virus and were uniformly negative (diamond symbols on the x axis), indicating that the virus resulted from reactivation from latency.
Immunoblotting.
Samples were analyzed as described previously (27). Briefly, equivalent numbers of cells were boiled in Laemmli buffer, resolved on SDS-12% PAGE gels, transferred to nitrocellulose, and incubated with 1:500 anti-p18 antibody (Santa Cruz) or 1:2,000 anti-β-actin (Sigma Chemical), followed by 1:6,000 donkey anti-rabbit or anti-mouse Ig-horseradish peroxidase (HRP) (Jackson ImmunoResearch) and development with ECL Plus (Amersham Pharmacia).
RT-PCR.
For reverse transcription (RT)-PCR for cell cycle analysis, on average, 2 × 106 to 4 × 106 peritoneal cells were collected per mouse. Total peritoneal cells from uninfected mice were directly subjected to RNA extraction. Infected peritoneal cell samples at 42 days postinfection were collected from 5 mice per group and pooled prior to plating for reactivation and limiting dilution (LD)-PCR analysis, with the remaining cells viably frozen. Total peritoneal cells from either uninfected p18-deficient or Black6 mice or mice infected with wild-type or cyclin-deficient GHV68 (cells corresponding to reactivation and genomic-DNA analyses are shown in Fig. 1A and B and I and J). RNA samples were prepared using TRIzol extraction (Life Technologies) with the manufacturer's protocol. RNA was treated with RQ1 DNase (Promega) for 1 h at 37°C to digest any residual DNA. RT-PCR was performed using a OneStep RT-PCR kit (Qiagen). Briefly, 200 ng of DNase-treated RNA was added per RT reaction. Expression of cellular p18INK4c, cyclin D3, cyclin D2, cyclin E, CDK6, or β-actin was measured using the transcript-specific primers listed below. To ensure no DNA contamination was present, PCR was also performed with no reverse transcriptase. PCRs were resolved on 2% agarose gels and visualized by ethidium bromide. The primers used were as follows: p18INK4c forward, 5′ GATTTGGGAGAACTGCGCTG; p18INK4c reverse, 5′ GCCTGGAACTCCAGCAAAGC; cyclinD3 forward, 5′ CAGGCAGACGGTACCTAGAAG; cyclinD3 reverse, 5′ CAGTGCGTGAAAAGGAGATC; cyclinD2 forward, 5′ GCTTGCGAAGGATGTGCTC; cyclinD2 reverse, 5′ CCTGGGTGCAGTGTGCATG; cyclinE1 forward, 5′ GAGCACTTTCTGCAGCGTCA; cyclinE1 reverse, 5′ GGCTGAAATCCCAATAAGCTGTAA; CDK6 forward, 5′ TACCCACAGAAACCATAAAGGAT; CDK6 reverse, 5′ ACGACCACCGAGGTAAGGGCCA; β-actin forward, 5′ GCCACCAGTTCGCCATGG; and β-actin reverse, 5′ CAATGCCATGTTCAATGGGGTA.
Flow cytometry.
Infected peritoneal cells were collected at 8 and 16 days postinfection and stained with fluorescently conjugated antibodies against CD19 (eBioscience; clone eBio1D3) and CD5 (eBioscience; clone 53-7.3). All staining was done in the presence of the Fc receptor-blocking antibody 24G2. Flow cytometric analysis was performed on an LSR II flow cytometer (BD Biosciences). Compensation values were calculated using DIVA software (BD Biosciences) and were based on fluorescence values obtained with antibody-stained compensation beads.
Plaque neutralization assay.
Virus neutralization assays were performed as described previously (28). Briefly, 104 PFU of virus in 45 μl was incubated with 5 μl serial dilutions of sera collected from preimmune and immune (42 days p.i.) p18+/+, p18−/−, or p18+/− mice. Virus and sera were incubated at 37°C for 1 h, followed by plaque assay to quantitate infectious virus. Internal controls included medium only and virus plaque standards, and the percent neutralization was measured based on the virus titer in the absence of antibody.
Ex vivo stimulation of infected cells.
PerCs from mice infected with WT GHV68 or vCycKO were harvested for ex vivo reactivation analysis. The cells were cultured in RPMI supplemented with 10% FBS, 100 U penicillin/ml, 100 mg streptomycin/ml, 2 mM l-glutamine, and 0.05 mM 2-mercaptoethanol. PerCs (2 × 106) were subjected to treatment with recombinant BAFF (B cell-activating factor) (50 ng/ml), anti-IgM (5 μg/ml; B76, rat IgG1), or both (29) or left untreated for 1 h at 37°C prior to plating on MEFs for ex vivo reactivation analysis, as described above. BAFF (10 ng/well) was added daily for the first 4 days of culture.
Statistical analysis.
All data were analyzed using GraphPad Prism software (GraphPad Software, San Diego, CA). The frequencies of reactivation were obtained by determining the number of cells at which 63% of wells scored positive, based on the Poisson distribution; the data were subjected to nonlinear-regression analysis to obtain the single-cell frequency for each limiting-dilution analysis, as indicated by the curve fit line. Statistical significance was determined using the paired Student t test.
RESULTS
V-cyclin is not required for virus reactivation in mice genetically deficient in the p27Kip1 and p18INK4c cell cycle inhibitor genes.
V-cyclin is critical for efficient reactivation from latency, a finding uniformly demonstrated across multiple mouse strains and genetic deficiencies (17, 19, 20). While recent studies identified certain features of v-cyclin that promote reactivation from latency (30), it remains unclear which host processes v-cyclin manipulates to promote reactivation from latency. In vitro, the v-cyclins have broadened partner and target specificity, confer increased kinase activity, and have decreased sensitivity to inhibitors relative to cellular cyclins (11).
One common feature of the v-cyclins is decreased sensitivity to the INK4 and Cip/Kip families of cell cycle inhibitors (11). Based on this, we hypothesized that a primary function of the v-cyclin is to bypass host CKIs. To test this, we generated mice with genetic insufficiency of the cell cycle inhibitors p27Kip1 and p18INK4c, two CKIs known to regulate B cell development and function (29). Due to infertility in p27−/− females, we analyzed p18−/− and p27+/− mice rather than full double knockouts. Previous work has shown that unmanipulated p27- and p18-deficient mice eventually succumb to pituitary tumors and display gigantism and organomegaly over time and hypercellularity in the spleen, despite normal B cell numbers (25). Mice were infected with either WT GHV68 or v-cyclin-deficient GHV68 (vCycKO), and at 42 days p.i., peritoneal cells were collected for limiting-dilution ex vivo analysis of virus reactivation. Consistent with previous reports, the v-cyclin was required for optimal reactivation in control C57BL/6 mice, with vCycKO virus having a >100-fold deficit in reactivation from latency (WT GHV68, 1 in 19,588, versus vCycKO, <1/1,000,000) (Fig. 1A) with no difference in the frequency of viral-genome-positive cells (Fig. 1B). In striking contrast, we found that the reactivation deficit of the vCycKO virus was partially restored following infection of p27Kip1+/− p18INK4c−/− mice, so that the vCycKO virus had only a 5-fold defect in the frequency of cells reactivating from latency (WT GHV68, 1 in 5,152 cells, versus vCycKO, 1 in 26,363 cells) (Fig. 1C). Importantly, the frequencies of infected cells were comparable in WT and vCycKO viruses in p27+/− p18−/− mice (Fig. 1D). These data indicate that the v-cyclin is largely dispensable for reactivation following genetic ablation of the p27Kip1 and p18INK4c cell cycle inhibitor genes.
Next, we dissected the contribution of either p27Kip1 or p18INK4c deficiency to the v-cyclin requirement in reactivation. In both p27+/− and p27−/− mice, we found that v-cyclin was required for reactivation, with vCycKO virus having a major defect in reactivation from latency (Fig. 1E and G) and p27Kip1 deficiency appearing instead to restrain overall reactivation. In striking contrast, when we analyzed reactivation in p18-deficient mice, we found a nearly equivalent frequency of reactivation between WT GHV68 and vCycKO virus (WT GHV68, 1 in 1,297 versus 1 in 3,162) (Fig. 1I). Notably, in p18-deficient mice, vCycKO has only a 2.4-fold reactivation defect compared to the >100-fold defect in p18-sufficient C57BL/6 mice. In both p27- and p18-deficient mice, the frequencies of latently infected cells were equivalent between WT GHV68 and vCycKO, indicating that changes in the reactivation frequency resulted from differences in reactivation rather than overall changes in the frequency of infected cells (Fig. 1F, H, and J). These reactivation and latent-cell frequencies are summarized in Table 1. In total, these data indicate that the genetic requirement for the v-cyclin in reactivation is relieved by ablation of the host cyclin-dependent kinase inhibitor p18INK4c.
TABLE 1.
Frequency of reactivation from latency as influenced by host and virus genotypes
| Host genotype | Frequency of reactivationa |
Reactivation defectb (vCycKO vs WT GHV68) | |
|---|---|---|---|
| WT GHV68 | vCycKO | ||
| C57BL/6J | 1/19,588 | <1/1,000,000 | >51.05 |
| p18−/− × p27+/− | 1/5,152 | 1/26,363 | 5.12 |
| p18+/− | 1/3,198 | 1/9,908 | 3.10 |
| p18−/− | 1/1,297 | 1/3,162 | 2.44 |
Frequency of virus reactivation from latency as measured from peritoneal cells explanted onto MEF monolayers, comparing wild-type and v-cyclin-deficient (vCycKO) GHV68 at day 42 p.i. as calculated from ex vivo reactivation analyses shown in Fig. 1.
The fold reactivation defect was calculated as the quotient of the frequency of reactivation for vCycKO divided by the frequency of reactivation for wild-type GHV68.
p18INK4c regulates virus reactivation in a dose-dependent manner.
The above-mentioned data indicate that a genetic deficiency in p18INK4c alleviates the requirement for the v-cyclin in reactivation from latency. To ensure that this phenotype was due to p18 deficiency rather than a potential background effect and to assess the consequence of p18 heterozygosity, we intercrossed C57BL/6 and p18−/− mice to make p18+/− mice and then intercrossed these F1 animals to generate p18+/+, p18+/−, and p18−/− mice. F2 progeny were genotyped for p18 and then infected with WT GHV68 or vCycKO. By comparing the frequencies of reactivation between WT GHV68 and v-cycKO in p18+/+, p18+/−, and p18−/− mice, we found that vCycKO had only a modest deficit relative to WT GHV68 in mice that were either p18−/− (Fig. 2A) or p18+/− (Fig. 2B) (WT, 1 in 3,198, versus vCycKO, 1 in 9,908).
FIG 2.

p18INK4c regulated virus reactivation in a dose-dependent manner. Shown is analysis of the frequencies of reactivation in PerCs from mice with different gene dosages of p18INK4c infected with either WT GHV68 or vCycKO and harvested at 42 days p.i. (A and B) Frequency of reactivation in p18+/+ and p18−/− mice (A) or p18+/− mice (B), comparing WT GHV68 and vCycKO. The data represent averages ± SEM of the results of 3 independent experiments, each with 4 or 5 pooled mice per sample. The frequency of reactivating cells was calculated by Poisson distribution, indicated by the dotted lines at 63%. (C) Western blot analysis of p18 protein expression from 5 × 106 splenocyte equivalents/sample from infected littermate p18+/+, p18+/−, and p18−/− mice. The positive-control sample was whole-cell lysate from p18-transfected 293T cells. (D) Splenocyte numbers from infected p18+/+, p18+/−, and p18−/− mice. The data represent averages ± SEM of the results of 3 independent experiments, each with 4 or 5 pooled mice. Within each genotype, there was a statistically significant difference in reactivation between WT virus and vCycKO, as calculated by paired t test (P < 0.05).
During infection, p18 heterozygous mice expressed an intermediate level of p18 protein in both splenocytes and peritoneal cells from mice at 42 days p.i. (Fig. 2C). Furthermore, p18 heterozygous mice demonstrated an intermediate splenocyte number compared to either p18+/+ or p18−/− mice (Fig. 2D). These data demonstrated a gene dosage effect, so p18INK4c haploinsufficiency significantly alleviates the requirement for the v-cyclin in reactivation.
p18INK4c-deficient mice mount an effective neutralizing-antibody response.
p18INK4c has been shown to be critical for the generation of T cell-dependent antibody responses using a model antigen system (31). Therefore, a potential explanation for increased vCycKO reactivation in p18-deficient mice could be defective antibody control. While our previous studies found that long-term infection of B cell-deficient (μMT) mice did not result in restoration of the vCycKO defect (19), μMT mice lack both B cells and antibodies. To test the requirement for p18INK4c in the antiviral neutralizing-antibody response, we directly assessed neutralizing antibody in preimmune and immune p18+/+, p18+/−, and p18−/− mice. As expected, uninfected (preimmune) mice had no detectable neutralizing antibodies (Fig. 3). In contrast, at 42 days p.i., neutralizing antibody against GHV68 was evident in all infected mice, with no significant differences between p18-sufficient and p18-deficient mice (Fig. 3). These data indicate that after virus infection, p18INK4c is dispensable for neutralizing-antibody formation. Further, these data, along with previous analysis of infected μMT mice (19), demonstrate that restoration of the vCycKO reactivation defect is specific to p18INK4c and was not due to a general defect in the antibody response.
FIG 3.
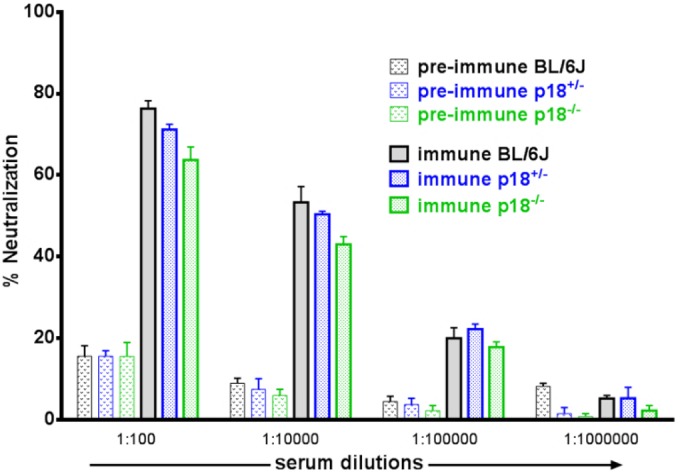
Restoration of v-cyclin reactivation in the absence of p18INK4c did not correspond to differences in neutralizing antibody. Shown is analysis of neutralizing-antibody production in C57BL/6, p18+/−, and p18−/− mice, comparing sera from uninfected (preimmune) or infected (immune; day 42 p.i.) mice, by plaque neutralization assay. WT GHV68 was incubated with or without the indicated dilutions of serum or medium for 1 h, and the percent neutralization was calculated as follows: (PFU in serum-treated samples/PFU in medium-treated controls) × 100. The data represent the means and SEM of the results of two independent experiments, with three independent mice per group.
p18INK4c-deficient peritoneal cells are not detectably altered in cell cycle status.
p18INK4c is a member of the INK4 family of cyclin-dependent kinase inhibitors that is typified by p16INK4a. The INK4 CKIs inhibit cell cycle progression by virtue of binding the G1 CDKs, preventing their binding and activation by the G1 cyclins and thus preventing cell cycle entry and progression to S phase. Therefore, one mechanism by which p18INK4c could impact virus reactivation is by inhibition of cell cycle progression, an effect that would be manifested as a greater number of cells or greater expression of cyclin E (S phase cyclin) in the absence of p18INK4c. We measured expression of cell cycle components in p18INK4C-sufficient and -deficient peritoneal cells. On average, C57BL/6 mice yielded 3.5 × 106 peritoneal cells, while p18−/− mice yielded 4.2 × 106 peritoneal cells per mouse. Although this analysis was limited in scope due to cell yield, expression of cellular cyclin E, indicative of cell cycle progression and entry into S phase, showed no significant difference in the presence or absence of p18INK4c (Fig. 4A). We also analyzed the expression of cell cycle-related genes in infected peritoneal cells (containing approximately 1/1,000 latently infected cells) remaining from the experiments shown in Fig. 1A and B and I and J (C57BL/6 or p18−/− mice infected with WT GHV68 or vCycKO for 42 days). This analysis also showed similar expression levels of cellular cyclins D2, D3, E1, and CDK6 regardless of host p18 or v-cyclin expression (Fig. 4B). Taken together, analysis of the total peritoneal cell population, containing B cells, macrophages, and other cell types, may conceal a cell-type-specific effect but overall suggests that the critical role of p18INK4C in reactivation is not in blocking cell cycle progression. This interpretation is consistent with published studies demonstrating that loss of p18 in B cells supports G1 phase entry, as demonstrated by subtle increases in cyclin D2 and CDK6, but does not support cell cycle progression (31).
FIG 4.

Expression analysis of cell cycle-associated genes. Peritoneal cells were collected for RNA isolation and RT-PCR analysis from uninfected C57BL/6 and p18-deficient (p18−/−) mice (A) and C57BL/6 and p18−/− mice infected with either WT GHV68 or vCycKO (KO) (B). Reverse transcription or no-RT control reactions are indicated on the left; the analysis shown represents at least two independent samples tested. Uninfected and infected samples showed no significant difference in cell cycle progression, as indicated by cyclin E levels, in the presence or absence of p18INK4C or in the presence or absence of the viral cyclin.
As previously reported, p18−/− mice display approximately 1.2-fold-increased cellularity, including increased homeostatic proliferation of peritoneal B1a cells, with accumulation over time (32). To determine whether p18 deficiency results in proliferation or skewing of potential reservoirs of infection, we quantified peritoneal B1a cells during infection. Peritoneal B1a cells (B220 and CD5 positive; n = 3 per group) were represented as follows: C57BL/6 mice infected with WT GHV68, 31.3%; C57BL/6 mice infected with vCycKO, 27.5%; p18KO mice infected with WT GHV68, 30.5%; p18KO mice infected with vCycKO, 28.4%. Together, these data exclude p18INK4C control of B1a cell proliferation as its critical role in reactivation.
Antagonism of p18 restored reactivation of vCycKO virus in normal C57BL/6 mice.
While the above-mentioned data indicate that p18INK4c genetic deficiency can alleviate the requirement for the v-cyclin in reactivation from latency, p18-deficient mice may have developmental or cell-extrinsic changes that could alter the nature of GHV68 infection. To address the role of p18INK4c in normal mice, we made use of the cytokine BAFF, a physiological antagonist of p18INK4c in the B lymphocyte cell cycle and differentiation (29). To do this, normal C57BL/6 mice were infected with WT GHV68 or vCycKO, and at 42 days p.i., peritoneal cells were treated with 50 ng/ml of BAFF, antibody to B cell receptor (BCR), or both prior to limiting-dilution analysis of reactivating cells. Notably, addition of BAFF alone was sufficient to increase vCycKO reactivation relative to WT GHV68 in C57BL/6 peritoneal cells. This effect was not dependent on, or augmented by, cross-linking of the BCR (Fig. 5A to C), consistent with the p18-specific effects of BAFF previously reported (29). This effect was dependent on antagonism of p18INK4c, as demonstrated by the lack of reactivation changes following BAFF treatment of infected p18-deficient cells (Fig. 5D). These data indicate that BAFF treatment of latently infected cells ex vivo was sufficient for partial restoration of the reactivation defect of vCycKO in normal C57BL/6 mice and provide independent evidence that the primary function of the conserved v-cyclin is to bypass the activity of the host CKI, p18INK4c.
FIG 5.

BAFF, a physiological antagonist of p18, restored vCycKO reactivation from infected C57BL/6 cells. Shown is analysis of the frequencies of reactivation in PerCs from C57BL/6 (A to C) or p18−/− (D) mice infected with either WT GHV68 or vCycKO and harvested at 42 days p.i. (A to C) Frequencies of reactivation in infected PerCs from C57BL/6 mice cultured either with medium or 50 ng/ml BAFF (A), with anti-IgM (B), or with anti-IgM plus 50 ng/ml BAFF (C). (D) Frequencies of reactivating cells in infected PerCs from p18−/− mice cultured with medium or 50 ng/ml BAFF. The data represent averages ± SEM of the results of 2 to 4 independent experiments, each with 4 or 5 pooled mice per sample. The frequency of reactivating cells was calculated by Poisson distribution, indicated by the dotted lines at 63%. Within each genotype, there was a statistically significant difference in reactivation between WT virus and vCycKO, as calculated by paired t test (P < 0.05).
DISCUSSION
The GHVs establish lifelong infection within their hosts, an intimate relationship that has coevolved over millions of years (33). Multiple virus-host interactions have evolved to facilitate virus infection in healthy hosts, a balance that is disrupted in immune-deficient individuals that develop GHV-associated chronic disease. Many GHVs express a v-cyclin that is homologous to host D-type cyclins. While studies have focused on the contributions of these v-cyclins to GHV pathogenesis and different stages of infection, there is still a relatively limited understanding of the in vivo relevance of v-cyclin interactions. One common property of v-cyclins is their relative resistance to CKIs (12). Given this common property, we hypothesized that a primary function of the v-cyclin is to bypass host CKIs and that ablation of the v-cyclin would reveal a CKI-dependent regulation of reactivation. To test this, we compared reactivation of WT and v-cyclin-deficient GHV68 in the presence or absence of the CKIs p27Kip1 and p18INK4c. These studies revealed that the v-cyclin was required for reactivation in p18INK4c-sufficient hosts but was completely dispensable for reactivation in p18INK4c-deficient hosts. These data show that the primary genetic requirement for the v-cyclin in promoting reactivation is to bypass p18INK4c and that in the absence of the v-cyclin, p18INK4c has a primary role in limiting reactivation.
These studies began with an analysis of reactivation in CKIlow mice, achieved by generating p18INK4c-deficient, p27 heterozygous mice. While p27Kip1 and p18INK4c are both categorized broadly as cell cycle inhibitors and are both expressed in lymphocytes, they regulate very different processes. On one hand, p27Kip1, as a member of the Cip/Kip protein family, was originally identified as a prominent inhibitor of CDKs and cell cycle progression. In contrast, p18INK4c, as a member of the INK4 protein family, was originally presumed to function primarily as an inhibitor of CDK4/6, akin to p16INK4a. Subsequent studies have revealed that p18INK4c has a number of important biochemical roles physiologically distinct from those of p16INK4a (34), and p18INK4c is now known to be a regulator of hematopoietic stem cell proliferation versus differentiation, plasma cell differentiation, and B cell homeostasis.
Though our initial hypothesis was that p18INK4c and p27Kip1 would together contribute to cell cycle inhibition and that ablation of these CKIs would obviate the requirement for the viral cyclin in reactivation, our data reveal unexpectedly divergent roles for p18INK4c and p27Kip1 in regulating reactivation. First, p18INK4c-deficient mice show enhanced reactivation from latency, a phenotype that is particularly exaggerated following infection with vCycKO virus. Conversely, p27-deficient mice show reduced reactivation from latency, with decreased reactivation of both wild-type and vCycKO viruses. This article has primarily focused on p18INK4c because of the profound and unexpectedly specific consequence of its deletion for reversing the reactivation defect of vCycKO virus.
While the molecular mechanism(s) behind the role of p27Kip1 in regulating reactivation from latency is not known, this result was completely unexpected. Indeed, biochemical studies of the v-cyclin have shown that the v-cyclin is capable of phosphorylating p27Kip1 for degradation (35), suggesting that one function of the v-cyclin in reactivation may be to degrade p27Kip1. Based on the inability of p27 deficiency to restore reactivation of the vCycKO virus, p27Kip1 does not demonstrate v-cyclin-specific effects during reactivation.
Beyond interactions between the v-cyclin and p27Kip1, why might p27 deficiency reduce reactivation? p27 deficiency may result in at least two alterations that could impair reactivation. First, p27Kip1 and the Cip/Kip proteins have a dual role in cell cycle regulation, where p27Kip1 promotes cell cycle progression by escorting G1 cyclins to the nucleus (36). Second, p27Kip1 has been shown to play a critical role in determining the balance between T cell anergy and differentiation (37). The relative contributions of these and other mechanisms remain subjects for future study. Despite the impaired reactivation of wild-type GHV68 in p27-insufficient mice, our data clearly indicate that p18 deficiency has a dominant effect when introduced on a p27-insufficient background, resulting in significantly increased reactivation of both wild-type GHV68 and vCycKO. The dominant effect of p18 deficiency over p27 deficiency has previously been observed in certain contexts (38).
Demonstration that the v-cyclin is dispensable for reactivation in p18INK4c-insufficient settings is notable, since previous studies uniformly found that v-cyclin-deficient viruses have a profound deficit in reactivation, regardless of genetic background or immune deficiency (17–20). The vCycKO reactivation defect is observed even in animals with high reactivation levels, such as mice unresponsive to the antiviral cytokine interferon gamma (IFN-γ) (20). The failure to reverse the reactivation defect of the vCycKO virus in these previous contexts demonstrates that increased reactivation is not sufficient to bypass the requirement for the v-cyclin. Instead, the specific alleviation of the v-cyclin requirement in reactivation in p18INK4c-deficient mice indicates that the primary function of the v-cyclin during reactivation is to bypass inhibition by p18INK4c or p18INK4c-dependent processes.
p18INK4c is one of four genes in the INK4 family of CKIs, all of which bind CDK6 and CDK4 (13), yet none of the other INK4 or Cip/Kip family members compensate for p18INK4c deficiency in GHV reactivation. This is particularly notable for p27Kip1, as it has been shown to be a target of v-cyclin-mediated phosphorylation (35); despite this, p27Kip1 does not compensate for p18INK4c deficiency in regard to v-cyclin-dependent reactivation. Rather, p27Kip1 deficiency resulted in an overall decrease in reactivation irrespective of the v-cyclin.
p18INK4c has been implicated in differentiation in multiple cell types, including B cells, in a genetically nonredundant manner (31, 32). p18INK4c plays distinct roles in hematopoietic development, autoimmunity, plasma cell differentiation, and tumorigenesis (32, 39, 40). Additionally, p18INK4c demonstrated dose dependence in regulation of viral reactivation. This is likely to be physiologically relevant, since not only can p18INK4c be antagonized by BAFF, but its expression can also be dynamically repressed by microRNA (miRNA), long noncoding RNA, transcription, and epigenetics (41–44).
It is interesting that while BAFF treatment reduces the difference in reactivation frequency between wild-type and vCycKO viruses, this outcome comes from a reduction in wild-type reactivation coincident with an increase in vCycKO reactivation. At this time, it is unclear why BAFF treatment does not phenocopy p18INK4c deficiency, in which both wild-type and vCycKO virus reactivation are increased. BAFF antagonism of p18 may differ from p18INK4c deficiency quantitatively, based on BAFF receptor status, or qualitatively, in that BAFF signaling also induces NF-κB (29), a known repressor of the lytic viral transactivator.
At this time, it has not been possible to measure the magnitude of BAFF antagonism of p18INK4c in our ex vivo cultures. Previous measurement of BAFF inhibition of p18INK4c relied on an indirect measure, induction of cell cycle entry, as reflected by increases in cyclin D2 and serine-phosphorylated retinoblastoma (Rb) protein (29). This effect was observed by comparing p18INK4c-sufficient and -deficient cells; to date, there is no evidence that BAFF directly influences the p18INK4c protein level, nor is there a specific p18INK4c protein modification known to be induced by BAFF treatment. We anticipate that future evaluation of this interaction will require the development of new tools to identify, analyze, and purify rare latently infected cells.
Our data highlight a specific, dose-dependent role for p18INK4c in control of viral reactivation, revealed by the absence of the v-cyclin. We and others previously described viral and host cyclin features required to promote reactivation from latency (30, 45). While both the GHV68 and KSHV cyclins can promote reactivation when expressed from the GHV68 v-cyclin locus, it is notable that host cyclin D3 (but not cyclin D2, E, or A) could also partially promote reactivation (30). Given that p18INK4c can restrict reactivation in the absence of the v-cyclin, this suggests that cyclin D3 may also be relatively resistant to p18INK4c inhibition. While this remains to be directly tested, cyclin D3/CDK6 complexes were shown to be less susceptible to CKI inhibition by p16INK4a, p27Kip1, and p21Cip1 (46, 47).
These studies establish the first genetic link between the GHV v-cyclin and host p18INK4c and raise a number of questions. First, is v-cyclin bypass of p18INK4c related to the cell cycle, or does the v-cyclin manipulate another CDK-dependent process (e.g., B cell differentiation [31, 32] or NF-κB activation [48])? Second, does the v-cyclin bypass p18INK4c in a CDK-dependent or -independent mechanism (45)? Third, while the KSHV cyclin can directly interact with the p18INK4c protein (49, 50), does the v-cyclin bypass p18INK4c by a direct protein-protein interaction or is it instead a functional bypass? We anticipate that use of a large panel of chimeric cyclins will afford unique insights into this process (30).
While our study focused on genetic and physiological antagonism of p18 by the v-cyclin, it is notable that the KSHV v-cyclin can form a ternary complex with p18 and CDK6 (49), and p18 was identified as the major protein interacting with the v-cyclin in a recently published genome interactome study (50). These studies suggest that the v-cyclin may directly physically interact with and modulate p18 function, something we are currently investigating.
At this time, our data do not support a role for p18 in inhibition of classical CDKs as the primary determinant of vCycKO restoration. This conclusion is based on the lack of a detectable difference in S phase progression between C57BL/6 and p18-deficient mice. One major caveat to this analysis, however, is that it is based on a mixed population of peritoneal cells, a very small number of which were latently infected. Future studies will require detailed analysis of cell cycle regulation within virus-infected cells in the presence or absence of the v-cyclin and in the presence or absence of p18. At this time, these studies are not technically feasible, due to both an inability to uniformly identify virus-infected cells and the relative paucity of primary latently infected cells (at most 1 in 100 cells at the peak of latency).
These studies provide genetic and physiological evidence that p18INK4c acts to repress reactivation and that the viral cyclin is able to overcome or bypass its repression. The mechanism(s) by which p18 limits reactivation remains unknown at this time. Although p18 is a member of the INK4 family of cell cycle inhibitors, the function of p18 appears to be primarily as a critical regulator of cellular differentiation and survival instead of as a canonical cell cycle inhibitor (34). Prominent roles of p18 include its unique role in promoting plasma cell differentiation (31) and homeostatic regulation of B1a cells (51); p18 is further lost in particular tumor cell types (52, 53). Given that B cells are a major latent reservoir for GHV68 (2) and that plasma cells are a critical mediator in virus reactivation (54), our current working model is that p18 restricts reactivation by modulating plasma cell differentiation. Future studies focused on the role of p18 in GHV68-infected B cells will be required to build further on our current genetic and physiological insights into the interactions between the viral cyclin p18 and reactivation.
In total, our data emphasize the importance of GHV v-cyclin resistance to host CKIs, a conserved feature of the GHV v-cyclins. Our data further provide concrete genetic evidence that a primary role of the v-cyclin is bypass of a single CKI, defining a new potential axis for intervention in GHV pathogenesis.
One potential point of intervention to antagonize the vCyclin could be either to enhance p18 expression (55) or to increase the functional capacity of p18, as previously described (24). These interventions would be predicted to limit GHV reactivation. Alternatively, deliberate disruption of p18 activity via small molecules (56) could be used to induce lytic reactivation, a therapeutic regimen that may be particularly useful in the context of latency-associated GHV malignancies (57).
ACKNOWLEDGMENTS
We thank the Selena Chen-Kiang laboratory for BAFF protocols, Gongyi Zhang for recombinant BAFF, John Cambier for anti-mouse mu heavy chain antibody, Ronen Marmorstein for the p18 plasmid, and the University of Colorado Center for Comparative Medicine and members of the van Dyk laboratory for critical discussions.
This research was funded by Burroughs Wellcome Investigators in Pathogenesis of Infectious Disease grant 10005532 and National Institutes of Health grants R01CA103632 and R01CA168558 to L.F.V.D. and 5T32AI052066 to B.F.N.
The contents of this report are our sole responsibility and do not necessarily represent official NIH views.
We have no competing financial interests to declare.
REFERENCES
- 1.Cesarman E. 2011. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett 305:163–174. doi: 10.1016/j.canlet.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barton E, Mandal P, Speck SH. 2011. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol 29:351–397. doi: 10.1146/annurev-immunol-072710-081639. [DOI] [PubMed] [Google Scholar]
- 3.Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. 2013. The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol 3:227–232. doi: 10.1016/j.coviro.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reese TA, Wakeman BS, Choi HS, Hufford MM, Huang SC, Zhang X, Buck MD, Jezewski A, Kambal A, Liu CY, Goel G, Murray PJ, Xavier RJ, Kaplan MH, Renne R, Speck SH, Artyomov MN, Pearce EJ, Virgin HW. 2014. Coinfection Helminth infection reactivates latent gamma-herpesvirus via cytokine competition at a viral promoter. Science 345:573–577. doi: 10.1126/science.1254517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jung JU, Stager M, Desrosiers RC. 1994. Virus-encoded cyclin. Mol Cell Biol 14:7235–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang Y, Moore PS, Talbot SJ, Boshoff CH, Zarkowska T, Godden K, Paterson H, Weiss RA, Mittnacht S. 1996. Cyclin encoded by KS herpesvirus. Nature 382:410. doi: 10.1038/382410a0. [DOI] [PubMed] [Google Scholar]
- 7.Virgin HW IV, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol 71:5894–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arvanitakis L, Yaseen N, Sharma S. 1995. Latent membrane protein-1 induces cyclin D2 expression, pRb hyperphosphorylation, and loss of TGF-beta 1-mediated growth inhibition in EBV-positive B cells. J Immunol 155:1047–1056. [PubMed] [Google Scholar]
- 9.Sinclair AJ, Palmero I, Holder A, Peters G, Farrell PJ. 1995. Expression of cyclin D2 in Epstein-Barr virus-positive Burkitt's lymphoma cell lines is related to methylation status of the gene. J Virol 69:1292–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannell EJ, Farrell PJ, Sinclair AJ. 1996. Epstein-Barr virus exploits the normal cell pathway to regulate Rb activity during the immortalisation of primary B-cells. Oncogene 13:1413–1421. [PubMed] [Google Scholar]
- 11.Verschuren EW, Jones N, Evan GI. 2004. The cell cycle and how it is steered by Kaposi's sarcoma-associated herpesvirus cyclin. J Gen Virol 85:1347–1361. doi: 10.1099/vir.0.79812-0. [DOI] [PubMed] [Google Scholar]
- 12.Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G, Jones N. 1997. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature 390:184–187. doi: 10.1038/36606. [DOI] [PubMed] [Google Scholar]
- 13.Sherr CJ, Roberts JM. 1999. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 14.Lim S, Kaldis P. 2013. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140:3079–3093. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- 15.van Dyk LF, Hess JL, Katz JD, Jacoby M, Speck SH, Virgin HI. 1999. The murine gammaherpesvirus 68 v-cyclin gene is an oncogene that promotes cell cycle progression in primary lymphocytes. J Virol 73:5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verschuren EW, Klefstrom J, Evan GI, Jones N. 2002. The oncogenic potential of Kaposi's sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell 2:229–241. doi: 10.1016/S1535-6108(02)00123-X. [DOI] [PubMed] [Google Scholar]
- 17.van Dyk LF, Virgin HW IV, Speck SH. 2000. The murine gammaherpesvirus 68 v-cyclin is a critical regulator of reactivation from latency. J Virol 74:7451–7461. doi: 10.1128/JVI.74.16.7451-7461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoge AT, Hendrickson SB, Burns WH. 2000. Murine gammaherpesvirus 68 cyclin D homologue is required for efficient reactivation from latency. J Virol 74:7016–7023. doi: 10.1128/JVI.74.15.7016-7023.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Dyk LF, Virgin HW IV, Speck SH. 2003. Maintenance of gammaherpesvirus latency requires viral cyclin in the absence of B lymphocytes. J Virol 77:5118–5126. doi: 10.1128/JVI.77.9.5118-5126.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gangappa S, van Dyk LF, Jewett TJ, Speck SH, Virgin HW IV. 2002. Identification of the in vivo role of a viral bcl-2. J Exp Med 195:931–940. doi: 10.1084/jem.20011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mora AL, Torres-Gonzalez E, Rojas M, Xu J, Ritzenthaler J, Speck SH, Roman J, Brigham K, Stecenko A. 2007. Control of virus reactivation arrests pulmonary herpesvirus-induced fibrosis in IFN-gamma receptor-deficient mice. Am J Respir Crit Care Med 175:1139–1150. doi: 10.1164/rccm.200610-1426OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tarakanova VL, Kreisel F, White DW, Virgin HW IV. 2008. Murine gammaherpesvirus 68 genes both induce and suppress lymphoproliferative disease. J Virol 82:1034–1039. doi: 10.1128/JVI.01426-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weck KE, Kim SS, Virgin HI, Speck SH. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol 73:3273–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venkataramani RN, MacLachlan TK, Chai X, El-Deiry WS, Marmorstein R. 2002. Structure-based design of p18INK4c proteins with increased thermodynamic stability and cell cycle inhibitory activity. J Biol Chem 277:48827–48833. doi: 10.1074/jbc.M208061200. [DOI] [PubMed] [Google Scholar]
- 25.Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, Su L, Xiong Y. 1998. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev 12:2899–2911. doi: 10.1101/gad.12.18.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. 1996. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 85:721–732. doi: 10.1016/S0092-8674(00)81238-6. [DOI] [PubMed] [Google Scholar]
- 27.Suarez AL, Kong R, George T, He L, Yue Z, van Dyk LF. 2011. Gammaherpesvirus 68 infection of endothelial cells requires both host autophagy genes and viral oncogenes for optimal survival and persistence. J Virol 85:6293–6308. doi: 10.1128/JVI.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gangappa S, Kapadia SB, Speck SH, Virgin HW IV. 2002. Antibody to a lytic cycle viral protein decreases gammaherpesvirus latency in B-cell-deficient mice. J Virol 76:11460–11468. doi: 10.1128/JVI.76.22.11460-11468.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang X, Di Liberto M, Cunningham AF, Kang L, Cheng S, Ely S, Liou HC, Maclennan IC, Chen-Kiang S. 2004. Homeostatic cell-cycle control by BLyS: induction of cell-cycle entry but not G1/S transition in opposition to p18INK4c and p27Kip1. Proc Natl Acad Sci U S A 101:17789–17794. doi: 10.1073/pnas.0406111101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee KS, Suarez AL, Claypool DJ, Armstrong TK, Buckingham EM, van Dyk LF. 2012. Viral cyclins mediate separate phases of infection by integrating functions of distinct mammalian cyclins. PLoS Pathog 8:e1002496. doi: 10.1371/journal.ppat.1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tourigny MR, Ursini-Siegel J, Lee H, Toellner KM, Cunningham AF, Franklin DS, Ely S, Chen M, Qin XF, Xiong Y, MacLennan IC, Chen-Kiang S. 2002. CDK inhibitor p18(INK4c) is required for the generation of functional plasma cells. Immunity 17:179–189. doi: 10.1016/S1074-7613(02)00364-3. [DOI] [PubMed] [Google Scholar]
- 32.Potula HH, Xu Z, Zeumer L, Sang A, Croker BP, Morel L. 2012. Cyclin-dependent kinase inhibitor Cdkn2c deficiency promotes B1a cell expansion and autoimmunity in a mouse model of lupus. J Immunol 189:2931–2940. doi: 10.4049/jimmunol.1200556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGeoch DJ, Dolan A, Ralph AC. 2000. Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J Virol 74:10401–10406. doi: 10.1128/JVI.74.22.10401-10406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gagrica S, Brookes S, Anderton E, Rowe J, Peters G. 2012. Contrasting behavior of the p18INK4c and p16INK4a tumor suppressors in both replicative and oncogene-induced senescence. Cancer Res 72:165–175. doi: 10.1158/0008-5472.CAN-11-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellis M, Chew YP, Fallis L, Freddersdorf S, Boshoff C, Weiss RA, Lu X, Mittnacht S. 1999. Degradation of p27(Kip) cdk inhibitor triggered by Kaposi's sarcoma virus cyclin-cdk6 complex. EMBO J 18:644–653. doi: 10.1093/emboj/18.3.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. 1999. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J 18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wells AD, Morawski PA. 2014. New roles for cyclin-dependent kinases in T cell biology: linking cell division and differentiation. Nat Rev Immunol 14:261–270. doi: 10.1038/nri3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Broxmeyer HE, Franklin DS, Cooper S, Hangoc G, Mantel C. 2012. Cyclin dependent kinase inhibitors differentially modulate synergistic cytokine responsiveness of hematopoietic progenitor cells. Stem Cells Dev 21:1597–1603. doi: 10.1089/scd.2011.0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. 2004. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol 6:436–442. doi: 10.1038/ncb1126. [DOI] [PubMed] [Google Scholar]
- 40.Chen-Kiang S. 2003. Cell-cycle control of plasma cell differentiation and tumorigenesis. Immunol Rev 194:39–47. doi: 10.1034/j.1600-065X.2003.00065.x. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Meyers C, Guo M, Zheng ZM. 2011. Upregulation of p18Ink4c expression by oncogenic HPV E6 via p53-miR-34a pathway. Int J Cancer 129:1362–1372. doi: 10.1002/ijc.25800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Du Y, Kong G, You X, Zhang S, Zhang T, Gao Y, Ye L, Zhang X. 2012. Elevation of highly up-regulated in liver cancer (HULC) by hepatitis B virus X protein promotes hepatoma cell proliferation via down-regulating p18. J Biol Chem 287:26302–26311. doi: 10.1074/jbc.M112.342113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hosokawa H, Tanaka T, Kato M, Shinoda K, Tohyama H, Hanazawa A, Tamaki Y, Hirahara K, Yagi R, Sakikawa I, Morita A, Nagira M, Poyurovsky MV, Suzuki Y, Motohashi S, Nakayama T. 2013. Gata3/Ruvbl2 complex regulates T helper 2 cell proliferation via repression of Cdkn2c expression. Proc Natl Acad Sci U S A 110:18626–18631. doi: 10.1073/pnas.1311100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sanchez-Aguilera A, Delgado J, Camacho FI, Sanchez-Beato M, Sanchez L, Montalban C, Fresno MF, Martin C, Piris MA, Garcia JF. 2004. Silencing of the p18INK4c gene by promoter hypermethylation in Reed-Sternberg cells in Hodgkin lymphomas. Blood 103:2351–2357. doi: 10.1182/blood-2003-07-2356. [DOI] [PubMed] [Google Scholar]
- 45.Upton JW, Speck SH. 2006. Evidence for CDK-dependent and CDK-independent functions of the murine gammaherpesvirus 68 v-cyclin. J Virol 80:11946–11959. doi: 10.1128/JVI.01722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin J, Jinno S, Okayama H. 2001. Cdk6-cyclin D3 complex evades inhibition by inhibitor proteins and uniquely controls cell's proliferation competence. Oncogene 20:2000–2009. doi: 10.1038/sj.onc.1204375. [DOI] [PubMed] [Google Scholar]
- 47.Faast R, White J, Cartwright P, Crocker L, Sarcevic B, Dalton S. 2004. Cdk6-cyclin D3 activity in murine ES cells is resistant to inhibition by p16(INK4a). Oncogene 23:491–502. doi: 10.1038/sj.onc.1207133. [DOI] [PubMed] [Google Scholar]
- 48.Buss H, Handschick K, Jurrmann N, Pekkonen P, Beuerlein K, Muller H, Wait R, Saklatvala J, Ojala PM, Schmitz ML, Naumann M, Kracht M. 2012. Cyclin-dependent kinase 6 phosphorylates NF-kappaB P65 at serine 536 and contributes to the regulation of inflammatory gene expression. PLoS One 7:e51847. doi: 10.1371/journal.pone.0051847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeffrey PD, Tong L, Pavletich NP. 2000. Structural basis of inhibition of CDK-cyclin complexes by INK4 inhibitors. Genes Dev 14:3115–3125. doi: 10.1101/gad.851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W, Jager S, Shales M, Horner J, Hernandez RD, Krogan NJ, Glaunsinger BA. 2015. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol Cell 57:349–360. doi: 10.1016/j.molcel.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Z, Potula HH, Vallurupalli A, Perry D, Baker H, Croker BP, Dozmorov I, Morel L. 2011. Cyclin-dependent kinase inhibitor Cdkn2c regulates B cell homeostasis and function in the NZM2410-derived murine lupus susceptibility locus Sle2c1. J Immunol 186:6673–6682. doi: 10.4049/jimmunol.1002544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bai F, Pei XH, Nishikawa T, Smith MD, Xiong Y. 2007. p18Ink4c, but not p27Kip1, collaborates with Men1 to suppress neuroendocrine organ tumors. Mol Cell Biol 27:1495–1504. doi: 10.1128/MCB.01764-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leone PE, Walker BA, Jenner MW, Chiecchio L, Dagrada G, Protheroe RK, Johnson DC, Dickens NJ, Brito JL, Else M, Gonzalez D, Ross FM, Chen-Kiang S, Davies FE, Morgan GJ. 2008. Deletions of CDKN2C in multiple myeloma: biological and clinical implications. Clin Cancer Res 14:6033–6041. doi: 10.1158/1078-0432.CCR-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. 2009. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog 5:e1000677. doi: 10.1371/journal.ppat.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim J, Kim J, Duong T, Lee G, Kim J, Yoon J, Kim J, Kim H, Ruley HE, El-Rifai W, Jo D. 2012. Antitumor activity of cell-permeable p18(INK4c) with enhanced membrane and tissue penetration. Mol Ther 20:1540–1549. doi: 10.1038/mt.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao Y, Yang P, Shen H, Yu H, Song X, Zhang L, Zhang P, Cheng H, Xie Z, Hao S, Dong F, Ma S, Ji Q, Bartlow P, Ding Y, Wang L, Liu H, Li Y, Cheng H, Miao W, Yuan W, Yuan Y, Cheng T, Xie XQ. 2015. Small-molecule inhibitors targeting INK4 protein p18(INK4C) enhance ex vivo expansion of haematopoietic stem cells. Nat Commun 6:6328. doi: 10.1038/ncomms7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kenney SC, Mertz JE. 2014. Regulation of the latent-lytic switch in Epstein-Barr virus. Semin Cancer Biol 26:60–68. doi: 10.1016/j.semcancer.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]